

Abstract
The Na+–Ca2+ exchanger (NCX) appears to play an important role in the regulation of the high K+-evoked Ca2+ transient in putative nociceptive dorsal root ganglion (DRG) neurons. The purpose of the present study was to (1) characterize the properties of NCX activity in subpopulations of DRG neurons, (2) identify the isoform(s) underlying NCX activity, and (3) begin to assess the function of the isoform(s) in vivo. In retrogradely labelled neurons from the glabrous skin of adult male Sprague–Dawley rats, NCX activity, as assessed with fura-2-based microfluorimetry, was only detected in putative nociceptive IB4+ neurons. There were two modes of NCX activity: one was evoked in response to relatively large and long lasting (∼325 nm for >12 s) increases in the concentration of intracellular Ca2+ ([Ca2+]i), and a second was active at resting [Ca2+]i > ∼150 nm. There also were two modes of evoked activity: one that decayed relatively rapidly (<5 min) and a second that persisted (>10 min). Whereas mRNA encoding all three NCX isoforms (NCX1–3) was detected in putative nociceptive cutaneous neurons with single cell PCR, pharmacological analysis and small interfering RNA (siRNA) knockdown of each isoform in vivo suggested that NCX2 and 3 were responsible for NCX activity. Western blot analyses suggested that NCX isoforms were differentially distributed within sensory neurons. Functional assays of excitability, action potential propagation, and nociceptive behaviour suggest NCX activity has little influence on excitability per se, but instead influences axonal conduction velocity, resting membrane potential, and nociceptive threshold. Together these results indicate that the function of NCX in the regulation of [Ca2+]i in putative nociceptive neurons may be unique relative to other cells in which these exchanger isoforms have been characterized and it has the potential to influence sensory neuron properties at multiple levels.
Introduction
We have previously demonstrated marked differences among subpopulations of dorsal root ganglia (DRG) neurons defined by cell body size, IB4 binding and capsaicin sensitivity with respect to the magnitude and decay of depolarization-evoked Ca2+ transients that were associated with differences in the relative contribution of many of the core components of the Ca2+ signalling toolkit (Berridge et al. 2000). These included voltage-gated Ca2+ channels, ryanodine receptor-mediated Ca2+-induced Ca2+ release, internal Ca2+ store operation mediated by sarco-endoplasmic reticulum ATPase (SERCA), mitochondria, and store-operated Ca2+ entry, as well as extrusion mechanisms such as the plasma membrane Ca2+ ATPase (PMCA) and the Na+–Ca2+ exchanger (NCX).
There are several reasons to focus on NCX for further analysis in DRG neurons. First, there are conflicting results in the literature regarding the extent to which NCX activity is detectable in sensory neurons. Whereas our data (Lu et al. 2006) and those of Verdru and colleagues (Verdru et al. 1997) suggest that the duration of depolarization-evoked Ca2+ transient in sensory neurons is determined, at least in part, by NCX activity, others have concluded that there is little if any detectable NCX activity in either the sensory neuron cell body (Werth & Thayer, 1994), central (Wan et al. 2012; Shutov et al. 2013) or peripheral terminals (Gover et al. 2007). Second, there has been no systematic analysis of the biophysical properties of NCX in sensory neurons. This is important as available evidence from other systems suggests that, while NCX has a higher extrusion rate than the PMCA, it should only be active in the presence of higher concentrations of intracellular [Ca2+]i due to a lower affinity for Ca2+ (Blaustein & Lederer, 1999; DiPolo & Beauge, 2006). This property could account for the apparent absence of NCX activity in sensory neurons, as the Ca2+ transients in the ‘negative’ studies were relatively small. Biophysical characterization of NCX activity in sensory neurons is also important in light of recent evidence that NCX can function in ‘reverse mode’ (Kuroda et al. 2013), a mode that has been suggested to contribute to axon injury observed in some forms of peripheral neuropathy (Ma, 2013). Third, there is not only evidence of the differential distribution of NCX activity among subpopulations of sensory neurons (we were only able to detect NCX activity in putative nociceptive DRG neurons; Lu et al. 2006), there is evidence of the differential distribution of NCX isoforms among sensory neurons, where NCX2, the only isoform detectable in sensory neurons, was restricted to those with a small cell body diameter (Persson et al. 2010). Thus, the selective distribution of NCX isoforms may also contribute to the conflicting results in the literature on NCX activity in sensory neurons. Finally, with the exception of data on the duration of evoked Ca2+ transients and recent evidence suggesting that NCX may act in reverse mode to contribute to peripheral nerve injury (Persson et al. 2013), there are no data on how NCX activity influences the normal function of sensory neurons. Therefore, the present study was designed to begin to address this relative dearth of information concerning the properties, distribution and function of NCX isoforms in sensory neurons.
A combination of approaches, ranging from the microfluorometric and polymerase chain reaction (PCR) analysis of isolated retrogradely labelled sensory neurons in vitro to behavioural analysis following targeted knockdown of NCX isoforms, were employed to address these issues. Our results suggest that NCX, more specifically NCX2 and 3, play a significant role in the regulation of decay of the evoked change in [Ca2+]i at the level of the cell body. Assays of NCX function in the context of excitability, action potential propagation, and nociceptive behaviour suggest NCX, in particular NCX3 activity, influences axonal [Ca2+]i levels, resting membrane potential, and nociceptive threshold via mechanisms probably secondary to the regulation of [Ca2+]i in axons and terminals.
Methods
Ethical approval
Adult male Sprague–Dawley rats (Harlan, 220–300 g) were used for all experiments. Animals were housed two per cage in a temperature and humidity controlled animal facility on a 12 h:12 h light–dark schedule with food and water freely available. All procedures were approved by the University of Pittsburgh Institutional Animal Care and Use Committee and performed in accordance with National Institutes of Health guidelines as well as the principles of United Kingdom regulation for the use of laboratory animals in research.
Tissue labelling
Fourteen to 17 days prior to tissue harvest, the retrograde tracer 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbo-cyanine perchlorate (DiI, Invitrogen, Carlsbad, CA, USA) was injected into the glabrous skin of the hindpaw to label cutaneous afferents. The tracer was dissolved at 170 mg ml−1 in dimethylsulfoxide (DMSO, diluted 1:10 in 0.9% sterile saline, and injected in 3–5 subcutaneous sites using a 30 g needle for a total volume of 10 μl per hindpaw under isoflurane (Abbott Laboratories, North Chicago, IL, USA) anaesthesia.
Tissue collection and isolation
Prior to tissue removal, rats were deeply anaesthetized with an intraperitoneal injection of a cocktail containing ketamine (55 mg kg−1), xylazine (5.5 mg kg−1) and acepromazine (1.1 mg kg−1). Following tissue removal, deeply anaesthetized rats were killed by cervical dislocation and/or bi-lateral thoracotomy. For studies involving isolated sensory neurons, the L4–L5 DRG were removed ipsilateral to labelling. Ganglia were enzymatically treated, mechanically dissociated, and neurons plated on laminin (Invitrogen; 1 mg ml−1) and poly-l-ornithine-coated (Sigma-Aldrich, St Louis, MO, USA; 1 mg ml−1) glass cover slips as previously described (Lu et al. 2006). All subsequent experiments were preformed within 8 h of tissue harvest and only neurons containing the retrograde label DiI were studied.
For isolated sciatic nerve recordings, rat sciatic nerves were harvested and prepared for compound action potential (CAP) recording as previously described (Yilmaz-Rastoder et al. 2012). Briefly, sciatic nerves (∼30 mm) were quickly dissected and transferred to a dish containing ice-cold Locke's solution of the following composition (in mm): 136 NaCl, 5.6 KCl, 14.3 NaHCO3, 1.2 NaH2PO4, 2.2 CaCl2, 1.2 MgCl2, 11 glucose, equilibrated continuously with 95% O2–5% CO2, pH 7.2–7.4. In ice-cold oxygenated Locke's solution nerves were trimmed of excess connective under a dissecting microscope. All subsequent experiments were preformed within 24 h of tissue harvest.
Ca2+ imaging
Neurons were incubated with 2.5 μm Ca2+ indicator fura-2 AM ester with 0.025 % Pluronic F-127 for 20 min at room temperature. Neurons were then labelled with FITC-conjugated IB4 (10 μg ml−1) for 10 min at room temperature. Labelled neurons were placed in a recording chamber and continuously superfused with normal bath solution (mm: 130 NaCl, 3 KCl, 2.5 CaCl2, 0.6 MgCl2, 10 Hepes, 10 glucose, pH 7.4, osmolality 325 mosmol kg–1) or a ‘Na+ free’ bath solution (mm: 130 Choline-Cl or LiCl, 3 KCl, 10 MgCl2, 2 EGTA, 10 Hepes, 10 glucose, pH 7.4, osmolality 325 mosmol kg–1). A Na+-free solution was used to block NCX activity by preventing NCX from fulfilling the Na+ binding requirement for exchange activity (Cook et al. 1998). Fluorescence data were acquired on a PC running Metafluor software (Molecular Devices, Sunnyvale, CA, USA) via a CCD camera (Roper Scientific; model RTE/CCD 1300). The ratio (R) of fluorescence emission (510 nm) in response to 340/380 nm excitation (controlled by a lambda 10–2 filter changer; Sutter Instruments, Novato, CA, USA) was acquired at 1 Hz during drug application. All drugs were applied through a computer-controlled peizo-driven perfusion system (switching time <20 ms; Warner Instruments, Hamden, CT, USA, Fast-Step Model SF-77B). [Ca2+]i was determined from fura-2 ratio following in situ calibration experiment as described in detail previously (Grynkiewicz et al. 1985; Kao, 1994; Scheff et al. 2013).
Patch-clamp electrophysiology
Gramicidin-perforated patch-clamp experiments were carried out using a HEKA EPC9 amplifier (HEKA Elektronik, Lambrecht/Rhineland-Pfalz, Germany). Glass electrodes (1–4 MΩ) were filled with (mm) 110 potassium methanesulfonate, 30 KCl, 5 NaCl, 1 CaCl2, 2 MgCl2, 10 Hepes, 11 EGTA, 2 Mg-ATP, and 1 Li-GTP for current-clamp excitability recordings. The pH was adjusted with Tris-base to 7.2 and osmolality was adjusted with sucrose to 320 mosmol kg–1.
To obtain whole cell access for patch-clamp experiments, a stock solution of gramicidin (1.5 mg (100 μl)–1, Sigma-Aldrich) was prepared in DMSO. This was diluted with electrode solution at 1:300 to give a final concentration of 50 μg ml−1. The gramicidin-containing electrode solution was vortexed for >15 s. No filtering was applied. The tip of the electrode was loaded with a small volume of gramicidin-free electrode solution in order to avoid interference of the antibiotic with seal formation. Gramicidin-containing electrode solution was subsequently back-loaded. The progress of perforation was monitored from a holding potential of −60 mV with a 10 ms step to −65 mV. Experiments were not started until access resistance was <7 MΩ.
Neuronal excitability was assessed in current clamp (Scheff & Gold, 2011) before and after a 4 s voltage step from −60 mV to 0 mV to drive an increase in [Ca2+]i, and consequently an increase in NCX activity. Resulting changes in [Ca2+]i were simultaneously monitored with fura-2 AM-based microfluorimetry. Series resistance compensation (>70%) was employed for all voltage-clamp recordings. Five distinct measures of excitability were used: the emergence of spontaneous activity, action potential (AP) threshold, rheobase, the response to suprathreshold current injection, and following frequency. Spontaneous activity was assessed at resting membrane potential (Vrest) for 30 s before and up to 90 s after the application of Na+-free bath. Neurons firing more than 1 AP during this period were considered spontaneously active. The subsequent three measures were determined with a 750 ms depolarizing square-pulse current injection. AP threshold was defined as the greatest depolarization reached before spike generation in response to depolarizing current injections. Rheobase was defined as the smallest amount of current needed to evoke a single AP. The result to suprathreshold current injection was determined by counting the number of action potentials evoked in response to current 2× and 3× rheobase. To mitigate the potential impact of cell body size on rheobase and facilitate comparisons between neurons, values were normalized with respect to membrane capacitance. Following frequency was assessed at 10, 20, and 40 Hz with 1 ms pulses 1.5-fold above threshold (determined as with rheobase but with the 1 ms pulse duration).
Single cell reverse transcriptase polymerase chain reaction (RT-PCR)
Isolated neurons were prepared for single cell RT-PCR experiments in a manner identical to that used for patch-clamp and microfluorimetry experiments. Single neurons were collected with large bore (30 μm) glass pipettes and expelled into reaction tube for subsequent cDNA synthesis via methods identical to those described previously (Nealen et al. 2003; Zhang et al. 2012). The cDNA generated from each neuron was used as a template for subsequent PCR reactions. To confirm successful cell collection and cDNA synthesis, 0.5 μl of the template cDNA was used for the amplification of the housekeeping gene cyclophilin. Negative controls for each round of cell collection included two neurons in which no reverse transcriptase was added to the reaction mixture and two tubes in which all other procedures were performed as if a neuron was collected, except no neuron was collected. Nested PCR primers (Table1) were used in two successive rounds of PCR amplification to assess the expression of NCX 1, 2 and 3, to increase the sensitivity and specificity of the PCR reaction. The ‘outer’ set of primers was used in the first round of amplification and 2 μl of this reaction product was used for a second round of amplification with the ‘inner’ set of primers. Thirty-five cycles were used for both rounds of amplification. Following agarose gel electrophoresis, PCR reaction products were visualized with ethidium bromide.
Table 1.
