

Abstract
Abstract
The molecular and cellular mechanisms governing cell motility and directed migration in response to the neuropeptide bradykinin are largely unknown. Here, we demonstrate that human glioma cells whose migration is guided by bradykinin generate bleb-like protrusions. We found that activation of the B2 receptor leads to a rise in free Ca2+ from internal stores that activates actomyosin contraction and subsequent cytoplasmic flow into protrusions forming membrane blebs. Furthermore Ca2+ activates Ca2+-dependent K+ and Cl− channels, which participate in bleb regulation. Treatment of gliomas with bradykinin in situ increased glioma growth by increasing the speed of cell migration at the periphery of the tumour mass. To test if bleb formation is related to bradykinin-promoted glioma invasion we blocked glioma migration with blebbistatin, a blocker of myosin kinase II, which is necessary for proper bleb retraction. Our findings suggest a pivotal role of bradykinin during glioma invasion by stimulating amoeboid migration of glioma cells.
Introduction
Glioma multiforme is an aggressive, fast expanding type of brain tumour that derives from glial cells. Resection of the tumour is typically not curative because single glioma cells invade the adjacent healthy brain parenchyma, where they can form secondary tumours. During invasion glioma cells move along blood vessels or white matter tracts (Farin et al. 2006; Zagzag et al. 2008). The factors that stimulate migration along these structures are not known.
Tumour cells migrate through dense tissue by degradation of extracellular matrix proteins. This type of migration is called mesenchymal migration and involves the release of matrix degrading enzymes by the tumour cell (Wolf et al. 2003; Markovic et al. 2009). Mesenchymal migration might be an important subtype of cell motion but pharmacological blockade of extracellular matrix degradation does not fully abolish the invasiveness of tumour cells (Wolf et al. 2003). Many groups have suggested an alternative, more basic migratory mechanism, called amoeboid migration. This migration type is associated with the formation of small bleb like protrusions at the plasma membrane (Paluch & Raz, 2013). Bleb formation was found in several malignancies and has been studied most intensively in melanoma cells (Charras et al. 2006). Interestingly, membrane blebs in zebra fish germ cells occur by Ca2+-dependent contraction of the actomyosin cortex which leads to cytoplasmatic pressure against the plasma membrane (Blaser et al. 2006). As a result blebs form in areas of the plasma membrane where the actomyosin cortex is destabilized. Subsequently the membrane can lift from its actomyosin cortex. Membrane blebs stabilize then transiently extend by the formation of new elements of the cytoskeleton like ezerin, actin and myosin (Fackler & Grosse, 2008). Finally membrane blebs retract by myosin II contraction mediated by myosin II kinase and the small GTPase Rho (Charras et al. 2006). Some authors suggest that the formation of membrane blebs could function as a mechanism for directed cell migration to chemoattractants (Blaser et al. 2006; Kapustina et al. 2013).
Membrane blebs can be evoked by ligands that bind to G-protein coupled receptors. Lysophosphatic acid for instance induces transient blebbing in NB2a rat neuroblastoma cells (Hagmann et al. 1999). Substance P, a neuropeptide evokes membrane blebbing in HEK and U373MG astrocytoma cells by activation of Rho-associated protein kinase (ROCK), another regulator of the actomyosin cortex (Meshki et al. 2009). The chemokine SDF-1 enhances blebbing in migrating primordial germ cells in zebra fish (Blaser et al. 2006).
In this study we sought to examine the chemotactic effects of bradykinin, a neuropeptide of the vasculature and a known mediator of inflammation. Bradykinin binds to the heterotrimeric G-protein coupled receptors B1 (B1R) and B2 (B2R). Migration of microglia and T cells is regulated by B1R (Ifuku et al. 2007; Schulze-Topphoff et al. 2009). Other cell types like endothelia cells, neurons and astrocytes express B2R. We recently showed that bradykinin attracts glioma cells chemotactically to blood vessels (Montana & Sontheimer, 2011). In glioma cells, bradykinin effects are mediated by B2R, via activation of inositol-1,4,5-trisphosphate (IP3), a molecule that triggers the release of Ca2+ from intracellular stores (Reiser et al. 1987).
