

Abstract
Using electrophysiological and optical techniques, we studied the mechanisms by which cholesterol depletion stimulates spontaneous transmitter release by exocytosis at the frog neuromuscular junction. We found that methyl-β-cyclodextrin (MCD, 10 mm)-mediated exhaustion of cholesterol resulted in the enhancement of reactive oxygen species (ROS) production, which was prevented by the antioxidant N-acetyl cysteine (NAC) and the NADPH oxidase inhibitor apocynin. An increase in ROS levels occurred both extra- and intracellularly, and it was associated with lipid peroxidation in synaptic regions. Cholesterol depletion provoked a rise in the intracellular Ca2+ concentration, which was diminished by NAC and transient receptor potential vanilloid (TRPV) channel blockers (ruthenium red and capsazepine). By contrast, the MCD-induced rise in [Ca2+]i remained unaffected if Ca2+ release from endoplasmic stores was blocked by TMB8 (8-(diethylamino)octyl-3,4,5-trimethoxybenzoate hydrochloride). The effects of cholesterol depletion on spontaneous release and exocytosis were significantly reduced by the antioxidant, intracellular Ca2+ chelation with BAPTA-AM and blockers of TRPV channels. Bath application of the calcineurin antagonist cyclosporine A blocked MCD-induced enhancement of spontaneous release/exocytosis, whereas okadaic acid, an inhibitor of phosphatases PP1 and PP2A, had no effect. Thus, our findings indicate that enhancement of spontaneous exocytosis induced by cholesterol depletion may depend on ROS generation, leading to an influx of Ca2+ via TRPV channels and, subsequently, activation of calcineurin.
Introduction
Evoked neurotransmitter release occurs by means of synaptic vesicle exocytosis and plays a crucial role in synaptic transmission. However, in the resting state, synaptic exocytosis also occurs spontaneously. Excitability of neurons, stability of synaptic connections and local synaptic protein synthesis may all depend on this spontaneous neurotransmitter release (Kaeser & Regehr, 2014). An increase in spontaneous exocytosis may lead to undesirable effects due to emptying of synaptic vesicle pools, desensitization of neurotransmitter receptors and a decrease in protein synthesis at synapses. Taken together, these factors could decrease the efficacy of synaptic transmission (Kavalali & Monteggia, 2012). The mechanisms of spontaneous exocytosis and its regulation are still poorly understood.
It is known that, in addition to fusion and docking proteins, the process of synaptic vesicle exocytosis depends on membrane cholesterol (Puchkov & Haucke, 2013). Cholesterol promotes the formation of lipid rafts at synaptic sites which contain many signalling molecules and ion channels (Jia et al. 2006; Petrov et al. 2011a). Methyl-β-cyclodextrin (MCD) has been shown to induce depletion of membrane cholesterol. This dramatically increases spontaneous release at neuromuscular and central synapses (Zamir & Charlton, 2006; Wasser & Kavalali, 2009; Dason et al. 2010, 2014; Tarakanova et al. 2011; Teixeira et al. 2012). The detailed mechanisms of this phenomenon have not yet been identified. It is possible that they include changes in interactions between lipids and exocytotic proteins and/or in fusion pore stability (Tong et al. 2009). An increase in the activity of signalling molecules (e.g. raft-resident enzymes), which can modulate spontaneous exocytosis, may also result from cholesterol depletion. This possibility is supported by data which demonstrate that the effect of MCD on spontaneous exocytosis is attenuated when several different protein kinases (in particular, type A, C and Ca2+/calmodulin-dependent kinases) are inhibited (Smith et al. 2010; Teixeira et al. 2012).
The production of reactive oxygen species (ROS) by different cell types is closely associated with cholesterol levels and the formation of lipid rafts. NADPH oxidase is one of the main ROS-generating enzymes and this is abundantly expressed at synapses, where it is involved in synaptic plasticity (Tejada-Simon et al. 2005). NADPH oxidase is present in lipid rafts and both cholesterol content, along with the activity of some protein kinases, as well as the concentration of cytosolic Сa2+, may influence its enzymatic activity (Jin et al. 2011). ROS also have a marked impact on Сa2+ homeostasis and protein phosphorylation, and can also contribute to modifications of the proteins involved in exocytosis (Giniatullin et al. 2006; Nishio et al. 2013). The activity of Сa2+-permeable ryanodine receptors and transient potential receptor (TRP) channels is also regulated by ROS. Сa2+ influx into the cytoplasm, mediated by ryanodine receptors and TRP channels, selectively enhances spontaneous exocytosis at several synapses (Shimizu et al. 2008; Song et al. 2011; Andresen et al. 2012; Nishio et al. 2013). Thus, it is possible that the increases in spontaneous exocytosis induced by cholesterol depletion, and ROS–Ca2+ signalling, might be linked.
In this study, we observed an NADPH oxidase-dependent rise in ROS levels in frog muscles, both extra- and intracellularly, under conditions of cholesterol depletion. Increased ROS production led to Ca2+ influx via transient receptor potential vanilloid (TRPV) channels, stimulating spontaneous exocytosis, perhaps through activation of Ca2+-dependent enzymes (phosphatase protein 2B (PP2B), specifically).
Methods
Ethical approval
All animal experiments were performed in accordance with the guidelines for the use of laboratory animals of Kazan State Medical University and conform to the principles of UK regulations, as described in Drummond (2009). The experimental protocol met the requirements of the European Communities Council Directive 86/609/EEC and was approved by the Ethical Committee of Kazan Medical University. Frogs (Rana ridibunda) were killed by decapitation and destruction of the brain and the spinal cord. Muscles were then quickly excised.
Solutions and chemicals
Experiments were performed on the isolated cutaneous pectoris muscles. The muscle with its nerve was pinned to the bottom of a Sylgard-lined glass chamber, which was superfused during the experiment with frog Ringer's solution, containing (in mm): NaCl 113, KCl 2.5, CaCl2 1.8, NaHCO3 2.4. pH was maintained at 7.2–7.4 and the bath temperature at 22–24°С. In all experiments, TTX (1 μm) was added to the external solution to prevent spontaneous generation of action potentials. MCD (10 mm) was used to deplete cell membranes of cholesterol. Complexed cholesterol–MCD (5 mм, cholesterol water soluble) was used for replenishment of membrane cholesterol after its depletion. The osmolarity of the solution was adjusted when MCD was added. In the present work, we used 200 μm N-acetyl cysteine (NAC, antioxidant), 200 μm apocynin (inhibitor of NADPH oxidase), 2 μm ruthenium red (blocker of ryanodine receptors and TRPV channels), 50 μm TMB8 (8-(diethylamino)octyl-3,4,5-trimethoxybenzoate hydrochloride, blocker of inositol trisphosphate and ryanodine receptors), 10 μm capsazepine (TRPV1 channel inhibitor), 200 μm BAPTA-AM (membrane-permeable Ca buffer of high affinity), 50 μm cyclosporine A (calcineurin blocker), 50 nm okadaic acid (inhibitor of phosphatase PP1 and PP2A). NAC, apocynin, ruthenium red, TMB8, capsazepine, cyclosporine A and okadaic acid were added to the external solution 40–50 min before MCD treatment and remained in the bath throughout the experiment. In some experiments, the preparation was pretreated with BAPTA-AM for 2 h, after which BAPTA-AM was washed out for 30 min prior to MCD treatment. All reagents were from Sigma (St Louis, MO, USA), except for FM1–43 and ADVASEP-7 (see below), which were purchased from Biotium (Hayward, CA, USA). The motor nerve was stimulated with a suction electrode and DS3 stimulator (Digitimer Ltd, Welwyn Garden City, UK), using supramaximal electrical pulses of 0.1 ms duration.
Electrophysiology
Recordings of miniature postsynaptic end-plate potentials (MEPPs) were made using standard intracellular microelectrodes (3–5 MΩ resistance) filled with 2.5 m KCl. The recordings were digitized at 50 kHz, stored on a PC and analysed off-line to calculate the frequency of MEPPs. Recording instrumentation consisted of an amplifier, Model 1600 (A-M System, Carlsborg, WA, USA) and LA II digital I/O board (Pushino, Russia) under the control of locally written software.
Fluorescence microscopy
Fluorescence images were acquired using an Olympus BX51WI microscope with a confocal attachment Disk Speed Unit and UPLANSapo 60хw, LumPlanPF 100xw objectives. Images were captured with DP71 (Olympus, Tokyo, Japan) and Orca R2 (Hamamatsu, Welwyn Garden City, UK) CCD cameras. Image analysis was performed using Cell^P (Olympus) and ImagePro software (Media Cybernetics, Bethesda, MD, USA). Multiple z axis optical sections were taken using a focus stepper (ECO-MOT, Märzhäuser Wetzlar GmbH & Co. KG, Wetzlar, Germany). Intensity analysis was made on regions of interest.
