

Abstract
High-throughput transcriptome sequencing allows identification of cancer-related changes that occur at the stages of transcription, pre-messenger RNA (mRNA), and splicing. In the current study, we devised a pipeline to predict novel alternative splicing (AS) variants from high-throughput transcriptome sequencing data and applied it to large sets of tumor transcriptomes from The Cancer Genome Atlas (TCGA). We identified two novel tumor-associated splice variants of matriptase, a known cancer-associated gene, in the transcriptome data from epithelial-derived tumors but not normal tissue. Most notably, these variants were found in 69% of lung squamous cell carcinoma (LUSC) samples studied. We confirmed the expression of matriptase AS transcripts using quantitative reverse transcription PCR (qRT-PCR) in an orthogonal panel of tumor tissues and cell lines. Furthermore, flow cytometric analysis confirmed surface expression of matriptase splice variants in chinese hamster ovary (CHO) cells transiently transfected with cDNA encoding the novel transcripts. Our findings further implicate matriptase in contributing to oncogenic processes and suggest potential novel therapeutic uses for matriptase splice variants.
Keywords: matriptase, alternative splicing, epithelial tumors, RNA sequencing, de novo assembly
Introduction
Alternative splicing (AS) allows a normal cell to generate multiple pre-messenger RNA (mRNA) transcripts of a gene, which can be translated into functionally diverse proteins. Similarly, cancer cells can usurp this mechanism to tailor functional transcripts that favor the malignant state. Splice variants have been identified in a variety of cancers, suggesting that widespread aberrant and AS may be a common consequence or even a cause of cancer.1 The biological activity of the majority of AS isoforms and, in particular, their contribution to cancer biology have yet to be elucidated. However, a number of studies have demonstrated that cancer-associated splice variants can serve as diagnostic or prognostic markers, or predict sensitivity to certain drugs.2–4 Treatments targeting these tumor-associated splice variants [eg, epidermal growth factor receptor (EGFR), CD44, and vascular endothelial growth factor (VEGF) receptor] are also showing promising results in preclinical studies and clinical trials.5,6
Massively parallel RNA sequencing (RNA-seq) allows the exploration of cancer-related changes at the level of transcription and splicing. In this study, we devised an AS-detection pipeline based on ABySS7 and Trans-ABySS8 software packages. ABySS is a de novo, parallel, and paired-end sequence assembler designed for short reads. It assembles a dataset multiple times using a De Bruijn graph-based approach. Trans-ABySS postprocesses ABySS assemblies to merge contigs and remove redundancy. This approach reconstructs transcripts from a broad range of expression levels, including those expressed at low levels. Using this approach, we performed a large-scale study to identify tumor-associated AS isoforms in the transcriptome of tumors available from The Cancer Genome Atlas (TCGA). We identified two highly frequent novel tumor-associated splice variants of matriptase with restricted expression to epithelial-derived tumors.
Matriptase (MT-SP1/TADG-15/ST14) is a type II transmembrane serine protease (TTSP) encoded by a gene located at human chromosome 11q24–25, and is localized to the cell surface.9 It has a multi-domain structure common for the TTSP family. The intracellular domain at its amino terminal contains a consensus phosphorylation site for protein kinase C, followed by a signal anchor transmembrane domain. At the extracellular region, matriptase contains a single SEA (sea urchin stem region, enteropeptidase, and agrin) domain, and two CUB repeats (complement C1r/C1s, Uegf, Bmp1) and four tandem repeats of an LDLRA domain (ligand-binding repeats of the low-density lipoprotein receptor class A).10 It is synthesized as an inactive, single-chain zymogen and catalyzes its own autoactivation.11 Once activated, matriptase cleaves and activates the hepatocyte growth factor/scattering factor (HGF/SF) and urokinase plasminogen activator (pro-uPA),12–14 suggesting that this protease functions as an epithelial membrane activator for other proteases and latent growth factors. Matriptase substrate proteins are known to play important roles in tumor development. Activated HGF/SF binds to its receptor, Met proto-oncogene (Met), and stimulates multiple downstream pathways including rat sarcoma viral oncogene-mitogen-activated protein kinase (Ras-MAPK), phosphoinositide-3-kinase (PI3K), Schmidt-Ruppin A-2 oncogene (Src), and signal transducer and activator of transcription 3 (Stat3). In turn, this leads to the activation of gene products required for invasive growth.15–17 uPA regulates cell/extracellular matrix (ECM) interactions as an adhesion receptor for vitronectin, and cell migration as a signal transduction molecule and by its intrinsic chemotactic activity, thereby promoting tumor invasion and metastasis.18 By controlling the activity of uPA and HGF/SF, matriptase becomes a prime constituent in the activation cascade for invasive growth and metastasis.
Matriptase activity is tightly regulated via antagonism from HGF activator inhibitor-1 (HAI-1). HAI-1 is a serine peptidase inhibitor encoded by Kunitz-type 1 gene (SPINT1).19 HAI-1 has not only an inhibitory function but is also required for matriptase activation, and regulates the proper expression and intracellular trafficking of matriptase.20,21 It has been shown that in the absence of HAI-1, matriptase biosynthesis is significantly lower because of autoproteolytic activation in the Golgi–endoplasmic reticulum apparatus. This event has a detrimental effect upon the trafficking of the matriptase protease and the cessation of further matriptase translation.21 The role of HAI-1 as both inhibitor and activator of matriptase provides a means to prevent unwanted proteolysis and the subsequent harmful effects of matriptase on cells.
Matriptase is widely expressed by the epithelia of almost all organs examined so far.22 Studies of matriptase-deficient mice have shown that matriptase is essential for postnatal survival, epidermal barrier function, hair follicle development, and thymic homeostasis.23 Matriptase has also been shown to be overexpressed in a variety of human cancers. In many cases, high matriptase expression levels are correlated with poor clinical outcome.24,25 In addition to matriptase overexpression, an imbalance in the ratio of matriptase to HAI-1 has been reported in late-stage tumors leading to the proposal that uninhibited matriptase activity may contribute to the development of advanced disease.25
Although many studies present matriptase as a promising potential therapeutic target in oncology,25,26 its therapeutic use is limited by its widespread expression and essential function in normal epithelial tissues. However, a unique form of matriptase within tumor cells could potentially overcome this limitation. Using our AS-detection pipeline, we identified two novel tumor-associated spliced isoforms of matriptase in the transcriptome of primary ovarian, breast, prostate, head and neck, lung, stomach, and bladder carcinoma that were not in normal transcriptomes from the adjacent non-tumor tissue. We confirmed quantitative mRNA expression of matriptase splice variants using quantitative reverse transcription PCR (qRT-PCR) on cDNA panels obtained from an orthogonal set of tumor tissues and cell lines. Using flow cytometry, we further demonstrated the presence of matriptase splice variants on the surface of transfected chinese hamster ovary (CHO) cells with cDNA encoding these variants. Tumor association and the high frequency of matriptase splice variants within and across epithelial tumors suggest that these mutant matriptase transcripts may be of potential therapeutic value. This is the first study reporting tumor-associated transcripts of matriptase in human cancers.