PCR primers
| Target | Accession # | Position | Start | Sequence |
|---|---|---|---|---|
| NCX1 | NM_001270778.1 | Outer | 1771 | F: AGGGGAGGACTTTGAGGACA |
| 2075 | R: TCCTCCTCCTCTTTGCTGGT | |||
| Inner | 1779 | F: GGAGGACTTTGAGGACACCTG | ||
| 2068 | R: TCCTCTTTGCTGGTCAGTGG | |||
| NCX2 | NM_078619.1 | Outer | 1954 | F: TTCTTGGGGAGAACTGTCGC |
| 2379 | R: GCAAACGTGTCAGGGATGGA | |||
| Inner | 2065 | F: CACTCATGGAGGGAGCAGTTT | ||
| 2270 | R: AGGATGCAGACACCAAAGCA | |||
| NCX3 | NM_078620.1 | Outer | 2681 | F: TGCCCTTGGTGAACCGAAAT |
| 3263 | R: CATACACATCCTGCAGGGCA | |||
| Inner | 2763 | F: ATGGAGGAAGAGGAGGCCAA | ||
| 3263 | R: CATACACATCCTGCAGGGCA |
Western blot
L4 and L5 DRG were homogenized with Teflon tube and mortar for less than 10 strokes in 400 μl of ice cold radioimmunoprecipitation assay (RIPA, Pierce Thermo Scientific, Rockford, IL, USA) buffer supplied with protease inhibitors (aprotinin, leupeptin, pepstatin, E-64, trypsin inhibitor, and phenylmethanesulfonyl fluoride (PMSF), all at a final concentration of 2 ng ml−1 except PMSF, which was used at a final concentration of 1 mm). All protease inhibitors were obtained from Sigma-Aldrich. Lysates were collected in 0.5-ml tubes. Teflon tubes were rinsed with RIPA buffer and the solutions were combined with the lysates previously collected. Lysates were centrifuged for 5 min at 10,000 rpm (9600 g) at 4°C. Protein concentration was determined via bicinchoninic acid (BCA) protein assay using a BCA assay kit (Thermo Scientific). Lysates were then mixed with Laemmli buffer (2×, 400 μl + 100 μl β-mercaptoethanol) and boiled for 5 min before loading. Protein (30 μg) from one animal was then loaded per lane and separated on a 7 % SDS–PAGE gel and transferred to nitrocellulose membrane. Membranes were blocked with 5% milk for 1 h at room temperature and then incubated with primary antibody at 4°C overnight (1:100 for R3F1 (anti-NCX1),1:200 for W1C3 (anti-NCX2), and 1:100 for anti-NCX3 (cat no. LS-B5775, LifeSpan Biosciences, Inc., Seattle, WA, USA), diluted with 5% milk–Tris-buffered saline with Tween 20 (TBST, Sigma-Aldrich). The blots were washed and then incubated with peroxidase-conjugated secondary antibody (1:2000 in 5% milk–TBST, Jackson ImmunoResearch Laboratories Inc. West Grove, PA, USA) for an hour at room temperature. An ECL kit (Amersham Biosciences, Piscataway, NJ, USA) was used for detection of immunoreactivity, luminescence data were collected on an LAS3000 imager (Fujifilm Inc., Japan), and analysed with ImageJ (NIH). Both antibodies used to detect NCX1 and NCX2 were gifts from Dr Kenneth Philipson of University of California Los Angeles. Both antibodies have been characterized by Dr Philipson and colleagues in heterologous expression systems and hippocampal cultures, where R3F1 (anti-NCX1) detects a prominent band at 120 kDa and minor bands at 70 kDa and 50 kDa, and W1C3 (anti-NCX2) detects only one prominent band at 60 kDa. There appears to be no cross-reactivity between isoforms (Thurneysen et al. 2002b; Papa et al. 2003). Furthermore, the specificity of the anti-NCX2 antibody was confirmed in NCX2 null mutant mice (Jeon et al. 2003).
Compound action potential (CAP) recordings
Isolated sciatic nerve recording was performed at room temperature. The isolated nerve was mounted in a recording chamber such that the distal end was laid over two platinum stimulating electrodes immersed in mineral oil and isolated from the central portion of the nerve with a grease-gap. A glass suction electrode connected to a differential preamplifier (WPI model DAM-80, Sarasota, FL, USA) was used to isolate the central end of the nerve. The remainder of the nerve (∼15–20 mm) was superfused continuously (2–5 ml min−1) with oxygenated Locke's solution with and without drugs delivered via a gravity-driven perfusion system. CAPs were evoked with electrical pulses 0.2–0.5 ms in duration applied at 0.1 Hz unless otherwise indicated, where the stimulus amplitude was ∼2× that needed to evoke a maximal amplitude C-fibre component of the CAP. CAP data were filtered at 2 kHz and sampled at 20 kHz via a CED 1401 Micro A/D converter, and acquired and analysed using CED Spike 2 version 5 for MS Windows (CED, Cambridge, UK). CAP data were rectified and the average of six consecutive CAPs, were integrated to quantify A- and C-fibre components as area under the curve (AUC). The A-fibre deflection of the CAP (A-wave) was easily distinguished from that associated with the C-fibre deflection (C-wave) because of the time delay between the arrivals of the two waves at the recording electrode.
siRNA and IB4–saporin injections
Slc8a ON-TARGETplus SMARTpool siRNAs (Thermo Scientific, Pittsburgh, PA, USA) were tested for their ability to knock down expression of individual isoforms of NCX in cultured L4-L5 DRG neurons where the extent of knockdown was assessed with PCR. Catalogue numbers for each isoform are as follows: NCX1 (LQ-091320-02), NCX2 (LQ-094819-02), NCX3 (LQ-094821-02). The siRNA sequence that produced the largest decrease in the expression of the targeted isoform was chosen for sciatic nerve injection with the transfection agent, lipofectamine (Invitrogen). At least 7–10 days after DiI injection, rats were deeply anaesthetized with isoflurane. The hair over the back of the thigh was shaved and blunt dissection was used to separate the hamstring muscle exposing the sciatic nerve. A subfasicular injection of 4–6 μl 1:1 lipofectamine to NCX-targeted siRNA or control siRNA was made using a 33 g needle. For Ca2+ imaging experiments, DRG were isolated as described above 6 days after siRNA injection. The extent of the knockdown was assessed with Western blot analysis of the sciatic nerve.
Methodology for the IB4–saporin and siRNA co-injections was adapted from Vulchanova et al. (2001). Under deep anaesthesia, the left sciatic nerve was exposed and a subfasicular injection of 10 μl consisting of either a mixture of 5 μl control or NCX3 siRNA (1:1 lipofectamine to siRNA) and 5 μl IB4-conjugated saporin (400 μg ml−1) or unconjugated IB4 and saporin (2.5 μl IB4 (280 μg ml−1, 71%) + 2.5 μl saporin (120 μg ml−1, 29%) was slowly infused using a 33 g needle at a rate of 10 μl min−1. IB4+ fibre loss was confirmed using IB4–FITC (Sigma-Aldrich) staining of 25 μm sections of the spinal cord lumbar enlargement.
Nociceptive behaviour
Testing for each group of rats was initiated at the same time of day with no more than two groups (12 rats) being tested on any given day. Rats from each experimental and control group were randomized between testing sessions. Rats were habituated to the testing procedure and experimenter for 3 days before the collection of baseline data. Habituation consisted of moving the rats from their home room to the testing room, handling the rats, and placing them in the testing apparatus for 30 min. The testing apparatus consisted of a row of six clear acrylic enclosures (4 in (∼10.2 cm) wide, 75/8 in (19.4 cm) long, and 5 inches (12.7 cm) high). Opaque dividers separated the enclosures. On testing days, rats were loaded in their enclosures and given 10 min of habituation before the initiation of data collection. All behavioural data were collected by an investigator blinded to the details of the nerve injection.
For the test of mechanical sensitivity, acrylic enclosures sat atop anodized aluminum mesh (¼ in (6.4 mm) waffle hole). An electronic Von Frey anaesthesiometer (IITC Plantar Test Analgesia Meter 2390; IITC Life Sciences Inc., Woodland Hills, CA, USA) fitted with a rigid tip (1.0 mm tip diameter) was used to assess changes in mechanical threshold. The tip was applied to the glabrous skin of the hindpaw with steady vertical pressure until the rat withdrew from the stimulus or until the paw was lifted off the mesh floor (at ∼70 g). The greatest force generated before withdrawal was recorded. This procedure was repeated three times, and the average of the three measures for each paw was considered the withdrawal threshold.
Thermal withdrawal latency was determined using a plantar analgesia meter (IITC Life Sciences). Acrylic enclosures sat atop a glass surface maintained at 30°C. A light beam was focused onto the middle of the ventral surface of each hindpaw and the amount of time lapsed until the animal withdrawals the paw cut-off (20 s) was reached.
Chemicals and reagents
DiI (1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbo-cyanine perchlorate) was purchased from Invitrogen (Carlsbad, CA, USA). Fura-2 acetoxymethyl (AM) ester (2.5 mm stock in DMSO) and Pluronic F-127 (0.025 % in water) were purchased from TEF Laboratories (Austin, TX, USA). FITC-conjugated isolectin B4 (FITC-IB4) was purchased from Sigma (St Louis, MO, USA). KB-R7943 and SN-6 were purchased from R&D Systems, Inc. (Minneapolis, MN, USA) and SEA0400 was purchased from ChemScene, LLC (Monmouth Junction, NJ, USA). All NCX inhibitors were dissolved to a 100 mm stock concentration in DMSO (Sigma-Aldrich, St Louis, MO, USA). Saporin (unconjugated and IB4 conjugated) was purchased from Advanced Targeting Systems (San Diego, CA, USA).
Statistical analysis
Data are expressed as means ± SEM. Student's t test was used for simple comparison between groups. For experiments involving the application of test compounds, vehicle controls were always included. Concentration–response data for NCX blockers were fitted with a modified Hill equation: Emax × Dn/(Dn + EC50n), where Emax is the maximal increase in the duration of the evoked Ca2+ transient, D is the concentration of NCX blocker, EC50 is the concentration of NCX blocker producing a response 50% of maximal, and n is the Hill coefficient. One- and two-way ANOVA was used for analysis of more than two groups with the Holm-Sidak test used for post hoc analysis. Statistical significance was assessed at P < 0.05.
Results
NCX contributes to the decay of the evoked Ca2+ transient in IB4+ small diameter cutaneous DRG neurons
We initially screened cutaneous (DiI+) DRG neurons for the presence of NCX activity by comparing high K+ (30 mm, 4 s)-evoked Ca2+ transients before and after blocking NCX using Na+-free bath solution (Fig.1A). A neuron was considered to have NCX activity if application of Na+-free bath produced an increase in the duration of the evoked Ca2+ transient, assessed as time to decay to 50% of the peak, or T50, >20% above baseline. Consistent with our previous results in unlabelled DRG neurons (Lu et al. 2006), NCX activity was only detected in a subpopulation of the cutaneous neurons: those with a small cell body diameter (≤30 μm), that were IB4+, and were, in general (29/34), responsive to 500 nm capsaicin (Cap). In this subpopulation of IB4+ (n = 34) neurons, blocking NCX activity had no effect on the magnitude of the evoked Ca2+ transient, but was associated with a significant increase in duration (529.22 ± 12.68 %, Fig.1B). In contrast, blocking NCX activity in IB4– neurons (n = 15) had no effect on either the magnitude or duration of the evoked Ca2+ transient (Fig.1B). Failure to detect NCX activity was not due to differences in the magnitude of Ca2+ transient used to assess the presence of activity (Fig.1B). Comparable results were obtained with Na+-free bath solution in which Na+ was replaced with Li+ or choline (Fig.1C), arguing against non-specific effects of Li+ on the regulation of [Ca2+]i. Lastly, there was no significant difference between IB4+ and IB4– neurons with respect to resting [Ca2+]i which was 104.02 ± 4.31 nm and 111.04 ± 3.98 nm, respectively.
Figure 1. NCX activity in isolated cutaneous DRG neurons.
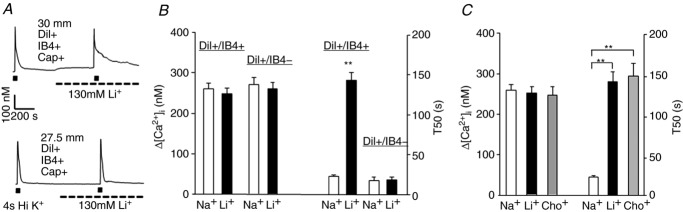
A, high K+ (30 mm for 4 s) evoked Ca2+ transients in small diameter (≤30 μm) retrogradely labelled (DiI+), capsaicin sensitive (Cap+), IB4+ (top trace) and IB4– (bottom trace) DRG neurons before and after NCX block with a bath solution in which Na+ was replaced with Li+. B, pooled magnitude (Δ[Ca2+]i) and decay (time to 50% decay from peak, or T50) of high K+ evoked Ca2+ transient data from the two populations of neurons illustrated in A (DiI+ small diameter Cap+ neurons that were either IB4+ (n = 34) or IB4– (n = 15)). The increase in the duration of the evoked transient was only observed in IB4+ neurons. C, pooled magnitude and decay of high K+ evoked Ca2+ transient data from DiI+/IB4+ neurons before (Na+) and after block of NCX with Li+ (n = 20) or choline (Cho+, n = 14) replacement. Pooled data in this and subsequent figures are plotted as means ± SEM unless otherwise indicated. **P < 0.01
Biophysical properties of NCX in putative nociceptive cutaneous neurons
NCX is described as a low affinity Ca2+ extrusion mechanism and evidence from other systems suggest that it is only activated with relatively large increases in [Ca2+]i (Blaustein & Lederer, 1999; Hilge, 2012). Furthermore, if NCX has a relatively high threshold for activation in sensory neurons, this biophysical property could explain the apparent inconsistencies among previous studies of sensory neurons as to whether NCX contributes to the regulation of evoked Ca2+ transients (Verdru et al. 1997; Thayer et al. 2002; Lu et al. 2006; Usachev et al. 2006). We therefore sought to characterize the relationship between [Ca2+]i and NCX activity in putative nociceptive cutaneous neurons. To address this issue, neurons (n = 20 to 40) were challenged with high K+ applied at durations increasing from 250 ms to 4 s, before and after Li+-induced block of NCX (Fig.2A). The stimulus duration-dependent increase in the peak of the evoked Ca2+ transient was well described by a single exponential with a time constant of 1.25 ± 0.08 s, and this was not changed by NCX block (Fig.2B). The stimulus duration-dependent increase in the decay of the evoked transient was more complex, with a marked increase in transient duration detectable as the duration of high K+ application was increased from 1 to 2 s. This transition was further highlighted by NCX block (Fig.2C), where a Li+-induced increase in transient duration was readily apparent in response to a 2 s high K+ application. Consistent with our previous results indicating that there are distinct mechanisms in sensory neurons responsible for the regulation of the magnitude and duration of evoked Ca2+ transients, the magnitude of the evoked transient was saturated by a 2 s high K+ application, at 403.7 ± 20.9 nm, while there was still a ∼60% increase in duration of the evoked transient between 2 and 4 s of application.
Figure 2. Properties of the Ca2+ transient necessary for the detection of NCX activity.
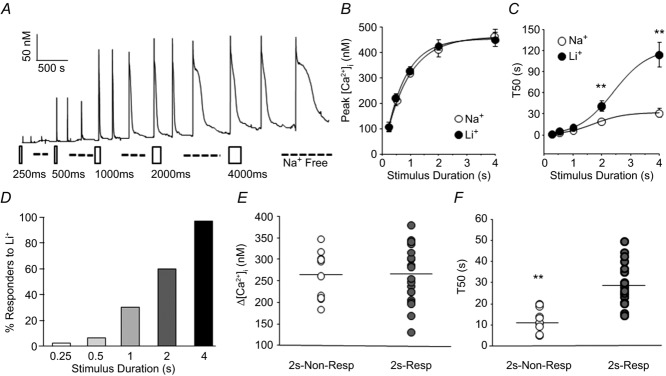
A, high K+ (30 mm) was applied to a cutaneous putative nociceptive neuron for stimulus durations ranging between 250 ms and 4 s, before and after NCX block with Na+-free bath solution (dashed line). The open rectangles indicate the first of three fixed-duration pulses of high K+ (30 mm). B, the peak evoked response from neurons (n = 40) stimulated as in A before (Na+) and after (Li+) NCX block, plotted as a function of the high K+ application duration. C, the duration, as assessed with the T50, of the evoked response of the neurons plotted in B, before (Na+) and after (Li+) block of NCX. D, a cumulative histogram of the neurons illustrated in B and C, analysed as a percentage of neurons studied (n = 32) in which NCX block resulted in an increase in the transient duration (% Responders to Li+) plotted as a function of the stimulus duration. A neuron was considered a responder if NCX block resulted in a 20% increase in the evoked transient duration (T50) over the baseline response. Because there appeared to be a group of neurons in which application of Li+ resulted in an increase in baseline [Ca2+]i and we were originally interested in evoked NCX activity, these neurons were not included in this analysis. Scatter plots of the magnitude (E) and decay (F) of the evoked Ca2+ transient in neurons with (2 s-Resp, n = 20) and without (2 s-Non-Resp, n = 14) evidence of NCX activity in response to a 2 s application of high K+.