Additionally bradykinin application in glioma cells activates Ca2+-dependent big conductance K+ (BK) channels (Ransom & Sontheimer, 2001) and intermediate conductance K+ (IK) channels in glioma cells (Cuddapah et al. 2013). Opening of these channels leads to a hyperpolarization of the membrane, and a depolarizing Cl− current finally terminates IK current-dependent hyperpolarization. Blocking these channels inhibits glioma cell migration, suggesting a pivotal role in tumour metastasis. Furthermore Ca2+-dependent ion channels have been proposed to facilitate regulation of cell volume in migratory cells.
This study examines the mechanism underlying bradykinin-induced glioma chemotaxis. To do so, we monitored intracellular Ca2+ concentrations ([Ca2+]i) in response to bradykinin while assessing cell movement and cell volume changes by fluorescence imaging. We found that bradykinin induces rises in [Ca2+]i resulting in the appearance of membrane blebs at the plasma membrane that are associated with small cell volume changes. This is the first study showing a correlation of [Ca2+]i increase and bleb formation in response to a ligand. Ca2+ triggers contractions of the actomyosin cortex with subsequent flow of cytoplasm into protrusions. We provide further evidence that Ca2+ targets Ca2+-dependent ion channels that participate in bleb regulation. Finally, we show that glioma cell migration was increased by bradykinin in situ, which was sensitive to the B2R blocker Hoe-140 and blebbistatin, a blocker of bleb retraction. Together these data provide evidence for an amoeboid type of movement by invading glioma cells.
Methods
Ethical approval
Experiments were performed in accordance with the University of Alabama Institutional Animal Care and Use Committee.
Cell lines
D54 cells, a human glioblastoma cell line classified by the World Health Organization as grade IV, was a gift from Dr Bigner from Duke University (Durham, NC, USA).
pN1-GCaMP3 vector was obtained from Addgene (Cambridge, MA, USA). pN1-EGFP and pN1-DsRed vectors were received from Clonetech (Mountain View, CA, USA). The transgenic cell line D54-EGFP was established as described (Watkins & Sontheimer, 2011). Additionally we created transgenic D54-GCaMP3/dsRed cells by transfecting D54 cells with Amax Cell Line Nucleofector Kit T from Lonza (Allendale, NJ, USA). Briefly, 1 × 106 D54 cells were washed with PBS pH 7.4 and centrifuged for 5 min at 6000 rpm at room temperature. After mixing the cells with transfection buffer and 3 μg of plasmid DNA the cells were electroporated with an Amaxa device. Afterwards cells were transferred into 6-well plates containing DMEM/F12. The medium was changed after 5 h to DMEM/F12 supplemented with 7% FBS (fetal bovine serum) and 1% geneticin from Invitrogen (Carlsbad, CA, USA). High expression clones were selected by fluorescence activated cell sorting and subsequently grown as individual clones for imaging and transplantation experiments.
For passage D54, D54-EGFP and D54-GCaMP3/dsRed cells were cultured in DMEM/F12 (Invitrogen) supplemented with 8% FBS. D54-EGFP and D54-GCaMP3/dsRed cells were kept under 0.5% geneticin.
Solutions
Hepes buffer contained (mm): NaCl 125, KCl 5.4, MgSO4 1, CaCl2 1.8, NaH2PO4 0.4, Na2HPO4 1.6, glucose 10.5 and Hepes acid 32.5. For experiments the buffer was warmed to 37°C and pH was adjusted to 7.4 with 10N NaOH. Osmolarity of this solution was 300 mosmol.
Artificial cerebrospinal fluid (ACSF) was composed of (mm): NaCl 124, KCl 2.5, MgCl2 1.3, CaCl2 2.0, K2HPO4 1.2, glucose 10.0, NaHCO3 26.0. Buffer was warmed to 37°C. Osmolarity was 300 mosmol.