Loading and unloading of FM1–43
Fluorescent dye FM1–43 (5 μm) was used to measure the rate of synaptic vesicle exocytosis. This dye reversibly binds to the presynaptic membrane and becomes sequestered in the recycled vesicles within the cytoplasm of nerve terminals during endocytosis. We refer to this as ‘dye loading’. Trains of 20 Hz stimulation for 3 min were used to load FM1–43 into the nerve terminals. The dye was present in the bath during, and 5 min after, stimulation. Following the loading procedure, the preparations were washed with physiological saline containing ADVASEP-7 (3 μm) for 30 min, to decrease background fluorescence (Petrov et al. b2011). This reagent accelerates dissociation of FM1–43 from the surface membranes and decreases non-specific fluorescence. TTX (1 μm) was added to the perfusion solution after dye loading. Stimulation of synaptic vesicle exocytosis (spontaneous or evoked) induces a decrease in fluorescence of dye-loaded nerve terminals (dye unloading). MCD itself can bind FM1–43 (Dason et al. 2010) and in this situation MCD acts as reagent that accelerates dissociation of FM1–43 from surface membranes. To prevent such an influence on the unloading curves we tried to minimize non-specific fluorescence by applying ADVASEP-7.
To record FM1–43 fluorescence intensity we used a 480/20 nm excitation filter, a 505 nm dichroic mirror and a 535/40 nm emission filter. Background fluorescence was estimated as mean fluorescence intensity in a 50 × 50 pixel region (2 × 2 μm2) beside the nerve terminal. Nerve terminal fluorescence intensity was defined the average pixel intensity in selected spots, after subtraction of background fluorescence (for details see Petrov et al. 2008). For analysis of the kinetics of unloading, the initial nerve terminal fluorescence (before MCD treatment) value was taken as 1.0.
Hydrogen peroxide assay (extracellular ROS detection)
The level of extracellular hydrogen peroxide (H2O2) generated in a preparation was estimated optically using an Amplex® Red Hydrogen Peroxide Kit (Molecular Probes, Carlsbad, CA, USA). This kit contains AmplexRed reagent (10-cetyl-3.7-dihydroxyphenoxazine) and HRP. In the presence of HRP, Amplex Red reacts with H2O2 in a 1:1 stoichiometry to produce the red-fluorescent oxidation product resorufin. Resorufin fluorescence was measured using excitation at 535 ± 10 nm and fluorescence detection at 590 ± 20 nm. Basal production of H2O2 in the neuromuscular preparation was determined by incubating the sample for 5 min in physiological solution (total volume 400 μl) containing Amplex Red (100 μm) and HRP (0.2 U ml−1). The preparation was then removed from this solution and immersed for 5 min in fresh solution containing MCD (10 mm) in addition to Amplex Red and HRP. This treatment was repeated and the preparation was then rinsed for 5 min in a MCD-free solution. Before recording the fluorescence intensity, the solutions were incubated at room temperature for 45 min, protected from light. Calibration was performed with a set of H2O2 concentrations (0.01, 0.025, 0.05, 0.1, 0.15, 0.2, 1 μm). Background fluorescence, determined in H2O2-free control medium, was subtracted from each value.
Intracellular ROS detection reagent
H2DCFDA (2′,7′-dichlorodihydrofluorescein diacetate, Molecular Probes) was used as a cell-permeant indicator for ROS. The dye was excited using a 1 s flash of light at 480/15 nm every minute and its emission was recorded using a band-pass filter of 515–590 nm. H2DCFDA was dissolved in DMSO. Pluronic F-127 (Molecular Probes) was used to enhance solution of the dye. The final concentration of DMSO and Pluronic F-127 in the working solution did not exceed 0.001%. The preparation was incubated with H2DCFDA (2 μm) at room temperature for 15 min and then perfused with physiological saline for 45 min before recording fluorescence intensity.
Lipid peroxidation
An Image-iT lipid peroxidation kit was used as a sensitive ratiometric reporter for lipid peroxidation (Molecular Probes). Upon oxidation in live cells, fluorescence shifts from red (∼590 nm) to green (∼510 nm), providing an indication of lipid peroxidation. The preparations were stained with Image iT-sensor (8 μm) for 30 min in physiological solution at room temperature, and then washed with physiological solution for 30 min and imaged with a BX51WI microscope, using a 100× objective and filters for fluorescein isothiocyanate and Texas Red. The ratio of signal intensities at 590 and 510 nm was used to quantify lipid peroxidation. Some preparations were pretreated with 10 mm MCD for 10 min. In other experiments, preparations were stained with Image iT-sensor before treatment with 10 mm MCD. In Fig.1, the normalized ratio of red/green fluorescence in fields of view containing nerve terminals is shown. The ratio before MCD application having been set to 1.0.
Figure 1. Kinetics of spontaneous release and synaptic vesicle exocytosis during cholesterol depletion.
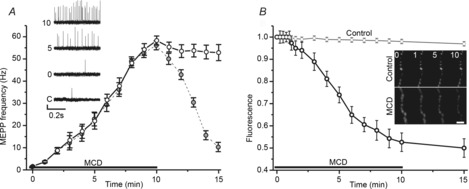
A, changes in MEPP frequency (Hz) induced by 10 mm MCD (open circles) and when MCD–cholesterol complex (5 mm) was applied immediately after MCD application (grey circles). Insets show traces illustrating MEPPs at different times before (C, control) and after the start of MCD application (0, 5 and 10 min in the presence of MCD). Vertical scale bar is 0.2 mV. B, time course of changes in nerve terminal fluorescence in response to MCD application. Nerve terminals were preloaded with FM1–43. Insets, fluorescence images of segments of the nerve terminals at rest and different times (in min) during MCD application. Scale bar is 3 μm. Fluorescence is normalized to the pre-MCD baseline fluorescence. Horizontal lines indicate the application of MCD. Data are mean ± SEM.
Cytosolic Ca2+ detection
The high-affinity cell-permeant Ca2+ indicator Fluo-4AM (Molecular Probes) was excited using short (about 1 s) light flashes at 488 ± 10 nm. Emission was recorded after passing through a 505–550 nm band-pass filter. Fluo-4AM was dissolved in DMSO and stored frozen (for up to a week) in the dark. Prior to the experiment, Pluronic F-127 was added to facilitate solution of Fluo-4-AM in aqueous (physiological) medium. In the working solution the final concentration of Fluo-4AM was 1 μm and the contents of DMSO and Pluronic F-127 were no more than 0.001%. Isolated preparations were stained with 1 μm Fluo-4AM for 10 min at room temperature. The specimens were then perfused with a physiological solution for 40 min, and measurements of fluorescence in regions containing nerve terminals were taken. The value of fluorescence before MCD application was set to 1.0.
Immunolabeling
For immunofluorescent staining, both control and MCD pre-treated neuromuscular preparations were fixed with 3.7% p-formaldehyde for 30 min at room temperature. After fixing, the preparations were permeabilized with 1% Triton X-100 in PBS at room temperature for 30 min followed by blocking with 2% normal goat serum, 0.1% Triton X-100, 0.05% Tween 20 and 1% BSA in PBS at room temperature for 1 h. The samples were subsequently incubated with primary rabbit polyclonal anti-TRPV1 antibody (1:200, Alomone Labs Ltd., Jerusalem, Israel) at 4°C overnight. Postsynaptic acetylcholine receptors were labelled by exposing the fixed tissue to 1 ng ml–1 of rhodamine-conjugated α-bungarotoxin (α-Btx, Molecular Probes) at 4°C for 4 h. Then the muscles were incubated with secondary AlexaFluor-488 labelled anti-rabbit antibody (1:1000, Abcam, Cambridge, MA, USA) for 1 h at room temperature in the dark. Stained tissue samples were then placed onto glass slides and sealed with mounting medium for confocal microscopy. Both the primary and the secondary antibodies were diluted in PBS containing 1% BSA, 0.05% Triton X-100 and 0.05% Tween 20. After each step of the labelling protocol the preparations were washed three times with PBS (in mm: 3.2 NaH2PO4, 0.5 K2HPO4, 1.3 KCl, 135 NaCl; pH 7.4) for 1 h. In some experiments, preparations were incubated simultaneously with the primary antibody and the blocking peptide (at 1:1 dilution), EDAEVFKDSMVPGEK, corresponding to amino acid residues 824–838 of rat TRPV1 (Alomone Labs). AlexaFluor-488/α-Btx fluorescence was excited by light of 488/555 nm wavelength and emission was recorded using band-pass filters of 505–545/610–650 nm. Images were acquired using an Olympus BX51WI microscope with a confocal attachment Disk Speed Unit and LumPlanPF 100xw objective, and captured with a DP71 CCD camera.