Interestingly, even within the subpopulation of putative nociceptive cutaneous neurons, there was heterogeneity with respect to the duration of high K+ application at which NCX activity was detectable. To illustrate this point, we plotted the percentage of neurons responsive to NCX block, as determined by an increase (>20%) in T50 of the high K+-evoked transient, as a function of the duration of high K+ application (Fig.2D). While NCX activity was detectable in all neurons following a 4 s high K+ application, NCX activity was only detectable in 62% of these with a 2 s high K+ application. Not surprisingly, there was no difference with respect to the magnitude of the Ca2+ transient evoked between neurons in which NCX activity was (2 s-responder) or was not (2 s-non-responder) detected with a 2 s high K+ application (Fig.2E; 267.25 ± 15.03 nm and 265.06 ± 17.61 nm, respectively). However, there was a significant difference between 2 s-responders and 2 s-non-responders with respect to the duration of the 2 s high K+-evoked transient (Fig.2F; 27.42 ± 2.11 s and 12.39 ± 1.74 s, respectively; P < 0.05).
We performed a more detailed analysis of the evoked Ca2+ transient in 2 s-responder and 2 s-non-responder neurons to determine whether it was possible to define a ‘threshold’ for NCX activation. Because the duration of the Ca2+ transient in 2 s-responders was so much longer than that in 2 s-non-responders (Fig.3A), we analysed Ca2+ transients for each neuron as a function of the duration at which the transient was at or above an [Ca2+]i ranging from 275 to 400 nm. Increments of 25 nm Ca2+ were used, as they appeared to provide sufficient resolution to detect a separation between subpopulations of neurons. For example, there was a separation in the time spent at or above 325 nm Ca2+ between 2 s-responders and 2 s-non-responders as the transient in 2 s-responders was at or above 325 nm Ca2+ for 13.4 ± 2.5 s (n = 20), compared to 7.00 ± 2.2 s (n = 14) in 2 s-non-responders (P < 0.01, Fig.3B). This separation was further increased in Na+-free bath. Since the 2 s-non-responders became responders with a 4 s high K+ application, we repeated this analysis for 2 s-non-responders comparing the response to 2 s and 4 s high K+ applications to determine if the ‘threshold’ of NCX activation was comparable in this group of neurons to that of the 2 s-responders (Fig.3C). An [Ca2+]i at or above 325 nm was again the separation point for the amplitude–duration plots (which increased from 4.0 ± 0.9 s to 12.0 ± 2.9 s (n = 10) with 2 and 4 s of high K+, respectively). This separation was again further augmented in the presence of Na+-free bath. Both sets of data suggest that NCX activity may be evoked in putative nociceptive cutaneous neurons with Ca2+ transients ≥325 nm for a duration of ≥12 s.
Figure 3. Ca2+ transient parameters associated with evoked NCX activity.
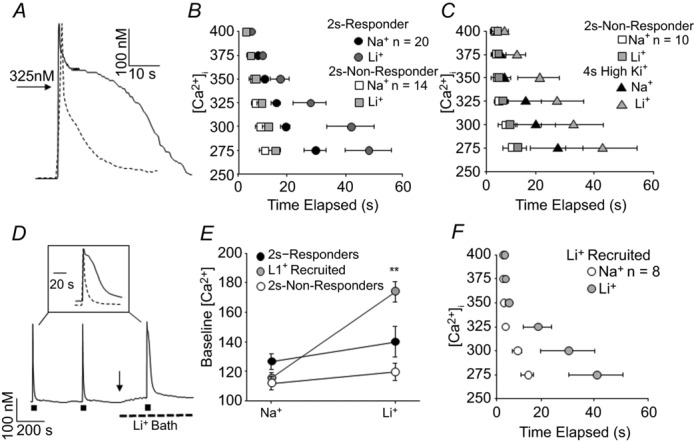
A, high K+ (30 mm, 4 s) evoked Ca2+ transients from an IB4+ (continuous trace) and an IB4– (dashed trace) cutaneous DRG neuron, in which a concentration of 325 nm Ca2+ is indicated to provide a benchmark to evaluate the amplitude–duration relationship. B, pooled transient amplitude–duration (time elapsed at or above a level [Ca2+]i) data for the 2 s-responders and 2 s-non-responders analysed in Fig.2E and F, where transient duration data before and after Li+ application are plotted. The evoked transient for each neuron was analysed as a function of the duration at which the transient was at or above concentrations ranging from 275 to 400 nm using increments of 25 nm Ca2+. C, data for the 2 s-non-responders in B (in response to the 2 s high K+ application) are replotted along with the average amplitude–duration data for these neurons in response to the 4 s high K+ application (4 s High K+), before (Na+) and after (Li+) NCX block. D, evoked NCX activity was detected in a third subset of neurons in which application if Li+ resulted in an increase in resting [Ca2+]i (arrow). Inset: evoked Ca2+ transients before (dashed line) and after (continuous line) NCX block. E, pooled data demonstrating the significant shift increase in resting [Ca2+]i with application of Li+ in a subset of neurons (Li+ Recruited, P < 0.01, n = 8) in which evoked NCX activity was subsequently detected. F, pooled amplitude–duration data for the subset of neurons in which the Li+-induced increase in resting [Ca2+]i resulted in the recruitment of evoked NCX activity.
In our initial analysis of the 2 s high K+ data set, we identified a subset of 2 s-responders (8/20) that had evoked Ca2+ transients in normal bath solution with parameters comparable to those of 2 s-non-responders (i.e. note the overlap in the scatter plot in Fig.2F). However, the resting [Ca2+]i appeared to be the unique feature in this group of 2 s-responders as it was increased following application of Li+ bath (Fig.3D). Pooled data indicate that this increase in resting [Ca2+]i was significantly (P < 0.01) greater than changes observed in the other 2 s-responders or 2 s-non-responders (Fig.3E). In this ‘Li+ recruited’ group of neurons, the evoked transient amplitude–duration plot again revealed a significant increase in the duration of the transient at or above 325 nm Ca2+ in the presence of Li+ (Fig.3F). In these Li+ recruited neurons, the 2 s-evoked Ca2+ transient resembled a ‘non-responder’, such that it did not fulfil the amplitude–duration criteria that appears to be necessary to engage NCX activity. However, the Li+-induced increase in [Ca2+]i enabled a 2 s depolarization to engage NCX. This group of neurons provided the first evidence of two modes of NCX activity: one evoked in response to relatively large and long-lasting Ca2+ transients and a second in which a resting NCX activity contributes to the maintenance of the baseline or resting [Ca2+]i.
We next sought to determine whether NCX activity persists once activated, and if so, for how long. We assumed that, as with PMCA, which appears to be constitutively active in sensory neurons to counter what appears to be a persistent Ca2+ leak (Rigaud et al. 2009), inhibition of NCX would result in an increase in [Ca2+]i. We could then monitor the duration of NCX activity following high K+ application with brief (60 s) applications of Li+ to temporally inactivate NCX activity if present (Fig.4A). Li+ bath was applied before and at 5 min intervals after two consecutive high K+-induced transients until Li+ application resulted in no change in [Ca2+]i. This protocol revealed two subsets of neurons: those with a Li+-induced increase in [Ca2+]i prior to 4 s depolarization (Resting NCX activity n = 14) and those without (n = 24). Furthermore, within the ‘no resting NCX activity’ subset, there was a group of neurons (14/24) in which the evoked NCX activity recovered relatively rapidly so that Li+ challenge before and after the high K+-evoked transient resulted in no change in [Ca2+]i. The remaining neurons in the ‘no resting activity’ subset (10/24), appeared to have evoked NCX activity that persisted well beyond full recovery to baseline [Ca2+]i levels so that Li+-evoked transients were detectable for up to 10 min after the 4 s stimulus (Fig.4A and B). Within the subset of neurons with resting NCX activity, there was a group of neurons (9/14) in which evoked NCX activity appeared to recover rapidly: a Li+-evoked Ca2+ transient was clearly detectable prior to high K+ challenge, but there was no change in the magnitude of the Li+-evoked transient after the 4 s stimulus (Fig.4B). Finally, in the remaining neurons in the subset with resting NCX activity (5/14), there was evidence of both resting NCX activity (i.e. a Li+ transient prior to high K+ challenge) and persistent evoked activity in which there was an increase in the magnitude of the Li+-evoked transient after the 4 s stimulus (Fig.4A and B). Analysis of the resting [Ca2+]i in these four subpopulations of neurons indicated that resting NCX activity was present in neurons with baseline [Ca2+]i > 150 nm (Fig.4C). Interestingly, despite the fact that the Li+-evoked transients were no longer detectable 15 min after high K+ challenge in neurons in which evoked NCX activity appeared to persist (Fig.4B), there was no evidence of recovery of resting [Ca2+]i to baseline levels in this subpopulation (Fig.4C). Similarly, there was no significant increase in resting [Ca2+]i in neurons with resting NCX activity in which there appeared to be a persistent evoked NCX activity. Taken together, these data lend further support to the suggestion that there are at least two distinct pools of NCX in putative nociceptive sensory neurons.
Figure 4. Characterization of ‘resting’ NCX activity in putative nociceptive cutaneous DRG neurons.
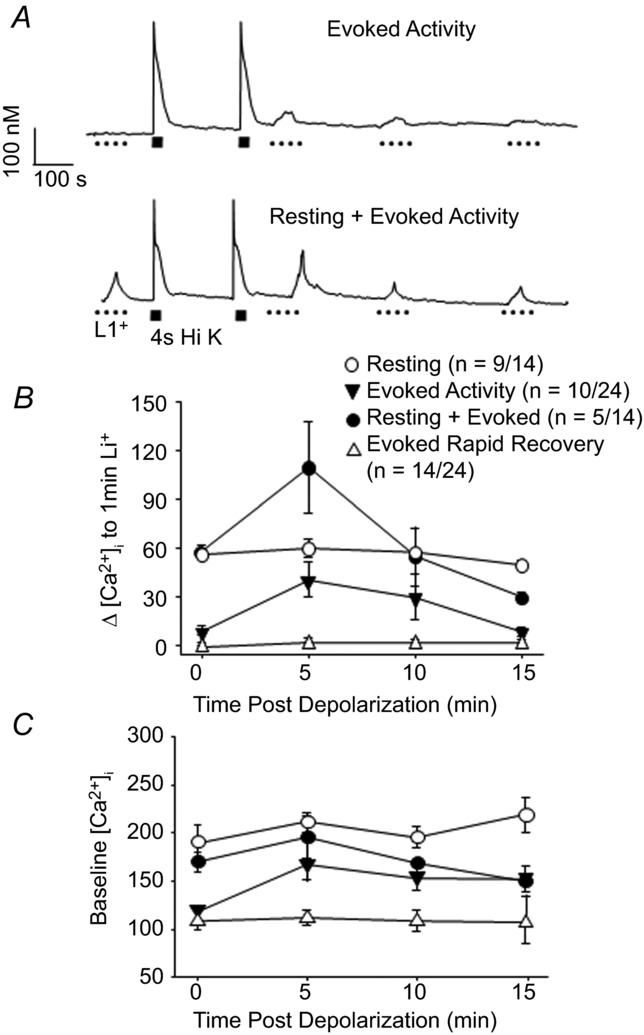
A, examples of neurons in which Ca2+ transients were evoked with Li+. In one neuron (top trace), Li+ (1 min) evoked transients were only detected after stimulating the neuron with high K+ and the amplitude of these transients decayed over time. In the second (bottom trace), Li+-evoked transients were evoked before and after stimulating the neuron with high K+. In this neuron, the amplitude of the Li+-evoked transient increased following high K+ stimulation, but decayed to baseline levels over time. B, the amplitude of the Li+-evoked transient in 4 types of putative nociceptive cutaneous DRG neurons are plotted as a function of time relative to high K+ stimulation as illustrated in A, where 0 is before and 15 indicates 15 min after high K+-induced depolarization. C, resting [Ca2+]i data for the four groups of neurons plotted in B, where resting [Ca2+]i was determined 10 s prior to Li+ application.
NCX expression in isolated cutaneous sensory neurons
The results of our biophysical characterization of NCX activity in putative nociceptive cutaneous neurons suggested that there is considerable heterogeneity in the properties of NCX both within and between putative nociceptive cutaneous neurons. Because of evidence suggesting that NCX isoforms have different properties (Thurneysen et al. 2002b; Jeon et al. 2003; Molinaro et al. 2011), we next sought to determine whether the heterogeneity in functional properties might be due, at least in part, to a differential expression of NCX isoforms. PCR analysis of NCX isoform expression of mRNA extracted for whole DRG indicated that not only are all three isoforms expressed in DRG, but at least two splice variants of NCX1 and NCX3 were detectable as well (Fig.5). To confirm NCX expression in putative nociceptive neurons, single cell PCR analysis was performed. Results of this analysis indicated that mRNA encoding all three NCX isoforms is present in the vast majority of DRG neurons from L4/L5 DRG (Fig.5) including two splice variants of NCX1 and two of the three splice variants of NCX3.
Figure 5. NCX isoform expression in DRG.
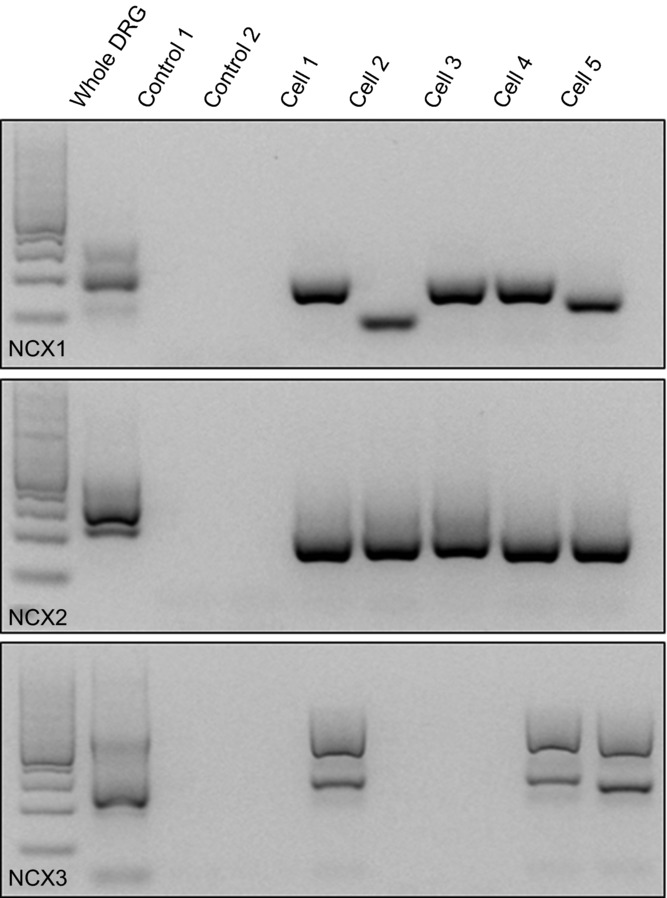
RT-PCR was used to screen for the presence of NCX1, 2 and 3 in mRNA extracted from whole ganglia and isolated putative nociceptive cutaneous DRG neurons. Images of ethidium bromide-stained agarose gel loaded with molecular weight marker (lane 1) and then as indicated where Control 1 is a no-cell control and Control 2 is a single neuron in which no reverse transcriptase was included in the reverse transcriptase reaction prior to PCR amplification, and Cells 1–5 are products from single putative nociceptive cutaneous DRG neurons.