Three-dimensional time lapse and Ca2+ imaging in vitro
D54-GCaMP3/dsRed cells were plated on 0.17 mm coverslips 1–3 days before the experiment at a density of 1 × 105 cells ml−1. Imaging experiments were performed in Hepes buffer at 37°C. Time-lapse images were acquired with a Hamamatsu IEEE1394 Digital CCD camera (325–6, Sunayama-Cho, Hamamatsu City, Japan) mounted on an Olympus IX81 motorized inverted microscope equipped with an Olympus Disk Scanning Unit from Olympus (Melville, NY, USA) and controlled by Slidebook software from Intelligent Imaging Innovations (Denver, CO, USA). The microscope was housed in a temperature-controlled incubator. Cells have been transferred to a closed recording chamber floated with prewarmed buffer. Fluorescence was recorded with filters from Semrock through a ×40 oil objective (numerical aperture 1.3) and the camera was binned to 4 × 4 pixels to increase light sensitivity. GCaMP3 fluorescence was recorded in one confocal z-plane, whereas dsRed fluorescence was recorded in 14 confocal planes (1 μm intervals). Intensity measurements for dsRed and GCaMP3 fluorescence were analysed with slide book software within regions of interest (ROIs). We used dsRed fluorescence to determine protrusion movement and bleb formation by analysing z-projections or performed volume rendering by acquiring mean pixel intensities of the 14 optical sections for each individual cell every 15 s. Values were normalized to the mean baseline intensity (F/F0). GCaMP3 fluorescence was normalized to appropriate dsRed fluorescence.
Transwell migration assay
Fluorescence blocking polyethylene terephthalate membrane filter inserts with 8 μm pores from Falcon by BD (Franklin Lakes, NJ, USA) have been coated with 10 μg ml−1 laminin from Sigma Aldrich (St. Louis, MO, USA) overnight and washed 3× with PBS. D54-EGFP cells were washed with PBS and suspended in DMEM/F12 supplemented with 0.1% fatty acid free fetal bovine serum (Sigma Aldrich). 4 × 104 cells were seeded to the top of each filter insert and allowed to settle for 30 min in the incubator. To start the assay drugs were added to the bottom of the filter. After 5 h incubation all solutions were removed and cells were fixed with 4% cold PBS for 10 min. Cells were washed with PBS and stained with DAPI for 10 min. Filter bottoms were photographed with a Zeiss axiovert M2 microscope connected to a LCD camera. Photos were taken at ×200 magnification.
Tumour implantation in vivo
D54-DsRed/GCaMP3 cells were washed 2× with PBS and centrifuged at 6000 rpm. Cells were diluted to a concentration of 5 × 107 cells ml−1 in methylcellulose and stored on ice until injection. BALB/c scid mice by Jackson Laboratory (Bar Harbor, ME, USA) were anaesthetized with 2–4% isoflurane. An incision was cut into the scalp and a hole was drilled in the skull. For tumour cell implantation a 20G needle was inserted at bregma −1.5 mm frontal, 1.5 mm lateral, 1.5 mm deep into the right frontal cortex, and then 250,000 cells were injected per mouse. The incision was closed using skin glue. Mice were killed after 3–4.5 weeks by cerebral dislocation. The tumour-bearing brains were removed and sliced in ice cold ACSF into 100 μm sections. Slices were recovered and stored in PBS at 28°C until measurement.
Three-dimensional time lapse and Ca2+ imaging in acute brain slices
Laser scanning confocal images were obtained using an Olympus Fluoview 1000 system equipped with a ×10/ and ×40/0.75 NA water-immersion lens from Olympus and diode lasers with excitation maxima at 405, 473, 559 and 635 nm. To separate emissions, dichroic mirrors separating at 560 nm and 640 nm were used. Appropriate emission filters from Semrock collected wavelength between 490–540 nm, 575–620 nm and 655–755 nm. Slices were transferred to a heated recording chamber and fixed with a grid. Single tumour cells were selected for time lapse imaging. Z-stacks with an interval of 2 μm every 15 s were recorded. Slices were recorded in ACSF continuously saturated with 5% CO2 and 95% O2. Image stacks were recorded with Olympus Fluoview software, exported and evaluated in Slide book software 5.1 (Intelligent Imaging Innovations, Inc., Denver, CO, USA).