Statistics
Statistical analysis was performed using Origin Pro software (OriginLab Corp., Northampton, MA, USA). Data are presented as mean ± SEM, where n is the number of independent experiments, with statistical significance assessed by Student's t test or ANOVA. Values of P < 0.05 were considered significant.
Results
Spontaneous release and exocytosis are increased by MCD treatment
To explore the effect of MCD treatment on spontaneous neurotransmitter release, we first recorded MEPPs under control conditions then following 10 min bath application of MCD. Control MEPP frequency was 1.6 ± 0.2 Hz (n = 6). The frequency increased dramatically following MCD treatment (Fig.1A; 58.3 ± 2.0 Hz after 10 min MCD application, n = 6, P < 0.001). Following washout of MCD the frequency of MEPPs decayed within 15 min to 10.7 ± 3.5 Hz (P < 0.01 vs. control) and reached the control level after about 1 h. If MCD–cholesterol complex was applied immediately after MCD application, to restore cholesterol to the muscle, then MEPP frequency decreased to the pre-MCD treatment baseline within about 10 min.
We did not observe significant decreases in the fluorescence nerve terminals preloaded with FM1-43 in normal Ringer solution in the absence of presynaptic stimulation, which suggests that weak dye unloading occurred during spontaneous exocytosis. However, addition of MCD (10 mm) to deplete cholesterol caused a rapid decrease in FM1–43 fluorescence (Fig.1B) to 0.52 ± 0.04 of the baseline value (P < 0.001 vs. pre-MCD baseline, n = 8) after 10 min. The relatively fast kinetics of FM1–43 unloading after MCD treatment suggest an increased rate of synaptic vesicle exocytosis when cholesterol is depleted from the surface membranes. As nerve terminals were loaded with FM1–43 using high frequency presynaptic stimulation, the observation of MCD-induced dye unloading suggests that the same synaptic vesicles could participate in exocytosis induced either by presynaptic action potentials or by cholesterol depletion. Note that lower doses of MCD (0.1 and 1 mm) did not change either the MEPP frequency or FM1–43 fluorescence at preloaded nerve terminals (data not shown).
Cholesterol depletion induces intra- and extracellular ROS generation, causing lipid peroxidation in synaptic regions
Cholesterol depletion may change production of ROS in some cell types (Yang et al. 2006; Han et al. 2008; Jin et al. 2011). Using a cell-permeant indicator for ROS (H2DCFDA), we determined that treatment of the neuromuscular preparations with MCD led to increased intracellular ROS generation (Fig.2A and B). H2DCF fluorescence was enhanced in synaptic regions where it rose to 1.21 ± 0.03 of the baseline pre-MCD value (Fig.2A; P < 0.001 vs. baseline, n = 8) after 10 min of MCD treatment. MCD-induced changes in H2DCF fluorescence were prevented when NADPH oxidase was blocked by apocynin (200 μm). In these conditions, fluorescence remained at 1.00 ± 0.03 of the baseline value (P > 0.05 vs. baseline, n = 8). Thus, exposure to MCD and the presumed decrease in membrane cholesterol content associated with it appear to trigger ROS production.
Figure 2. ROS production and cholesterol depletion at frog neuromuscular synapses.
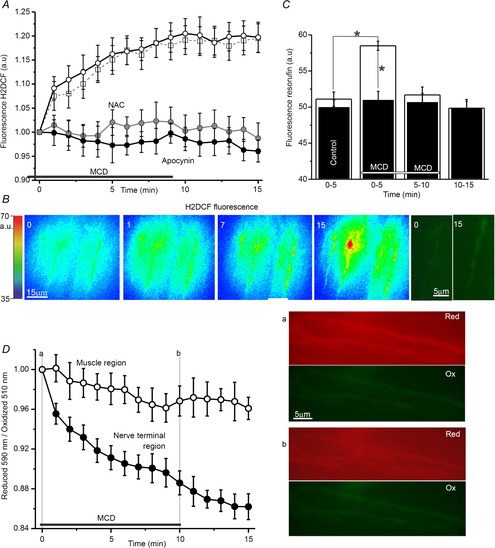
A, changes in H2DCF fluorescence in synaptic areas induced by MCD (open circles) and the effects on those changes of ROS antagonists (NAC/antioxidant, grey circles; apocynin/NADPH oxidase inhibitor, filled circles) or TRPV1 antagonist (capsazepine, open squares). B, pseudo-colour images of H2DCF fluorescence captured at time points 0, 1, 7, 15 min from the onset of MCD application. The corresponding intensity scale is provided to the left (a.u.). Right, magnified native fluorescence images of synaptic regions (0 before and 15 min after cholesterol depletion). C, fluorescence of resorufin (indicator of extracellular H2O2 level generated within 5 min). Open columns display the fluorescence before (from 0 to 5 min), during MCD treatment (from 0 to 5, from 5 to 10) and after washing (from 10 to 15 min) in control, whereas filled columns illustrate similar conditions but with NADPH oxidase blocked by apocynin. Asterisks denote significant differences (*P < 0.05). D, detection of membrane lipid peroxidation using fluorescence iT-sensor in synaptic (filled circles) and extrasynaptic muscular (open circles) regions. Values are the normalized ratio of the intensities of red to green fluorescence of the Image-iT lipid peroxidation sensor with time. To the right are images of red (reduced form, Red) and green (oxidized form, Ox) iT-sensor fluorescence in the synaptic region. The images illustrate the shift of fluorescence before (top, a) and after (bottom, b) MCD treatment. The horizontal line in A, C and D indicates the period of MCD application. Data are mean ± SEM.
It has been shown previously that activity of NADPH oxidase may be coupled to a rise in extracellular levels of H2O2, which eventually enters the cytosol (Miller et al. 2010). We estimated the amount of extracellular H2O2 based on the oxidation of Amplex Red reagent in the fluorescent product, resorufin (Fig.2C). After 5 min incubation in solutions containing Amplex Red and HRP resorufin fluorescence was 51.1 ± 1.0 a.u. (n = 6; corresponding to production of about 8.0 × 10−5 μg H2О2 per 1 mg of raw muscle weight). During the first 5 min of MCD treatment, fluorescence was increased to 58.5 ± 0.6 a.u. (P < 0.05, ∼9.4 × 10−5 μg H2О2 mg–1). Subsequently, with MCD treatment from 5 to 10 min and without MCD treatment from 10 to 15 min, fluorescence of the solution fell to 51.6 ± 1.1 (P = 0.83 vs. control) and 49.9 ± 1.2 a.u. (P = 0.13 vs. control), respectively. Thus, a rise in extracellular ROS concentration (by ∼17%) was observed during the initial 5 min of MCD treatment. This increase in fluorescence was prevented by adding the NADPH oxidase inhibitor apocynin to the solution (Fig.2C). When NADPH oxidase was blocked, resorufin fluorescence during the first 5 min of MCD treatment was 50.9 ± 1.2 a.u. (n = 5, P = 0.25 vs. pre-MCD values: 49.9 ± 1.0 a.u.).
The increase in ROS production associated with cholesterol depletion could affect membrane lipids by inducing their oxidation. Consistent with this, we observed lipid oxidation in synaptic regions using the lipid peroxidation sensor Image-iT (Fig.2D). Ratios of fluorescence (reduced 590 nm/oxidized 510 nm) were 3.2 ± 0.1 and 3.6 ± 0.2 (n = 5) in synaptic and non-synaptic regions, respectively. After MCD addition to the external medium, the ratio decreased in synaptic regions so that the normalized ratio was reduced to 0.89 ± 0.01 (P < 0.05, n = 5) after 10 min of MCD application. The decrease in the normalized ratio was almost undetectable in non-synaptic areas (0.97 ± 0.02, n = 5, P > 0.05, vs. pre-MCD baseline). When preparations were pretreated with 10 mm MCD and subsequently stained with Image-iT sensor, the ratios were 2.6 ± 0.2 (n = 4, P < 0.05 vs. control preparation prior to MCD treatment) and 3.4 ± 0.3 (P = 0.68 vs. control preparation prior to MCD treatment) in synaptic and extrasynaptic regions, respectively. This indicates that lipid oxidation occurred predominantly in synaptic regions.
Together, these results show that cholesterol depletion may result in increased ROS levels in extra- and intracellular space through NAPDH oxidase activation. This agrees with an earlier observation that maintaining the activity of NADPH oxidase in proximal renal tube cells requires cholesterol (Han et al. 2008).
ROS are involved in MCD-mediated enhancement of spontaneous release and exocytosis
To test the involvement of ROS in the effects of cholesterol depletion on exocytosis, we applied an antioxidant, NAC (200 μm) to the external solution. NAC effectively prevented MCD-induced increase of H2DCF fluorescence (Fig.2A), which remained at 0.99 ± 0.03 of the baseline value (P > 0.05, n = 8) after 10 min application of MCD when ROS were sequestrated by NAC.