Because the presence of mRNA does not necessarily indicate the presence of functional protein, we next sought to determine which isoforms were functional within the isolated cell body. High K+ was applied for 4 s to putative nociceptive cutaneous neurons before and after application of three different NCX inhibitors, KB-R7943, SEA0400, and SN-6 (Fig.6A). These inhibitors were chosen based on their relative selectivity for individual isoforms. In transfected fibroblasts, when NCX is functioning in the reverse mode, KB-R7943 has been shown to block NCX3 with a higher potency than NCX1 or 2, whereas SEA0400 and SN-6 have been shown to block NCX1 with higher potency than NCX2 and 3 (Iwamoto & Shigekawa, 1998; Watanabe et al. 2006). There is currently no commercially available NCX2 selective blocker. KB-R7943 produced a concentration-dependent increase in the duration of the high K+-evoked transient (Emax = 107.25 ± 20.36 s; EC50 = 0.045 ± 0.01 μm, Fig.6B). The impact of KB-R7943 on the duration of the evoked Ca2+ transient appeared to be specific for NCX block as the application of KB-R7943 (100 nm) in the presence of Li+ bath resulted in no further change in the duration of the evoked transient (−2.26 ± 1.58 %, n = 8). While 100 nm was not quite a saturating concentration of KB-R7943 in cutaneous putative nociceptive neurons, this concentration was used in subsequent experiments for two main reasons. First, because NCX blocker ‘selectivity’ is concentration dependent, we wanted to use a concentration that was likely to provide the best combination of block and selectivity based on results previously reported by Kuroda and colleagues (Kuroda et al. 2013). Second, we observed what appeared to be non-specific effects of all three blockers at concentrations >5 μm, as had been described by others (Iwamoto et al. 1996; Birinyi et al. 2005; Niu et al. 2007). Thus, we also sought to minimize the potential for non-specific actions of the blocker. To estimate the fraction of total NCX activity blocked by KB-R943, we compared the maximal increase in the decay of the evoked Ca2+ transient to that produced by Na+-free block. At 100 nm, KB-R7943 inhibited 67.4 ± 0.1 % of total NCX (Fig.6C). Neither SEA0400 nor SN-6 produced a detectable change in resting Ca2+ (Fig.6A) or the evoked (Fig.6B) Ca2+ transient at concentrations between 1 nm and 1 μm, at and above those thought to be specific for NCX1.
Figure 6. Pharmacological analysis of NCX activity in putative nociceptive cutaneous DRG neurons.
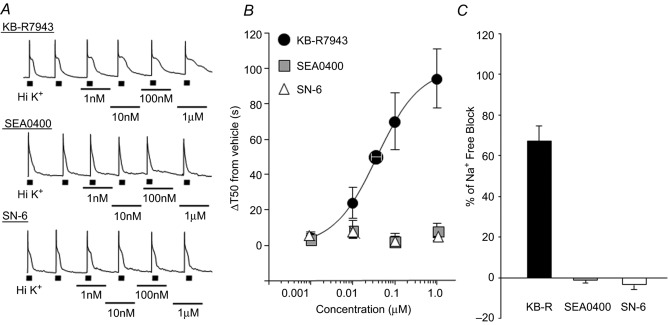
A, high K+ evoked Ca2+ transients in putative nociceptive cutaneous neurons before and in the presence of increasing concentrations of the NCX3 selective blocker KB-R7943 (top trace), the NCX1 selective blocker SEA0400 (middle trace) and a second NCX1 selective blocker SN-6 (bottom trace). B, pooled concentration–response data from neurons treated with KB-R7943 (n = 25), SEA0400 (n = 11) or SN-6 (n = 10). Data were analysed as an increase in transient duration (T50) relative to the transient duration in the presence of vehicle. KB-R7943 data were well fitted with a Hill equation, yielding an Emax = 107.25 ± 20.36 s and EC50 = 0.045 ± 0.01 μm. C, the maximal change in transient duration in the presence of blocker was analysed as a percentage of the total block of NCX activity represented by the maximal change in duration observed in Na+-free bath. Data for each blocker was pooled and plotted.
Previous studies suggest that Li+ bath activates NCX in the reverse mode (Annunziato et al. 2004). However, as suggested earlier, we attributed the change in baseline resting [Ca2+]i with application of Li+ bath (Fig.3D) to inhibition of resting NCX activity and a subsequent build-up of Ca2+ from a persistent Ca2+ leak. In support of this hypothesis, application of 100 nm KB-R7943 also resulted in a ≥20% increase in baseline resting [Ca2+]i (Δ[Ca2+]i = 31.16 ± 4.8 nm) in 15 of the 29 neurons tested (Fig.7).
Figure 7. NCX contributes to the regulation of resting [Ca2+]i in a subpopulation of putative nociceptive cutaneous neurons.
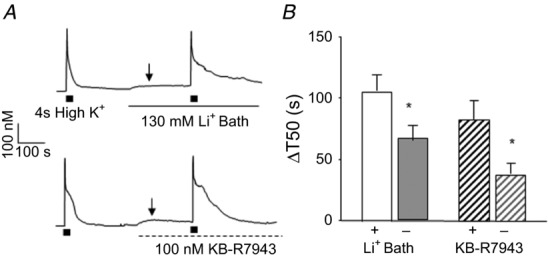
A, examples of neurons in which application of Li+ bath (top trace) or KB-R7943 (bottom trace) resulted in an increase in resting [Ca2+]i (indicated by arrow). B, pooled change in T50 (ΔT50) data, calculated as the difference between the T50 in the presence of NCX block and the baseline response, for neurons in which NCX was blocked with Li+ bath or KB-R7943 (100 nm). Data for each group of neurons was separated according to whether NCX block was associated with an increase in resting [Ca2+]i (+, n = 14 and 15 for Li+ and KB-R7943 groups, respectively), relative to those in which there was no detectable change in resting [Ca2+]i associated with NCX block (–, n = 24 and 14 for Li+ and KB-R7943 groups, respectively) following NCX block. *P < 0.05.
To further assess the relative contribution of NCX isoforms to resting and evoked NCX activity, we used siRNA to selectively knock down expression of each isoform. Targeted or control siRNA was injected into the sciatic nerve 6 days prior to the harvest and dissociation of DRG neurons for assessment of NCX activity. The extent of the knockdown was estimated using Western blot analysis of the sciatic nerve (Fig.8A). A single intraneural injection of targeted siRNA resulted in a decrease in NCX protein of 57.44 ± 3.36%. Importantly, Western blot analysis confirmed the specificity of each siRNA construct, where the only significant change in each isoform was in response to the appropriately targeted siRNA (Fig.8A). Consistent with results obtained with SEA0400 and SN-6, NCX1 knockdown was associated with no detectable changes in resting or evoked Ca2+ transients, or in the changes observed in response to Na+-free bath or 100 nm KB-R7943 application (T50 = 100.55 ± 11.8 s and 65.9 ± 12.1 s, respectively) compared to the effect of NCX inhibition in neurons from naive or control-targeted siRNA-treated rats (Fig.8B). Results with NCX3 knockdown were also consistent with pharmacological results, where, in contrast to NCX1 siRNA-treated rats, evidence of evoked NCX activity in putative nociceptive cutaneous neurons was significantly attenuated (Fig.8B). NCX3 knockdown was also associated with a significant suppression of the effects of 100 nm KB-R7943 (Fig.8B). Strikingly, in contrast to assumptions made about the concentration-dependent specificity of inhibitors, there was a significant suppression of the increase in T50 associated with both Li+ bath and KB-R7943 in neurons from NCX2 siRNA-treated rats (Fig.8B).
Figure 8. Targeted siRNA knockdown of NCX isoforms on high K+-evoked Ca2+ transients in putative nociceptive cutaneous neurons.
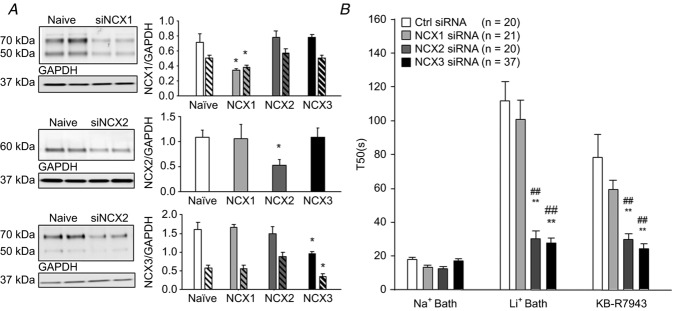
A, Western blot analysis of protein extracted from the sciatic nerve proximal to the site of siRNA injection was used to quantify the extent of the knockdown 6 days after siRNA treatment. Typical blots from animals treated with siRNA targeted to NCX1, 2 and 3. Pooled data from 4 animals per condition demonstrate specificity of siRNA sequences, where relative density of each isoform is plotted for tissue obtained under 4 conditions: naive, NCX1 siRNA, NCX2 siRNA and NCX3 siRNA treated. Antibodies for NCX1 and NCX3 detected multiple bands and all were quantified. Filled bars represent pooled data for higher molecular weight (70 kDa) band and hatched bars represent pooled data for lower molecular weight (50 kDa) band. Data were normalized with respect to GAPDH and plotted. B, pooled high K+-evoked transient duration (T50) data for neurons from control and NCX isoform targeted siRNA-treated rats, before (Na+ bath) and after block of NCX with Na+-free bath (Li+ Bath) or KB-R7943 (100 nm). **P < 0.01 relative to control siRNA treated; ##P < 0.01 relative to NCX1 siRNA treated.
Differential distribution of NCX isoforms within sensory tissues
The presence of mRNA encoding all three NCX isoforms in sensory neurons and evidence that only NCX3 and/or both NCX2 and NCX3 are functional in the sensory neuron cell body raises the possibility that NCX isoforms are differentially distributed within sensory neurons. To begin to address this possibility, we assessed the relative protein density of NCX isoforms in the spinal cord, dorsal root, DRG, and sciatic nerve (Fig.9). Consistent with the literature (Schulze et al. 2002; Quednau et al. 2004), antibodies for NCX1 and NCX3 detected multiple bands. Only the most robust bands were quantified across tissues. There was substantially more NCX1-like immunoreactivity (LI) in the spinal cord (n = 4) compared to peripheral tissues (Fig.9A). NCX2 appeared to be equally distributed among peripheral tissues, but only weakly expressed in the spinal cord (Fig.9B). Finally, NCX3-LI was greatest in the DRG and sciatic nerve relative to the spinal cord suggesting trafficking of NCX3 towards the periphery (Fig.9C).
Figure 9. Distribution of NCX isoform-like immunoreactivity (LI) in protein extracted from spinal cord, central segment of the sensory nerve (central nerve), DRG and sciatic nerve.
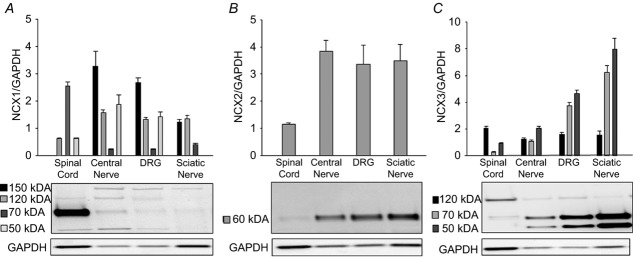
Pooled data (n = 4) and representative blots demonstrating the relative density of expression of NCX1 (A), NCX2 (B) and NCX3 (C) in spinal cord, central nerve, DRG and sciatic nerve tissues. Band intensity at each molecular weight was normalized to that for GAPDH. For the representative blots, different protein amounts were loaded in each lane to facilitate visualization of the bands in each tissue type.
NCX plays a limited role in the regulation of primary afferent excitability
NCX activity may influence excitability directly and/or indirectly. Because the exchanger is electrogenic, a direct influence of NCX on excitability would be through depolarizing inward Na+ current associated with exchanger activity in forward mode. The most proximal indirect influence of NCX activity on excitability would be via an influence on [Ca2+]i, which may influence the activity of other ion channels. We therefore sought to assess the impact of NCX activity on the excitability of putative nociceptive cutaneous DRG neurons. To minimize the impact of recording on the regulation of intracellular Ca2+ and consequently, NCX activity, we used gramicidin perforated-patch to obtain whole cell access for current-clamp recording. Neurons were first loaded with fura-2 AM to enable simultaneous recording of membrane potential and [Ca2+]i. The impact of NCX on the excitability was assessed with direct current injection protocols designed to enable determination of AP threshold, rheobase, and the response to suprathreshold stimuli (2× and 3× rheobase) in which each parameter was assessed under four different conditions: in the absence and presence of Li+ bath, and before and after activating NCX with a 4 s voltage-step to 0 mV to drive an increase in [Ca2+]i. The Ca2+ transient evoked with a voltage step was comparable to that evoked with a 4 s application of high K+ (the peak magnitude was 467.77 ± 23.3 nm and T50 of decay was 24.57 ± 6.5 s; n = 9). Furthermore, as evidence that this Ca2+ transient was sufficient to activate NCX, block of NCX with Li+ resulted in a 276.96 ± 75% increase in T50 of the depolarization-evoked transient. Excitability data under each of these four experimental conditions are summarized in Table2 and Fig.10A–C. Analysing excitability data with a two-way ANOVA revealed a main effect associated with NCX activation (depolarization) on rheobase (P < 0.05; Fig.10A) and the response to suprathreshold stimulation (P < 0.05; Fig.10C). However, there was no significant effect of NCX block (Li+ bath) on these parameters, nor was there an interaction between depolarization and Li+ bath (P > 0.05). There was, however, a significant (P < 0.05) main effect of Li+ bath on action potential threshold (Fig.10B), but no significant influence of depolarization, or an interaction between depolarization and Li+ bath, on this parameter. Comparable analysis of passive and active electrophysiological properties (Table2) revealed a main effect of Li+ bath on resting membrane potential (RMP), and AP overshoot. Additionally, there was a main effect of depolarization on afterhyperpolarization (AHP) and AP overshoot (Table2). However, there was no interaction between depolarization and Li+ bath on any parameter assessed.
Table 2.