Material and Methods
Obtaining transcriptome data from TCGA
Raw RNA-seq data (Table 1) and clinicopathological data were downloaded from the TCGA data portal (http://cancergenome.nih.gov). Permission to access TCGA data was obtained from the Data Access Committee of the National Center for Biotechnology Information’s Genotypes and Phenotypes Database (dbGAP) at the National Institute of Heath. Sample collection, library preparation, and RNA-seq were described by TCGA previously.27 TCGA transcriptomes were generated from specimens that have not received any systemic treatment.
Table 1.
Number of individual tumor and corresponding adjacent non-cancerous tissue samples investigated in this study.
| TUMOR TYPE | TUMOR TISSUE | #TUMOR | #ADJACENT NORMAL |
|---|---|---|---|
| Ovarian serous cystadenocarcinoma (OV) | Primary | 420 | N/A |
| Triple Negative Breast Cancer (TNBC) | Primary | 109 | 10 |
| Stomach adenocarcinoma (STAD) | Primary | 285 | 33 |
| Skin Cutaneous Melanoma (SKCM) | Primary | 260 | N/A |
| Head and Neck squamous cell carcinoma (HNSC) | Primary | 181 | 27 |
| Glioblastoma multiforme (GBM) | Primary | 73 | N/A |
| Prostate adenocarcinoma (PRAD) | Primary | 166 | 41 |
| Bladder Urothelial Carcinoma (BLCA) | Primary | 116 | 15 |
| Lung squamous cell carcinoma (LUSC) | Primary | 305 | 41 |
| Lung adenocarcinoma (LUAD) | Primary | 185 | 55 |
| Acute Myeloid Leukemia (AML) | Primary | 161 | N/A |
AS-detection pipeline
The AS-detection pipeline starts with raw RNA-seq data (fastq files). Fastq files were either directly downloaded from the TCGA data portal or extracted from downloaded Binary Alignment/Map (BAM) files using SamToFastq.28 The pipeline core steps include de novo transcriptome assembly, identifying tumor-associated events, assessing the quality of assembled transcripts, quantifying predicted transcripts, and prediction of protein sequence and domains (Fig. 1). The key steps are described below:
Figure 1.

An overview of AS-detection pipeline.
De novo transcriptome construction: The de novo transcriptome assembly leverages the redundancy of short-read sequencing to find overlaps between the reads and assembles them into transcripts. We assembled short RNA-seq reads into contigs using ABySS version 1.3.4 for multiple values of K-mer. K-mer is all the possible subsequences (of length K) from a read obtained through sequencing of RNA. TCGA RNA-seq libraries are paired end, and the read length is 48 bp. We assembled each library for 13 different values of K-mer from 24 to 48 (increasing by 2). This approach captures transcripts from a broad range of expression levels, thus allowing low-expressing transcripts to be constructed. Trans-ABySS (version 1.4.4) was then used to merge ABySS assemblies, removing redundancy and reconstructing transcripts. The de novo transcriptome construction captures major splice rearrangements and novel variations that occur in the transcriptome, including exon skipping, novel exons, retained introns, and AS at 3′-acceptor and 5′-donor sites. As this approach does not rely on a reference genome, it can assemble novel AS as well as trans-spliced transcripts. Constructed transcripts were then annotated by mapping them to the human reference genome (hg19).
Identification of tumor-associated transcripts: In order to identify and remove tissue-specific splicing variants, we compared predicted transcripts from tumor libraries with the ones present in available corresponding normal data from TCGA, as well as Illumina BodyMap 2.0 project (Supplementary Table S1). BodyMap consists of 19 normal transcriptomes from 16 different tissue types, making it an invaluable source for studying tissue-specific transcript models. Tissue-specific AS events were also predicted using ABySS/Trans-ABySS software package as described above. Transcript variants not detected by the de novo transcriptome assembly approach are considered as not being expressed.
Transcript quality assessment: Predicted AS transcripts were evaluated by their contig size, number of reads supporting predicted novel junction, and their alignment quality. Transcripts with contigs smaller than 200 bp and less than four supporting reads to predicted novel junction were removed from the analysis. Misassembly of transcriptome reads may occur as a result of mutation, low quality and low complexity of the reads, as well as presence of repeats. This could lead to the prediction of a false junction. In order to identify such cases, we aligned predicted AS transcripts back to the human genome (hg19) using stand-alone BLAT from UCSC (http://hgdownload.cse.ucsc.edu/admin/exe/) and evaluated the alignment quality of sequences that span predicted novel junctions. BLAT was run using default parameters. If sequences that span a novel junction were also aligned to a different part of genome with similarity greater than 70%, we labeled such transcripts as unreliable and removed them from further analysis. Transcripts that passed quality assessment were visualized by UCSC Genome Browser (https://genome.ucsc.edu/) or Integrative Genomics Viewer (IGV; http://www.broadinstitute.org/igv/).
Quantifying predicted transcripts: Only the reads that align to a novel junction are isoform informative. Trans-ABySS estimates the number of these reads, which allows quantifying the novel AS isoforms. Assuming each unique read spanning a novel junction is generated from a transcript uniformly, each exon in an isoform was assigned an equal number of reads as the number of spanning reads, and estimated fragments per kilobase of transcript per million mapped reads (FPKM) values (Supplementary Method).
Prediction of protein sequence and domain: Open reading frame (ORF) prediction was performed using NCBI ORF Finder (http://www.ncbi.nlm.nih.gov/projects/gorf/) to identify the longest ORF in each transcript. Protein domains were predicted by RPS-BLAST at NCBI (http://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi).
qRT-PCR validation
Reverse transcription reaction was performed using commercially available sets of human normal tissue or ovarian, breast, lung, and bladder cancer cDNA (OriGene Technologies); as well as cDNA synthesized from RNA isolated from ovarian cell lines, including OvCAR3, CaOV3, UACC-1598, Ov-90, and triple negative breast cell lines, including MDA-MB-231, MDA-MB-468, and HCC 1937. All cells lines were cultured under ATCC re commended culture conditions (Supplementary Method). PCR amplification was performed for 40 cycles with matriptase A1 forward 5′-GAC ACC GGC TTC TTA GCT GAA T-3′ and A1 reverse 5′-GAA GAG GGG CTT GCA GAA CTT G-3′, as well as A3 forward 5′-GAA CGA CTG CGG AGA CAA CA-3′ and A3 reverse 5′-TGC TCA AGC AGA GCC CAT T-3′ primers. For each target, relative levels of expression were normalized against housekeeping gene beta-glucuronidase signal (GusB), generating ∆Ct value for each reaction (Supplementary Method). GusB was found to be stably expressed across ovarian samples for use as a reference gene by Li et al.29 The relative fold change for each sample was calculated similar to the approach taken by Beillard et al.30 Samples were grouped by cell lines, cancer subtypes, or normal tissues, and graphed using GraphPad Prism software version 5.0 (GraphPad Software Inc.).
