

Abstract
Neurodegeneration has been correlated with mitochondrial DNA (mtDNA) damage and exposure to environmental toxins, but causation is unclear. We investigated the ability of several known environmental genotoxins and neurotoxins to cause mtDNA damage, mtDNA depletion, and neurodegeneration in Caenorhabditis elegans. We found that paraquat, cadmium chloride and aflatoxin B1 caused more mitochondrial than nuclear DNA damage, and paraquat and aflatoxin B1 also caused dopaminergic neurodegeneration. 6-hydroxydopamine (6-OHDA) caused similar levels of mitochondrial and nuclear DNA damage. To further test whether the neurodegeneration could be attributed to the observed mtDNA damage, C. elegans were exposed to repeated low-dose ultraviolet C radiation (UVC) that resulted in persistent mtDNA damage; this exposure also resulted in dopaminergic neurodegeneration. Damage to GABAergic neurons and pharyngeal muscle cells was not detected. We also found that fasting at the first larval stage was protective in dopaminergic neurons against 6-OHDA-induced neurodegeneration. Finally, we found that dopaminergic neurons in C. elegans are capable of regeneration after laser surgery. Our findings are consistent with a causal role for mitochondrial DNA damage in neurodegeneration, but also support non mtDNA-mediated mechanisms.
Introduction
The possible mitochondrial toxicity of environmental pollutants has attracted increasing interest in recent years [1]–[4], and mitochondrial DNA (mtDNA) may represent a particularly critical target. The human health significance of the mitochondrial genome as a target of genotoxins has recently received greater attention [5]–[10]. mtDNA is potentially more prone to damage than nuclear DNA (nDNA) due to the tendency of lipophilic and certain charged chemicals to accumulate in the mitochondria [11]–[14], the proximity of mtDNA to electron transport chain-mediated production of reactive oxygen species (ROS), and the absence of chromatin packing and many DNA repair pathways [15]–[17]. Furthermore, non-genotoxic mitochondrial toxins may indirectly cause mtDNA damage by disrupting oxidative phosphorylation [18]–[20]. A number of genotoxins cause more mtDNA than nDNA damage [21]–[24] but the opposite is true for other chemicals [23], [25], [26], and relatively few chemicals have been tested for their potency in damaging the mitochondrial genome (reviewed in Meyer et al. [4]).
Mitochondria play an important role in multiple neurological disorders [18], [27]. Neurons are high energy-use cells that rely on mitochondria for their supply of energy [6]. The high metabolic activity of neurons leads to the production of ROS, and the brain is particularly susceptible to oxidative stress due to its low supply of antioxidant enzymes and high lipid content [28], [29]. mtDNA damage and mutation have been correlated with neurodegeneration [30]–[37]. A recent study showed dopaminergic neurodegeneration in mice exhibiting mtDNA doublestrand breaks produced by a mitochondrial-targeted restriction enzyme [38]. Another recent study detected oxidative mtDNA lesions in the brain of Parkinson's disease (PD) patients, and also in vivo and in vitro after mitochondrial impairment by rotenone [39]. Furthermore, mutations in the only mtDNA polymerase, DNA polymerase γ, can result in parkinsonism in humans [40], [41].
A significant portion of neurodegenerative disease, especially PD, may result from environmental exposures [42]. Epidemiological studies have identified an association between neurodegeneration and exposure to environmental chemicals including pesticides and heavy metals [43]–[48], and laboratory studies support the ability of some of these chemicals to cause neurodegeneration [49], [50]. These chemicals, however, have not been tested for their relative genotoxicity in the nuclear and mitochondrial genomes.
Finally, there is growing evidence that neurodegeneration can result from early lifestage exposures [51]–[53]. Environmental genotoxins that target the mtDNA are strong candidates for acting in this fashion. Since mtDNA copies in somatic cells are all amplified from a smaller pool of mtDNA in the embryo [54], the mtDNA damage resulting from environmental exposure in early life stages may impact physiological functions in a later stage of life.
Thus, mtDNA is particularly vulnerable to many environmental pollutants, mtDNA damage can cause neurodegeneration, and some neurodegenerative diseases are associated with exposure to environmental chemicals. These associations suggest the possibility that environmental pollutants that cause mtDNA damage (i.e., “environmental mito-genotoxins”) could also cause neurodegeneration.
We carried out a series of experiments to examine whether or not (a) important environmental genotoxins and neurotoxins could cause mtDNA damage or depletion, (b) mitochondrial genotoxins could cause dopaminergic neurodegeneration, and if (c) the observed dopaminergic neurodegeneration could be attributed specifically to mtDNA damage. The chemicals that we tested include chemicals associated in the experimental and/or epidemiological literature with PD (paraquat, rotenone, maneb, and manganese) as well as chemicals that are known genotoxins and mitochondrial poisons (aflatoxin B1 and cadmium). We utilized 6-OHDA as a positive control for chemical-induced dopaminergic neurodegeneration [55].
Our findings support a potential causal role for mtDNA damage resulting from exposure to environmental chemicals in neurodegeneration. Our experiments also led us to the observations that fasting early in life was protective against 6-OHDA-induced dopaminergic neurodegeneration, and that dopaminergic neurons in C. elegans are capable of regeneration.
Materials and Methods
C. elegans culture
Populations of C. elegans were maintained on K agar plates [56] seeded with OP50 bacteria. Synchronized populations of nematodes were obtained by bleach-sodium hydroxide isolation of eggs. L1 growth-arrested (starved) larvae were obtained by hatching eggs in K+ medium, previously referred to as “complete K-medium” [57]. All transfers were made by washing nematodes off of agar plates and rinsing (after centrifugation at 2200 g for 2 min) in K medium.
The germline-deficient JK1107 strain (glp-1) of C. elegans was obtained from the Caenorhabditis Genetics Center (University of Minnesota). The transgenic strains BY250 (vtIs7[Pdat-1::GFP], expressing GFP only in dopaminergic neurons) and CZ1200 (unc-25::GFP, expressing GFP only in GABAergic neurons) were generously provided by Michael Aschner (Vanderbilt University). Strain XE1311 (vtIs7[Pdat-1::GFP];mkk-4(ju91)) was generated by crossing the mkk-4(jk91) mutation into the vtIs7 background.
Chemical exposures
Aflatoxin B1 (AFB1), paraquat, rotenone, maneb, manganese chloride (MnCl2), cadmium chloride (CdCl2) and 6-OHDA HCl were purchased from Sigma (St Louis, MO). Paraquat, MnCl2, and CdCl2 were dissolved into K+ medium for L1 larvae or K medium for young adults. 6-OHDA was dissolved into a solution of ascorbic acid in K+ medium. AFB1, rotenone, and maneb were dissolved in dimethyl sulfoxide (DMSO) to prepare stock solutions (100x the final treatment concentration). C. elegans were treated with the solutions in 12-well plates. Stock solutions were added to treatment wells at a maximum amount of 1 % (v/v) in K+ or K medium. Each well contained 1 ml of the treatment solution, 1000 L1 larvae, or 300 adults, and OP50 in the case of the adults. 1% DMSO did not affect nematode growth or survival (data not shown).
Adult lethality assay
The acute toxicities of paraquat, rotenone, maneb, MnCl2, and CdCl2 were determined in glp-1 nematodes prior to the DNA damage assay. Synchronized glp-1 nematodes used in this assay were grown at 15°C for 40 h and sterilized at 25°C for 18 h. The worms were then exposed to AFB1, paraquat, rotenone, maneb, MnCl2, and CdCl2 for 24 h. 50 animals were examined in two separate experiments with lethality defined as a lack of response to gentle probing with platinum wire. AFB1 was previously found to cause no acute lethality in adult C. elegans to its solubility limit (100 µM) [58].
DNA damage assay
Synchronized glp-1 worms, grown at 15°C for 40 h and sterilized for 18 h at 25°C, were exposed to 3, 30 and 100 µM AFB1, 0.6, 6 and 20 mM paraquat, 0.6, 6 and 21 µM rotenone, 23, 226 and 754 µM maneb, 0.75, 7.5, and 25 mM MnCl2, and 0.03, 0.3 and 1 mM CdCl2 in K medium in the presence of OP50 for 48 h. The highest exposure level chosen for each chemical was the one that resulted in no or minimal lethality, as observed in our 24 h adult lethality assay. No mortality was observed during the 48 h exposures. The solubility limit was chosen if lethality was not detected. The worms were also exposed to 50, 100 mM and 150 mM 6-OHDA. 6-OHDA exposures were constrained to 1 h due to the short half-life of 6-OHDA in aqueous solution, and no mortality was observed during this exposure. Six worms were picked and pooled in a single tube per biological replicate, and eight biological replicates were taken per treatment (three replicates for 6-OHDA) in two experiments separated in time.
nDNA and mtDNA damage were evaluated using a QPCR-based method as previously described [59], [60]. This assay defines the control samples as undamaged and determines a lesion frequency in experimental samples based on any decrease in amplification efficiency relative to the control samples [16]. Two nuclear genome targets (unc-2 and small nuclear; 9.3 and 0.2 kb) and two mitochondrial genome targets (10.9 and 0.2 kb) were amplified. The amount of long PCR product provides a measurement of lesion frequency, while the amount of short PCR product provides normalization to DNA concentration as well as a relative measure of mitochondrial genome copy number [59].