Changes in passive and active electrophysiological properties of cutaneous sensory neurons with NCX inhibition
| Baseline |
Depolarization |
|||
|---|---|---|---|---|
| Na+ bath | Li+ bath | Na+ bath | Li+ bath | |
| RMP (mV) | −67.47 ± 2.0 | −61.67 ± 1.6 | −70.14 ± 2.5 | −63.50 ± 3.0 |
| Rin (MΩ) | 1143.06 ± 179.2 | 1050.40 ± 333.6 | 603.70 ± 180.4 | 445.08 ± 42.6 |
| AP overshoot | 50.54 ± 1.6 | 47.11 ± 2.1 | 42.18 ± 1.6 | 35.35 ± 2.5 |
| AP duration (ms) | 3.02 ± 0.3 | 2.72 ± 0.03 | 2.61 ± 0.2 | 2.67 ± 0.2 |
| AHP magnitude (mV) | 21.56 ± 0.7 | 20.47 ± 1.9 | 18.74 ± 1.1 | 15.97 ± 2.1 |
| AHP τ decay (ms) | 70.90 ± 7.4 | 54.40 ± 8.5 | 48.90 ± 6.5 | 38.24 ± 9.5 |
Values are expressed as means ± SEM. Depolarization consisted of a 4 s voltage step from −60 mV to 0 mV. Na+ bath values were collected in the presence of Na+ in the bath solution. Li+ bath values were collected in the absence of Na+ and presence of Li+ in the bath solution. RMP, resting membrane potential; Rin, input resistance; AP, action potential; AHP, afterhyperpolarization; τ, time constant. Data were analysed with a two-way ANOVA to assess the impact of NCX block (with Li+ bath) and depolarization-induced activation of NCX (Depolarization), or the interaction between the two. Results revealed a significant influence of Li+ bath on both RMP (P < 0.05) and AP overshoot (P < 0.05), but no significant influence of depolarization (P > 0.05) or a significant interaction between Li+ bath and depolarization (P > 0.05). There was a main effect of depolarization on AHP and AP overshoot (P < 0.05) but no significant impact of Li+ bath on these parameters (P > 0.05), or a significant interaction between depolarization and Li+ bath (P > 0.05).
Figure 10. The impact of NCX on the excitability of putative nociceptive cutaneous DRG neurons.
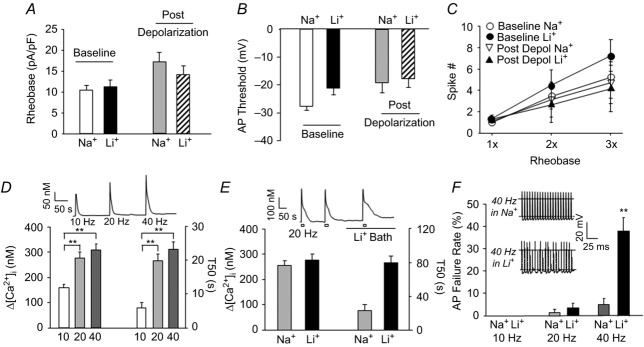
Using gramicidin-perforated patch configuration, excitability was assessed with a square-wave current injection as described in Methods to enable determination of rheobase (A), action potential threshold (B) and the response to suprathreshold current injection (at 2 and 3× rheobase) (C). Excitability was assessed for each neuron (n = 6) before (Na+) and after (Li+) NCX block under resting [Ca2+]i (Baseline) and after a depolarization (4 s to 0 mV)-induced increase in [Ca2+]i. The impact of depolarization was significant (P < 0.05, two-way ANOVA) on both rheobase and AP threshold (i.e. decrease in excitability). However, there was no significant (P > 0.05) effect of NCX block on these parameters, nor was there a significant (P > 0.05) interaction between depolarization and NCX block. D, representative Ca2+ trace (inset) and pooled data showing the significant increase in both the average magnitude ([Ca2+]i) and decay (T50) of Ca2+ transients evoked with 20 pulses of depolarizing current injection delivered at frequencies increasing from 10 to 40 Hz. E, representative Ca2+ trace (inset) and pooled data (n = 8) for the change in the magnitude (Δ[Ca2+]i) and duration of the Ca2+ transient evoked with 20 pulses of depolarizing current injection delivered at 20 Hz before (Na+) and after (Li+) block of NCX. There was a significant (P < 0.01) interaction between stimulation frequency and NCX block on the duration of the evoked transient, where post hoc analysis indicated that the increase in duration for both the 20 Hz and 40 Hz (not shown), but not the 10 Hz stimulation frequency (not shown), were significant. F, the 20 pulse current injection protocol was used to assess following frequency. Inset: typical voltage traces of a neuron stimulated at 40 Hz before (in Na+) and after (in Li+) block of NCX. Pooled following frequency data (n = 8), where the failure rate (number of failed action potentials/train of 20 stimuli) before (Na+) and after (Li+) block of NCX is plotted for stimulation frequencies of at 10, 20 and 40 Hz. There was a significant (P < 0.01, two-way ANOVA) interaction between stimulation frequency and NCX block on the failure rate, where post hoc analysis confirmed the increase in failure rate observed at 40 Hz was significant. **P < 0.01.
As a fifth measure of excitability, we assessed following frequency of neurons in the presence or absence of Li+ bath. Following frequency was assessed with a train of 20 depolarizing current injections (1 ms at 1.5× current threshold) delivered at 10, 20 and 40 Hz. It was not necessary to use the 4 s depolarization protocol for this measure, as a 20 Hz stimulus was able to evoke a Ca2+ transient (Fig.10D; peak magnitude was 412.66 ± 23.1 nm and T50 of decay was 22.33 ± 5.5 s) similar to a 4 s high K+-evoked transient (P > 0.05). Furthermore, a 20 Hz stimulus was sufficient to evoke NCX activity as indicated by the impact of Li+ which resulted in a significant 254.30 ± 84.3 % increase in the T50 of decay (Fig.10E).
Current threshold was determined for each neuron in the presence and absence of Li+ for a 1 ms depolarizing current step in a manner identical to that used for determination of rheobase. Li+ bath was associated with a 16.59 ± 4.7 % increase in the amount of current necessary to evoke an AP relative to that in Na+ bath (P < 0.05, data not shown). Nevertheless, there was no detectable influence of Li+ on the following frequency at 10 and 20 Hz stimulation. However, at 40 Hz, there was a significant increase in the failure rate following NCX block from 5.0 ± 2.5% to 38.1 ± 6.0% (Fig.10F, P < 0.01, paired t test).
Compound action potentials and NCX activity
Given recent evidence suggesting that NCX functioning in reverse mode contributes to axon damage in small fibre peripheral neuropathy (Persson et al. 2013), we next sought to determine whether the NCX protein observed in the peripheral nerve might be functional in the absence of tissue injury. To do this, compound action potential (CAP) recording was performed on isolated sciatic nerves in the presence and absence of Li+ or 100 nm KB-R7943. The CAP was evoked at 0.1 Hz to assess ‘resting’ NCX activity and following 100 pulses at 5 Hz and 2× the intensity required to evoke a maximal C-wave, to assess ‘evoked’ NCX activity (Fig.1A). Li+ bath had no significant effect on the AUC (a measure of the number of fibres contributing to the CAP) or conduction velocity (CV) of the CAP A-wave (0.36 ± 1.6% and 1.91 ± 0.8 % of baseline, respectively). Similarly, at 0.1 Hz stimulation, neither Li+ nor KB-R7943 had a significant influence on the AUC of the CAP C-wave. However, the application of KB-R7942 (Fig.1A), but not Li+, resulted in a significant increase (20.31 ± 5.3 %, P < 0.05, n = 7) in the CV of the C-wave (C-CAP; Fig.1B). Stimulation at 5 Hz, used to assess ‘evoked activity,’ resulted in a decrease in CV in the CAP C-wave. This decrease was significantly (P < 0.05) attenuated in Li+ bath (Fig.1C). KB-R7943 further reduced the activity-dependent decrease in CV such that there was no net change in CV in the presence of KB-R7943 at 5 Hz stimulation. Control nerves sampled every 10 min for 1 h (Na+ bar, Fig.1B) showed very little change in CV with time (0.78 + 4.25 %). Because the impact of KB-R7943 on CV was roughly comparable at 0.1 and 5 Hz, we performed an additional experiment to determine whether the NCX contribution to the regulation of CV was already saturated at 0.1 Hz stimulation. Two nerves were stimulated at 0.017 Hz before and after Li+ application. With this frequency of stimulation, Li+ bath resulted in a 24.9% decrease in CV (data not shown).
Figure 11. Impact of NCX on the compound action potential (CAP) in the sciatic nerve.
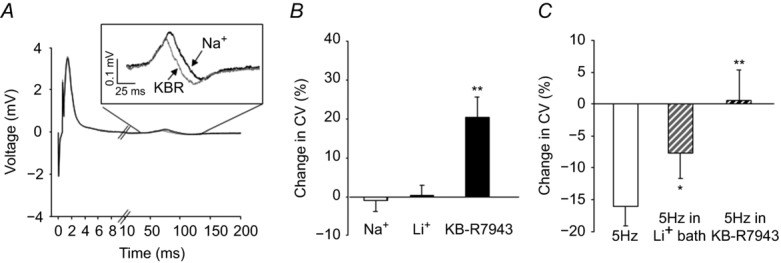
A, compound action potentials were evoked in the sciatic nerve before (black trace) and after (grey trace) block of NCX with Li+. The sciatic nerve is long enough that the A- and C-waves are easily distinguished. Inset: C-wave re-plotted on a more appropriate scale. B, no changes in the A-wave or the area of the C-wave were detected. However, there appeared to be an increase in the conduction velocity of the C-wave following block of NCX. Pooled data analysed as a percentage change from baseline were analysed over time (Na+) relative to that associated with NCX block with Li+ bath (Li+) or KB-R79423 (100 nm). Control nerves were sampled every 10 min for 1 h (Na+ bar). C, pooled data indicating that 5 Hz stimulation resulted in activity-dependent slowing of the nerve, which was significantly attenuated in the presence of Li+ (n = 6) and 100 nm KB-R7943 (n = 6). *P < 0.05; **P < 0.01.
Loss of NCX3 in IB4+ fibres decreases nociceptive thresholds
The restricted distribution of NCX activity in putative nociceptive cutaneous afferents suggests that this exchanger may play a particularly important role in nociceptive processing. Therefore, as a final test of NCX function, we assessed the impact of targeted siRNA knockdown of NCX isoforms on nociceptive threshold. A single intra-sciatic nerve injection of targeted or control siRNA was used to knock down NCX in the afferents innervating the glabrous skin of the hindpaw. Changes in mechanical nociceptive threshold were assessed with an electronic Von Frey and changes in thermal nociceptive threshold were assessed with a Hargreaves apparatus (Hargreaves et al. 1988). The extent of NCX knockdown was assessed with Western blot as shown in Fig.8. Interestingly, knockdown of NCX3, but not NCX1 or NCX2 resulted in a significant reduction in both mechanical (Fig.2A) and thermal (Fig.2B) threshold. Changes in threshold were detectable at 5 days and returned to baseline levels by 10 days post siRNA injection. Western blot analysis of nerves harvested at day 10 revealed protein compared to baseline levels (data not shown, P > 0.05), suggesting that there was recovery from siRNA-induced knockdown.
Figure 12. Nociceptive threshold changes in response to knockdown of NCX isoforms.
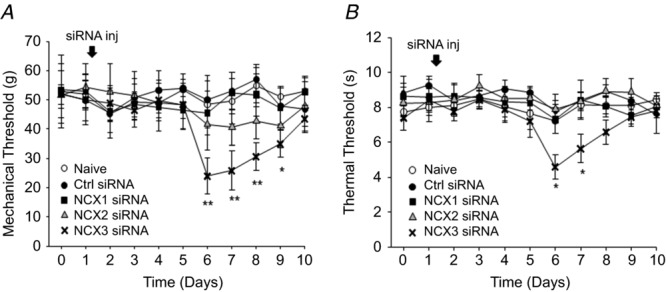
A, mechanical nociceptive threshold as determined with an electronic Von Frey test, was assessed once daily in naive animals (n = 6) and in animals before and after a single sciatic nerve injection with control (n = 6) or targeted siRNA against NCX1 (n = 12), 2 (n = 12) or 3 (n = 18). B, thermal nociceptive threshold was determined with a Hargreaves device in the same groups of animals. Knockdown was confirmed using Western blot analysis as indicated in Fig.8. (*P < 0.05; **P < 0.01).
Lastly, we sought to determine if the significant reduction in both mechanical and thermal thresholds were due to loss of NCX3 in IB4+ fibres, based on our in vitro results indicating that NCX activity is only present IB4+ neurons. Using the cytotoxin, saporin, conjugated to IB4 (IB4–SAP) to specifically ablate the IB4+ fibre population (Vulchanova et al. 2001; Tarpley et al. 2004). We injected the sciatic nerve with a combination of the unconjugated or conjugated form of saporin (IB4–SAP) with either NCX3-targeted or control non-targeted siRNA. The co-injection of IB4–SAP and control siRNA (IB4–SAP + Ctrl siRNA) or NCX3 siRNA (IB4–SAP + NCX3 siRNA) significantly elevated mechanical (Fig.3A) and thermal (Fig.3B) thresholds (P < 0.05, n = 6). Unconjugated saporin with control siRNA (Unconj + Ctrl siRNA) had no significant effect on nociceptive behaviour compared to naive animals (P > 0.05, n = 6). Unconjugated saporin co-injected with NCX3 siRNA (Unconj + NCX3 siRNA) resulted in decrease in both mechanical (Fig.3A) and thermal (Fig.3B) thresholds, consistent with NCX3 siRNA alone (n = 6). The change in nociceptive threshold over time was analysed as an area under the curve (AUC) where data on day 0 was used to assess the change in threshold for each animal on days 0 through 10. The results confirms that there was no significant difference between IB4–SAP + Ctrl siRNA and IB4–SAP + NCX3 siRNA (Fig.3C) suggesting that loss of IB4+ fibres attenuates the siRNA-induced hypersensitivity seen in the Unconj + NCX3 siRNA-treated group. To confirm the loss of IB4 fibres following IB4–SAP treatment, we analysed the pattern of IB4 staining in the spinal cord of IB4–SAP- and control-treated rats. Consistent with previous studies using IB4–SAP (Vulchanova et al. 2001; Tarpley et al. 2004), there was a dramatic decrease in IB4+ staining in the superficial dorsal horn of L4 and L5 spinal segments in rats treated with the conjugated saporin (IB4–SAP + Ctrl siRNA and IB4–SAP + NCX3 siRNA) (Fig.3D). The loss of IB4 staining was restricted to the medial portion of the dorsal horn thought to be the zone of innervation of the sciatic nerve. No significant loss of IB4 staining was detected in those rats treated with the unconjugated saporin (Fig.13D).
Figure 13. Assessment of the relative contribution of IB4+ fibres to the changes in nociceptive behaviour associated with NCX3-targeted knockdown.

The IB4+ population of sensory neurons in the sciatic nerve was ablated with an intra-sciatic injection of IB4 conjugated to the toxin, saporin (IB4–SAP). The control for this was an injection of unconjugated IB4 and saporin (Unconj). Groups of rats (n = 6 per group) received injections of either IB4–SAP or Unconj with either control siRNA (Ctrl) or NCX3-siRNA (NCX3). Mechanical (A) and thermal (B) nociceptive thresholds were determined as in Fig.2 before and after sciatic nerve injections. C, the change from baseline over time in each group was analysed as an area under the curve (AUC) for both mechanical (top graph) and thermal (bottom graph) data. D, the extent of IB4 fibre ablation was assessed in spinal cord sections stained with IB4–FITC as described in Methods. Consistent with the termination pattern of afferents in the sciatic nerve, there is almost a complete loss of IB4 binding in the medial superficial dorsal horn of IB4–SAP-treated animals. Scale bar = 100 μm. **P < 0.01 relative to Unconj + Ctrl; ##P < 0.01 relative to Unconj + NCX3.