NAC application itself had no effect on MEPP frequency (1.5 ± 0.3 Hz, n = 6, P > 0.05 vs. pre-application baseline), but suppressed the facilitating effect of MCD on spontaneous release (Fig.3A). After 10 min of treatment with MCD and in the presence of NAC, MEPP frequency was only 16.0 ± 3.1 Hz (n = 6, P < 0.001 vs. MCD application alone) after 10 min of MCD treatment. The administration of NAC also delayed MCD-induced FM1–43 unloading from nerve terminals (Fig.3B). After MCD treatment (10 min), FM1–43 fluorescence was reduced to only 74 ± 2% of the baseline value (n = 8, P < 0.01 vs. control MCD effect). This suggests that the cholesterol depletion-induced production of ROS may promote spontaneous neurotransmitter release and synaptic vesicle exocytosis. Note that antioxidant treatment attenuates, but does not fully block, the MCD effect, implying that the enhancement of release by MCD is only partially ROS-dependent.
Figure 3. ROS involvement in the MCD effects on spontaneous release and synaptic vesicle exocytosis.
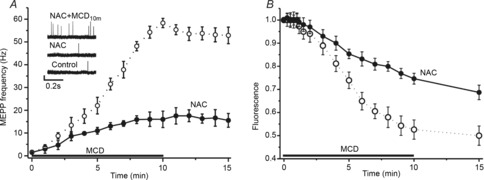
A, MCD-induced changes in MEPP frequency in control conditions (open circles, from Fig. 1) and when ROS were chelated by the antioxidant, NAC (filled circles). Insets show traces of MEPPs in control, 40 min after perfusion with NAC and 10 min after MCD application in the presence of NAC. B, time course of FM1–43 dye unloading from pre-loaded nerve terminals in response to MCD application (from Fig. 1) and in the presence of antioxidant. Other details are as in Fig 1. Data are mean ± SEM.
The rise in intracellular Ca2+ depends on MCD-induced ROS production
Synaptic vesicle exo/endocytosis and vesicular traffic at nerve terminals are tightly regulated by the level of cytosolic Ca2+. That level is mainly dependent on the influx of Ca2+ from the extracellular space via Ca2+ channels and/or Ca2+ release from an endoplasmic reticulum, due to activation of ryanodine and inositol trisphosphate receptors (Zefirov & Petrov, 2012; Kaeser & Regehr, 2014). It is possible that the increased ROS production after cholesterol depletion may interfere with Ca2+ homeostasis in nerve terminals. Indeed, we found (using Fluo4 dye) that MCD treatment triggered a rise in intracellular [Ca2+] (Fig.4A and B). Fluo4 fluorescence reached 1.17 ± 0.02 of the baseline value (P < 0.01, n = 8) 10 min after application of MCD. This increase in Fluo4 fluorescence was not observed when the actions of ROS were prevented with antioxidant. Under these conditions, fluorescence was 0.93 ± 0.02 of the baseline value (P > 0.05, n = 8) after 10 min of MCD treatment. Similar changes of intracellular Ca2+ concentration were observed when TRPV channels and ryanodine receptors were blocked with ruthenium red (2 μm) (Xu et al. 1999; García-Martínez et al. 2000). In our experiments, Fluo4 fluorescence was 1.05 ± 0.02 of the baseline value (P > 0.05, n = 8) after 10 min treatment with MCD. However, inhibition of ryanodine and inositol trisphosphate receptors with 50 μm TMB8 (Simizu et al. 2008) did not diminish the effect of cholesterol depletion on dye fluorescence (Fig.4C), which increased to 1.20 ± 0.02 of the baseline value (P < 0.01, n = 6).
Figure 4. MCD effects on the intracellular Ca2+ level in the nerve terminal region: dependence of MCD action on ROS production and Ca2+ channels.
A and C, changes in Fluo4 fluorescence at neuromuscular synapses under the influence of MCD alone (open circles, A); and in conditions of ROS chelation by NAC (filled circles, A); or inhibition of different types of Ca2+ channels (C). TMB-8 was used to block inositol trisphosphate and ryanodine receptors in the endoplasmatic reticulum (filled squares). Ruthenium red was applied to inhibit both ryanodine receptors and TRPV channels (open squares, RR). TRPV channels were blocked with capsazepine (open triangles, CP). The grey line (+CP) indicates the experiments in which capsazepine was applied 5 min after the onset of MCD. Values are normalized fluorescence (the initial fluorescence before MCD addition was taken as 1.0). Data are mean ± SEM. B, pseudo-colour images of Fluo4 fluorescence in synaptic regions at time points 0, 1, 5, 10 and 15 min from onset of MCD application in control solution (top) and in the presence of the ROS chelator NAC (bottom). Nerve terminal regions are marked by arrows. The corresponding intensity scale is shown on the left of the image (a.u.). D, immunolocalization of TRPV1 channels at control (left) and MCD-pretreated (right) neuromuscular junctions. Fluorescent images of preparations double-labelled with anti-TRPV1 antibody (green channel, top) and rhodamine-conjugated α-Btx (Btx, red channel, second row). The images in the green and red channels were merged (overlap) to illustrate the relative disposition of TRPV1 channels and postsynaptic acetylcholine receptors (third row). The lowest row shows images of a control preparation immunolabelled with TRPV1-specific antibody (left) and Btx (right) in the presence of a specific blocking peptide (see text for further details).
Therefore, it is plausible that an increase in ROS production may lead to the elevated intracellular Ca2+ concentration by activating TRPV channels in the plasma membranes. We examined this possibility using a specific inhibitor of TRPV1 channels, capsazepine (10 μm). Capsazepine application for 30 min had no effect on Fluo4 fluorescence, but it prevented the increase in fluorescence in response to cholesterol depletion (Fig.4C). Fluo4 fluorescence remained at 0.96 ± 0.02 of the baseline value, P > 0.05, n = 6). If the application of capsazepine was started 5 min after the onset of MCD action (Fig.4C), any further increase in Fluo4 fluorescence was prevented and fluorescence gradually returned to the baseline level after 11–13 min (P > 0.05, n = 6).
To test whether ROS generation occurred first and then led to calcium influx, we measured H2DCF fluorescence (ROS indicator) in the presence of a TRPV inhibitor. Capsazepine did not prevent the MCD-induced increase in H2DCF fluorescence, which remained at 1.19 ± 0.03 of the baseline value (P < 0.05 vs. baseline, n = 6) after MCD treatment (Fig.2A).
Immunolabeling of TRPV1 channels
To confirm that TRPV1 channels are expressed at frog neuromuscular junctions, we used immunolabelling with specific anti-TRPV1 antibody against the intracellular C-terminal region. Figure 4D shows immunochemical detection of TRPV1 channels in the nerve terminal region. Fluorescence had an irregular pattern of distribution within the synaptic region. These fluorescence spots were mainly located at intervals between the bands of the postsynaptic nicotinic acetylcholine receptors (marked by α-Btx), indicating their location in areas near the active zone. Pre-treatment with MCD did not disrupt the staining of nicotinic acetylcholine receptors, but immunolabelling of TRPV1 channels extended over a larger synaptic area and overlapping of TRPV1 channels with the bands of the postsynaptic receptor became visible. When the anti-TRPV1 antibody was applied along with a specific blocking peptide, the green nerve terminal fluorescence was almost undetectable (Fig.4D). The mean green fluorescence intensity was also dramatically reduced in the presence of the blocking peptide. Fluorescence labelling of TRPV1 channels was not observed when either primary or secondary antibodies were not added (data not shown).
In sum, immunofluorescence analysis indicates that the endogenous expression of TRPV1 in the frog neuromuscular junction is not uniform with preferential staining in synaptic regions. It also shows that MCD treatment leads to an enlargement of the TRPV1-positive areas.
An increase in intracellular Ca2+ is required for MCD effects on spontaneous release and exocytosis
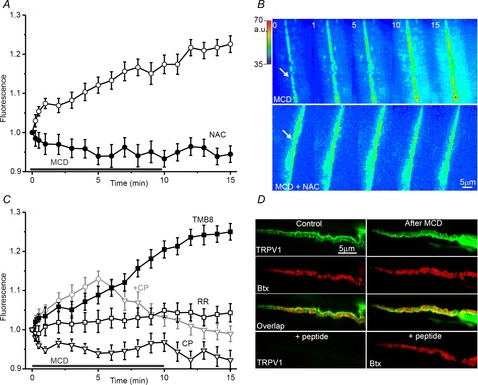
Pretreatment of neuromuscular preparations with the cell-permeant high-affinity Ca2+ chelator, BAPTA-AM (200 μm), slightly decreased MEPP frequency to 1.1 ± 0.1 Hz (P < 0.05, n = 6) but sharply reduced the effects of MCD on MEPP frequency (Fig.5A and В). Thus, in the BAPTA-AM-pretreated preparations, MEPP frequency was 13.1 ± 1.6 Hz (n = 6, P < 0.01 vs. control MCD action) after 10 min of MCD treatment. A similar effect was observed when the TRPV channels were blocked with ruthenium red or capzasepine (Fig.5A and В), when MEPP frequency decreased to 0.2 ± 0.1 Hz (n = 6, P < 0.001) and 0.6 ± 0.2 Hz (n = 6, P < 0.01), respectively. Also, in preparations treated with ruthenium red or capzasepine, MCD treatment (10 min) induced increases in MEPP frequency, to 15.2 ± 1.7 Hz (P < 0.01, n = 6) or 9.1 ± 1.3 Hz (P < 0.01, n = 6), respectively.