Preparation of brain slice cultures
Brains of P13–P16 BALB/c scid mice (Jackson Laboratory) were dissected and cut with a Vibratome 3000 into 300 μm coronal brain sections. Brain slices were transferred onto the polycarbonate membrane of a filter insert with a pore size of 3 μm (Falcon, BD). Filters were placed into 6-well plates containing 1 ml DMEM supplemented with 8% FBS, 0.2 mm glutamine, 100 U ml−1 penicillin and 100 mg ml−1 streptomycin. After resting overnight the medium was changed to cultivation medium containing 25% heat-inactivated horse serum, 50 mm sodium bicarbonate, 2% glutamine, 25% Hank's balanced salt solution, 1 mg ml−1 insulin (all from Invitrogen), 2.46 mg ml−1 glucose (Sigma Aldrich), 0.8 mg ml−1 vitamin C (Sigma Aldrich), 100 U ml−1 penicillin, 100 mg ml−1 streptomycin (Sigma Aldrich), and 5 mm tris-hydroxymethylaminomethane in DMEM without phenol red (Invitrogen).
Tumour implantation into brain slice cultures
After 3 days of slice culturing, 3000 D54-EGFP tumour cells in PBS (final volume 1 μl) were implanted in each brain slice (Fig.8A). Cells were injected using a 1 μl Hamilton syringe (Reno, NV, USA) mounted to a micromanipulator. The cell suspension was inoculated into the right cortex. The left side of the brain slice was used for control purposes. Some brain slices were used to determine tumour growth after treatment with various concentrations of bradykinin or Hoe-140 by Tocris (Bristol, UK). Drug treatment was started 1 day after D54 implantation. The slice medium was exchanged every 2 days for medium containing fresh drugs. Live brain slices were photographed at 1 and 8 days with a Leica MZ 120 Microscope. The tumour area within the organotypic brain slice was determined by fluorescence measurements at 488nm excitation using Image J software. tumour size (%) was calculated by normalizing the tumour area of day 8 to the tumour area of day 1. Some brain slices were fixed with 4% paraformaldehyde after each experiment and used for fluorescence staining. Alternatively live brain slice cultures with D54-EGFP tumours were used for time lapse imaging at days 2–5.
Figure 8. Bradykinin facilitates glioma growth in cultured brain slices by increasing migration speed along blood vessels.
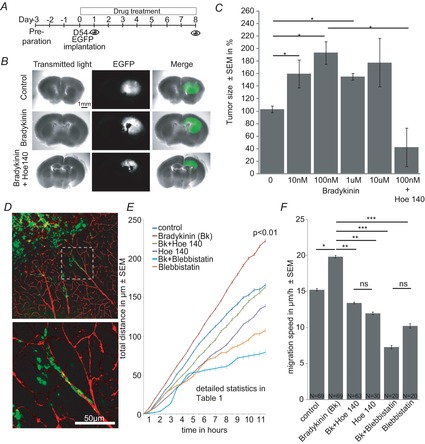
A, time line of experimental set up. Brain slices were prepared 3 days before implantation of D54-EGFP cells. The brain slices were photographed on days 1 and 8. B, examples of brain slices bearing D54-EGFP tumours at 8 days after implantation treated with 0 (control) and 0.1 μm bradykinin or 0.1 μm bradykinin + 5 μm Hoe-140. C, tumour size (%) after treatment with 0, 0.01, 0.1, 1 and 10 μm bradykinin or 0.1 μm bradykinin + 5 μm Hoe-140 (N = 4 per group). D, fixed brain slices stained with anti-EGFP (green) and anti-laminin (red) antibodies. Lower picture is a magnification of the area in the white dashed box in the upper picture. E, in situ migration assay (N = 4 per group with n = 10/N). Control slices did not receive drugs. Drugs were used in the following concentrations: 100 nm bradykinin, 5 μm Hoe-140, 2.5 μm blebbistatin (see Supporting information videos S4–8). F, averaged migration speed of the experiments in E.