Discussion
The purpose of this study was to characterize the biophysical properties of NCX among subpopulations of sensory neurons, identify the NCX isoform(s) underlying NCX activity, and to begin to determine the function of this exchanger in sensory neurons. We have confirmed our previous results indicating NCX activity is restricted to a subpopulation of putative nociceptive neurons, which extends to cutaneous neurons innervating the glabrous skin of the hindpaw. There appear to be two modes of NCX activity: one evoked in response to relatively large and long-lasting (∼325 nm for >12 s) increases in [Ca2+]i, and a second that is active at resting [Ca2+]i >∼150 nm. There also appeared to be two modes of evoked activity: one that decayed relatively rapidly (<5 min) and a second that persisted (>10 min). While NCX activity was detected in all IB4+ small diameter cutaneous neurons, there was heterogeneity in this subpopulation of neurons with respect to the different modes of NCX activity present. Pharmacological data suggest that NCX3 accounts for ∼70% of the NCX activity in sensory neurons with NCX2 accounting for the remaining 30%. Knockdown using targeted siRNA of NCX isoforms yielded results consistent with the pharmacological data, but suggested a larger role of NCX2. Additionally, mRNA encoding all three isoforms was detectable in putative nociceptive cutaneous neurons, with evidence for the differential distribution of splice variants of NCX1 and 3. Western blot analysis confirmed the presence of all three isoforms in sensory neuron and spinal cord tissue and isoforms appeared to be differentially distributed among these tissues. While NCX appears to contribute to the regulation of resting [Ca2+]i in a small subpopulation of neurons, it appears to play a much more dominant role in regulating the decay rate of evoked Ca2+ transients. Additionally, NCX appears to have a minor role in the regulation of neuronal excitability, where the only detectable influence was on the following frequency of neurons at relatively high (40 Hz) rates of stimulation. We also obtained evidence of NCX activity in C-fibre axons, where block of the exchanger resulted in a small increase in conduction velocity. Furthermore, knockdown of NCX3, but not NCX1 or 2, resulted in a significant decrease in nociceptive threshold. Using IB4 conjugated to the toxin saporin, we obtained evidence consistent with the suggestion that the shift in nociceptive thresholds is due to the loss of NCX3 in the IB4+ fibre population. Together, these data show that NCX, most likely NCX3, plays an important role in the regulation of [Ca2+]i within the primary sensory neurons.
Biophysical properties: evoked activity
The primary approaches used to assess the presence of NCX activity in the first half of this study were largely indirect, inferred from changes in [Ca2+]i secondary to the removal of extracellular Na+ needed for exchange activity (Cook et al. 1998) or the application of NCX inhibitors. Li+ was used in the majority of experiments as the replacement for Na+ because it is voltage-gated Na+ channel permeable (Hille, 1972; Gold & Thut, 2001), and does not block the mitochondrial Na+–Ca2+ exchanger (Palty et al. 2010). We acknowledge, however, that the use of Li+ is potentially problematic for the assessment of NCX activity, particularly with the indirect approach used due to the variety of other proteins that may be affected and result in a secondary influence on [Ca2+]i including other pumps (Tolkovsky & Richards, 1987; Hermans et al. 1997), exchangers (Herbert et al. 2004; Palty et al. 2010), and ion channels (Vaughn & Gold, 2010; Zhang et al. 2010). Nevertheless, while there may have been off-target effects of Li+ in these experiments, we propose that the Li+-induced changes in resting and evoked Ca2+ transients were due predominantly to block of NCX, and therefore reflect NCX activity for two reasons. First, comparable changes in resting and evoked Ca2+ transients were observed with Li+, choline, and the relatively NCX-specific blocker, KB-R7943. Second, siRNA-induced knockdown of NCX occluded the Li+-induced changes to the evoked Ca2+ transient.
In most tissues, NCX is considered a low affinity exchanger requiring a high [Ca2+]i for activation. The present results were generally consistent with this model, where, in putative nociceptive neurons, an increase in [Ca2+]i to ∼325 nm for at least 12 s was required to evoke NCX activity. That is, despite evidence that NCX was present and functional in all putative nociceptive neurons, NCX activity was only detected in these neurons if the evoked transient met this threshold. At first pass, it appeared paradoxical that the evoked transient in neurons, such as the 2 s-responders, in which NCX had been activated, decayed more slowly than the 2 s-non-responders in which NCX had not been activated. The implication of this observation is that 2 s-responders are less able to handle large Ca2+ loads with other Ca2+ regulatory mechanisms, such that Ca2+ influx associated with the 2 s high K+ application overwhelms the available buffering capacity, and NCX is engaged.
In a subpopulation of neurons, evoked NCX activity persisted for >10 min, far longer than the time required for full recovery to baseline [Ca2+]i. One mechanism to account for this persistent activation is suggested by data from non-neuronal cells where the rate of NCX deactivation appeared to be far slower than the activation rate (Hilge et al. 2009). There are at least two other possible explanations for the persistent NCX activity. First, while it is thought that a drop in Ca2+ below the Kd for the NCX Ca2+ binding domain (CBD1) is sufficient to terminate NCX activity (Hilge, 2013), it is possible that microdomains of elevated Ca2+ not detected with the relatively low resolution whole cell imaging used persist for over 10 min in some neurons. Consistent with this suggestion, Acsai et al. (2011) demonstrated divergence between microdomain and global Ca2+ signals suggesting that for different global Ca2+ signals, microdomain levels can be 20–30 times higher (Acsai et al. 2011). Second, the persistent activation may reflect second messenger-mediated modulation of NCX as phosphorylation-dependent changes in NCX activity have been demonstrated (Formisano et al. 2008; Sirabella et al. 2012).
Biophysical properties: resting activity
We were also able to detect resting NCX activity in 35% of the putative nociceptive neurons tested. There are at least two likely explanations for the presence of resting NCX activity in a subpopulation of sensory neurons. First, as suggested above for the persistent evoked activity, microdomains of elevated [Ca2+]i above the threshold of activation could account for the presence of resting NCX activity in a subpopulation of neurons. Second, there is evidence that the Ca2+ affinity for NCX activation depends on splice variants within the intracellular loop that makes up two distinct Ca2+ binding domains (CBD1 and 2). The primary Ca2+ sensor responsible for Ca2+ activation, CBD1, has a higher affinity for binding (∼Kd = 100–600 nm; Hilgemann et al. 1992), whereas CBD2 has a low affinity for Ca2+ (∼Kd = 250 nm–20 μm). Thus, a splice variant with only CBD1 may have activity at an [Ca2+]i comparable to that observed in the present study. Similarly, a combination of CBD1 and CBD2 in some neurons may result in a pattern of NCX activity that requires a high [Ca2+]i for full activity, as well as partial activity at considerably lower [Ca2+]i. Consistent with this suggestion, data from cardiomyocytes indicates that ∼5% of the maximal NCX current is detected at resting [Ca2+]i levels, whereas full activity requires a the rise of [Ca2+]i to 1–2 μm (Boyman et al. 2011).
Our biophysical characterization of NCX as a low affinity exchanger is consistent with Verdru et al. (1997) who found that removal of external Na+ dramatically slowed the [Ca2+]i decay in a subpopulation of DRG neurons in which the average peak was >400 nm (Verdru et al. 1997). Furthermore, they also detected an increase in baseline [Ca2+]i with the block of NCX activity in 85% of the DRG neurons tested (Verdru et al. 1997). Shutov and colleagues found that inhibition of NCX in synaptic terminals resulted in a small but significant increase in [Ca2+]i (Shutov et al. 2013), suggesting there is some resting NCX activity in the central terminals as well as the cell body. In contrast, others have failed to detect a significant contribution of NCX to the regulation of either resting or evoked Ca2+ (Thayer & Miller, 1990; Usachev et al. 2006; Gemes et al. 2012), leading the authors of these studies to conclude that NCX does not contribute to the regulation [Ca2+]i in sensory neurons. We suggest, however, that the presence of NCX was probably missed in these previous studies because of the relatively small Ca2+ transients evoked (all less than 300 nm) and/or because of the relatively limited number of neurons in which NCX appears to function.
Another possible explanation for the presence of ‘resting’ NCX activity is that it is actually NCX working in ‘reverse’ mode where the exchanger is thought to extrude Na+ in exchange for an increase in [Ca2+]i. This was the suggested explanation for the small increase in resting [Ca2+]i observed after removal of extracellular Na+ in rat and mouse DRG (Duchen, 1990; Verdru et al. 1997). NCX exchange depends on the driving force on the exchanged ions defined as the difference between the membrane potential and the ‘equilibrium potential’ for Na+ and Ca2+ across the exchanger (ENa–Ca). ENa–Ca is estimated by the Nernst potentials for the Na+ and Ca2+ and the net flux of ions by the exchanger: three Na+ in one direction for every one Ca2+ in the other (Fang et al. 1998). Under physiological conditions ENa–Ca is ∼ −26 mV, resulting in a driving force that enables the exchanger to function in forward mode at membrane potentials more negative than −26 mV. Theoretically, with increases in intracellular Na+, as may occur with high levels of neural activity, ENa–Ca may hyperpolarize to a potential less than the resting membrane potential. As a result, the driving force on the exchanger would enable it to function in reverse mode. By this reasoning, the Na+-free bath used to assess the presence of resting NCX activity should result in an ENa–Ca that is very negative to the resting membrane potential, generating a tremendous driving force on the exchanger to work in reverse mode. However, there are at least three observations that argue against the possibility that NCX functioning in reverse mode accounts for the increase in [Ca2+]i observed following application of Na+-free bath. First, we failed to detect the evidence of resting NCX activity in the majority of putative nociceptive neurons tested despite evidence of functional NCX in all of these neurons. Second, we observed ‘resting activity’ that was not only activated, but resolved in neurons with evidence of persistent evoked activity. Third, as shown in Fig.7, we detected an increase in [Ca2+]i in the presence of the NCX specific blocker, KB-R7943 comparable to that observed in Na+-free bath. Thus, rather than NCX acting in reverse mode, we would suggest that the increase in [Ca2+]i observed in the presence of 0 Na+ bath or KB-R7943 is due to Ca2+ leak that is no longer being attenuated by the NCX activity in forward mode. Nevertheless, conclusively ruling out the possibility of NCX functioning in reverse mode in sensory neurons will require more direct measurements of NCX function under conditions in which the concentrations of Na+ and Ca2+ on either side of the membrane are better controlled.
Isoform(s) underlying NCX activity in the isolated neuron
We used complementary strategies to determine which isoforms contribute to resting and evoked NCX activity in sensory neurons. Consistent with the suggestion that differential expression of the three NCX isoforms accounts for the heterogeneity in the biophysical properties of NCX in sensory neurons, pharmacological analysis of NCX suggested that NCX2 and 3 contributed to both resting and evoked NCX activity. Also consistent with this finding, PCR analysis indicated the presence of mRNA encoding both NCX2 and NCX3 in putative nociceptive sensory neurons and our siRNA data indicating that knockdown of NCX2 or 3 was sufficient to occlude the actions of 0 Na+ bath.
Contrary to our PCR analysis suggesting NCX1 mRNA is expressed in putative nociceptive neurons, we failed to detect evidence of NCX1 activity in the isolated sensory neuron cell body with the relatively selective NCX1 selective blocker SEA0400 (Iwamoto & Shigekawa, 1998) or with siRNA-induced knockdown of NCX1. This was even more surprising in light of evidence that NCX1 appears to be ubiquitous with splice variants found in brain, heart, kidney and pancreas (Philipson et al. 1993). And while incomplete knockdown could explain the negative results with NCX1 targeted siRNA, inhibition of [Na+]i-dependent 45Ca2+ uptake (i.e. reverse mode) in fibroblasts (Iwamoto & Shigekawa, 1998) suggests that the concentration of SEA0400 used here should have been sufficient to enable detection of NCX1 activity in sensory neurons. We therefore suggest that the failure to detect evidence of functional NCX1 activity in sensory neurons is due to one of three likely possibilities: (1) the [Ca2+]i generated in response to high K+ was insufficient to activate NCX1 in the cell body, (2) the protein is synthesized and trafficked out of the cell body consistent with the Western blot data discussed below and/or (3) the mRNA does not result in functional protein, although this is least likely given our Western blot analysis. Additional experiments will be needed to distinguish between these possibilities.
There was also an apparent discrepancy between results obtained with the relatively selective NCX3 blocker KB-R7943 and siRNA-induced knockdown of NCX2 and NCX3. While there is evidence to suggest that the potency of KB-R7943-induced block of NCX is comparable when the exchanger is functioning in forward or reverse mode under conditions enabling bi-directional exchanger activity (Watanabe et al. 2006), the relative potency of these compounds was determined in reverse mode (Iwamoto & Shigekawa, 1998). Thus, it is possible that even the limited selectivity for NCX3 over NCX1 or NCX2 reported for KB-R7943 in reverse mode is lost in forward mode. Furthermore, with only three times higher potency for NCX3 over NCX1 and 2, there should be at least some block of NCX2 at all concentrations of KB-R7943. Therefore, we have probably underestimated the relative contribution of NCX2 with the combination of blockers employed. More perplexing, however, was the observation that knocking down either NCX2 or NCX3 rendered the T50 insensitive to KB-R7943. This observation raises the possibility that full NCX activity requires the interaction of both NCX2 and NCX3 isoforms. It is also possible that there are compensatory changes associated with siRNA knockdown of a protein such as NCX that is critical for cell homeostasis which masked our ability to fully characterize the relative contribution of NCX isoforms. Consistent with this last possibility was the observation that following siRNA knockdown of NCX2 or 3, there was no increase in the duration of the evoked transient as is observed following acute block of NCX activity. This suggests an increase in other Ca2+ regulatory mechanisms such as PMCA or SERCA may have been recruited to compensate for the loss of NCX activity.
Identification of NCX isoforms within sensory tissues
The results of our PCR experiments are consistent with the presence of splice variants for both NCX1 and NCX3 in sensory neurons. In contrast, there was no evidence of NCX2 splice variants at either the mRNA or protein level, consistent with previous results (Kofuji et al. 1994; Quednau et al. 2004). The PCR analysis focused on exons A and B, which have previously been shown to be mutually exclusive, whereas the four other exons (C, D, E and F) in the splicing region are thought to be cassette exons (Kofuji et al. 1994). The present data are consistent with the expression of at least six out of the 17 possible variants that have been identified for NCX1. Three of the possible five NCX3 variants identified in brain are denoted NCX3-AC, NCX3-B, and NCX3-BC. The PCR product sizes we detected at the single cell level are consistent with the presence of NCX3-AC and NCX3-B splice variants (Quednau et al. 1997; Michel et al. 2014).
The size of the splicing region within the intracellular loop is relatively small, consisting of ∼250 amino acids, and the mutually exclusive exons A and B do not differ much in the relative number of base pairs. Therefore, it would be very difficult to differentiate between isoform splice variants at the protein level. Nevertheless, multiple NCX1- and NCX3-, but not NCX2-LI bands were detected with Western blot analysis of peripheral neural tissue. The additional bands larger than the predicted molecular weight of NCX1 suggest the possibility of tissue-dependent post-translational modifications, as a number of putative glycosylation sites are present on putative extracellular regions of all three isoforms. The bands below the predicted NCX molecular weight are probably cleavage products, as the bands appear to be the size of the large intracellular loop, a region particularly prone to proteolytic cleavage by endogenous proteases (Linck et al. 1998; Thurneysen et al. 2002b). While confirmation that these additional bands are in fact NCX protein will ultimately be necessary, it is important to note that the bands detected in the present study are comparable to those previously reported with the antibodies used for all three isoforms and at least NCX2 and NCX3 specificity has been confirmed in knockout mice (Jeon et al. 2003; Molinaro et al. 2011). Thus, if reflective of differential processing and/or protein cleavage, results from the present analysis of tissue from the dorsal horn to the peripheral nerve suggest that there is a differential distribution of NCX isoforms.