Utilizing a germline-deficient mutant (glp-1) for this experiment allowed us to study DNA damage in adults composed entirely of non-dividing cells. Young adult C. elegans have a rapidly proliferating germ line, so that DNA damage caused by chemical exposure could be readily “diluted” by the new DNA produced by dividing germ cells, reducing the apparent level of DNA damage [61]. The confounding effect of DNA replication can be minimized by using a glp-1 mutant strain since outside of the germ line, no cell divisions occur in adult C. elegans [62].
L1 development assay
The growth inhibitory effects of DMSO, AFB1, CdCl2, and paraquat were determined on L1 larvae prior to neurodegeneration studies. 500 L1 larvae were exposed to DMSO, AFB1, CdCl2, and paraquat for 48 h in the absence of food, and then transferred to seeded K agar plates. Nematodes from the exposed populations were scored every 24 h after the treatment for 3 days, and developmental delay was evaluated in comparison to the untreated control. The experiment was repeated once (i.e. two to four biological replicates in total). The highest concentration that resulted in minimal decrease in size as compared to the control was used as a basis to choose highest exposure level in neurodegeneration studies.
Dopaminergic and GABAergic neurodegeneration
Synchronized BY250 and CZ1200 L1s were exposed to AFB1, paraquat, and CdCl2 in K+ medium in the absence of OP50 for 48 h. BY250 L1 larvae were exposed to 6-OHDA for 1 h. The worms were washed with K medium twice, transferred to seeded K agar plates, and sampled 24, 48, and 96 h after the exposure.
Treated C. elegans were picked onto a 2% agar pad and immobilized with 15 µl of 1% sodium azide (Sigma-Aldrich). Nematodes were examined using a Zeiss Axioskop microscope and neuronal morphology was assessed by individual observation of each cephalic (CEP) neuron. Neurons were assigned a score from 0 to 2 based on the amount of morphological abnormalities present. Each experiment was repeated three or four times with ∼100 worms analyzed per treatment at each time point. For the 6-OHDA exposures, 10–15 worms were analyzed per treatment for each time point, and the experiment was repeated twice. CZ1200 worms were analyzed similarly, focusing on damage to the ventral motor neural cord. All scoring was double-blinded.
UVC and ethidium bromide treatment
C. elegans were exposed to serial UVC doses over 48 h as described in Bess et al. [63]. This exposure protocol is based on the fact that UVC-induced DNA damage is quickly repaired in the nuclear but not mitochondrial genome [61], resulting in accumulation of mtDNA damage while permitting repair of nDNA [63], [64]. This protocol results in larval growth delay that is mild at the doses used in this study [63]. Ethidium bromide exposures were performed on K agar plates [65] using doses leading to mild larval growth delay [63].
Laser surgery
The dendrites of the CEP neurons were severed with a pulsed laser essentially as described [66]. L4 stage BY250 vtIs7[Pdat-1::GFP] and XE1311 vtIs7[Pdat-1::GFP];mkk-4(ju91) worms were used. 2 CEP neurons were cut in each worm, at the point just anterior to the curve made around the anterior pharyngeal bulb. Worms were recovered onto NGM plates at 20°C for 24 h before being remounted and scored for regeneration. Regeneration was defined as anterior growth beyond the cut site. Representative images were collected on a Zeiss LSM710 point scanner and analyzed with ImageJ (U. S. National Institutes of Health, Bethesda, Maryland, USA).
Statistical analysis
DNA damage and relative mtDNA copy number data were analyzed with Statview for Windows (Version 5.0.1, SAS Institute Inc., Cary, NC) and JMP Pro for Windows (Version 11.0.0, SAS Institute Inc., Cary, NC). One- or two-factor analysis of variance (ANOVA) was used and each chemical was analyzed as an independent experiment. A p-value of less than 0.05 was considered statistically significant.
Dopaminergic neurodegeneration data were analyzed using the statistical software R version 2.12.0 (R Foundation for Statistical Computing, Vienna, Austria), Statview or JMP. The nonparametric Kruskal-Wallis test was used to test for differences between dosage levels for each chemical at each time point. Due to sparseness in the cross-tabulations of dosage levels and scores, Fisher's exact test (FET) was used when testing independence of chemical dosage levels and scores at each time point. FET was also used to analyze laser axotomy data. A p-value of less than 0.05 was considered statistically significant.
Results
Paraquat, AFB1 and CdCl2 caused more mitochondrial than nuclear DNA damage
We first tested the ability of known neurotoxins or genotoxins to cause either mitochondrial or nuclear DNA damage. The doses used were sublethal (S1 Table), chosen as described in Materials and Methods. Paraquat, AFB1, and 6-OHDA exposure resulted in dose-dependent DNA damage (p<0.0001, <0.0001, and <0.0001 respectively, for main effects of dose, Fig. 1) in C. elegans. Exposure to CdCl2 also resulted in DNA damage (p = 0.028), although this damage was not dose-dependent (p>0.05 for all pairwise comparisons except each dose compared to control). Rotenone, maneb, and MnCl2 did not cause detectable DNA damage (p>0.05 for the effect of dose in all cases, Fig. 1). mtDNA was more susceptible than nDNA to damage due to AFB1, paraquat or CdCl2 exposure (p<0.0001, <0.0001, and p = 0.0125 respectively for main effect of genome).
Figure 1. Exposures to aflatoxin B1, paraquat, cadmium chloride and 6-OHDA caused detectable DNA damage.

p<.0001, p<.0001, p = 0.028, p<.0001 respectively for main effects of dose. Aflatoxin B1, paraquat and cadmium chloride caused more damage to the mitochondrial than nuclear genomes (p<.0001, p<.0001, and p = 0.0125 respectively for main effect of genome). Data analyzed by Two-way ANOVA. Bars ± SEM.
MnCl2 and CdCl2 caused a decrease in mtDNA:nDNA ratio
Some environmental exposures are known to result in depletion [67] or proliferation [68] of the mitochondrial genome. To test for such effects, a relative mtDNA:nDNA ratio was calculated by comparing the amount of PCR products amplified from small nDNA and mtDNA targets [59], [69]. The mtDNA:nDNA ratio was defined as 100% in controls. Exposures to MnCl2 and CdCl2 resulted in a dose-dependent change in the mtDNA:nDNA ratio (Fig. 2. For MnCl2, one-way ANOVA, effect of concentration p = 0.03; Fisher's PLSD, p = 0.0231 for 7.5 mM and p = 0.0015 for 25 mM. For CdCl2, one-way ANOVA, effect of concentration p = 0.0091; Fisher's PLSD, p = 0.0032 for 1 mM).
Figure 2. Manganese chloride and cadmium chloride resulted in a decrease in the relative mtDNA:nDNA ratio.

Ratio defined as 100% in controls. For MnCl2, one-way ANOVA, effect of concentration p = 0.03; Fisher's PLSD, p = 0.0231 for 7.5 mM and p = 0.0015 for 25 mM. For CdCl2, one-way ANOVA, effect of concentration p = 0.0091; Fisher's PLSD, p = 0.0032 for 1 mM. Bars ± SEM.
Establishment of a positive control-based scoring system for dopaminergic neurodegeneration
6-OHDA is a known selective dopaminergic neurodegenerative agent. We carried out dose-response studies (S1 Figure, Fig. 3) to establish a scoring system for dopaminergic neurodegeneration. GFP-tagged CEP neurons were observed and assigned a numerical score based on neuronal morphology. The scoring system is as follows: 0 = No observable damage, 0.5 = Dendrite has <5 blebs, no observable breaks, 1 = Dendrite has ≥5 blebs, <5 breaks, 1.5 = Dendrite has ≥5 blebs and ≥5 breaks, 2 = Dendrite shows same amount of damage as in a score of 1.5, plus the neuronal cell body is rounded, 2.5 = Neuron structure is unrecognizable. Interestingly, the four CEP neurons damaged in an individual often responded very differently, with affected individuals frequently displaying a combination of highly damaged and entirely intact morphologies.
Figure 3. Representation of progressive damage in dopaminergic neurons after 6-OHDA exposure.
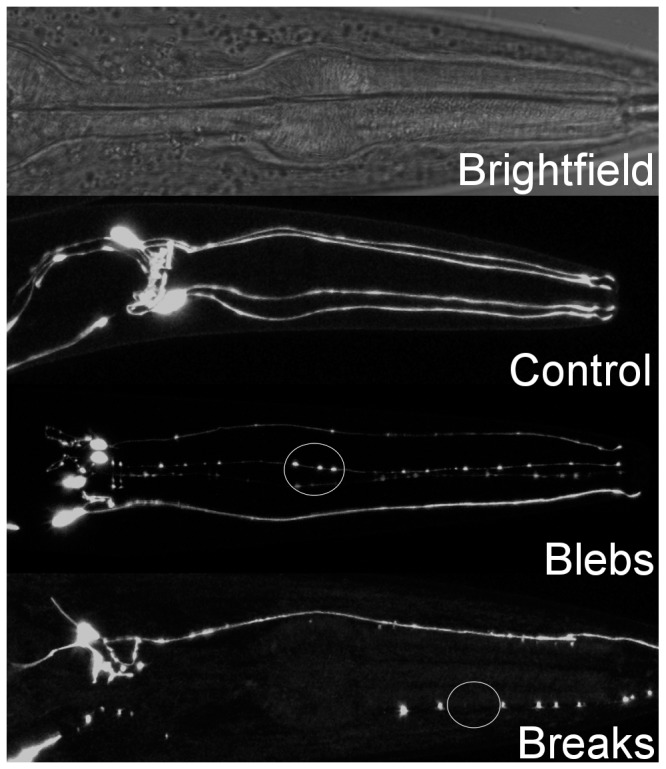
The circled areas include typically observed abnormalities, referred to as blebs and breaks in this publication.