Figure 5. Intracellular Ca2+ and increase in spontaneous release and exocytosis due to cholesterol depletion.
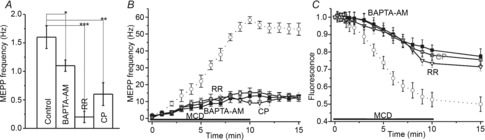
A, MEPP frequency (Hz) in control solution and after pre-treatment with BAPTA-AM, ruthenium red (RR) or capsazepine (CP) prior to MCD application (see text for details). Asterisks indicate significant differences (*P < 0.05, **P < 0.01, ***P < 0.001). B, changes in the MCD-induced MEPP frequency in control conditions (open circles, dotted line, from Fig. 1) and in conditions of intracellular Ca2+ chelation by BAPTA-AM (filled squares), or of TRPV channel inhibition using ruthenium red (RR, open triangles), or capsazepine (CP, open squares). C, influence of cytosolic Ca2+ chelation and TRPV channel inhibition on MCD-induced FM1–43 unloading. Other details are as in Fig 1.
Both BAPTA-AM and TRPV blockers (ruthenium red and capzasepine) slowed FM1–43 dye unloading (Fig.5С) evoked by cholesterol depletion. After 10 min of MCD treatment, FM1–43 fluorescence decreased to 0.81 ± 0.03 of the baseline value (P < 0.01, n = 6) in the BAPTA-AM pretreatment preparations, and to 0.78 ± 0.03 of the baseline value (P < 0.01, n = 6) and 0.73 ± 0.022 of the baseline value (P < 0.01, n = 6) in preparations where TRPV channels were blocked by ruthenium red or capzasepine, respectively. These values were significantly different (P < 0.01) from the decrease in FM1–43 fluorescence when MCD was applied alone. The kinetics of FM1–43 unloading under conditions of chelation of the intracellular Ca2+ and TRPV channel blockade were similar to those of MCD-induced FM1–43 destaining in the presence of antioxidant. Thus, it is possible that cholesterol depletion triggers ROS production and that the ROS molecules generated could, in turn, activate TRPV channels through which Ca2+ may enter the cytoplasm and facilitate spontaneous exocytosis of synaptic vesicles. As with antioxidant treatment, the Ca2+ antagonists only partially reduced the effects of MCD on transmitter release and dye unloading.
Ca2+-dependent dephosphorylation is coupled to the effects of cholesterol depletion on release and exocytosis
Inhibition of calcineurin with cyclosporine A (50 μm) decreased spontaneous release at rest to 0.7 Hz (n = 6, P < 0.05) and significantly reduced the enhancement of MEPP frequency induced by cholesterol depletion (Fig.6A). Under these conditions, MEPP frequency was 22.1 ± 3.0 Hz (P < 0.01, n = 6) after a 10 min application of MCD. Inhibition of phosphatase 2B also slowed FM1–43 dye unloading resulting from cholesterol depletion (Fig.6B). Thus, FM1–43 fluorescence was reduced to 0.76 ± 0.03 of the baseline value (P < 0.01, n = 8). We also tested the effects of okadaic acid (50 nm), which effectively blocks phosphatases PP1 and PP2A (Guatimosim et al. 2002). After perfusion for 40 min with okadaic acid, MEPP frequency was 1.7 ± 0.2 (n = 6, P > 0.05 vs. control). Under conditions of cholesterol depletion, okadaic acid did not suppress either the increase in MEPP frequency or FM1–43 dye unloading (Fig.6A and B). When PP1 and PP2A were blocked, MEPP frequency after MCD treatment was increased to 54.5 ± 3.0 Hz (n = 6, P < 0.001 vs. cyclosporine A action) and FM1–43 fluorescence decreased to 0.57 ± 0.04 of the baseline value (n = 6, P < 0.001 vs. cyclosporine A action). Thus, the rise in cytosolic Ca2+, mediated by cholesterol depletion, may activate calcineurin, which could in turn promote spontaneous synaptic vesicle exocytosis.
Figure 6. Role of phosphatases in MCD modulation of spontaneous release and synaptic vesicle exocytosis.
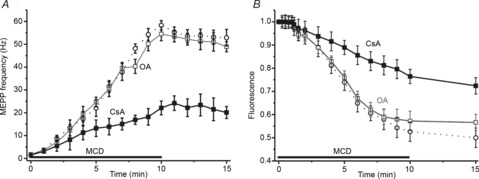
A, changes in MCD effects on MEPP frequency (open circles, data from Fig 1) under conditions of calcineurin (filled squares) or phosphatases 1/2A (open squares) inhibition by cyclosporine A (CsA) or okadaic acid (OA), respectively (see text for details). B, effects of cyclosporine A or okadaic acid on cholesterol depletion-mediated FM1–43 dye unloading. Other details are as in Fig 1. Data are mean ± SEM.
Discussion
MCD at a concentration of 10 mm is widely used for cholesterol depletion and lipid raft disruption (Zamir & Charlton, 2006; Wasser & Kavalali, 2009; Dason et al. 2010,2014; Smith et al. 2010; Teixeira et al. 2012; Rodrigues et al. 2013). At this dose, MCD reduced cholesterol in isolated crayfish nerve and muscle by 20–30% but had no effect on either the input resistance of muscle fibres (Zamir & Charlton, 2006; Ormerod et al. 2012) or nicotinic receptor clustering at neuromuscular junctions (Rodrigues et al. 2013).
Brain cholesterol is maintained at a relatively constant level and is mainly regulated by the amount of cholesterol itself (Brown & Goldstein, 1986). However, cholesterol concentration may be reduced in the course of normal ageing, in response to strong synaptic activation, or under certain pathological conditions (Sodero et al. 2011). Decreases in membrane cholesterol may interfere with the mechanisms of synaptic transmission, affecting both pre- and postsynaptic functions. Membrane cholesterol is essential for ‘clamping’ of spontaneous release and facilitation of evoked exocytosis at different neuromuscular junctions, as well as at central synapses (e.g. in the cerebellum or hippocampus) (Zamir & Charlton, 2006; Wasser & Kavalali, 2009; Tarakanova et al. 2011; Petrov et al. 2010, 2011b; Teixeira et al. 2012; Rodrigues et al. 2013). It has been suggested that cholesterol may be required for formation of fusion pores formation, and is thus essential for any type of exocytosis (Tong et al. 2009; Puchkov & Haucke, 2013). Alternatively, it is possible that cholesterol may control different types of exocytosis specifically through interaction with distinct signalling molecules.
Cholesterol depletion might have multifactorial effects on ROS production (Jin et al. 2011). It has been found that MCD treatment leads to the enhancement of NADPH oxidase activity in proximal renal tube cells (Han et al. 2008) and to the reduction of H2O2 production in aortic endothelial cells (Yang et al. 2006). By contrast, loss of cholesterol during ageing due to neurotransmission in cultured rat hippocampal neurons involves upregulation of the cholesterol hydroxylation enzyme Cyp46 as a result of production of ROS by NADPH oxidase (Sodero et al. 2011). In our study, we demonstrated that cholesterol depletion is associated with a rise in intra- and extracellular ROS levels. It is possible that NADPH oxidase generates H2O2, which is released into the extracellular space; and may then pass through cell membranes to enter the cytoplasm (Jin et al. 2011). Notably, NADPH oxidase (particularly the Nox2 isoform) has been found in mice synaptosomes where it is co-localized with the presynaptic protein synaptophysin (Tejada-Simon et al. 2005). Recently, using immunofluorescence staining, we found evidence of the Nox2 isoform located in synaptic regions of the mouse neuromuscular junction (A. M. Petrov and A. R. Giniatullin, unpublished results). It is possible that ROS production may occur non-uniformly along the length of nerve terminals, resulting in formation of ROS pools. Thus, in conditions of cholesterol depletion, H2DCF fluorescence increased in distinct regions of the frog nerve terminal. Inhibition of NADPH oxidase by apocynin and ROS chelation with NAC prevented the increase in ROS levels in response to cholesterol depletion. Additionally, the antioxidant significantly reduced the effect of MCD on spontaneous exocytosis.