The expression and distribution of NCX has yet to be extensively characterized in the peripheral nervous system. Consistent with the results of the present study, previous expression analysis indicates mRNA for all three NCX isoforms is detectable in sensory neurons (Kuroda et al. 2013; Shutov et al. 2013). Other immunohistochemical (IHC) data indicating that NCX1-LI protein is only detectable in satellite cells within the DRG are also consistent with the results of the present study (Persson et al. 2010). In contrast, however, previous IHC data on the distribution of NCX2 and 3 suggests that NCX2 is the dominant isoform in putative nociceptive neurons and NCX3 is more widely distributed but at much lower levels (Persson et al. 2010). To begin to explore the basis for the apparent difference between these previous results and those of the present study, we probed protein from DRG, sciatic nerve and skin with the anti-NCX2 antibody used in this previous study. However, we were only able to detect a band at 36 kDa (not shown), suggestive of an NCX ancillary protein (Michaelis et al. 1992) as the estimated size of NCX2 protein is 102 kDa. Nonetheless, the present results with an antibody obtained from Dr Philipson at UCLA were consistent with previous results in brain tissue (Thurneysen et al. 2002a; Papa et al. 2003).
Functional implications: soma excitability, CAP propagation and nociceptive behaviour
The results of our functional analysis of NCX suggests that beyond the regulation of the Ca2+ transient and possibly resting Ca2+ in the soma of a small subpopulation of neurons, NCX contributes minimally to the regulation of excitability. As noted above, there are two primary ways NCX is likely to influence neuronal excitability. We have previously demonstrated that Ca2+-dependent K+ currents are present at relatively high density in putative nociceptive cutaneous neurons (Zhang et al. 2010), suggesting that, as with the direct influence of NCX activity in forward mode, an indirect influence would also be excitatory, secondary to the net suppression of these K+ channels. Consistent with the suggestion that Ca2+-dependent K+ channels have a dominant influence on the excitability of putative nociceptive neurons following an increase in [Ca2+]i, the depolarization-induced increase in [Ca2+]i was associated with a decrease in excitability. Our largely negative results in the cell body excitability experiments suggest that mechanisms other than those influenced by NCX play a much more dominant role in the regulation of the excitability of the isolated cell body. Consistent with this suggestion, it was only under relatively extreme conditions (40 Hz stimulation), that a weak excitatory influence of NCX could be detected.
The impact of NCX on action potential propagation in the isolated sciatic nerve was consistent with the mechanisms described in the cell body, but suggested that NCX is active even at relatively low levels of activity. That is, if both direct and indirect actions of NCX activity are associated with membrane depolarization, this depolarization would lead to the inactivation of voltage-gated Na+ channels, and a concomitant decrease in CV. Conversely, block of NCX should result in membrane hyperpolarization, the relief of Na+ channel inactivation and an increase in CV. Li+ bath was associated with a relatively large (∼24%) decrease in the C-wave CV when the CAP was evoked at a very low frequency (i.e. 0.017 Hz). We suggest that this decrease in CV is due not to NCX block, but instead to what is equivalent to a partial block of voltage-gated Na+ channels that reflects the slightly lower permeability (85%) of Li+ compared to Na+ through voltage-gated Na+ channels (Gold & Thut, 2001). There is also evidence that there may be CV slowing due to a block of the Na+–K+ ATPase (De Col et al. 2008), resulting in further membrane depolarization and a further reduction in Na+ channel density due to Na+ channel inactivation. Thus, in the face of Li+-induced CV slowing via mechanisms independent of an action on NCX, we suggest that absence of a change in CV with Li+ at 0.1 Hz stimulation, actually reflects a significant increase in CV due to NCX block. That this effect appears to be attenuated at 5 Hz stimulation, would suggest that NCX activity is close to maximal even at 0.1 Hz stimulation. Consistent with this suggestion, KB-R7943 produced a significant increase in CV at 0.1 Hz but only reversed the activity-dependent slowing observed at 5 Hz.
Lastly, given evidence in support of a contribution of both NCX2 and 3 to the regulation of [Ca2+]i in putative nociceptive cutaneous neurons, it was surprising that hypersensitivity to mechanical and thermal stimuli was only observed following knockdown of NCX3. While this observation is consistent with our pharmacological data implicating a dominant role for NCX3 in putative nociceptive neurons, this result may also suggest that the functional role of these isoforms in sensory neurons may be dissociable. Importantly, the timing of the changes observed is consistent with previous studies using similar siRNA based strategies (Xie et al. 2013; Acosta et al. 2014), and recovery of the behaviour corresponded with recovery of NCX3 protein in the ganglia.
The decrease in nociceptive threshold observed following NCX3 knockdown was larger than expected given the relatively small number of fibres in which our results suggested NCX3 should be functional. However, the observation that nociceptive behaviour of rats treated with both NCX3 siRNA and IB4 conjugated to saporin (IB4–SAP) resembled the rats treated with control siRNA and IB4–SAP suggests that the loss of IB4 fibres attenuated the NCX3 siRNA-induced hypersensitivity. If the hypersensitivity had been due to dysregulation of fibres in addition to the IB4+ population, one would have expected an additive effect in which the increase in nociceptive threshold observed with IB4–SAP would have been significantly attenuated by the NCX3 siRNA. Nevertheless, it is possible that the antinociception associated with the loss of IB4+ fibres caused a ceiling effect, masking the NCX3 siRNA-induced hypersensitivity. We consider this possibility unlikely, however, as the IB4–SAP-induced thermal antinociception detected was far from 20 s cut off employed.
There are several possible explanations for the hypersensitivity observed following knockdown of NCX3. However, we suggest the simplest explanation is a loss of NCX-mediated regulation of [Ca2+]i in the central terminals. This would result in an increase in the duration of evoked Ca2+ transients and, consequently, an increase in transmitter release, and thus hypersensitivity. While such a mechanism is often proposed in the literature (Maggi et al. 1990; Evans et al. 1996; Neher & Sakaba, 2008; Xie et al. 2013; Acosta et al. 2014), there is, unfortunately little direct evidence for a role of NCX in synaptic transmission. However, in the only study of which we are aware addressing a role of NCX in the central terminals of putative nociceptive afferents, it was demonstrated that mitochondria and PMCA, but not NCX, are involved in presynaptic Ca2+ regulation; NCX appeared to contribute to the regulation of resting Ca2+ levels and only 12% of the decay of evoked transients (Shutov et al. 2013). Unfortunately, the impact of NCX block on post-synaptic responses was not determined in this study, so the contribution of NCX to transmitter release from central terminals of nociceptive afferents has yet to be determined.
In summary, the results of the present study indicate that NCX isoforms contribute to the regulation of the duration of the evoked Ca2+ transient in putative nociceptive neurons from naive animals, and in a subpopulation of these neurons NCX contributes to the regulation of resting [Ca2+]i. In the absence of tissue injury, NCX appears to have little influence on excitability per se, but contributes to both determination of the action potential conduction velocity in C-fibre axons as well as the establishment of nociceptive threshold. Our characterization of this protein in tissue from naive animals suggests that it is poised to play an important role in tissue injury and repair. Indeed, it has been suggested the NCX contributes to nerve injury observed in small fibre polyneuropathies (Ma, 2013) and there is evidence to suggest it is important for neurite outgrowth during regeneration (Persson et al. 2013). Now, with a better mechanistic understanding of NCX in sensory neurons, there is a framework with which to interpret changes observed following injury. Future experiments will need to assess the function of NCX in the presence of injury and/or chronic pain states.
Acknowledgments
We thank Drs Ken Philipson and Michela Ottolia at the Cedars-Sinai Heart Institute for the very generous supply of NCX antibodies as well as insight into the execution of these experiments. We thank Mr Parthasarathy Kesavaramanujam for his expert technical assistance. We also thank Drs Gerald Gebhart, Brian Davis, Steve Meriney and William de Groat for their constructive feedback during the preparation of this manuscript.
Glossary
- AP
action potential
- Cap
capsaicin
- CAP
compound action potential
- [Ca2+]i
the concentration of intracellular Ca2+
- CV
conduction velocity
- DMSO
dimethyl sulfoxide
- DRG
dorsal root ganglia
- IB4
isolectin B4
- NCX
Na+–Ca2+ exchanger
- PMCA
plasma membrane ATPase
- RT-PCR
reverse transcriptase polymerase chain reaction
- SAP
saporin
- SERCA
sarco-endoplasmic reticulum Ca2+ ATPase
Additional information
Competing interests
The authors have no conflicts of interest.
Author contributions
Conception and design of the experiments was done by N.N.S. and M.S.G. All data were collected and analysed by N.N.S. and E.Y. The manuscript was prepared and written by N.N.S. and M.S.G. All authors read and approved the submitted version of this manuscript.
Funding
This work is supported by the NIH grant F31-NS08466-1 and the Virginia Kaufman Fund.
Key points
There has been little if any systematic analysis of the Na+–Ca2+ exchanger (NCX) in sensory neurons despite conflicting results in the literature regarding the extent to which it contributes to the regulation of intracellular Ca2+ in these neurons.
While the differential distribution and/or biophysical properties of NCX isoforms may contribute to these conflicting results, only indirect evidence points to the consequences of NCX activity on afferent function.
NCX activity is restricted to a subpopulation of putative nociceptive neurons.
All three NCX isoforms are expressed in putative nociceptive afferents and appear to be differentially distributed along major neuron compartments: central axon, cell body and peripheral axon.
NCX 3 plays a dominant role in the regulation of the duration of the evoked Ca2+ transient in the cell body, regulation of the action potential conduction velocity, and establishment of nociceptive threshold.
References
- Acosta C, Djouhri L, Watkins R, Berry C, Bromage K. Lawson SN. TREK2 expressed selectively in IB4-binding C-fiber nociceptors hyperpolarizes their membrane potentials and limits spontaneous pain. J Neurosci. 2014;34:1494–1509. doi: 10.1523/JNEUROSCI.4528-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acsai K, Antoons G, Livshitz L, Rudy Y. Sipido KR. Microdomain [Ca2+] near ryanodine receptors as reported by L-type Ca2+ and Na+–Ca2+ exchange currents. J Physiol. 2011;589:2569–2583. doi: 10.1113/jphysiol.2010.202663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annunziato L, Pignataro G. Di Renzo GF. Pharmacology of brain Na+/Ca2+ exchanger: from molecular biology to therapeutic perspectives. Pharmacol Rev. 2004;56:633–654. doi: 10.1124/pr.56.4.5. [DOI] [PubMed] [Google Scholar]
- Berridge MJ, Lipp P. Bootman MD. The versatility and universality of calcium signalling. Nat Rev Molec Cell Biol. 2000;1:11–21. doi: 10.1038/35036035. [DOI] [PubMed] [Google Scholar]
- Birinyi P, Acsai K, Banyasz T, Toth A, Horvath B, Virag L, Szentandrassy N, Magyar J, Varro A, Fulop F. Nanasi PP. Effects of SEA0400 and KB-R7943 on Na+/Ca2+ exchange current and L-type Ca2+ current in canine ventricular cardiomyocytes. Naunyn Schmiedebergs Arch Pharmacol. 2005;372:63–70. doi: 10.1007/s00210-005-1079-x. [DOI] [PubMed] [Google Scholar]
- Blaustein MP. Lederer WJ. Sodium/calcium exchange: its physiological implications. Physiol Rev. 1999;79:763–854. doi: 10.1152/physrev.1999.79.3.763. [DOI] [PubMed] [Google Scholar]
- Boyman L, Hagen BM, Giladi M, Hiller R, Lederer WJ. Khananshvili D. Proton-sensing Ca2+ binding domains regulate the cardiac Na+/Ca2+ exchanger. J Biol Chem. 2011;286:28811–28820. doi: 10.1074/jbc.M110.214106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook O, Low W. Rahamimoff H. Membrane topology of the rat brain Na+–Ca2+ exchanger. Biochim Biophys Acta. 1998;1371:40–52. doi: 10.1016/s0005-2736(97)00272-1. [DOI] [PubMed] [Google Scholar]
- De Col R, Messlinger K. Carr RW. Conduction velocity is regulated by sodium channel inactivation in unmyelinated axons innervating the rat cranial meninges. J Physiol. 2008;586:1089–1103. doi: 10.1113/jphysiol.2007.145383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiPolo R. Beauge L. Sodium/calcium exchanger: influence of metabolic regulation on ion carrier interactions. Physiol Rev. 2006;86:155–203. doi: 10.1152/physrev.00018.2005. [DOI] [PubMed] [Google Scholar]
- Duchen MR( Effects of metabolic inhibition on the membrane properties of isolated mouse primary sensory neurones. J Physiol. 1990;424:387–409. doi: 10.1113/jphysiol.1990.sp018073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans AR, Nicol GD. Vasko MR. Differential regulation of evoked peptide release by voltage-sensitive calcium channels in rat sensory neurons. Brain Res. 1996;712:265–273. doi: 10.1016/0006-8993(95)01447-0. [DOI] [PubMed] [Google Scholar]
- Fang Y, Condrescu M. Reeves JP. Regulation of Na+/Ca2+ exchange activity by cytosolic Ca2+ in transfected Chinese hamster ovary cells. Am J Physiol Cell Physiol. 1998;275:C50–C55. doi: 10.1152/ajpcell.1998.275.1.C50. [DOI] [PubMed] [Google Scholar]
- Formisano L, Saggese M, Secondo A, Sirabella R, Vito P, Valsecchi V, Molinaro P, Di Renzo G. Annunziato L. The two isoforms of the Na+/Ca2+ exchanger, NCX1 and NCX3, constitute novel additional targets for the prosurvival action of Akt/protein kinase B pathway. Mol Pharmacol. 2008;73:727–737. doi: 10.1124/mol.107.042549. [DOI] [PubMed] [Google Scholar]