AFB1 and paraquat exposures result in dopaminergic neurodegeneration
We next tested the hypothesis that chemical exposures causing mtDNA damage can also result in neurodegeneration in C. elegans. The doses used were the highest doses that resulted in no or minimal growth inhibition as determined in the L1 development assay (S2 Table). We detected a dose-dependent increase in lesions in dopaminergic CEP neurons after paraquat and AFB1 exposure, but no damage after CdCl2 exposure (Table 1, S2 Figure). Although the 24 h p value for CdCl2 is borderline statistically significant, the biological effect is very small if it is real. We only assessed neurodegeneration in chemicals that caused mtDNA damage in our damage assay, but it is important to mention that manganese, maneb, and rotenone have already been shown to cause dopaminergic neurodegeneration in C. elegans [70]–[74]. Neither significant lesions in the GABAergic dorsal nerve cord nor any sign of pharyngeal necrosis (measured as described in Samokhvalov et al. [75]) were detected (data not shown). Interestingly, the amount of damage detected in paraquat- and AFB1-treated animals was lower at 96 than 48 h (this was not the result of death of more-injured individuals, since there was no lethality).
Table 1. Dopaminergic neurodegeneration caused by exposure to paraquat, aflatoxin B1 and cadmium chloride in early stage C. elegans.
| Chemical and Time after Exposure | Concentration and Damage Score (Sample Size) | ||||
| Paraquat | 0 µM | 54 µM | 180 µM | p value | |
| 24 hr | 0.13 (124) | 0.16 (84) | 0.33 (82) | 0.050* | |
| 48 hr | 0.01 (135) | 0.05 (87) | 0.10 (67) | 0.007* | |
| 96 hr | 0.03 (172) | 0.04 (94) | 0.03 (95) | 0.864 | |
| Aflatoxin B1 | 0 µM | 30 µM | 100 µM | p value | |
| 24 hr | 0.10 (83) | 0.17 (75) | 0.26 (100) | 0.002* | |
| 48 hr | 0.07 (94) | 0.07 (75) | 0.10 (93) | 0.098 | |
| 96 hr | 0.07 (93) | 0.06 (93) | 0.06 (95) | 0.717 | |
| Cadmium chloride | 0 µM | 240 µM | 800 µM | p value | |
| 24 hr | 0 (45) | 0 (45) | 0.01 (45) | 0.0492* | |
| 48 hr | 0 (45) | 0 (45) | 0 (45) | 1.000 | |
| 96 hr | 0 (45) | 0 (45) | 0 (45) | 1.000 | |
Neuronal damage was scored from 0 (lowest) to 2.5 (highest) and assessed statistically using the Kruskal-Wallis test. Number of worms scored in parentheses, asterisks (*) denote statistical significance.
Fasting is protective in the context of chemical-induced dopaminergic neurodegeneration in C. elegans
Since we exposed L1 nematodes to chemicals for a 48 h period without food, and caloric restriction is known to increase stress resistance in some contexts [76]–[78], we tested whether this could have had a neuroprotective effect by assessing neuronal damage in BY250 C. elegans exposed to 15 mM and 50 mM 6-OHDA. We found that animals exposed to 6-OHDA after a 48 h period of fasting were significantly more resistant to dopaminergic neurodegeneration than animals exposed immediately after the overnight liquid hatch (Fig. 4); others have made the same observation [79]. This suggests that our chemical neurodegeneration results are conservative compared to what would be observed in nematodes that had not been fasted. To test if starvation was protective by reducing the degree of DNA damage incurred, we repeated our young adult C. elegans DNA damage assay after paraquat exposure, but included a 48 h starvation period prior to dosing. We measured slightly (∼25%) less mtDNA damage in starved worms dosed with 6 mM and 20 mM paraquat (S3 Figure), but this was not statistically significant (three-way ANOVA, p = 0.1411 for the interaction between dose, genome, and starvation status). This result suggests that the protective effect of absence of food was mediated by the biological response to damage, rather than by protection against the damage that was initially incurred. We note that DNA damage measurements (Fig. 1) were made in fed, not starved, nematodes.
Figure 4. Fasting protects against 6-OHDA–induced dopaminergic neurodegeneration.
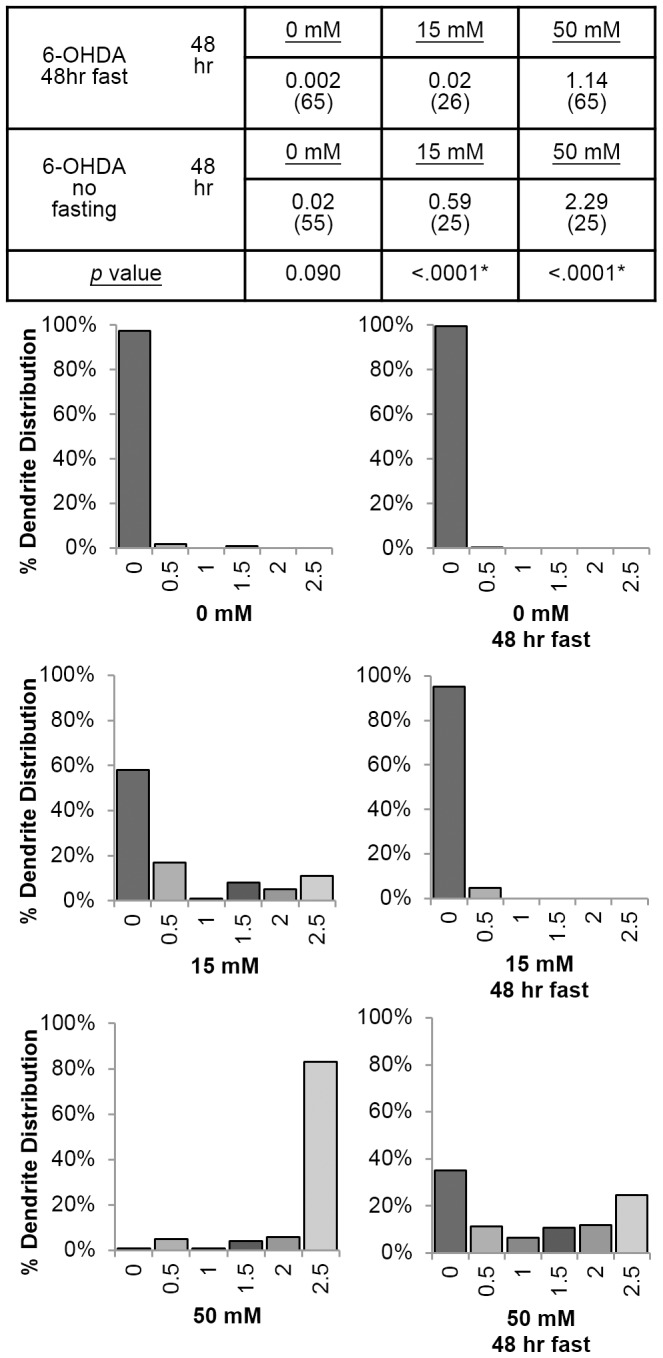
Neuronal damage was scored from 0 (lowest) to 2.5 (highest) and assessed statistically using the Fisher's Exact Test.
Dopaminergic neuron damage can be regenerated in C. elegans
Damage to the dopamine neurons after paraquat and AFB1 exposure decreased over time, from a peak at 24 hours down to undetectable levels at 96 hours post-exposure (Table 1). One possible explanation for this decrease is that regenerative processes restore normal morphology to injured neurons. However, the regenerative potential of the CEP dendrites has not been determined. To assess whether these neurons are capable of regenerating after damage, we tested their ability to respond to pulsed laser surgery. Many neurons in C. elegans can respond to laser surgery by initiating regeneration [80] but the dendrites of the dopamine neurons have not been tested. We found that the CEP dendrites are capable of injury-induced growth after laser surgery: 13 out of 16 severed dendrites exhibited anterior growth beyond the cut site (Fig. 5). Next, we asked whether injury-induced growth of the CEP dendrites requires the critical dlk-1 MAP kinase pathway, which is important for regeneration in a variety of neuron types in C. elegans [81], [82]. We found that animals defective for dlk-1 pathway signaling (mkk-4 mutants) have reduced CEP regeneration (Fig. 5), suggesting that regeneration of the CEP dendrite has some molecular similarities to axon regeneration.
Figure 5. Damage to dopaminergic neurons caused by laser ablation is repaired.