ROS may influence numerous cellular processes, including assembly of the SNARE complex and exocytosis (Giniatullin et al. 2005,). We attempted to identify the possible molecular targets of ROS that could stimulate spontaneous exocytosis. ROS may act on Ca2+ channels, including ryanodine receptors of the endoplasmic reticulum and certain types of TRP channels in plasma membranes (Song et al. 2011). At the frog neuromuscular junction, it appears that voltage-gated Ca2+ channels are located near ryanodine receptors on internal stores, and sustained high-frequency presynaptic activation of voltage-gated Ca2+ channels can trigger Ca2+-induced Ca2+ release from the internal store that, in turn, promotes asynchronous vesicle fusion (Narita et al. 2000). However, other studies have reported that preventing release from internal stores has a minimal effect on presynaptic Ca2+ signals and asynchronous release, indicating that release from internal stores is not essential (Carter et al. 2002). TRPV1 channels have been found on presynaptic nerve terminals of C-fibres in the nucleus solitarius. Ca2+ influx through these channels has been shown to increase spontaneous exocytosis, whereas it inhibited synchronous evoked synaptic vesicle exocytosis (Andersen et al. 2012). Moreover, the administration of a tert-butyl hydroperoxide, an ROS donor, markedly increased the frequency of spontaneous excitatory postsynaptic currents in substantia gelationosa neurons, and this enhancement was suppressed by the TRPV1 channel antagonist capsazepine (Nishio et al. 2013). It has been previously shown using immunohistochemistry that motor nerve terminals express the TRP protein of the vanilloid subfamily, TRPV1, the capsaicin receptor (Thyagarajan et al. 2009). In this study, immunofluorescence analysis demonstrated that frog nerve terminals expresses TRPV1 channels mostly in the regions around active zones lying opposite the postsynaptic acetylcholine receptor clusters (Robitaille et al. 1996). Furthermore, the distribution of TRPV1 channels becomes enlarged after cholesterol depletion. This could reflect a perturbation in channel clustering and/or insertion of additional channels in the plasma membrane due to exocytosis of vesicles containing TRPV1 channels (Goswami et al. 2010).
Our findings show that cholesterol depletion-induced increases in the cytosolic Ca2+ concentration were suppressed in the presence of antioxidant or TRPV channel blockers. Moreover, both pharmacological blockage of TRPV channels and Ca2+ chelation reduced the cholesterol depletion-dependent increase in spontaneous release. Together, these results indicate that membrane cholesterol may inhibit ROS production and, thus, preventing the Ca2+ influx through TRPV channels that normally promotes spontaneous exocytosis. It has been shown previously that exogenous H2O2 in low micromolar concentrations has no effect on MEPP frequency at frog neuromuscular junctions, whereas higher concentrations of H2O2 (100–300 μm) reduced spontaneous release (Giniatullin & Giniatullin, 2003). This observation may be explained by direct actions of exogenous ROS on fusion proteins (particularly SNAP-25), reducing their functional activity (Giniatullin et al. 2006). It may be that when membrane cholesterol is depleted, an effective concentration of ROS might be achieved only in synaptic areas containing TRPV channels. We cannot rule out that, at normal levels of membrane cholesterol, TRPV channels could have a weak susceptibility to ROS, whereas cholesterol depletion could lead to the increase in sensitivity of these channels to ROS. For instance, capsaicin-induced currents through the TRPV1 channels are markedly decreased in TRPV1-expressing HEK cells enriched with cholesterol, but not with its diastereoisomer, epicholesterol (Picato-Juares et al. 2011).
Note that while we found only a transient increase in ROS following application of MCD (mainly in the first 5 min of exposure), the intracellular Ca2+ level and spontaneous exocytosis continued to rise during the whole period of MCD application. This suggests that ROS are required for an initial induction phase, but not afterwards. Presumably, once elevated, ROS activate TRPV channels, they continue to mediate an increase in intracellular Ca2+ for a substantial period of time. This is supported by the decreasing level of cytosolic Ca2+ after acute addition of TRPV blocker during the period of MCD action. A transient ROS signal might sensitize TRPV1 by covalent cysteine modification, which produces sustained modulation of the activity of these channels (Chuang & Lin, 2009). However, the temporal profile of intracellular Ca2+ dynamics does not exactly match that of release (in the first few minutes after MCD treatment, Ca2+ continues to rise slightly whereas transmitter release does not change significantly), indicating that other factors along with cytosolic Ca2+ determine transmitter release due to cholesterol depletion. In addition, increasing intracellular Ca2+ concentration may slow down, or inhibit, synaptic vesicle endocytosis (von Gersdorff & Matthews, 1994; Leitz & Kavalali, 2011). Finally, prolonged MCD treatment might impair recycling of synaptic vesicles, limiting the size of the pool of vesicles available for release (Dason et al. 2010; Petrov et al. 2010).
Several previous studies demonstrated a relationship between an effect of MCD on spontaneous transmitter release, and the activity of several protein kinases (in particular, protein kinases A and C, Ca2+ calmodulin-dependent kinase) (Smith et al. 2010; Teixeira et al. 2012). However, the role of Ca2+/calmodulin-activated phosphatase (calcineurin) in the cholesterol depletion-induced enhancement of spontaneous exocytosis has not been examined previously. Calcineurin participates in triggering endocytosis and may dephosphorylate proteins such as dynamin, amphyphisin, synaptojanin, epsin, Eps15, GTPase Arf6, phosphatidylinositol 4-phosphate 5-kinase, that are all involved in the presynaptic vesicle cycle (Cheung & Cousin, 2013). The possibility that calcineurin is activated via TRPV1 channels has been demonstrated recently at hippocampal synapses, where calcineurin induces long-term depression (Jensen & Edwards, 2012). In our experiments, we found that enhancement of spontaneous exocytosis due to cholesterol depletion was substantially less when calcineurin was blocked, whereas inhibition of phosphatases 1 and 2A with okadaic acid did not change the effect of MCD on spontaneous release. Therefore, calcineurin activity might be important for the maintenance of spontaneous exocytosis at high levels when membrane cholesterol content is decreased. It is possible that calcineurin activation may increase the number of synaptic vesicles involved in exocytosis induced by cholesterol depletion. For example, inhibition of calcineurin at hippocampal synapses decreased the pool of functionally active synaptic vesicles (Marra et al. 2012), whereas genetic knockdown of calcineurin increased the size of the resting pool (Kim & Ryan, 2010). Another possibility is that calcineurin activity may be required for efficient endocytosis, leading to the formation of ‘hot’ vesicles with an increased ability to fuse (Rose et al. 2013).
Based on our findings, we hypothesize that a decrease in membrane cholesterol content activates the ROS-generating enzyme NADPH oxidase. In turn, ROS act on TRPV channels, admitting Ca2+ into the cytoplasm. Subsequently, the complex of Ca2+ with calmodulin may stimulate calcineurin-dephosphorylating proteins involved in maintenance of spontaneous exocytosis. Calcineurin might then facilitate spontaneous release by regulating both the delivery of synaptic vesicles to active zones and compensatory endocytosis (Fig.7).
Figure 7. Putative pathways for ROS-dependent enhancement of spontaneous exocytosis due to cholesterol depletion.
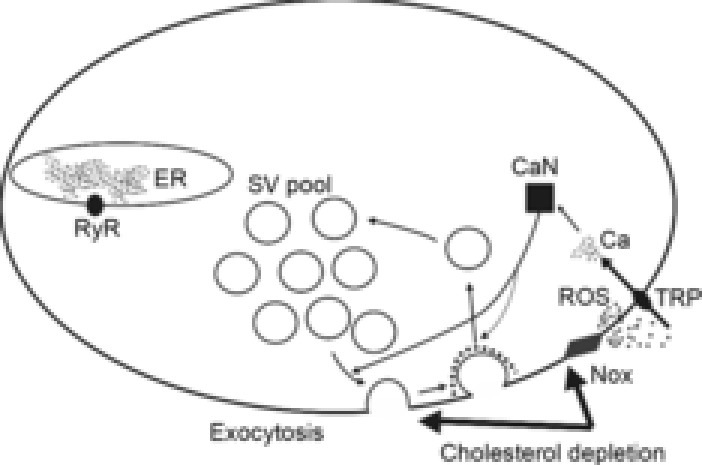
Decreases in cholesterol cause increases in the activity of membrane-bound NADPH oxidase (Nox), resulting in increased ROS production. Consequently, the amount of ROS is enhanced both in the extra- and in the intracellular spaces. This leads to stimulation of TRPV channels, thus admitting Ca2+ to the cytoplasm. The rise in intracellular Ca2+ may maintain spontaneous exocytosis at high levels. Ca2+ may activate phosphatase 2B (calcineurin, CaN), which has effects on proteins involved in the synaptic vesicle cycle, and may thus facilitate both recruitment of synaptic vesicles from pools for exocytosis and compensatory endocytosis. This pathway could be recruited along with others, as cholesterol depletion potentiated spontaneous exocytosis much less when any of the components of the NADPH oxidase–ROS–TRPV–Ca2+–calcineurin pathway were blocked.