Transfection constructs
Total RNA was isolated with the RNeasy Mini Kit (Qiagen) from MDA-MB-468 and HCC 1937 cells to generate cDNA encoding HAI-1 and wild-type matriptase, respectively. cDNA was generated as per manufacturer’s instructions using SuperScript® III Reverse Transcriptase (Life Technologies) and Oligo(dT)18 primer (Thermo Fisher Scientific). HAI-1 and wild-type matriptase were amplified from the above cDNA using Q5® Hot Start High-Fidelity DNA Polymerase (New England Biolabs). SPINT1 (GenBank accession no.: NM_181642.2) encoding HAI-1 was cloned into the pTT5 vector (National Research Council of Canada, Biotechnology Research Institute) using Gibson Assembly® (New England Biolabs) as per manufacturer’s instructions. The HAI-1 forward primer was 5′-aaacggatctctagcgaattcgccaccATGGCCCCTGCGAGGA CG-3′ and the reverse primer was 5′-aggtcgaggtcgggggatccTCAGAGGGGCCGCGTGGT-3′. Lower case letters correspond to the pTT5 vector and upper case to HAI-1, and the start/stop codons are underlined. Wild-type matriptase (aka ST14; GenBank accession no.: NM_021978) was cloned into the pTT5 vector using 5′ EcoRI and 3′ BamHI restriction sites (in bold) incorporated into the ST14 forward primer (5′-GATCGAATTCGCCACCATGGGGAGCGATCGGGCCCGCAA-3′) and ST14 reverse primer (5′-GATCGGATCCCTATACCCCAGTGTTCTCTTTGATCCAGTCCC-3′). The exon 12 deletion (variant A1) was introduced by amplifying the regions 5′ and 3′ to the exon deletion from the wild-type matriptase cDNA using the ST14 forward and reverse primers. The ST14 forward primer was paired with the variant A1 reverse primer (5′-CCGGCGTCGCAACGGTCACTGGAGTCGTAGGAGAG-3′) to amplify the region 5′ to the exon deletion, and the ST14 reverse primer was paired with the variant A1 forward primer (5′-ACTCCAGTGACCGTTGCGACGCCGGCCACCAGTT-3′) to amplify the region 3′ to the exon deletion. The variant A1 primers introduced an overhang depicted by the underlined sequence. Equimolar amounts of the above 5′ and 3′ PCR products were added to a PCR reaction as the template, along with the ST14 forward and reverse primers to produce a full-length construct with the region corresponding to exon 12 deleted. The exon 14 deletion (variant A3) was constructed the same way using the variant A3 forward primer (5′-AGCAGGGGTGCATGAACGTCGTCACTTGTACCAA-3′) and the variant A3 reverse primer (5′-TGACGACGTTCATGCACCCCTGCTCGTCGCTGTT-3′). All constructs were verified by DNA sequencing.
Cell culture conditions and transfection
CHO-K1 cells (ATCC) were maintained in Ham’s F-12 media (Life Technologies) supplemented with 10% fetal bovine serum (FBS; Life Technologies) at 37 °C and 5% CO2. The day before transfection, 2.5 × 106 cells per plate were seeded in the above media on four 10-cm plates for each transfection. The four transfections consisted of empty pTT5 vector alone, wild type plus HAI-1, variant A1 plus HAI-1, and variant A3 plus HAI-1. Twenty-four hours later, each transfection was performed by mixing a total of 10 μg of cDNA into 500 μL of Opti-MEM® (Life Technologies), and 30 μg of poly ethylenimine (PEI) Max (Polysciences, Inc.) into another tube with 500 μL of Opti-MEM®. The two tubes were incubated at room temperature for five minutes, and then, the PEI Max solution was added to the cDNA solution followed by a 25-minute incubation. PEI/cDNA complexes were added dropwise to the 10-cm plate while swirling/rocking to mix, and the cells were returned to the incubator.
Flow cytometry
Twenty-four hours after transfection, the plates were washed once with phosphate buffered saline (PBS, Life Technologies), and the cells were dissociated from the plate with non-enzymatic cell dissociation solution (Sigma-Aldrich). After 15 minutes at 37 °C, the cells were collected by pipetting up and down in PBS plus 1% FBS (PBS/FBS), counted on a Vi-Cell™, and resuspended in PBS/FBS. Cells were added to a 96-well plate, spun at 400 × g for five minutes, and resuspended in 5 μg/mL of human antimatriptase or 10 μg/mL of mouse anti-SPINT1 (OriGene Technologies). Isotype controls were also prepared for each transfection with 5 μg/mL of human IgG1 kappa (Sigma-Aldrich) or 10 μg/mL of mouse IgG1 kappa (eBioscience). After an one-hour incubation on ice, cells were washed two times in ice-cold PBS/FBS and resuspended in PBS/FBS containing 2.5 μg/mL of 7-aminoactinomycin D (7-AAD; Sigma-Aldrich) plus 2 μg/mL of either Alexa Fluor® 647 Goat Anti-Human IgG-Fc (Jackson ImmunoResearch Labs, Inc.) or Alexa Fluor® 647 Goat Anti-Mouse IgG-Fc (Jackson ImmunoResearch Labs, Inc.). Cells were incubated for 30 minutes on ice in the dark, then washed two times in PBS/FBS, and resuspended in PBS/FBS. Data were acquired with an Intelli-Cyt® high-throughput flow cytometer (HTFC) that consisted of an Accuri® C6 Flow Cytometer® (BD Biosciences), CFlow® Software (version 1.0.227.4), HyperCyt® CFlow Automator (version 3.4.0.0), and HyperView iDM® Client Edition 4.0 (R2 version 4.0.4395). Analysis was carried out using the CFlow® Software (version 1.0.227.4) and FCS Express 4 Professional Standalone Research Edition with histogram smoothing set to 1 (De Novo Software™, version 4.07.0014).