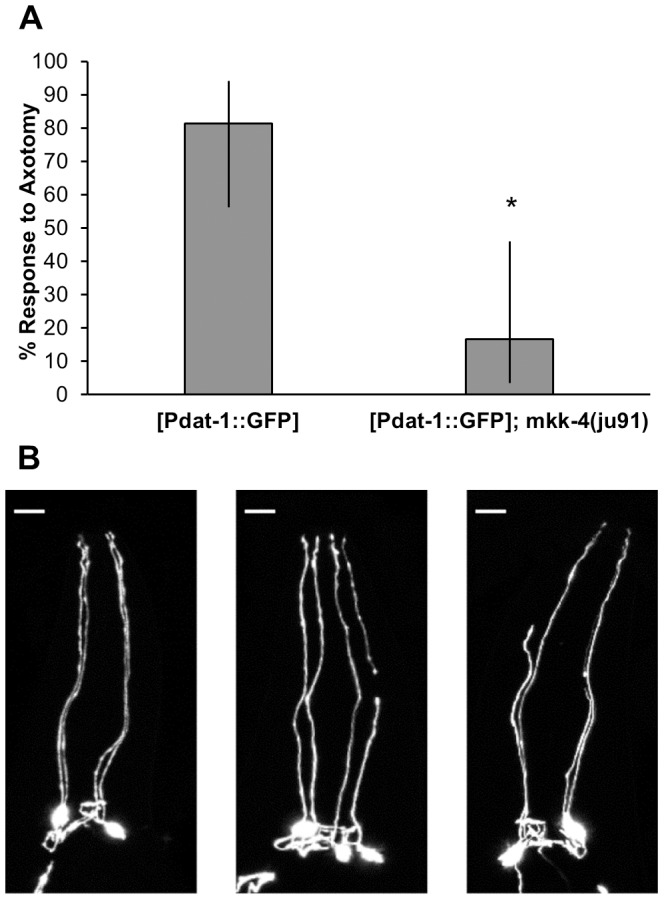
A, After laser ablation, 81% of vtIs7[Pdat-1::GFP] worms showed neuronal anterior growth, compared to 17% of the vtIs7[Pdat-1::GFP];mkk-4(ju91) worms tested (p = 0.0016, Fisher's Exact Test, bars represent 95% CI). B, From left to right, images are: uncut (with dashed line representing cut site), no response, and response. Scale bars are 10 µm.
We then explored the effect of the dlk-1 MAP kinase pathway in the response to chemical-induced neurodegeneration, as chemically induced neuronal damage could be different from that caused by laser surgery and therefore trigger a different regenerative response. We tested the effect of the mkk-4 mutation on the response to 6-OHDA, as this chemical causes the widest range of neuronal damage scores. The XE1311 worms (mkk-4 KO) were more sensitive to 6-OHDA than wild type BY250 at almost every time point and dose as determined by the Fisher's exact test (S4 Figure). However, we did not observe regeneration in the BY250 worms; damage worsened over time for both strains of C. elegans. These results suggest that the MAP kinase pathway response to neuronal damage depends on the type of damage induced, as the responses we saw to chemical injury and laser surgery were different. Furthermore, exposure to different chemicals may or may not trigger regenerative pathways in response to neuronal damage, with damage caused by some toxins (paraquat and AFB1) but not others (6-OHDA) exhibiting apparent regeneration over time.
Together, these data suggest that recovery of CEP dendrite morphology after paraquat and AFB1 exposure could be mediated by injury response pathways, possibly involving the conserved dlk-1 MAP kinase pathway.
UVC and ethidium bromide exposures result in dopaminergic neurodegeneration
Although paraquat, AFB1, and CdCl2 caused more mtDNA than nDNA damage, we could not rule out that the nDNA damage and other forms of macromolecular damage caused by these chemicals might also have caused the observed neurodegeneration. To further explore the possibility that mtDNA damage on its own could lead to dopaminergic neurodegeneration we exposed C. elegans to (a) repeated low-dose UVC radiation that results in persistent DNA damage in the mitochondria but not in the nucleus [63], [64]; and/or (b) ethidium bromide that blocks mtDNA replication [83] and exacerbates some effects of persistent mtDNA damage in this model organism, but not the induction of DNA damage per se [63], [64]. We note that we did not employ UVC and ethidium bromide because of exposure relevance in the context of PD (UVC does not reach dopaminergic neurons in people, and ethidium bromide is not a relevant chemical from the perspective of human neurodegeneration), but rather as mechanistic tools.
Exposures to both UVC and combined UVC and ethidium bromide treatment resulted in neuronal lesions 48 and 96 h after the exposure (Table 2, S2 Figure). Exposure to ethidium bromide alone resulted in a low level of lesions, and only at one time, 48 h after the exposure (Table 2). The combined UVC and ethidium bromide treatment did not result in a higher level of lesions as compared to the UVC or ethidium bromide treatment alone.
Table 2. Dopaminergic neurodegeneration caused by exposure to ultraviolet C radiation, ethidium bromide, and both in early stage C. elegans.
| Chemical and Time after Exposure | Concentration and Damage Score (Sample Size) | ||||
| Ultraviolet C radiation (UVC) | 0 J/m2 | 10 J/m2 | p value | ||
| 24 hr | 0.13 (124) | 0.23 (96) | 0.137 | ||
| 48 hr | 0.01 (135) | 0.27 (139) | <0.001* | ||
| 96 hr | 0.03 (172) | 0.11 (135) | 0.003* | ||
| Ethidium bromide (EtBr) | 0 µM | 5 µM | 10 µM | p value | |
| 24 hr | 0.13 (124) | 0.16 (83) | 0.19 (118) | 0.414 | |
| 48 hr | 0.01 (135) | 0.08 (96) | 0.11 (130) | 0.011* | |
| 96 hr | 0.03 (172) | 0.02 (95) | 0.07 (172) | 0.082 | |
| UVC + EtBr | 0 µM | 5 µM+5 J/m2 | 5 µM+10 J/m2 | p value | |
| 24 hr | 0.13 (124) | 0.26 (116) | 0.16 (104) | 0.082 | |
| 48 hr | 0.01 (135) | 0.07 (130) | 0.16 (123) | <0.001* | |
| 96 hr | 0.03 (172) | 0.07 (157) | 0.19 (167) | <0.001* | |
Neuronal damage was scored from 0 (lowest) to 2.5 (highest) and assessed statistically using the Kruskal-Wallis test. Number of worms scored in parentheses, asterisks (*) denote statistical significance.
Discussion
Our results show that the important environmental pollutants paraquat, AFB1 and CdCl2 can cause more mtDNA than nDNA damage in the nematode C. elegans. Early developmental exposure to paraquat and AFB1 also caused dopaminergic neurodegeneration. Our experiments also showed that fasting can have a neuroprotective effect, and that the dendrites of dopaminergic CEP neurons can regenerate in this model organism. It will be important to consider the possibility of regeneration, and effects of starvation, when interpreting results from chemical exposures and neuronal damage assessment assays performed with this model organism.
We report for the first time that paraquat, a heavily used herbicide, selectively targets mtDNA over nDNA. This could be due to the fact that paraquat is a potent redox cycler, readily accepting electrons and generating superoxide anions [84], [85] and several studies have shown that mtDNA is three to five times more susceptible than nDNA to oxidative damage caused by hydrogen peroxide, organic peroxide, and peroxynitrite [21], [86]–[89]. Exposure to paraquat also disrupts mitochondrial electron transport chain function [18], increases mitochondrial production of ROS [90], [91], and increases oxidative DNA damage in mammalian species in vivo [92], [93].
We also found that AFB1, a fungal genotoxin widely found in different agricultural products, selectively targets mtDNA in C. elegans. This finding is consistent with previous work by Niranjan and colleagues [94], [95] who identified a higher level of AFB1 bound to hepatic mtDNA than nDNA in several rodent species. The relative susceptibility of mtDNA versus nDNA was within the same order of magnitude in all these studies. Mitochondrial effects of AFB1 are well documented and include alteration of mitochondrial morphology, disruption of respiratory function, and reduction of energy production [96]–[98]. However, AFB1 is best known as a nDNA mutagen and hepatic carcinogen [3], [99]. The clinical significance of the mitochondrial genotoxicity of AFB1 intoxication is not well understood, as is the case for many other mitochondrial toxins [2], [4].
Paraquat and AFB1 exposures result in different types of DNA damage, which are repaired by different repair mechanisms. Paraquat exposure generates oxidative DNA damage [100], much of which is known to be repaired by base excision repair in both the nucleus and mitochondria [15]. Interestingly, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), another chemical that causes production of ROS and dopaminergic neurodegeneration, also causes more mtDNA than nDNA damage [101]. AFB1 exposure, in contrast, generates bulky DNA adducts [3] which are repaired by nucleotide excision repair, a repair mechanism that has not been identified in mitochondria [102]. Thus, AFB1 adducts are expected to be highly persistent in mtDNA. Interestingly, there is a subset of oxidative DNA damage that is probably repaired by nucleotide excision repair [103], and therefore also likely to be persistent in mtDNA.
Cadmium, a heavy metal of considerable occupational and environmental concern, is a known carcinogen that induces DNA damage through multiple mechanisms, including alteration of gene expression, inhibition of DNA repair, and induction of oxidative stress [104]. Cadmium accumulates in mitochondria and disrupts the functions of the organelles through a variety of targets [105], [106]. Our findings show that cadmium causes greater levels of mtDNA than nDNA damage.
We also report that 6-OHDA causes detectable levels of DNA damage in vivo. There are several reports in the literature suggesting the presence of damage to DNA after 6-OHDA exposure [107]–[109]. Here we present, to our knowledge for the first time, direct evidence of DNA lesions in both mitochondrial and nuclear genomes after in vivo 6-OHDA exposure, further strengthening the association between oxidative DNA damage and neurodegeneration.