Importantly, pharmacological manipulations did not fully eliminate the cholesterol depletion-induced increases in spontaneous exocytosis. Thus, under conditions of ROS or Ca2+ chelation, or blockade of TRPV channels or calcineurin, the effects of MCD on MEPP frequency and FM1–43 unloading were reduced by only 65–75%. This indicates that cholesterol depletion may activate other exocytosis-intensifying mechanisms. It is possible that specific contributions to the effects of cholesterol depletion may be made by changes in the activity of protein kinases, ion channels and pumps and membrane fluidity (Zamir & Charlton, 2006; Wasser & Kavalali, 2009; Smith et al. 2010; Teixeira et al. 2012), warranting future investigation.
Acknowledgments
We thank Professors Clarke Slater, Vadim Bolshakov and Richard Ribchester for helpful comments on the manuscript.
Glossary
- α-Btx
α-bungarotoxin
- H2DCFDA
2′,7′-dichlorodihydrofluorescein diacetate
- MCD
methyl-β-cyclodextrin
- MEPP
miniature postsynaptic end-plate potential
- NAC
N-acetyl cysteine
- PP
protein phosphatase
- ROS
reactive oxygen species
- TMB8
8-(diethylamino)octyl-3,4,5-trimethoxybenzoate hydrochloride
- TRP
transient receptor potential
- TRPV
transient receptor potential vanilloid.
Additional Information
Competing interests
We declare no competing interests.
Author contributions
All experiments were performed in the laboratory of the Department of Normal Physiology at Kazan State Medical University. A.M.P. and A.A.Y. performed all experiments, data collection and analysis. A.M.P. was responsible for the conception and design of the study. A.M.P. and A.L.Z. were involved in the interpretation of data and drafting the article. All authors read and approved the final version of the manuscript.
Funding
This study was supported by grants from the Russian Foundation for Basic Research (#14-04-00094 and #12-04-33195), and a grant for Support Young Doctors of Philosophy (#108.2013.4).
Key points
Cholesterol depletion increases reactive oxygen species (ROS) levels in extra- and intracellular space through NAPDH oxidase activation, which is accompanied by synaptic lipid oxidation.
ROS production due to extraction of cholesterol involves both an enhancement of synaptic vesicle exocytosis and an increase in cytosolic [Ca2+]i.
An ROS-dependent rise in [Ca2+]i is suppressed by inhibitors of the transient receptor potential vanilloid channel and leads to an increase in synaptic vesicle exocytosis.
The ROS–calcium pathway might influence synaptic vesicle exocytosis via activation of phosphatase protein 2B (calcineurin).
The results help us better understand why a decrease of membrane cholesterol increases spontaneous synaptic vesicle exocytosis.
References
- Andresen MC, Hofmann ME. Fawley JA. The unsilent majority-TRPV1 drives “spontaneous” transmission of unmyelinated primary afferents within cardiorespiratory NTS. Am J Physiol Regul Integr Comp Physiol. 2012;303:R1207–1216. doi: 10.1152/ajpregu.00398.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MS. Goldstein JL. A receptor-mediated pathway for cholesterol homeostasis. Science. 1986;232:34–47. doi: 10.1126/science.3513311. [DOI] [PubMed] [Google Scholar]
- Carter AG, Vogt KE, Foster KA. Regehr WG. Assessing the role of calcium-induced calcium release in short-term presynaptic plasticity at excitatory central synapses. J Neurosci. 2002;22:21–28. doi: 10.1523/JNEUROSCI.22-01-00021.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung G. Cousin MA. Synaptic vesicle generation from activity-dependent bulk endosomes requires calcium and calcineurin. J Neurosci. 2013;33:3370–3379. doi: 10.1523/JNEUROSCI.4697-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang HH. Lin S. Oxidative challenges sensitize the capsaicin receptor by covalent cysteine modification. Proc Natl Acad Sci U S A. 2009;106:20097–20102. doi: 10.1073/pnas.0902675106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dason JS, Smith AJ, Marin L. Charlton MP. Vesicular sterols are essential for synaptic vesicle cycling. J Neurosci. 2010;30:15856–15865. doi: 10.1523/JNEUROSCI.4132-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dason JS, Smith AJ, Marin L. Charlton MP. Cholesterol and F-actin are required for clustering of recycling synaptic vesicle proteins in the presynaptic plasma membrane. J Physiol. 2014;592:621–633. doi: 10.1113/jphysiol.2013.265447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond GB. Reporting ethical matters in The Journal of Physiology: standards and advice. J Physiol. 2009;587:713–719. doi: 10.1113/jphysiol.2008.167387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Martínez C, Morenilla-Palao C, Planells-Cases R, Merino JM. Ferrer-Montiel A. Identification of an aspartic residue in the P-loop of the vanilloid receptor that modulates pore properties. J Biol Chem. 2000;275:32552–32558. doi: 10.1074/jbc.M002391200. [DOI] [PubMed] [Google Scholar]
- Giniatullin AR, Darios F, Shakirzyanova A, Davletov B. Giniatullin R. SNAP25 is a pre-synaptic target for the depressant action of reactive oxygen species on transmitter release. J Neurochem. 2006;98:1789–1797. doi: 10.1111/j.1471-4159.2006.03997.x. [DOI] [PubMed] [Google Scholar]
- Giniatullin AR. Giniatullin RA. Dual action of hydrogen peroxide on synaptic transmission at the frog neuromuscular junction. J Physiol. 2003;552:283–293. doi: 10.1113/jphysiol.2003.050690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giniatullin AR, Grishin SN, Sharifullina ER, Petrov AM, Zefirov AL. Giniatullin RA. Reactive oxygen species contribute to the presynaptic action of extracellular ATP at the frog neuromuscular junction. J Physiol. 2005;565:229–242. doi: 10.1113/jphysiol.2005.084186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goswami C, Rademacher N, Smalla KH, Kalscheuer V, Ropers HH, Gundelfinger ED. Hucho T. TRPV1 acts as a synaptic protein and regulates vesicle recycling. J Cell Sci. 2010;123:2045–2057. doi: 10.1242/jcs.065144. [DOI] [PubMed] [Google Scholar]
- Guatimosim C, Hull C, Von Gersdorff H. Prado MA. Okadaic acid disrupts synaptic vesicle trafficking in a ribbon-type synapse. J Neurochem. 2002;82:1047–1057. doi: 10.1046/j.1471-4159.2002.01029.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han W, Li H, Villar VA, Pascua AM, Dajani MI, Wang X, Natarajan A, Quinn MT, Felder RA, Jose PA. Yu P. Lipid rafts keep NADPH oxidase in the inactive state in human renal proximal tubule cells. Hypertension. 2008;51:481–487. doi: 10.1161/HYPERTENSIONAHA.107.103275. [DOI] [PubMed] [Google Scholar]
- Jensen T, Edwards JG. Calcineurin is required for TRPV1-induced long-term depression of hippocampal interneurons. Neurosci Let. 2012;510:82–87. doi: 10.1016/j.neulet.2012.01.006. [DOI] [PubMed] [Google Scholar]
- Jia JY, Lamer S, Schümann M, Schmidt MR, Krause E. Haucke V. Quantitative proteomics analysis of detergent-resistant membranes from chemical synapses: evidence for cholesterol as spatial organizer of synaptic vesicle cycling. Mol Cell Proteomics. 2006;5:2060–2071. doi: 10.1074/mcp.M600161-MCP200. [DOI] [PubMed] [Google Scholar]
- Jin S, Zhou F, Katirai F. Li PL. Lipid raft redox signaling: molecular mechanisms in health and disease. Antioxid Redox Signal. 2011;15:1043–1083. doi: 10.1089/ars.2010.3619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaeser PS. Regehr WG. Molecular mechanisms for synchronous, asynchronous and spontaneous neurotransmitter release. Annu Rev Physiol. 2014;76:333–363. doi: 10.1146/annurev-physiol-021113-170338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavalali ET. Monteggia LM. Synaptic mechanisms underlying rapid antidepressant action of ketamine. Am J Psychiatry. 2012;169:1150–1156. doi: 10.1176/appi.ajp.2012.12040531. [DOI] [PubMed] [Google Scholar]