Immunoprecipitation and Western blot analysis
The immunoprecipitation was performed as described31 with the following modifications. Unless otherwise stated, all reagents were purchased from Sigma. As outlined above for the flow cytometry experiment, HAI-1 plus either wild type, A1, or A3 transfected CHO-K1 cells were dissociated from 10-cm plates with non-enzymatic dissociation solution, and collected by pipetting up and down in PBS alone. Cells were spun for five minutes at 400 × g, and the supernatant was aspirated. Pellets were resuspended in 0.5–1 mL of ice-cold lysis buffer [50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% Triton X-100, 0.1% sodium dodecyl sulfate (SDS), 1 mM CaCl2, 1 mM MgCl2, and one complete mini ethylenediaminetetra-acetic acid (EDTA)-free protease inhibitor cocktail tablet (Roche) per 10 mL of buffer]. While on ice, the cells were broken open with 10 strokes of the pestle using a pestle and microtube set (VWR), and then the lysate was passed through a 26-gage syringe 10 times to shear the DNA. DNase was added to 10 μg/mL, and the lysates were gently rotated at 4 °C for 30 minutes. Lysates were clarified by centrifugation at 20,000 × g for 10 minutes at 4 °C, and supernatant was subjected to bicinchoninic acid (BCA) protein concentration assay (Pierce). Clarified lysates were adjusted to 1 mg/mL in 1 mL (Fig. 5G “start”). In all, 40 μL of a 50% slurry of Protein G Sepharose Fast Flow beads (GE Healthcare) preequilibrated in lysis buffer was added followed by rotation at 4 °C for one to two hours to preclear the lysate. The beads were removed by centrifugation at 2,500 × g for 2.5 minutes at 4 °C, and the precleared lysate was transferred to a new 1.7 mL tube. In all, 1.5 μg of human anti-matriptase antibody was added followed by rotation for 14–16 hours at 4 °C. Matriptase-antibody complexes were then rotated with 40 μL of the above Sepharose bead preparation for another two hours at 4 °C. The beads were washed three times in 1 mL of ice-cold lysis buffer by centrifuging at 2,500 × g for 2.5 minutes at 4 °C followed by supernatant aspiration. The beads were resuspended in non-reducing Laemmli sample buffer,32 and heated at 95 °C for five minutes to dissociate the matriptase-antibody bead complex. The beads were removed by centrifugation using a custom- made spin column, and the proteins (Fig. 5G “elution”) were separated by SDS-polyacrylamide gel electrophoresis in 1× Tris/glycine/SDS buffer (Bio-Rad). The resolved proteins were electrotransferred to 0.45 μm nitrocellulose membrane (Bio-Rad) at 100 V for 90 minutes in 1× Tris/glycine buffer with 20% methanol33 (Bio-Rad). The nitrocellulose was air dried to fix the proteins, and then subjected to Western blot analysis as described.31 The primary rabbit anti-matriptase antibody was used at 1:2,000 (Millipore), and the secondary anti-rabbit conjugated horseradish peroxidase was used at 1:50,000 (GE Healthcare). Proteins were detected with Super-Signal West Dura Chemiluminescent Substrate (Pierce) and exposed to Amersham Hyperfilm (GE Healthcare).
Figure 5.

Flow cytometric analysis reveals surface expression of matriptase splice variants. Cells were transfected with 10 μg of empty vector alone (pTT5) or 5 μg of each matriptase variant plus 5 μg of HAI-1 (A–G). The next day, duplicate wells containing 100,000 cells per well were stained with either human anti-matriptase or mouse anti-SPINT1 (HAI-1) antibodies (data not shown) followed by species-specific secondary Alexa Fluor® 647 Goat anti-IgG-Fc antibodies plus the live/dead cell discriminator 7-AAD followed by flow cytometric analysis. The gating tree is as follows: (A) SSC versus FSC depicts the distribution of cells as oppose to the debris that was excluded; to (B) living cells not stained with 7-AAD. (C) Wild-type matriptase, (D) matriptase variant A1, and (E) matriptase variant A3 (F) graph depicting the mean fluorescent intensity plus/minus the standard error of mean of matriptase expressed on the surface of CHO cells. These data are representative of three independent experiments analyzed with a student’s t-test (P < 0.05). Flow cytometry data were acquired on an IntelliCyt® HTFC, which uses an Accuri® C6 Flow Cytometer® (BD Biosciences) with the sip time set at three seconds. Laser lines for this instrument are 488 and 640 nm. FL3 emission detection for 7-AAD is >670 nm, and FL4 emission detection for Alexa Fluor® 647 is 675/25 nm. (G) Recombinant wild type, A1, and A3 variants were immunoprecipitated with 1.5 μg of human anti-matriptase antibody, followed by Western blot analysis on the clarified start lysates (20 μg each) and elutions (15 μL each). The arrow shows the bands corresponding to the expected size of each matriptase variant.
Statistical and survival analysis
For each tumor set, Kaplan–Meier survival curve of patients was prepared according to the presence status of matriptase A1 and A3 transcripts, with differences in overall survival rates determined by the log-rank test. Overall survival time was defined as the period between initial pathologic diagnosis and the time of death. Survival time of patients who were still alive was noted with the data of the most recent follow-up appointment. The Fisher’s exact test was used to compare the two categorical variables. The Mann–Whitney t-test was used to determine significant differences in gene expression between groups. A statistically significant P-value was defined as P-value ≤0.05. We have used R for statistical and survival analysis.
Results
Epithelial-derived tumors express novel splicing variants of matriptase
De novo assembly of matriptase transcripts revealed two novel splice variants in epithelial-derived tumors. As depicted in Figure 2, these variants contain an in-frame exon skipping of the LDLRA1 or LDLRA3 domain. The novel transcripts were, therefore, denoted as A1 (skipping LDLRA1), and A3 (skipping LDLRA3). Similar analysis for transcriptomes derived from melanoma, leukemia, and glioblastoma tumors did not identify A1 and A3 variants. This is consistent with the observation that matriptase is predominantly expressed by the epithelial tissue (P = 0.006 and P = 0.0242, respectively).
Figure 2.
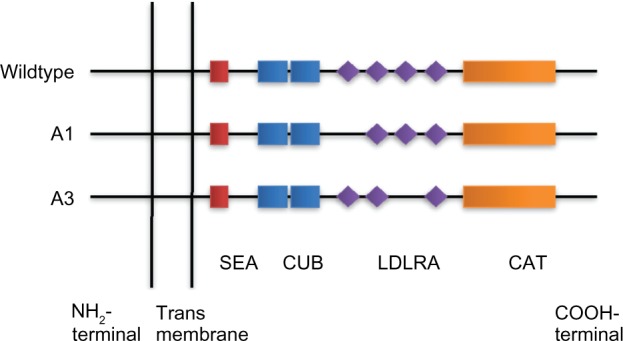
Schematic presentation of novel matriptase AS transcripts. Four LDL receptor class A domains are found in matriptase (LDLRA1: residues 452–486, LDLRA2: residues 487–523, LDLRA3: residues 524–561, and LDLRA4: residues 566–604). A1 and A3 are produced by skipping exon 12 (encoding LDLRA1) and exon 14 (encoding LDLRA3), resulting in in-frame deletion of 105 and 114 bp, respectively. CAT: serine protease catalytic domain.
An estimation of A1 and A3 transcript abundances using the number of reads supporting the novel exon–exon junction from Trans-ABySS indicated higher expression for A1 compared to the A3 transcript in all tumors studied (Supplementary Figs. S1 and S2). We observed a wide range in the frequency of epithelial tumors displaying these matriptase splice variants, from 3% in prostate adenocarcinoma (PRAD) to 69% in lung squamous cell carcinoma (LUSC) (Fig. 3). Matriptase variant A1 was found more frequent than A3 across all tumors studied (P = 0.01). In addition, A3 variant was not detected in the transcriptomes from the PRAD. Among samples with matriptase splice variant-positive cancer, we observed cases that either express one or both splice variants of matriptase (Fig. 3).