Paraquat, AFB1, UVC, and the UVC + ethidium bromide combination all result in more mtDNA than nDNA damage, as well as dopaminergic neurodegeneration. However, rotenone and maneb did not result in detectable mtDNA damage despite being known dopaminergic neurotoxins. It may be that such damage occurred, but at later timepoints, at levels that our assay could not detect, or in a subset of cells (e.g. neurons) such that the damage signal was diluted because our assay only detects damage in whole-organism extracts. It is also worth noting that different C. elegans strains including glp-1 [64], [110], [111] can have different responses to the same exposure conditions, so performing these experiments in the glp-1 background rather than wild type could in principle have influenced the level of DNA damage detected. However, we have previously observed similar levels of UVC-induced DNA damage in N2 and glp-1 nematodes [61], as well as similar rates of nDNA repair [61]. It seems unlikely that exposure to a complex I inhibitor would not lead to production of ROS and some level of mtDNA damage [20]. Zhou et al. [74] recently reported that exposure to rotenone and Mn led to decreases in mtDNA content; we observed the same after Mn exposure, but not rotenone exposure (Fig. 2). Overall, these results suggest that some mitochondrial genotoxins can also cause neurodegeneration. Whether or not the genotoxicity is directly responsible for the neurodegeneration cannot be determined from our experiments. Furthermore, other toxins capable of causing neurodegeneration did not cause mtDNA damage, suggesting a different mechanism of neurotoxicity. These results are consistent with the idea that PD results from a combination of multiple different factors [112].
We found that dopaminergic neurons were more susceptible than GABAergic neurons and pharyngeal muscle cells to chemical-induced degeneration, although both of those cell types are presumably also dependent on mitochondrial function. The reason for this difference is unclear. Dopamine readily oxidizes to react with proteins, lipids, and nucleic acids and produces neurotoxic derivatives, including 6-OHDA [113], which in turn leads to greater ROS formation. We hypothesize that for mitochondrial genotoxins that cause dopaminergic neuron damage, mtDNA damage and depletion may synergize with the endogenous toxicity of dopaminergic metabolism and result in selective dopaminergic lesions.
We carried out our exposures during early larval stages because we hypothesize that mtDNA is likely to be a particularly susceptible target in early stages of life. Early life stages of mammals exhibit a “bottleneck” characterized by a much lower mtDNA copy number [114], and this is likely true in C. elegans as well: although germ cell-specific mtDNA copy numbers have not been reported in C. elegans, mtDNA copy number per cell is high in freshly laid eggs, decreases on average (across all cells) during development, and is then increased again in newly produced oocytes [64], [115], [116]. The presence of a bottleneck means that early life-stage mtDNA damage may be converted to mutations and amplified during mtDNA replication [117], [118]. Furthermore, some types of mtDNA damage may also inhibit mitochondrial transcription and replication. All of these effects could lead to mitochondrial dysfunction [63], [64], which is likely to be especially problematic in cells highly dependent on mitochondrial function such as neurons. This is also consistent with the neurodevelopmental basis of PD in which dopaminergic neurons sustain damage in early life stage, resulting in clinical symptoms while their number drops below a critical limit as a part of the aging process [51].
The neurodegeneration that we observed is likely ameliorated in our experimental conditions by the protective effects of fasting and by the ability of C. elegans to regenerate damaged dopaminergic neurons. Numerous studies have established a link between caloric restriction and protection from oxidative stress, a likely mechanism of neurotoxicity for many relevant environmental toxins [77], [119]. It also has been shown that caloric restriction or intermittent fasting can ameliorate symptoms of induced neurological disease in animal models [120], [121]. These findings are in agreement with our results, since 6-OHDA exerts its toxicity through auto-oxidation and generation of ROS [122], and it is feasible that a 48 h period of fasting could protect from such damage. While previous work on dopaminergic neurodegeneration in C. elegans has not typically including a fasted stage, that work has very likely identified less damage than actually occurred, due to regeneration.
Limitations of our study include the fact that we measured DNA damage at the level of the whole organism, rather than specifically in the dopaminergic neurons, because it is unfortunately not currently logistically possible to measure DNA damage only in the eight dopaminergic neurons present in C. elegans. However, we note that UVC is likely to penetrate and cause photodimer formation relatively uniformly, so whole-organism measures of DNA damage are likely highly reflective of neuronal levels at least for this stressor. A second limitation is that while Nass et al. [55] showed that dat-1::GFP image-based analysis of neurodegeneration was supported by transmission electron microscopy analysis for 6-OHDA, this has not been tested for other environmental stressors (or genetic deficiencies) to our knowledge.
PD has very strong environmental components [46], [47], [112], [123], but it remains unclear which environmental exposures are the most important. The relationship between environmental factors associated epidemiologically with PD [46], [124], [125] and the mechanistic pathways illustrated in controlled experiments [126], [127] is not entirely clear. Our results support the hypothesis that mtDNA damage plays an important role in the development of some cases of PD. These results also suggest an intriguing mechanism-based hypothesis for epidemiology studies to investigate: environmental exposure to mitochondrial genotoxins during early development may predispose to dopaminergic neurodegeneration later in life.
Supporting Information
Establishment of a 6-hydroxydopamine-based scoring system for dopaminergic neurodegeneration. Neuronal damage was scored from 0 (lowest) to 2.5 (highest) and assessed statistically using the Kruskal-Wallis test.
(TIF)
Representative dopaminergic neuron damage after treatment with 180 µM paraquat, 10 J/m2 UVC, and 50 mM 6-hydroxydopamine. Visualized via confocal microscopy.
(TIF)
Starvation did not have a protective effect against paraquat exposure. Worms were dosed to 54 µM and 180 µM paraquat after a 48 hr starvation period.
(TIF)
A mutation in the mkk-4 gene required for neuronal regeneration worsens dopaminergic neurodegeneration in 6-OHDA-exposed nematodes.
(TIF)
Lethality caused by toxins of interest in young adult C. elegans . No lethality was detected in both blank and 1% dimethyl sulfoxide (carrier). n = 100 per chemical per dose.
(TIF)
Growth inhibition caused by exposure to paraquat, aflatoxin B1 and cadmium chloride in C. elegans . The development of L1 BY250 (n = 4) was compared to control and scored on a 4 point scale: 1: mostly dead and dying; 2: obvious decrease in size and motility; 3: slight decrease in size and motility; 4: similar to control.
(TIF)
Acknowledgments
We thank Michael Aschner for advice and Avery M. Berkowitz for experimental assistance.
Data Availability
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data files are available from the Dryad digital repository at the following DOI: 10.5061/dryad.dd84g.
Funding Statement
This work was supported by the Society of Toxicology Colgate-Palmolive Award for Student Research Training in Alternative Methods (MCKL), American Foundation of Aging Research GlaxoSmithKline Foundation Award (MCKL), RJR-Duke Leon Golberg Toxicology Training Program (JNM and RB), National Institute of Neurological Disorders and Stroke (R21 NS065468 to JNM), and National Institute of Environmental Health Sciences (R01-ES017540-01A2 to JNM). Some strains were provided by the CGC, which is funded by the National Institutes of Health Office of Research Infrastructure Programs (P40 OD010440). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Schmidt CW (2010) Mito-Conundrum: Unraveling Environmental Effects on Mitochondria. Environ Health Perspect 118. [Google Scholar]
- 2. Shaughnessy DT, Worth L Jr, Lawler CP, McAllister KA, Longley MJ, et al. (2010) Meeting report: Identification of biomarkers for early detection of mitochondrial dysfunction. Mitochondrion 10:579–581. [DOI] [PubMed] [Google Scholar]
- 3. Bedard LL, Massey TE (2006) Aflatoxin B1-induced DNA damage and its repair. Cancer Lett 241:174–183. [DOI] [PubMed] [Google Scholar]
- 4. Meyer JN, Leung MCK, Rooney JP, Sendoel A, Hengartner MO, et al. (2013) Mitochondria as a Target of Environmental Toxicants. Toxicological Sciences 134:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wallace DC (2005) A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu Rev Genet 39:359–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Weissman L, De Souza-Pinto NC, Stevnsner T, Bohr VA (2007) DNA repair, mitochondria, and neurodegeneration. Neuroscience 145:1318–1329. [DOI] [PubMed] [Google Scholar]