- Kim SH. Ryan TA. CDK5 serves as a major control point in neurotransmitter release. Neuron. 2010;67:797–809. doi: 10.1016/j.neuron.2010.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitz J. Kavalali ET. Ca2+ influx slows single synaptic vesicle endocytosis. J Neurosci. 2011;31:16318–16326. doi: 10.1523/JNEUROSCI.3358-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marra V, Burden JJ, Thorpe JR, Smith IT, Smith SL, Hausser M, Branco T. Staras K. A preferentially segregated recycling vesicle pool of limited size supports neurotransmission in native central synapses. Neuron. 2012;76:579–589. doi: 10.1016/j.neuron.2012.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller EW, Dickinson BC. Chang CJ. Aquaporin-3 mediates hydrogen peroxide uptake to regulate downstream intracellular signaling. Proc Natl Acad Sci U S A. 2010;107:15681–15686. doi: 10.1073/pnas.1005776107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narita K, Akita T, Hachisuka J, Huang S, Ochi K. Kuba K. Functional coupling of Ca2+ channels to ryanodine receptors at presynaptic terminals: amplification of exocytosis and plasticity. J Gen Physiol. 2000;115:519–532. doi: 10.1085/jgp.115.4.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishio N, Tanicuchi W, Sugimura YK, Takiguchi N, Yamanaka M, Kiyoyuki Y, Yamada H, Miyazaki N, Yoshida M. Nakatsuka T. Reactive oxygen species enhance excitatory synaptic transmission in rat spinal dorsal horn neurons by activating TRPA1 and TRPV1 channels. Neurosci. 2013;247:201–212. doi: 10.1016/j.neuroscience.2013.05.023. [DOI] [PubMed] [Google Scholar]
- Ormerod KG, Rogasevskaia TP, Coorssen JR. Mercier AJ. Cholesterol-independent effects of methyl-β-cyclodextrin on chemical synapses. PLoS One. 2012;7:e36395. doi: 10.1371/journal.pone.0036395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrov AM. Giniatullin AR, Sitdikova GF. Zefirov AL. The role of cGMP-dependent signaling pathway in synaptic vesicle cycle at the frog motor nerve terminals. J Neurosci. 2008;28:13216–13222. doi: 10.1523/JNEUROSCI.2947-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrov AM, Kasimov MR, Giniatullin AR, Tarakanova OI. Zefirov AL. The role of cholesterol in the exo- and endocytosis of synaptic vesicles in frog motor nerve endings. Neurosci Behav Physiol. 2010;40:894–901. doi: 10.1007/s11055-010-9338-9. [DOI] [PubMed] [Google Scholar]
- Petrov AM, Kudryashova KE, Odnoshivkina YG. Zefirov AL. Сholesterol and lipid rats in the membrane of nerve terminal and membrane of synaptic vesicle. Neurochem J. 2011;5:13–19. [Google Scholar]
- Petrov AM, Naumenko NV, Uzinskaya KV, Giniatullin AR, Urazaev AK. Zefirov AL. Increased non-quantal release of acetylcholine after inhibition of endocytosis by methyl-β-cyclodextrin: the role of vesicular acetylcholine transporter. Neuroscience. 2011;186:1–12. doi: 10.1016/j.neuroscience.2011.04.051. [DOI] [PubMed] [Google Scholar]
- Picato-Juares G, Romero-Suarez S, Nieto-Posadas A, Llorente I, Jara-Oseguera A, Biggs M, Mclntosh TJ, Simon SA, Ladron-de-Guevara E, Islas L. Rosenbaum T. Identification of a binding motif in the S5 helix that confers cholesterol sensitivity to the TRPV1 ion channel. J Biol Chem. 2011;286:24966–24976. doi: 10.1074/jbc.M111.237537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puchkov D. Haucke V. Greasing the synaptic vesicle cycle by membrane lipids. Trends Cell Biol. 2013;23:493–503. doi: 10.1016/j.tcb.2013.05.002. [DOI] [PubMed] [Google Scholar]
- Rodrigues HA, Lima RF, Fonseca MD, Amaral EA, Martinelli PM, Naves LA, Gomez MV, Kushmerick C, Prado MA. Guatimosim C. Membrane cholesterol regulates different modes of synaptic vesicle release and retrieval at the frog neuromuscular junction. Eur J Neurosci. 2013;38:2978–2987. doi: 10.1111/ejn.12300. [DOI] [PubMed] [Google Scholar]
- Rose T, Schoenenberger P, Jezek K. Oertner TG. Developmental refinement of vesicle cycling at Shaffer collateral synapses. Neuron. 2013;77:1109–1121. doi: 10.1016/j.neuron.2013.01.021. [DOI] [PubMed] [Google Scholar]
- Robitaille R, Bourque MJ. Vandaele S. Localization of L-type Ca2+ channels at perisynaptic glial cells of the frog neuromuscular junction. J Neurosci. 1996;16:148–158. doi: 10.1523/JNEUROSCI.16-01-00148.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu H, Fukaya M, Yamasaki M, Watanabe M, Manabe T. Kamiya H. Use-dependent amplification of presynaptic Ca2+ signaling by axonal ryanodine receptors at the hippocampal mossy fiber synapse. Proc Natl Acad Sci U S A. 2008;105:11998–12003. doi: 10.1073/pnas.0802175105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sodero AO, Weissmann C, Ledesmac MD. Dotti CG. Cellular stress from excitatory neurotransmission contributes to cholesterol loss in hippocampal neurons aging in vitro. Neurobiol Aging. 2011;32:1043–1053. doi: 10.1016/j.neurobiolaging.2010.06.001. [DOI] [PubMed] [Google Scholar]
- Song MY, Makino A. Yuan JX. Role of reactive oxygen species and redox in regulating the function of transient receptor potential channels. Antioxid Redox Signal. 2011;15:1549–1565. doi: 10.1089/ars.2010.3648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AJ, Sugita S. Charlton MP. Cholesterol-dependent kinase activity regulates transmitter release from cerebellar synapses. J Neurosci. 2010;30:6116–6121. doi: 10.1523/JNEUROSCI.0170-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarakanova OI, Petrov AM. Zefirov AL. The role of membrane cholesterol in neurotransmitter release from motor nerve terminals. Dokl Biol Sci. 2011;438:138–140. doi: 10.1134/S0012496611030070. [DOI] [PubMed] [Google Scholar]
- Teixeira G, Vieira LB, Gomez MV. Guatimosim C. Cholesterol as a key player in the balance of evoked and spontaneous glutamate release in rat brain cortical synaptosomes. Neurochem Int. 2012;61:1151–1159. doi: 10.1016/j.neuint.2012.08.008. [DOI] [PubMed] [Google Scholar]
- Tejada-Simon MV, Serrano F, Villasana LE, Kanterewicz BI, Wu GY, Quinn MT. Klann E. Synaptic localization of a functional NADPH oxidase in the mouse hippocampus. Mol Cell Neurosci. 2005;29:97–106. doi: 10.1016/j.mcn.2005.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thyagarajan B, Krivitskaya N, Potian JG, Hognason K, Garcia CC. McArdle JJ. Capsaicin protects mouse neuromuscular junctions from the neuroparalytic effects of botulinum neurotoxin A. J Pharmacol Exp Ther. 2009;331:361–71. doi: 10.1124/jpet.109.156901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong J, Borbat PP, Freed JH. Shin Y-K. A scissors mechanism for stimulation of SNARE-mediated lipid mixing by cholesterol. Proc Natl Acad Sci U S A. 2009;106:5141–6. doi: 10.1073/pnas.0813138106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Gersdorff H, Matthews G. Inhibition of endocytosis by elevated internal calcium in a synaptic terminal. Nature. 1994;370:652–655. doi: 10.1038/370652a0. [DOI] [PubMed] [Google Scholar]
- Wasser CR. Kavalali ET. Leaky synapses: regulation of spontaneous neurotransmission in central synapses. Neuroscience. 2009;158:177–188. doi: 10.1016/j.neuroscience.2008.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Tripathy A, Pasek DA. Meissner G. Ruthenium red modifies the cardiac and skeletal muscle Ca2+ release channels (ryanodine receptors) by multiple mechanisms. J Biol Chem. 1999;274:32680–32691. doi: 10.1074/jbc.274.46.32680. [DOI] [PubMed] [Google Scholar]
- Yang B, Oo TN. Rizzo V. Lipid rafts mediate H2O2 prosurvival effects in cultured endothelial cells. FASEB J. 2006;20:1501–1503. doi: 10.1096/fj.05-5359fje. [DOI] [PubMed] [Google Scholar]
- Zamir O. Charlton MP. Cholesterol and synaptic transmitter release at crayfish neuromuscular junctions. J Physiol. 2006;571:83–99. doi: 10.1113/jphysiol.2005.098319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zefirov AL. Petrov AM. Lipids in the processes of exo- and endocytosis of synaptic vesicles. Neurosci Behav Physiol. 2012;42:144–152. [Google Scholar]