Figure 3.
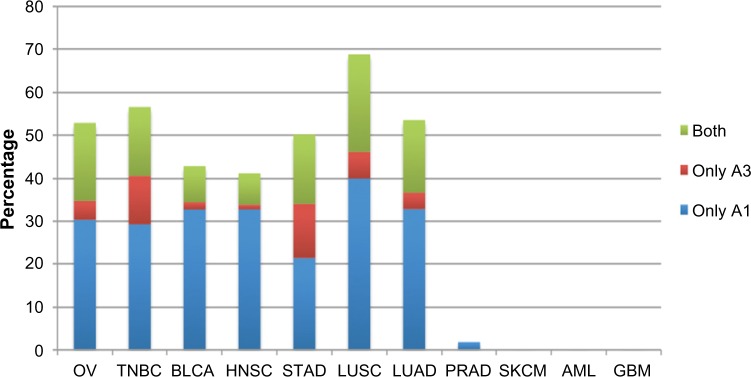
Frequency of novel matriptase AS transcripts across tumors studied. Samples expressing matriptase novel transcripts were divided into three groups: (1) expressing transcript A1, (2) expressing transcript A3, and (3) expressing both A1 and A3 transcripts. Matriptase transcript A1 is more frequent than A3 (P = 0.01). Transcript A3 was not detected in prostate cancer samples. Expression of matriptase splice variants is restricted to epithelial-derived tumors (P < 0.05); therefore, AML, SKCM, and GBM do not express these variants. Abbreviations are described in Table 1.
The human matriptase is located on chromosome 11q24–25, spanning a genomic region of 50 kb. It is composed of 19 exons (NCBI reference sequence GenBank: NM_021978), and encodes a protein containing 855 amino acids. Our nucleo tide sequence analysis revealed that A1 was produced as a result of skipping exon 12. Similarly, the A3 deletion occurred by skipping exon 14 (Fig. 2). Analysis of predicted protein sequences revealed that both matriptase variants contain fully functional ORFs, suggesting the possibility of expressing two novel proteins (Supplementary Sequence S2). Protein domain prediction further demonstrated that matriptase variants A1 and A3 lack LDLRA1 and LDLRA3 domains, respectively. Pairwise protein sequence alignment versus wild-type matriptase showed that the predicted protein for A1 transcript skips amino acids 452–487 followed by occurrence of an amino acid arginine (R) through the resultant of a novel exon–exon junction (Supplementary Sequence S3.1). The protein product of A1 transcript contains 820 amino acids. The A3 transcript encodes a protein of 817 amino acids, which is the result of skipping amino acids 524–562 followed by substitution of methionine (M) as a result of the formation of a novel exon–exon junction (Supplementary Sequence S3.2).
Matriptase splice variants are novel and tumor associated
To search AS information for matriptase, we performed literature searches using PubMed, online mendelian inheritance in man (OMIM), and other databases of AS, including the AS and Transcript Discovery (ASTD) database.34 In addition, we searched publicly available expressed sequence tag (EST) and mRNA databases including GenBank, Ensembl, dbEST, and UniGene. Our search did not find these novel matriptase variants. We only found three AS transcripts of matriptase, which are formed as a result of an intron retention event (Ensembl IDs: ENST00000530532, ENST00000524718, and ENST00000530376). Furthermore, we did not detect the novel transcripts of matriptase in adjacent non-cancerous tissue from TCGA or in the transcriptome data available from the BodyMap 2.0 project, thus suggesting these variants are tumor associated.
qRT-PCR analysis confirms differential expression of novel matriptase transcripts in epithelial-derived tumors
To validate the expression of matriptase splice variants in epithelial tumors, we designed matriptase wild type or splice variant-specific probes for qRT-PCR (Material and methods). qRT-PCR was carried out on orthogonal panels of cell lines, and human primary and metastatic tumor tissues from ovarian, breast, lung, and bladder cancer and a panel of normal tissues. The normal panel included 48 healthy tissues (Supplementary Table S2) from normal ovary, lung, bladder, and breast. We measured changes in the gene expression by comparing the threshold cycle (Ct) of PCR product detection normalized against a reference gene transcript. The expression levels detected by qRT-PCR for wild-type matriptase and its splice variants showed that wild-type matriptase was the predominant transcript in both tumor and normal tissues (P < 0.0001). A1 transcript was overexpressed in tumor samples compared to normal tissues of ovarian (P < 0.0001) and lung panels (P = 0.0082). However, this did not apply to the bladder (P = 0.6414) and breast (P = 0.6466) panels. We also investigated the expression level of A3 splice variant in a panel of ovarian tissues and cell lines. A3 was overexpressed in ovarian tumors compared to normal samples (P = 0.0004). However, we observed lower expression of A3 transcript compared to A1 transcript in ovarian tumors (P = 0.0004).
We further tested the expression of matriptase splice variants in a panel of normal tissue samples including 48 normal tissues from across the human body. Both matriptase splice variants A1 and A3 showed higher expression in tumor samples compared to the normal tissue panel (P < 0.0001). In fact, the majority of tissues in the normal tissue panel did not express matriptase A1 and A3 transcript variants at all, while a small number showed a much lower expression compared to tumor samples (Fig. 4). That is, we detected the presence of the A1 and A3 transcripts in only 16 and 17 out of the 48 normal tissues in the normal tissue panel, respectively. The ∆Ct values and detailed figures from qRT-PCR experiments are available in Supplementary Figures S3–S11 and Supplementary Tables S2–S6.
Figure 4.

qRT-PCR validation. qRT-PCR was carried out on orthogonal panels of cell lines and human primary and metastatic tumor tissues from ovarian, breast, lung, and bladder cancer and a panel of normal tissues. Mann–Whitney t-test was used to determine significant differences in gene expression between groups. The resulting P-values are summarized below the x-axis. The x-axis labels from left to right are (1) wild type in normal ovary, (2) wild type in ovarian cancer, (3) A1 in normal ovary, (4) A1 in ovarian cancer, (5) A3 in normal ovary, (6) A3 in ovarian cancer, (7) wild type in normal tissue panel, (8) A1 in normal tissue panel, (9) A3 in normal tissue panel, (10) wild type in normal breast, (11) wild type in breast cancer, (12) A1 in normal breast, (13) A1 in breast cancer, (14) wild type in normal bladder, (15) wild type in bladder cancer, (16) A1 in normal bladder, (17) A1 in bladder cancer, (18) wild type in normal lung, (19) wild type in lung cancer, (20) A1 in normal lung, and (21) A1 in lung cancer. The y-axis is log scaled.