- 7. Druzhyna NM, Wilson GL, LeDoux SP (2008) Mitochondrial DNA repair in aging and disease. Mech Ageing Dev 129:383–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. DiMauro S, Davidzon G (2005) Mitochondrial DNA and disease. Ann Med 37:222–232. [DOI] [PubMed] [Google Scholar]
- 9. Howell N, Elson JL, Chinnery PF, Turnbull DM (2005) mtDNA mutations and common neurodegenerative disorders. Trends Genet 21:583–586. [DOI] [PubMed] [Google Scholar]
- 10. Kohler JJ, Lewis W (2007) A brief overview of mechanisms of mitochondrial toxicity from NRTIs. Environ Mol Mutagen 48:166–172. [DOI] [PubMed] [Google Scholar]
- 11. Wunderlich V, Schutt M, Bottger M, Graffi A (1970) Preferential alkylation of mitochondrial deoxyribonucleic acid by N-methyl-N-nitrosourea. Biochem J 118:99–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wunderlich V, Tetzlaff I, Graffi A (1972) Studies on nitrosodimethylamine: preferential methylation of mitochondrial DNA in rats and hamsters. Chem Biol Interact 4:81–89. [DOI] [PubMed] [Google Scholar]
- 13. Backer JM, Weinstein IB (1982) Interaction of benzo(a)pyrene and its dihydrodiol-epoxide derivative with nuclear and mitochondrial DNA in C3H10T 1/2 cell cultures. Cancer Res 42:2764–2769. [PubMed] [Google Scholar]
- 14. Cohen BH (2010) Pharmacologic effects on mitochondrial function. Developmental Disabilities Research Reviews 16:189–199. [DOI] [PubMed] [Google Scholar]
- 15. Larsen NB, Rasmussen M, Rasmussen LJ (2005) Nuclear and mitochondrial DNA repair: similar pathways? Mitochondrion 5:89–108. [DOI] [PubMed] [Google Scholar]
- 16. Meyer JN (2010) QPCR: a tool for analysis of mitochondrial and nuclear DNA damage in ecotoxicology. Ecotoxicology 19:804–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Van Houten B, Woshner V, Santos JH (2006) Role of mitochondrial DNA in toxic responses to oxidative stress. DNA Repair (Amst) 5:145–152. [DOI] [PubMed] [Google Scholar]
- 18. Gomez C, Bandez MJ, Navarro A (2007) Pesticides and impairment of mitochondrial function in relation with the parkinsonian syndrome. Front Biosci 12:1079–1093. [DOI] [PubMed] [Google Scholar]
- 19. Ishiguro H, Yasuda K, Ishii N, Ihara K, Ohkubo T, et al. (2001) Enhancement of oxidative damage to cultured cells and Caenorhabditis elegans by mitochondrial electron transport inhibitors. IUBMB Life 51:263–268. [DOI] [PubMed] [Google Scholar]
- 20. Li N, Ragheb K, Lawler G, Sturgis J, Rajwa B, et al. (2003) Mitochondrial Complex I Inhibitor Rotenone Induces Apoptosis through Enhancing Mitochondrial Reactive Oxygen Species Production. Journal of Biological Chemistry 278:8516–8525. [DOI] [PubMed] [Google Scholar]
- 21. Yakes FM, Van Houten B (1997) Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc Natl Acad Sci U S A 94:514–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Allen JA, Coombs MM (1980) Covalent binding of polycyclic aromatic compounds to mitochondrial and nuclear DNA. Nature 287:244–245. [DOI] [PubMed] [Google Scholar]
- 23. Ayala-Torres S, Chen Y, Svoboda T, Rosenblatt J, Van Houten B (2000) Analysis of gene-specific DNA damage and repair using quantitative polymerase chain reaction. Methods 22:135–147. [DOI] [PubMed] [Google Scholar]
- 24. Brar SS, Meyer JN, Bortner C, Van Houten B, Martin WJII (2012) Mitochondrial DNA-depleted A549 cells are resistant to bleomycin. American Journal of Physiology - Lung Cellular and Molecular Physiology [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Boamah EK, Brekman A, Tomasz M, Myeku N, Figueiredo-Pereira M, et al. (2010) DNA adducts of decarbamoyl mitomycin C efficiently kill cells without wild-type p53 resulting from proteasome-mediated degradation of checkpoint protein 1. Chem Res Toxicol 23:1151–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hunter SE, Gustafson MA, Margillo KM, Lee SA, Ryde IT, et al. (2012) In vivo repair of alkylating and oxidative DNA damage in the mitochondrial and nuclear genomes of wild-type and glycosylase-deficient Caenorhabditis elegans. DNA Repair (Amst) 11:857–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. DiMauro S, Schon EA (2008) Mitochondrial disorders in the nervous system. Annu Rev Neurosci 31:91–123. [DOI] [PubMed] [Google Scholar]
- 28. Pinto M, Pickrell AM, Moraes CT (2012) Regional susceptibilities to mitochondrial dysfunctions in the CNS. Biol Chem 393:275–281. [DOI] [PubMed] [Google Scholar]
- 29. Wang X, Michaelis EK (2010) Selective neuronal vulnerability to oxidative stress in the brain. Frontiers in Aging Neuroscience 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fukui H, Moraes CT (2008) The mitochondrial impairment, oxidative stress and neurodegeneration connection: reality or just an attractive hypothesis? Trends Neurosci 31:251–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pinto M, Moraes CT (2013) Mitochondrial genome changes and neurodegenerative diseases. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kraytsberg Y, Kudryavtseva E, McKee AC, Geula C, Kowall NW, et al. (2006) Mitochondrial DNA deletions are abundant and cause functional impairment in aged human substantia nigra neurons. Nat Genet 38:518–520. [DOI] [PubMed] [Google Scholar]
- 33. Bender A, Krishnan KJ, Morris CM, Taylor GA, Reeve AK, et al. (2006) High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat Genet 38:515–517. [DOI] [PubMed] [Google Scholar]
- 34. Wang J, Xiong S, Xie C, Markesbery WR, Lovell MA (2005) Increased oxidative damage in nuclear and mitochondrial DNA in Alzheimer's disease. Journal of Neurochemistry 93:953–962. [DOI] [PubMed] [Google Scholar]
- 35. Alam ZI, Jenner A, Daniel SE, Lees AJ, Cairns N, et al. (1997) Oxidative DNA Damage in the Parkinsonian Brain: An Apparent Selective Increase in 8-Hydroxyguanine Levels in Substantia Nigra. Journal of Neurochemistry 69:1196–1203. [DOI] [PubMed] [Google Scholar]
- 36. Polidori MC, Mecocci P, Browne SE, Senin U, Beal MF (1999) Oxidative damage to mitochondrial DNA in Huntington's disease parietal cortex. Neuroscience Letters 272:53–56. [DOI] [PubMed] [Google Scholar]
- 37. Mecocci P, MacGarvey U, Beal MF (1994) Oxidative damage to mitochondrial DNA is increased in Alzheimer's disease. Ann Neurol 36:747–751. [DOI] [PubMed] [Google Scholar]
- 38. Pickrell AM, Pinto M, Hida A, Moraes CT (2011) Striatal Dysfunctions Associated with Mitochondrial DNA Damage in Dopaminergic Neurons in a Mouse Model of Parkinson's Disease. The Journal of Neuroscience 31:17649–17658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sanders LH, McCoy J, Hu X, Mastroberardino PG, Dickinson BC, et al. (2014) Mitochondrial DNA damage: Molecular marker of vulnerable nigral neurons in Parkinson's disease. Neurobiology of Disease 70:214–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Luoma P, Melberg A, Rinne JO, Kaukonen JA, Nupponen NN, et al. (2004) Parkinsonism, premature menopause, and mitochondrial DNA polymerase γ mutations: clinical and molecular genetic study. The Lancet 364:875–882. [DOI] [PubMed] [Google Scholar]
- 41. Graziewicz MA, Bienstock RJ, Copeland WC (2007) The DNA polymerase γ Y955C disease variant associated with PEO and parkinsonism mediates the incorporation and translesion synthesis opposite 7,8-dihydro-8-oxo-2′-deoxyguanosine. Human Molecular Genetics 16:2729–2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bronstein J, Carvey P, Chen H, Cory-Slechta D, DiMonte D, et al. (2009) Meeting report: consensus statement-Parkinson's disease and the environment: collaborative on health and the environment and Parkinson's Action Network (CHE PAN) conference 26–28 June 2007. Environ Health Perspect 117:117–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Dinis-Oliveira RJ, Remiao F, Carmo H, Duarte JA, Navarro AS, et al. (2006) Paraquat exposure as an etiological factor of Parkinson's disease. Neurotoxicology 27:1110–1122. [DOI] [PubMed] [Google Scholar]
- 44. Guilarte TR (2010) Manganese and Parkinson's disease: a critical review and new findings. Environ Health Perspect 118:1071–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bronstein J, Carvey P, Chen H, Cory-Slechta D, DiMonte D, et al. (2008) Meeting Report: Consensus Statement—Parkinson's Disease and the Environment: Collaborative on Health and the Environment and Parkinson's Action Network (CHE PAN) Conference 26–28 June 2007. Environ Health Perspect 117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tanner CM, Kamel F, Ross GW, Hoppin JA, Goldman SM, et al. (2011) Rotenone, paraquat, and Parkinson's disease. Environ Health Perspect 119:866–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Cannon JR, Greenamyre JT (2011) The Role of Environmental Exposures in Neurodegeneration and Neurodegenerative Diseases. Toxicological Sciences 124:225–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. van der Mark M, Brouwer M, Kromhout H, Nijssen P, Huss A, et al. (2012) Is pesticide use related to Parkinson disease? Some clues to heterogeneity in study results. Environ Health Perspect 120:340–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Benedetto A, Au C, Aschner M (2009) Manganese-induced dopaminergic neurodegeneration: insights into mechanisms and genetics shared with Parkinson's disease. Chem Rev 109:4862–4884. [DOI] [PubMed] [Google Scholar]