- Gemes G, Oyster KD, Pan B, Wu HE, Bangaru ML, Tang Q. Hogan QH. Painful nerve injury increases plasma membrane Ca2+-ATPase activity in axotomized sensory neurons. Mol Pain. 2012;8:46. doi: 10.1186/1744-8069-8-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold MS. Thut PD. Lithium increases potency of lidocaine-induced block of voltage-gated Na+ currents in rat sensory neurons in vitro. J Pharmacol Exp Ther. 2001;299:705–711. [PubMed] [Google Scholar]
- Gover TD, Moreira TH, Kao JP. Weinreich D. Calcium regulation in individual peripheral sensory nerve terminals of the rat. J Physiol. 2007;578:481–490. doi: 10.1113/jphysiol.2006.119008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M. Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- Hargreaves K, Dubner R, Brown F, Flores C. Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32:77–88. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
- Herbert CP, Wright JM, Maclure M, Wakefield J, Dormuth C, Brett-MacLean P, Legare J. Premi J. Better Prescribing Project: a randomized controlled trial of the impact of case-based educational modules and personal prescribing feedback on prescribing for hypertension in primary care. Fam Pract. 2004;21:575–581. doi: 10.1093/fampra/cmh515. [DOI] [PubMed] [Google Scholar]
- Hermans AN, Glitsch HG. Verdonck F. Activation of the Na+/K+ pump current by intra- and extracellular Li ions in single guinea-pig cardiac cells. Biochim Biophys Acta. 1997;1330:83–93. doi: 10.1016/s0005-2736(97)00143-0. [DOI] [PubMed] [Google Scholar]
- Hilge M( Ca2+ regulation of ion transport in the Na+/Ca2+ exchanger. J Biol Chem. 2012;287:31641–31649. doi: 10.1074/jbc.R112.353573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilge M( Ca2+ regulation in the Na+/Ca2+ exchanger features a dual electrostatic switch mechanism. Adv Exp Med Biol. 2013;961:27–33. doi: 10.1007/978-1-4614-4756-6_3. [DOI] [PubMed] [Google Scholar]
- Hilge M, Aelen J, Foarce A, Perrakis A. Vuister GW. Ca2+ regulation in the Na+/Ca2+ exchanger features a dual electrostatic switch mechanism. Proc Natl Acad Sci U S A. 2009;106:14333–14338. doi: 10.1073/pnas.0902171106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilgemann DW, Matsuoka S, Nagel GA. Collins A. Steady-state and dynamic properties of cardiac sodium–calcium exchange. Sodium-dependent inactivation. J Gen Physiol. 1992;100:905–932. doi: 10.1085/jgp.100.6.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hille B. The permeability of the sodium channel to metal cations in myelinated nerve. J Gen Physiol. 1972;59:637–658. doi: 10.1085/jgp.59.6.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwamoto T. Shigekawa M. Differential inhibition of Na+/Ca2+ exchanger isoforms by divalent cations and isothiourea derivative. Am J Physiol Cell Physiol. 1998;275:C423–C430. doi: 10.1152/ajpcell.1998.275.2.C423. [DOI] [PubMed] [Google Scholar]
- Iwamoto T, Watano T. Shigekawa M. A novel isothiourea derivative selectively inhibits the reverse mode of Na+/Ca2+ exchange in cells expressing NCX1. J Biol Chem. 1996;271:22391–22397. doi: 10.1074/jbc.271.37.22391. [DOI] [PubMed] [Google Scholar]
- Jeon D, Yang YM, Jeong MJ, Philipson KD, Rhim H. Shin HS. Enhanced learning and memory in mice lacking Na+/Ca2+ exchanger 2. Neuron. 2003;38:965–976. doi: 10.1016/s0896-6273(03)00334-9. [DOI] [PubMed] [Google Scholar]
- Kao JP( Practical aspects of measuring [Ca2+] with fluorescent indicators. In: Nuccitelli, editor. A Practical Guide to the Study of Calcium in Living Cells. San Diego, CA, USA: Academic Press; 1994. pp. 155–181. [DOI] [PubMed] [Google Scholar]
- Kofuji P, Lederer WJ. Schulze DH. Mutually exclusive and cassette exons underlie alternatively spliced isoforms of the Na/Ca exchanger. J Biol Chem. 1994;269:5145–5149. [PubMed] [Google Scholar]
- Kuroda H, Sobhan U, Sato M, Tsumura M, Ichinohe T, Tazaki M. Shibukawa Y. Sodium–calcium exchangers in rat trigeminal ganglion neurons. Mol Pain. 2013;9:22. doi: 10.1186/1744-8069-9-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linck B, Qiu Z, He Z, Tong Q, Hilgemann DW. Philipson KD. Functional comparison of the three isoforms of the Na+/Ca2+ exchanger (NCX1, NCX2, NCX3) Am J Physiol Cell Physiol. 1998;274:C415–C423. doi: 10.1152/ajpcell.1998.274.2.C415. [DOI] [PubMed] [Google Scholar]
- Lu SG, Zhang X. Gold MS. Intracellular calcium regulation among subpopulations of rat dorsal root ganglion neurons. J Physiol. 2006;577:169–190. doi: 10.1113/jphysiol.2006.116418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma M. Role of calpains in the injury-induced dysfunction and degeneration of the mammalian axon. Neurobiol Dis. 2013;60:61–79. doi: 10.1016/j.nbd.2013.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maggi CA, Tramontana M, Cecconi R. Santicioli P. Neurochemical evidence for the involvement of N-type calcium channels in transmitter secretion from peripheral endings of sensory nerves in guinea pigs. Neurosci Lett. 1990;114:203–206. doi: 10.1016/0304-3940(90)90072-h. [DOI] [PubMed] [Google Scholar]
- Michaelis ML, Nunley EW, Jayawickreme C, Hurlbert M, Schueler S. Guilly C. Purification of a synaptic membrane Na+/Ca2+ antiporter and immunoextraction with antibodies to a 36-kDa protein. J Neurochem. 1992;58:147–157. doi: 10.1111/j.1471-4159.1992.tb09290.x. [DOI] [PubMed] [Google Scholar]
- Michel LY, Verkaart S, Koopman WJ, Willems PH, Hoenderop JG. Bindels RJ. Function and regulation of the Na+–Ca2+ exchanger NCX3 splice variants in brain and skeletal muscle. J Biol Chem. 2014;289:11293–11303. doi: 10.1074/jbc.M113.529388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molinaro P, Viggiano D, Nistico R, Sirabella R, Secondo A, Boscia F, Pannaccione A, Scorziello A, Mehdawy B, Sokolow S, Herchuelz A, Di Renzo GF. Annunziato L. Na+–Ca2+ exchanger (NCX3) knock-out mice display an impairment in hippocampal long-term potentiation and spatial learning and memory. J Neurosci. 2011;31:7312–7321. doi: 10.1523/JNEUROSCI.6296-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nealen ML, Gold MS, Thut PD. Caterina MJ. TRPM8 mRNA is expressed in a subset of cold-responsive trigeminal neurons from rat. J Neurophysiol. 2003;90:515–520. doi: 10.1152/jn.00843.2002. [DOI] [PubMed] [Google Scholar]
- Neher E. Sakaba T. Multiple roles of calcium ions in the regulation of neurotransmitter release. Neuron. 2008;59:861–872. doi: 10.1016/j.neuron.2008.08.019. [DOI] [PubMed] [Google Scholar]
- Niu CF, Watanabe Y, Iwamoto T, Yamashita K, Satoh H, Urushida T, Hayashi H. Kimura J. Electrophysiological effects of SN-6, a novel Na+/Ca2+ exchange inhibitor on membrane currents in guinea pig ventricular myocytes. Ann N Y Acad Sci. 2007;1099:534–539. doi: 10.1196/annals.1387.037. [DOI] [PubMed] [Google Scholar]
- Palty R, Silverman WF, Hershfinkel M, Caporale T, Sensi SL, Parnis J, Nolte C, Fishman D, Shoshan-Barmatz V, Herrmann S, Khananshvili D. Sekler I. NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proc Natl Acad Sci U S A. 2010;107:436–441. doi: 10.1073/pnas.0908099107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papa M, Canitano A, Boscia F, Castaldo P, Sellitti S, Porzig H, Taglialatela M. Annunziato L. Differential expression of the Na+–Ca2+ exchanger transcripts and proteins in rat brain regions. J Comp Neurol. 2003;461:31–48. doi: 10.1002/cne.10665. [DOI] [PubMed] [Google Scholar]
- Persson AK, Black JA, Gasser A, Cheng X, Fischer TZ. Waxman SG. Sodium–calcium exchanger and multiple sodium channel isoforms in intra-epidermal nerve terminals. Mol Pain. 2010;6:84. doi: 10.1186/1744-8069-6-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persson AK, Kim I, Zhao P, Estacion M, Black JA. Waxman SG. Sodium channels contribute to degeneration of dorsal root ganglion neurites induced by mitochondrial dysfunction in an in vitro model of axonal injury. J Neurosci. 2013;33:19250–19261. doi: 10.1523/JNEUROSCI.2148-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philipson KD, Nicoll DA. Li Z. The cardiac sodium–calcium exchanger. Soc Gen Physiol Ser. 1993;48:187–191. [PubMed] [Google Scholar]
- Quednau BD, Nicoll DA. Philipson KD. Tissue specificity and alternative splicing of the Na+/Ca2+ exchanger isoforms NCX1, NCX2, and NCX3 in rat. Am J Physiol Cell Physiol. 1997;272:C1250–C1261. doi: 10.1152/ajpcell.1997.272.4.C1250. [DOI] [PubMed] [Google Scholar]
- Quednau BD, Nicoll DA. Philipson KD. The sodium/calcium exchanger family-SLC8. Pflugers Arch. 2004;447:543–548. doi: 10.1007/s00424-003-1065-4. [DOI] [PubMed] [Google Scholar]
- Rigaud M, Gemes G, Weyker PD, Cruikshank JM, Kawano T, Wu HE. Hogan QH. Axotomy depletes intracellular calcium stores in primary sensory neurons. Anesthesiology. 2009;111:381–392. doi: 10.1097/ALN.0b013e3181ae6212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheff NN. Gold MS. Sex differences in the inflammatory mediator-induced sensitization of dural afferents. J Neurophysiol. 2011;106:1662–1668. doi: 10.1152/jn.00196.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheff NN, Lu SG. Gold MS. Contribution of endoplasmic reticulum Ca2+ regulatory mechanisms to the inflammation-induced increase in the evoked Ca2+ transient in rat cutaneous dorsal root ganglion neurons. Cell Calcium. 2013;54:46–56. doi: 10.1016/j.ceca.2013.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulze DH, Polumuri SK, Gille T. Ruknudin A. Functional regulation of alternatively spliced Na+/Ca2+ exchanger (NCX1) isoforms. Ann N Y Acad Sci. 2002;976:187–196. doi: 10.1111/j.1749-6632.2002.tb04740.x. [DOI] [PubMed] [Google Scholar]
- Shutov LP, Kim MS, Houlihan PR, Medvedeva YV. Usachev YM. Mitochondria and plasma membrane Ca2+-ATPase control presynaptic Ca2+ clearance in capsaicin-sensitive rat sensory neurons. J Physiol. 2013;591:2443–2462. doi: 10.1113/jphysiol.2012.249219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirabella R, Secondo A, Pannaccione A, Molinaro P, Formisano L, Guida N, Di Renzo G, Annunziato L. Cataldi M. ERK1/2, p38, and JNK regulate the expression and the activity of the three isoforms of the Na+–Ca2+ exchanger, NCX1, NCX2, and NCX3, in neuronal PC12 cells. J Neurochem. 2012;122:911–922. doi: 10.1111/j.1471-4159.2012.07838.x. [DOI] [PubMed] [Google Scholar]
- Tarpley JW, Kohler MG. Martin WJ. The behavioral and neuroanatomical effects of IB4–saporin treatment in rat models of nociceptive and neuropathic pain. Brain Res. 2004;1029:65–76. doi: 10.1016/j.brainres.2004.09.027. [DOI] [PubMed] [Google Scholar]
- Thayer SA. Miller RJ. Regulation of the intracellular free calcium concentration in single rat dorsal root ganglion neurones in vitro. J Physiol. 1990;425:85–115. doi: 10.1113/jphysiol.1990.sp018094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thayer SA, Usachev YM. Pottorf WJ. Modulating Ca2+ clearance from neurons. Front Biosci. 2002;7:d1255–1279. doi: 10.2741/A838. [DOI] [PubMed] [Google Scholar]
- Thurneysen T, Nicoll DA, Philipson KD. Porzig H. Immunohistochemical detection of the sodium–calcium exchanger in rat hippocampus cultures using subtype-specific antibodies. Ann N Y Acad Sci. 2002a;976:367–375. doi: 10.1111/j.1749-6632.2002.tb04763.x. [DOI] [PubMed] [Google Scholar]
- Thurneysen T, Nicoll DA, Philipson KD. Porzig H. Sodium/calcium exchanger subtypes NCX1, NCX2 and NCX3 show cell-specific expression in rat hippocampus cultures. Brain Res Mol Brain Res. 2002b;107:145–156. doi: 10.1016/s0169-328x(02)00461-8. [DOI] [PubMed] [Google Scholar]
- Tolkovsky AM. Richards CD. Na+/H+ exchange is the major mechanism of pH regulation in cultured sympathetic neurons: measurements in single cell bodies and neurites using a fluorescent pH indicator. Neuroscience. 1987;22:1093–1102. doi: 10.1016/0306-4522(87)92984-8. [DOI] [PubMed] [Google Scholar]
- Usachev YM, Marsh AJ, Johanns TM, Lemke MM. Thayer SA. Activation of protein kinase C in sensory neurons accelerates Ca2+ uptake into the endoplasmic reticulum. J Neurosci. 2006;26:311–318. doi: 10.1523/JNEUROSCI.2920-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughn AH. Gold MS. Ionic mechanisms underlying inflammatory mediator-induced sensitization of dural afferents. J Neurosci. 2010;30:7878–7888. doi: 10.1523/JNEUROSCI.6053-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdru P, De Greef C, Mertens L, Carmeliet E. Callewaert G. Na+–Ca2+ exchange in rat dorsal root ganglion neurons. J Neurophysiol. 1997;77:484–490. doi: 10.1152/jn.1997.77.1.484. [DOI] [PubMed] [Google Scholar]
- Vulchanova L, Olson TH, Stone LS, Riedl MS, Elde R. Honda CN. Cytotoxic targeting of isolectin IB4-binding sensory neurons. Neuroscience. 2001;108:143–155. doi: 10.1016/s0306-4522(01)00377-3. [DOI] [PubMed] [Google Scholar]
- Wan QF, Nixon E. Heidelberger R. Regulation of presynaptic calcium in a mammalian synaptic terminal. J Neurophysiol. 2012;108:3059–3067. doi: 10.1152/jn.00213.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe Y, Koide Y. Kimura J. Topics on the Na+/Ca2+ exchanger: pharmacological characterization of Na+/Ca2+ exchanger inhibitors. J Pharmacol Sci. 2006;102:7–16. doi: 10.1254/jphs.fmj06002x2. [DOI] [PubMed] [Google Scholar]
- Werth JL. Thayer SA. Mitochondria buffer physiological calcium loads in cultured rat dorsal root ganglion neurons. J Neurosci. 1994;14:348–356. doi: 10.1523/JNEUROSCI.14-01-00348.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie W, Strong JA, Ye L, Mao JX. Zhang JM. Knockdown of sodium channel NaV1.6 blocks mechanical pain and abnormal bursting activity of afferent neurons in inflamed sensory ganglia. Pain. 2013;154:1170–1180. doi: 10.1016/j.pain.2013.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yilmaz-Rastoder E, Gold MS, Hough KA, Gebhart GF. Williams BA. Effect of adjuvant drugs on the action of local anesthetics in isolated rat sciatic nerves. Reg Anesth Pain Med. 2012;37:403–409. doi: 10.1097/AAP.0b013e3182485965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X-L, Mok L, Charbonnet M, Lee K-Y. Gold MS. Inflammation-induced changes in BKCa currents in cutaneous dorsal root ganglion neurons from the adult rat. Mol Pain. 2012;8 doi: 10.1186/1744-8069-8-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XL, Mok LP, Katz EJ. Gold MS. BKCa currents are enriched in a subpopulation of adult rat cutaneous nociceptive dorsal root ganglion neurons. Eur J Neurosci. 2010;31:450–462. doi: 10.1111/j.1460-9568.2009.07060.x. [DOI] [PMC free article] [PubMed] [Google Scholar]