Matriptase splice variants can be translocated to the surface of transfected CHO cells
To address the question of whether matriptase A1 and A3 transcripts yield protein variants that are capable of being translocated to the cell surface, we transiently transfected CHO cells with cDNA encoding these genes followed by flow cytometric analysis of surface matriptase proteins (wild type, variant A1, and variant A3). For this experiment, we used a human anti-matriptase antibody that binds to the catalytic domain of all three matriptase variants and is not variant specific. Co-expression of the matriptase variants with HAI-1 resulted in a significant increase in the mean fluorescent intensity for wild type, variant A1, and variant A3 (P < 0.05; Fig. 5C–F), whereas expression of matriptase variants alone showed modest increases in surface expression (data not shown). So to verify that the recombinant proteins detected by flow cytometry were the expected molecular weight for each variant, matriptase variants were immunoprecipitated from transfected CHO cells using the same human anti-matriptase antibody and analyzed by Western blot (Fig. 5G). As observed in the flow cytometry experiment, endogenous matriptase was not detected in the elution from CHO cells transfected with the empty vector alone. In contrast, bands corresponding to the expected molecular weight for each variant were detected in the respective elutions. These results support the assertion that proteins corresponding to the expected molecular weight of matriptase variants A1 and A3 are trafficked to the cell surface of transiently transfected cells, despite the deletion of the LDLRA domains.
Discussion
AS is a widespread mechanism for the generation of diverse protein products and regulation of protein expression. Tumor cells exploit this mechanism to favor the malignant state.1,35,36 In the past decade, cancer-associated splice variants of genes that control mechanisms such as DNA damage and proliferation [EGFR, fibroblast growth factor receptor 3 (FGFR3), breast cancer 1 (BRCA1)], adhesion and invasion [CD44, macrophage stimulating 1 receptor (MST1R)], angiogenesis (VEGF), and apoptosis [B-cell lymphoma/leukemia 10 (BCL10), caspase 2 (CASP2)] have been reported.37 Among these, AS transcripts with altered protein structure localized to the cell surface are of particular interest as they represent potential biomarkers for discrimination between healthy and cancerous cells. That is, monoclonal antibodies can be produced to selectively target cancerous cells expressing such protein isoforms. An antibody against a tumor-associated surface-localized variant of EGFR (EGFRvIII) with exons 2–7 deleted has shown effective anti-tumor activity in preclinical studies,6 and is now in phase I clinical trials.
With the advent of massively parallel RNA-seq, the large-scale exploration of cancer-related changes at the stage of transcription and posttranscriptional splicing has the potential to determine many more tumor-associated or enriched targets. In the current study, we devised an AS-detection pipeline from high-throughput RNA-seq data. The AS-detection pipeline allowed us to mine large sets of tumor transcriptomes to identify novel tumor-associated AS variants. Most notably, we identified two novel tumor- associated splicing variants of matriptase through analysis of more than 2,200 tumor transcriptome data available from TCGA. The variant designated A1 has an in-frame skipping of exon 12, and variant A3 is generated as a result of skipping exon 14. Our analysis revealed a high frequency of these variants across epithelial-derived tumors, which were absent or expressed at extremely low levels in transcriptomes derived from normal tissues. Novel matriptase isoforms appear to form 2–8% of the overall matriptase gene expression in tumor samples, with wild type being the dominantly expressed form (Supplementary Figs. S1, S2, and S12). qRT-PCR confirmed mRNA expression of matriptase variants, and revealed differential higher expression of variant A1 in ovarian and lung tumor tissues and cell lines compared to low or no expression in normal samples. Similarly, the A3 transcript was overexpressed in ovarian tumor tissues and cells. We then investigated variants A1 and A3 in cDNA panels derived from 48 healthy tissue types from across the body, such as brain, heart, kidney, and lung. We observed no mRNA expression of matriptase variants in more than two-thirds of normal samples and a low level of expression in the remainder. Sequence analysis indicates that the transcript variants can produce two fully functional ORFs. Our immunoprecipitation results show that these two novel proteins are being produced in CHO cells transiently transfected with cDNA encoding matriptase splice variants. With matriptase localized to the cell surface, there is a possibility that these novel isoforms of matriptase are also present on the cell surface. We tested this hypothesis by performing flow cytometry on CHO cells expressing these recombinant proteins. This analysis demonstrated the presence of these novel proteins on the surface of CHO cells, where wild-type matriptase surface expression predominated followed by variant A1 and then variant A3. Thus, protein expression of matriptase splice variants on the surface of CHO cells supports the notion that A1 and A3 protein products can localize on the surface of tumor cells as well.
The LDL receptor class A domain is an ~40-amino-acid-long structure. The prototype structure of the LDLRA domain is found in the LDL receptor itself, which contains seven such domains. The crystal structure of the fifth LDLRA domain in the LDL receptor revealed that this domain contains six amino acids that bind calcium in an octahedral arrangement (calcium cage).38 Point mutations at critical residues in this calcium cage have been found to potently inhibit the LDLRA ligand binding.39 Oberst et al showed that mutations in the Ca2+-binding motifs of any or all of the four LDLRA domains of matriptase prevent its activation.20 Interestingly, however, the complete deletion of all four LDLRA domains allows constitutive activation of this enzyme. Additional experiments are required to demonstrate the impact of deleting LDLRA1 and LDLRA3 domains as observed in the A1 and A3 variants. Although these two deletions may have variable effects on matriptase activity, our results demonstrate that they do not affect the ability of their protein products to form a complex with HAI-1 and traffic to the cell surface.
We identified no splice-site mutation associated with skipping exons 12 and 14 of matriptase in TCGA mutation analysis data derived from matching whole-exome sequencing dataset. We predicted RNA-binding proteins (RBPs) that possibly bind to matriptase mRNA using RBPmap online web server40 (Supplementary Table S7) and compared the expression of these RBPs according to the expression status of matriptase variants. This analysis revealed significant change (P < 0.05) in the expression of several RBPs (Supplementary Tables S8–S17). We found no correlation between expression of matriptase variants and patient’s survival time (P > 0.05, Supplementary Figs. S13–S14), age, tumor size, tumor clinical stage, and histological grade (P > 0.05, Supplementary Tables S18–S24) where the data were available. However, the high frequency of matriptase novel splice variants among patients with epithelial tumors and low or no occurrence in normal tissue, as well as cell surface localization could offer a potential use in selective therapy of cancer for these variants.
In the current study, we devised an AS-detection pipeline and performed our discovery analysis on a large number of tumors from TCGA. Our analysis revealed two novel tumor-associated splice variants of matriptase, which were confirmed in an orthogonal set of tumor tissues and cell lines. This approach highlights high frequency of matriptase variants among patients with epithelial-derived tumors as well as low or no occurrence in normal tissue. In addition to gene expression data, our flow cytometric analysis confirmed protein expression of both matriptase variants on the surface of CHO cells, suggesting matriptase variants as potential biomarkers of tumor cells. Clinical validation would prove valuable in confirming the utility of matriptase variants for therapeutic use.
Supplementary Data
Supplementary_Method.doc. Includes additional details of RNA-seq transcript quantification and qRT-PCR experiment.
Supplementary_Figures.doc. Includes supporting RNA-seq and qRT-PCR expression plots as well as survival analysis.