- 50. Franco R, Li S, Rodriguez-Rocha H, Burns M, Panayiotidis MI (2010) Molecular mechanisms of pesticide-induced neurotoxicity: Relevance to Parkinson's disease. Chem Biol Interact 188:289–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Barlow BK, Cory-Slechta DA, Richfield EK, Thiruchelvam M (2007) The gestational environment and Parkinson's disease: evidence for neurodevelopmental origins of a neurodegenerative disorder. Reprod Toxicol 23:457–470. [DOI] [PubMed] [Google Scholar]
- 52. Borenstein AR, Copenhaver CI, Mortimer JA (2006) Early-life risk factors for Alzheimer disease. Alzheimer Dis Assoc Disord 20:63–72. [DOI] [PubMed] [Google Scholar]
- 53. Miller DB, O'Callaghan JP (2008) Do early-life insults contribute to the late-life development of Parkinson and Alzheimer diseases? Metabolism 57 Suppl 2S44–49. [DOI] [PubMed] [Google Scholar]
- 54.Shoubridge EA, Wai T (2007) Mitochondrial DNA and the Mammalian Oocyte. In: Justin CSJ, editor. Current Topics in Developmental Biology: Academic Press. pp. 87–111. [DOI] [PubMed] [Google Scholar]
- 55. Nass R, Hall DH, Miller DM 3rd, Blakely RD (2002) Neurotoxin-induced degeneration of dopamine neurons in Caenorhabditis elegans. Proc Natl Acad Sci U S A 99:3264–3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Williams PL, Dusenbery DB (1988) Using the nematode Caenorhabditis elegans to predict mammalian acute lethality to metallic salts. Toxicol Ind Health 4:469–478. [DOI] [PubMed] [Google Scholar]
- 57. Boyd WA, Smith MV, Kissling GE, Rice JR, Snyder DW, et al. (2009) Application of a Mathematical Model to Describe the Effects of Chlorpyrifos on Caenorhabditis elegans Development. PLoS ONE 4:e7024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Leung MC, Goldstone JV, Boyd WA, Freedman JH, Meyer JN (2010) Caenorhabditis elegans generates biologically relevant levels of genotoxic metabolites from aflatoxin B1 but not benzo[a]pyrene in vivo. Toxicol Sci 118:444–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hunter SE, Jung D, Di Giulio RT, Meyer JN (2010) The QPCR assay for analysis of mitochondrial DNA damage, repair, and relative copy number. Methods 51:444–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Furda AM, Bess AS, Meyer JN, Van Houten B (2012) Analysis of DNA damage and repair in nuclear and mitochondrial DNA of animal cells using quantitative PCR. Methods Mol Biol 920:111–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Meyer JN, Boyd WA, Azzam GA, Haugen AC, Freedman JH, et al. (2007) Decline of nucleotide excision repair capacity in aging Caenorhabditis elegans. Genome Biol 8:R70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sulston JE (1988) Cell Lineage. In The Nematode Caenorhabditis elegans: Cold Spring Harbor Laboratory Press. [Google Scholar]
- 63. Bess AS, Crocker TL, Ryde IT, Meyer JN (2012) Mitochondrial dynamics and autophagy aid in removal of persistent mitochondrial DNA damage in Caenorhabditis elegans. Nucleic Acids Res. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Leung M, Rooney J, Ryde I, Bernal A, Bess A, et al. (2013) Effects of early life exposure to ultraviolet C radiation on mitochondrial DNA content, transcription, ATP production, and oxygen consumption in developing Caenorhabditis elegans. BMC Pharmacology and Toxicology 14:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Tsang WY, Sayles LC, Grad LI, Pilgrim DB, Lemire BD (2001) Mitochondrial respiratory chain deficiency in Caenorhabditis elegans results in developmental arrest and increased life span. J Biol Chem 276:32240–32246. [DOI] [PubMed] [Google Scholar]
- 66. Byrne AB, Edwards TJ, Hammarlund M (2011) In vivo Laser Axotomy in C. elegans. e2707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Larosche I, Letteron P, Berson A, Fromenty B, Huang TT, et al. (2010) Hepatic mitochondrial DNA depletion after an alcohol binge in mice: probable role of peroxynitrite and modulation by manganese superoxide dismutase. J Pharmacol Exp Ther 332:886–897. [DOI] [PubMed] [Google Scholar]
- 68. Korr H, Botzem B, Schmitz C, Enzmann H (2001) N-Nitrosomorpholine induced alterations of unscheduled DNA synthesis, mitochondrial DNA synthesis and cell proliferation in different cell types of liver, kidney, and urogenital organs in the rat. Chem Biol Interact 134:217–233. [DOI] [PubMed] [Google Scholar]
- 69. Santos JH, Meyer JN, Mandavilli BS, Van Houten B (2006) Quantitative PCR-based measurement of nuclear and mitochondrial DNA damage and repair in mammalian cells. Methods Mol Biol 314:183–199. [DOI] [PubMed] [Google Scholar]
- 70. Benedetto A, Au C, Avila DS, Milatovic D, Aschner M (2010) Extracellular Dopamine Potentiates Mn-Induced Oxidative Stress, Lifespan Reduction, and Dopaminergic Neurodegeneration in a BLI-3–Dependent Manner in Caenorhabditis elegans . PLoS Genet 6:e1001084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Negga R, Rudd DA, Davis NS, Justice AN, Hatfield HE, et al. (2011) Exposure to Mn/Zn ethylene-bis-dithiocarbamate and glyphosate pesticides leads to neurodegeneration in Caenorhabditis elegans. Neurotoxicology 32:331–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Ruan Q, Harrington AJ, Caldwell KA, Caldwell GA, Standaert DG (2010) VPS41, a protein involved in lysosomal trafficking, is protective in Caenorhabditis elegans and mammalian cellular models of Parkinson's disease. Neurobiology of Disease 37:330–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Saha S, Guillily MD, Ferree A, Lanceta J, Chan D, et al. (2009) LRRK2 Modulates Vulnerability to Mitochondrial Dysfunction in Caenorhabditis elegans. The Journal of Neuroscience 29:9210–9218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Zhou S, Wang Z, Klaunig JE (2013) Caenorhabditis elegans neuron degeneration and mitochondrial suppression caused by selected environmental chemicals. Int J Biochem Mol Biol 4:191–200. [PMC free article] [PubMed] [Google Scholar]
- 75. Samokhvalov V, Scott BA, Crowder CM (2008) Autophagy protects against hypoxic injury in C. elegans. Autophagy 4:1034–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Yu BP, Chung HY (2001) Stress Resistance by Caloric Restriction for Longevity. Annals of the New York Academy of Sciences 928:39–47. [DOI] [PubMed] [Google Scholar]
- 77. Sohal RS, Weindruch R (1996) Oxidative stress, caloric restriction, and aging. Science 273:59–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Martin B, Mattson MP, Maudsley S (2006) Caloric restriction and intermittent fasting: Two potential diets for successful brain aging. Ageing Research Reviews 5:332–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Nass R, Hamza I (2007) The Nematode C. elegans as an Animal Model to Explore Toxicology In Vivo: Solid and Axenic Growth Culture Conditions and Compound Exposure Parameters. Current Protocols in Toxicology: John Wiley & Sons, Inc [DOI] [PubMed] [Google Scholar]
- 80. Bejjani RE, Hammarlund M (2012) Neural Regeneration in Caenorhabditis elegans. Annual Review of Genetics 46:499–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Hammarlund M, Nix P, Hauth L, Jorgensen EM, Bastiani M (2009) Axon regeneration requires a conserved MAP kinase pathway. Science 323:802–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Yan D, Wu Z, Chisholm AD, Jin Y (2009) The DLK-1 kinase promotes mRNA stability and local translation in C. elegans synapses and axon regeneration. Cell 138:1005–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Gaines G, Attardi G (1984) Intercalating drugs and low temperatures inhibit synthesis and processing of ribosomal RNA in isolated human mitochondria. J Mol Biol 172:451–466. [DOI] [PubMed] [Google Scholar]
- 84. Jones GM, Vale JA (2000) Mechanisms of toxicity, clinical features, and management of diquat poisoning: a review. J Toxicol Clin Toxicol 38:123–128. [DOI] [PubMed] [Google Scholar]
- 85. Yumino K, Kawakami I, Tamura M, Hayashi T, Nakamura M (2002) Paraquat- and diquat-induced oxygen radical generation and lipid peroxidation in rat brain microsomes. J Biochem 131:565–570. [DOI] [PubMed] [Google Scholar]
- 86. Salazar JJ, Van Houten B (1997) Preferential mitochondrial DNA injury caused by glucose oxidase as a steady generator of hydrogen peroxide in human fibroblasts. Mutat Res 385:139–149. [DOI] [PubMed] [Google Scholar]
- 87. Santos JH, Hunakova L, Chen Y, Bortner C, Van Houten B (2003) Cell sorting experiments link persistent mitochondrial DNA damage with loss of mitochondrial membrane potential and apoptotic cell death. J Biol Chem 278:1728–1734. [DOI] [PubMed] [Google Scholar]
- 88. Cover C, Mansouri A, Knight TR, Bajt ML, Lemasters JJ, et al. (2005) Peroxynitrite-induced mitochondrial and endonuclease-mediated nuclear DNA damage in acetaminophen hepatotoxicity. J Pharmacol Exp Ther 315:879–887. [DOI] [PubMed] [Google Scholar]
- 89. Hollins DL, Suliman HB, Piantadosi CA, Carraway MS (2006) Glutathione regulates susceptibility to oxidant-induced mitochondrial DNA damage in human lymphocytes. Free Radic Biol Med 40:1220–1226. [DOI] [PubMed] [Google Scholar]