Supplementary_Tables.doc. Includes data from qRT-PCR, RBP, and correlation with clinicopathological data analysis.
Supplementary_Sequences.doc. Includes mRNA and protein sequences of wildtype and novel alternatively spliced transcripts of matriptase.
Acknowledgments
The results published here are in whole or part based upon data generated by TCGA pilot project established by national cancer institute (NCI) and national human genome research institute (NHGRI). The authors would like to thank TCGA group for making these data publically available. Information about TCGA can be found at http://cancergenome.nih.gov. They would also like to thank Mitacs-Accelerate for a PhD fellowship to DD. This work was carried out in facilities supported by the Centre for Drug Research and Development (CDRD), CDRD Ventures Inc. (CVI), and Genome Sciences Centre.
Footnotes
ACADEMIC EDITOR: JT Efird, Editor in Chief
FUNDING: This work was supported by the Genome British Columbia Strategic Opportunities Fund (grant number SOF5-014-R16) and a MITACS-Accelerate PhD fellowship to DD (award number IT02518). The authors confirm that the funder had no influence over the study design, content of the article, or selection of this journal.
COMPETING INTERESTS: DD, JSB and SJMJ have filed a patent application relevant to the current study, relating to matriptase variants associated with tumors. JSB, EMD, RD, LY, PJB, RDS and BJH are employed at CDRD. CDRD is a non-profit company focused on the identification of genetic alterations in human cancer for diagnostic and therapeutic purposes. Other authors disclose no competing interests.
Paper subject to independent expert blind peer review by minimum of two reviewers. All editorial decisions made by independent academic editor. Upon submission manuscript was subject to anti-plagiarism scanning. Prior to publication all authors have given signed confirmation of agreement to article publication and compliance with all applicable ethical and legal requirements, including the accuracy of author and contributor information, disclosure of competing interests and funding sources, compliance with ethical requirements relating to human and animal study participants, and compliance with any copyright requirements of third parties. This journal is a member of the Committee on Publication Ethics (COPE).
Author Contributions
Conceived and designed the experiments: DD, JSB, SJMJ. Analyzed the data: DD, RDS, LY, PJB, BJH, EMD, AHM, RD, JA. Wrote the first draft of the manuscript: DD. Contributed to the writing of the manuscript: DD, RDS, LY, PJB, BJH, AHM. Agreed with manuscript results and conclusions: DD, RDS, LY, PJB, BJH, EMD, AHM, RD, JA, JSB, SJMJ. Jointly developed the structure and arguments for the paper: DD, RDS, SJMJ. Made critical revisions and approved the final version: DD, RDS, LY, PJB, BJH, EMD, AHM, RD, JA, JSB, SJMJ. All authors reviewed and approved the final manuscript.
REFERENCES
- 1.Venables JP. Aberrant and alternative splicing in cancer. Cancer Res. 2004;64(21):7647–54. doi: 10.1158/0008-5472.CAN-04-1910. [DOI] [PubMed] [Google Scholar]
- 2.Pajares MJ, Ezponda T, Catena R, Calvo A, Pio R, Montuenga LM. Alternative splicing: an emerging topic in molecular and clinical oncology. Lancet Oncol. 2007;8(4):349–57. doi: 10.1016/S1470-2045(07)70104-3. [DOI] [PubMed] [Google Scholar]
- 3.Venables JP, Klinck R, Bramard A, et al. Identification of alternative splicing markers for breast cancer. Cancer Res. 2008;68(22):9525–31. doi: 10.1158/0008-5472.CAN-08-1769. [DOI] [PubMed] [Google Scholar]
- 4.Griffith M, Mwenifumbo JC, Cheung PY, et al. Novel mRNA isoforms and mutations of uridine monophosphate synthetase and 5-fluorouracil resistance in colorectal cancer. Pharmacogenomics J. 2012;13:148–58. doi: 10.1038/tpj.2011.65. [DOI] [PubMed] [Google Scholar]
- 5.Heider KH, Kuthan H, Stehle G, Munzert G. CD44v6: a target for antibody-based cancer therapy. Cancer Immunol Immunother. 2004;53(7):567–79. doi: 10.1007/s00262-003-0494-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sampson JH, Archer GE, Mitchell DA, Heimberger AB, Bigner DD. Tumor-specific immunotherapy targeting the EGFRvIII mutation in patients with malignant glioma. Semin Immunol. 2008;20(5):267–75. doi: 10.1016/j.smim.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Simpson JT, Wong K, Jackman SD, Schein JE, Jones SJ, Birol I. ABySS: a parallel assembler for short read sequence data. Genome Res. 2009;19(6):1117–23. doi: 10.1101/gr.089532.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Birol I, Jackman SD, Nielsen CB, et al. De novo transcriptome assembly with ABySS. Bioinformatics. 2009;25(21):2872–7. doi: 10.1093/bioinformatics/btp367. [DOI] [PubMed] [Google Scholar]
- 9.Lin CY, Wang JK, Torri J, Dou L, Sang QA, Dickson RB. Characterization of a novel, membrane-bound, 80-kDa matrix-degrading protease from human breast cancer cells. monoclonal antibody production, isolation, and localization. J Biol Chem. 1997;272(14):9147–52. [PubMed] [Google Scholar]
- 10.Tanimoto H, Underwood LJ, Wang Y, Shigemasa K, Parmley TH, O’Brien TJ. Ovarian tumor cells express a transmembrane serine protease: a potential candidate for early diagnosis and therapeutic intervention. Tumour Biol. 2001;22(2):104–14. doi: 10.1159/000050604. [DOI] [PubMed] [Google Scholar]
- 11.Lee MS, Tseng IC, Wang Y, et al. Autoactivation of matriptase in vitro: requirement for biomembrane and LDL receptor domain. Am J Physiol Cell Physiol. 2007;293(1):95–105. doi: 10.1152/ajpcell.00611.2006. [DOI] [PubMed] [Google Scholar]
- 12.Lee SL, Dickson RB, Lin CY. Activation of hepatocyte growth factor and urokinase/plasminogen activator by matriptase, an epithelial membrane serine protease. J Biol Chem. 2000;275(47):36720–5. doi: 10.1074/jbc.M007802200. [DOI] [PubMed] [Google Scholar]
- 13.Takeuchi T, Harris JL, Huang W, Yan KW, Coughlin SR, Craik CS. Cellular localization of membrane-type serine protease 1 and identification of protease-activated receptor-2 and single-chain urokinase-type plasminogen activator as substrates. J Biol Chem. 2000;275(34):26333–42. doi: 10.1074/jbc.M002941200. [DOI] [PubMed] [Google Scholar]
- 14.Unterholzner L, Keating SE, Baran M, et al. IFI16 is an innate immune sensor for intracellular DNA. Nat Immunol. 2010;11(11):997–1004. doi: 10.1038/ni.1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kang JY, Dolled-Filhart M, Ocal IT, et al. Tissue microarray analysis of hepatocyte growth factor/met pathway components reveals a role for met, matriptase, and hepatocyte growth factor activator inhibitor 1 in the progression of node-negative breast cancer. Cancer Res. 2003;63(5):1101–5. [PubMed] [Google Scholar]
- 16.Matsumoto K, Nakamura T. Emerging multipotent aspects of hepatocyte growth factor. J Biochem. 1996;119(4):591–600. doi: 10.1093/oxfordjournals.jbchem.a021283. [DOI] [PubMed] [Google Scholar]
- 17.Trusolino L, Comoglio PM. Scatter-factor and semaphorin receptors: cell signalling for invasive growth. Nat Rev Cancer. 2002;2(4):289–300. doi: 10.1038/nrc779. [DOI] [PubMed] [Google Scholar]
- 18.Sidenius N, Blasi F. The urokinase plasminogen activator system in cancer: recent advances and implication for prognosis and therapy. Cancer Metastasis Rev. 2003;22(2–3):205–22. doi: 10.1023/a:1023099415940. [DOI] [PubMed] [Google Scholar]
- 19.Shimomura T, Denda K, Kitamura A, et al. Hepatocyte growth factor activator inhibitor, a novel Kunitz-type serine protease inhibitor. J Biol Chem. 1997;272(10):6370–6. doi: 10.1074/jbc.272.10.6370. [DOI] [PubMed] [Google Scholar]
- 20.Oberst MD, Williams CA, Dickson RB, Johnson MD, Lin CY. The activation of matriptase requires its noncatalytic domains, serine protease domain, and its cognate inhibitor. J Biol Chem. 2003;278(29):26773–9. doi: 10.1074/jbc.M304282200. [DOI] [PubMed] [Google Scholar]
- 21.Oberst MD, Chen LY, Kiyomiya K, et al. HAI-1 regulates activation and expression of matriptase, a membrane-bound serine protease. Am J Physiol Cell Physiol. 2005;289(2):462–70. doi: 10.1152/ajpcell.00076.2005. [DOI] [PubMed] [Google Scholar]
- 22.Oberst MD, Singh B, Ozdemirli M, Dickson RB, Johnson MD, Lin CY. Characterization of matriptase expression in normal human tissues. J Histochem Cytochem. 2003;51(8):1017–25. doi: 10.1177/002215540305100805. [DOI] [PubMed] [Google Scholar]
- 23.List K, Haudenschild CC, Szabo R, et al. Matriptase/MT-SP1 is required for postnatal survival, epidermal barrier function, hair follicle development, and thymic homeostasis. Oncogene. 2002;21(23):3765–79. doi: 10.1038/sj.onc.1205502. [DOI] [PubMed] [Google Scholar]
- 24.List K, Szabo R, Molinolo A, et al. Deregulated matriptase causes ras-independent multistage carcinogenesis and promotes ras-mediated malignant transformation. Genes Dev. 2005;19(16):1934–50. doi: 10.1101/gad.1300705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Oberst MD, Johnson MD, Dickson RB, et al. Expression of the serine protease matriptase and its inhibitor HAI-1 in epithelial ovarian cancer: correlation with clinical outcome and tumor clinicopathological parameters. Clin Cancer Res. 2002;8(4):1101–7. [PubMed] [Google Scholar]
- 26.Wu SR, Cheng TS, Chen WC, et al. Matriptase is involved in ErbB-2-induced prostate cancer cell invasion. Am J Pathol. 2010;177(6):3145–58. doi: 10.2353/ajpath.2010.100228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cancer Genome Atlas Research Network Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474(7353):609–15. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li H, Handsaker B, Wysoker A, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25(16):2078–9. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li YL, Ye F, Hu Y, Lu WG, Xie X. Identification of suitable reference genes for gene expression studies of human serous ovarian cancer by real-time polymerase chain reaction. Anal Biochem. 2009;394(1):110–6. doi: 10.1016/j.ab.2009.07.022. [DOI] [PubMed] [Google Scholar]
- 30.Beillard E, Pallisgaard N, van derVelden VH, et al. Evaluation of candidate control genes for diagnosis and residual disease detection in leukemic patients using ‘real-time’ quantitative reverse-transcriptase polymerase chain reaction (RQ-PCR)–a Europe against cancer program. Leukemia. 2003;17(12):2474–86. doi: 10.1038/sj.leu.2403136. [DOI] [PubMed] [Google Scholar]
- 31.Swayze RD, Braun AP. A catalytically inactive mutant of type I cGMP- dependent protein kinase prevents enhancement of large conductance, calcium- sensitive K+ channel by sodium nitroprusside and cGMP. J Biol Chem. 2001;276(23):19729–37. doi: 10.1074/jbc.M005711200. [DOI] [PubMed] [Google Scholar]
- 32.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227(5259):680–5. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 33.Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci U S A. 1979;76(9):4350–4. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Koscielny G, Le Texier V, Gopalakrishnan C, et al. ASTD: the alternative splicing and transcript diversity database. Genomics. 2009;93(3):213–20. doi: 10.1016/j.ygeno.2008.11.003. [DOI] [PubMed] [Google Scholar]
- 35.Ghigna C, Valacca C, Biamonti G. Alternative splicing and tumor progression. Curr Genomics. 2008;9(8):556–70. doi: 10.2174/138920208786847971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang J, Manley JL. Misregulation of pre-mRNA alternative splicing in cancer. Cancer Discov. 2013;3(11):1228–37. doi: 10.1158/2159-8290.CD-13-0253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brinkman BM. Splice variants as cancer biomarkers. Clin Biochem. 2004;37(7):584–94. doi: 10.1016/j.clinbiochem.2004.05.015. [DOI] [PubMed] [Google Scholar]
- 38.Fass D, Blacklow S, Kim PS, Berger JM. Molecular basis of familial hypercholesterolaemia from structure of LDL receptor module. Nature. 1997;388(6643):691–3. doi: 10.1038/41798. [DOI] [PubMed] [Google Scholar]
- 39.Esser V, Limbird LE, Brown MS, Goldstein JL, Russell DW. Mutational analysis of the ligand binding domain of the low density lipoprotein receptor. J Biol Chem. 1988;263(26):13282–90. [PubMed] [Google Scholar]
- 40.Paz I, Kosti I, Ares M, Jr, Cline M, Mandel-Gutfreund Y. RBPmap: a web server for mapping binding sites of RNA-binding proteins. Nucleic Acids Res. 2014;42(Web Server issue):W361–7. doi: 10.1093/nar/gku406. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary_Method.doc. Includes additional details of RNA-seq transcript quantification and qRT-PCR experiment.
Supplementary_Figures.doc. Includes supporting RNA-seq and qRT-PCR expression plots as well as survival analysis.
Supplementary_Tables.doc. Includes data from qRT-PCR, RBP, and correlation with clinicopathological data analysis.
Supplementary_Sequences.doc. Includes mRNA and protein sequences of wildtype and novel alternatively spliced transcripts of matriptase.