- 90. Cocheme HM, Murphy MP (2008) Complex I is the major site of mitochondrial superoxide production by paraquat. J Biol Chem 283:1786–1798. [DOI] [PubMed] [Google Scholar]
- 91. Drechsel DA, Patel M (2009) Chapter 21 Paraquat-induced production of reactive oxygen species in brain mitochondria. Methods Enzymol 456:381–393. [DOI] [PubMed] [Google Scholar]
- 92. Suzuki J, Inoue Y, Suzuki S (1995) Changes in the urinary excretion level of 8-hydroxyguanine by exposure to reactive oxygen-generating substances. Free Radic Biol Med 18:431–436. [DOI] [PubMed] [Google Scholar]
- 93. Tokunaga I, Kubo S, Mikasa H, Suzuki Y, Morita K (1997) Determination of 8-hydroxy-deoxyguanosine formation in rat organs: assessment of paraquat-evoked oxidative DNA damage. Biochem Mol Biol Int 43:73–77. [DOI] [PubMed] [Google Scholar]
- 94. Niranjan BG, Bhat NK, Avadhani NG (1982) Preferential attack of mitochondrial DNA by aflatoxin B1 during hepatocarcinogenesis. Science 215:73–75. [DOI] [PubMed] [Google Scholar]
- 95. Niranjan BG, Schaefer H, Ritter C, Avadhani NG (1986) Protection of mitochondrial genetic system against aflatoxin B1 binding in animals resistant to aflatoxicosis. Cancer Res 46:3637–3641. [PubMed] [Google Scholar]
- 96. Sajan MP, Satav JG, Bhattacharya RK (1996) Alteration of energy-linked functions in rat hepatic mitochondria following aflatoxin B1 administration. J Biochem Toxicol 11:235–241. [DOI] [PubMed] [Google Scholar]
- 97. Sajan MP, Satav JG, Bhattacharya RK (1997) Effect of aflatoxin B in vitro on rat liver mitochondrial respiratory functions. Indian J Exp Biol 35:1187–1190. [PubMed] [Google Scholar]
- 98. Ergun E, Ergun L, Essiz D (2006) Light and electron microscopic studies on liver histology in chicks fed aflatoxin. Dtsch Tierarztl Wochenschr 113:363–368. [PubMed] [Google Scholar]
- 99. Groopman JD, Kensler TW (2005) Role of metabolism and viruses in aflatoxin-induced liver cancer. Toxicol Appl Pharmacol 206:131–137. [DOI] [PubMed] [Google Scholar]
- 100. Loft S, Deng XS, Tuo J, Wellejus A, Sorensen M, et al. (1998) Experimental study of oxidative DNA damage. Free Radic Res 29:525–539. [DOI] [PubMed] [Google Scholar]
- 101. Mandavilli BS, Ali SF, Van Houten B (2000) DNA damage in brain mitochondria caused by aging and MPTP treatment. Brain Research 885:45–52. [DOI] [PubMed] [Google Scholar]
- 102. Kazak L, Reyes A, Holt IJ (2012) Minimizing the damage: repair pathways keep mitochondrial DNA intact. Nat Rev Mol Cell Biol 13:659–671. [DOI] [PubMed] [Google Scholar]
- 103. Brooks PJ (2007) The case for 8,5′-cyclopurine-2′-deoxynucleosides as endogenous DNA lesions that cause neurodegeneration in xeroderma pigmentosum. Neuroscience 145:1407–1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Joseph P (2009) Mechanisms of cadmium carcinogenesis. Toxicol Appl Pharmacol 238:272–279. [DOI] [PubMed] [Google Scholar]
- 105. Cannino G, Ferruggia E, Luparello C, Rinaldi AM (2009) Cadmium and mitochondria. Mitochondrion 9:377–384. [DOI] [PubMed] [Google Scholar]
- 106. Kurochkin IO, Etzkorn M, Buchwalter D, Leamy L, Sokolova IM (2011) Top-down control analysis of the cadmium effects on molluscan mitochondria and the mechanisms of cadmium-induced mitochondrial dysfunction. Am J Physiol Regul Integr Comp Physiol 300:R21–31. [DOI] [PubMed] [Google Scholar]
- 107. Bernstein A, Garrison S, Zambetti G, O'Malley K (2011) 6-OHDA generated ROS induces DNA damage and p53- and PUMA-dependent cell death. Molecular Neurodegeneration 6:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Blum D, Torch S, Lambeng N, Nissou M-F, Benabid A-L, et al. (2001) Molecular pathways involved in the neurotoxicity of 6-OHDA, dopamine and MPTP: contribution to the apoptotic theory in Parkinson's disease. Progress in Neurobiology 65:135–172. [DOI] [PubMed] [Google Scholar]
- 109. Kobayashi H, Oikawa S, Umemura S, Hirosawa I, Kawanishi S (2008) Mechanism of metal-mediated DNA damage and apoptosis induced by 6-hydroxydopamine in neuroblastoma SH-SY5Y cells. Free Radical Research 42:651–660. [DOI] [PubMed] [Google Scholar]
- 110.Rooney JP, Luz AL, Gonzalez-Hunt CP, Bodhicharla R, Ryde IT, et al. (2014) Effects of 5′-Fluoro-2-deoxyuridine on Mitochondrial Biology in Caenorhabditis elegans. Exp Gerontol. [DOI] [PMC free article] [PubMed]
- 111. Mendenhall AR, LeBlanc MG, Mohan DP, Padilla PA (2009) Reduction in ovulation or male sex phenotype increases long-term anoxia survival in a daf-16-independent manner in Caenorhabditis elegans. Physiol Genomics 36:167–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Schapira AHV (2008) Mitochondria in the aetiology and pathogenesis of Parkinson's disease. The Lancet Neurology 7:97–109. [DOI] [PubMed] [Google Scholar]
- 113. Sulzer D (2007) Multiple hit hypotheses for dopamine neuron loss in Parkinson's disease. Trends Neurosci 30:244–250. [DOI] [PubMed] [Google Scholar]
- 114. Cao L, Shitara H, Horii T, Nagao Y, Imai H, et al. (2007) The mitochondrial bottleneck occurs without reduction of mtDNA content in female mouse germ cells. Nat Genet 39:386–390. [DOI] [PubMed] [Google Scholar]
- 115. Bratic I, Hench J, Henriksson J, Antebi A, Burglin TR, et al. (2009) Mitochondrial DNA level, but not active replicase, is essential for Caenorhabditis elegans development. Nucleic Acids Res 37:1817–1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Tsang WY, Lemire BD (2002) Mitochondrial genome content is regulated during nematode development. Biochem Biophys Res Commun 291:8–16. [DOI] [PubMed] [Google Scholar]
- 117. Graziewicz MA, Sayer JM, Jerina DM, Copeland WC (2004) Nucleotide incorporation by human DNA polymerase gamma opposite benzo[a]pyrene and benzo[c]phenanthrene diol epoxide adducts of deoxyguanosine and deoxyadenosine. Nucleic Acids Res 32:397–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Cline SD, Lodeiro MF, Marnett LJ, Cameron CE, Arnold JJ (2010) Arrest of human mitochondrial RNA polymerase transcription by the biological aldehyde adduct of DNA, M1dG. Nucleic Acids Res 38:7546–7557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Gredilla R, Barja G (2005) Minireview: The Role of Oxidative Stress in Relation to Caloric Restriction and Longevity. Endocrinology 146:3713–3717. [DOI] [PubMed] [Google Scholar]
- 120. Maalouf M, Rho JM, Mattson MP (2009) The neuroprotective properties of calorie restriction, the ketogenic diet, and ketone bodies. Brain Research Reviews 59:293–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Maswood N, Young J, Tilmont E, Zhang Z, Gash DM, et al. (2004) Caloric restriction increases neurotrophic factor levels and attenuates neurochemical and behavioral deficits in a primate model of Parkinson's disease. Proceedings of the National Academy of Sciences 101:18171–18176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Sachs C, Jonsson G (1975) Mechanisms of action of 6-hydroxydopamine. Biochem Pharmacol 24:1–8. [DOI] [PubMed] [Google Scholar]
- 123. Landrigan PJ, Sonawane B, Butler RN, Trasande L, Callan R, et al. (2005) Early environmental origins of neurodegenerative disease in later life. Environ Health Perspect 113:1230–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Liou HH, Tsai MC, Chen CJ, Jeng JS, Chang YC, et al. (1997) Environmental risk factors and Parkinson's disease: a case-control study in Taiwan. Neurology 48:1583–1588. [DOI] [PubMed] [Google Scholar]
- 125. Priyadarshi A, Khuder SA, Schaub EA, Priyadarshi SS (2001) Environmental risk factors and Parkinson's disease: a metaanalysis. Environ Res 86:122–127. [DOI] [PubMed] [Google Scholar]
- 126. Melrose H, Lincoln S, Tyndall G, Farrer M (2006) Parkinson's disease: a rethink of rodent models. Experimental Brain Research 173:196–204. [DOI] [PubMed] [Google Scholar]
- 127. Cicchetti F, Drouin-Ouellet J, Gross RE (2009) Environmental toxins and Parkinson's disease: what have we learned from pesticide-induced animal models? Trends in Pharmacological Sciences 30:475–483. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Establishment of a 6-hydroxydopamine-based scoring system for dopaminergic neurodegeneration. Neuronal damage was scored from 0 (lowest) to 2.5 (highest) and assessed statistically using the Kruskal-Wallis test.
(TIF)
Representative dopaminergic neuron damage after treatment with 180 µM paraquat, 10 J/m2 UVC, and 50 mM 6-hydroxydopamine. Visualized via confocal microscopy.
(TIF)
Starvation did not have a protective effect against paraquat exposure. Worms were dosed to 54 µM and 180 µM paraquat after a 48 hr starvation period.
(TIF)
A mutation in the mkk-4 gene required for neuronal regeneration worsens dopaminergic neurodegeneration in 6-OHDA-exposed nematodes.
(TIF)
Lethality caused by toxins of interest in young adult C. elegans . No lethality was detected in both blank and 1% dimethyl sulfoxide (carrier). n = 100 per chemical per dose.
(TIF)
Growth inhibition caused by exposure to paraquat, aflatoxin B1 and cadmium chloride in C. elegans . The development of L1 BY250 (n = 4) was compared to control and scored on a 4 point scale: 1: mostly dead and dying; 2: obvious decrease in size and motility; 3: slight decrease in size and motility; 4: similar to control.
(TIF)
Data Availability Statement
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data files are available from the Dryad digital repository at the following DOI: 10.5061/dryad.dd84g.