Time lapse imaging in brain slice cultures
Brain slice cultures with D54-EGFP tumours were placed in a 6-well plate under a Zeiss Axiovert 200M microscope equipped with a motorized stage and filter cube turret from Carl Zeiss (Thornwood, NY, USA). The microscope was housed in a temperature- and CO2-controlled humidified incubator maintained at 37°C, 10% CO2. The slices were imaged in their culture media. Maximally six cultured slices were recorded at a time. Drugs (obtained from Tocris) were added 1 h before recording. Time lapse videos were recorded by taking a picture every 30 min with an Axiocam MRm camera. As light source an X-cite 120 mercury lamp was used. Time lapse video recordings of migrating glioma cells were evaluated by the Cell Tracker plugin in Image J software.
Staining
D54 cells were seeded 2 days before staining on coverslips at a density of 1 × 105 cells ml−1. Cells were treated for 25 s with 100 nm bradykinin (Tocris) and fixed subsequently with 3% cold PFA in PBS for 15 min. After washing with PBS, cells were incubated with Alexa Fluor 488 Phalloidin, CellMask and DAPI according to the manual (Molecular Probes). Alternatively fixed cells were incubated with antibodies overnight. We used primary antibodies against IK channels from Abcam (ab79259) at a dilution of 1:300, against BK channels at 1:200 (APC 0021), against TMEM16A channels at 1:200 (ACL-011) and against ClC3 channels at 1:250 all from Alomone (Jerusalem, Israel). Secondary antibodies were purchased from Jackson Laboratory and applied at a dilution of 1:2000 for 2 h at room temperature. Stained D54 cells were mounted after several washing steps on glass slides with Aqua-Mount (Thermo Scientific) and confocal pictures were taken with an Olympus Fluoview F1000 microscope.
Cultured brain slices were fixed with 4% PFA overnight. Slices were washed 3× for 20 min in TBS, 0.1% Triton-X 100. Slices were permeabilized with TBS, 1% Triton-X 100 for 1 h. Unspecific binding was blocked by TBS supplemented with 5% donkey serum and 0.1% Trition-X 100. The primary antibody was incubated in 10% block buffer for 48 h at 4°C. Anti-laminin antibody was used at 1:200 and anti-GFP antibody at 1:1000, both purchased from Sigma Aldrich. Slices were washed with TBS, 0.1% Triton-X 100 and incubated with secondary antibodies from Invitrogen. Slices were mounted on glass slides with Aqua-Mount (Thermo Scientific). Confocal z-stacks were recorded on a F1000 Olympus microscope as described above.
Statistics
Data have been tested with GraphStat Prism6 software. Data are displayed as means ± SEM. P values are indicated as: *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001 or n.s. (not significant) P > 0.05.
Results
Bradykinin induces transient Ca2+ signals in glioma cells that are accompanied by protrusion movement and volume changes
Previous studies have shown that bradykinin induces migration of glioma cells (Montana & Sontheimer, 2011; Cuddapah et al. 2013). We have been interested in how bradykinin might lead to tumour cell migration. Bradykinin increases [Ca2+]i in glioma cells in vitro (Reetz & Reiser, 1996). Therefore we expected that bradykinin-induced Ca2+ signals are associated with changes in cell shape. To observe [Ca2+]i in parallel with cell shape we created a D54 tumour cell line expressing GCaMP3 (green fluorescent protein-based Ca2+ probe) and dsRed (red fluorescent protein of Discosoma sp.). We monitored [Ca2+]i with the genetically encoded Ca2+ sensor GCaMP3 and observed changes in cell shape by analysing dsRed fluorescence. Both proteins were expressed in the cytoplasm of the cells. Figure1A shows two example cells before, during and after bath application of 100 nm bradykinin for 2 min. In response to bradykinin application a clear increase in GCaMP3 fluorescence was visible throughout the cell, indicating a global rise in [Ca2+]i. At about the same time we observed changes in dsRed fluorescence. DsRed fluorescence was analysed in two ways: z-stacks were used to determine process movement and additionally we visualized volume changes by 3D rendering.
Figure 1. Bradykinin induces an increase of intracellular [Ca2+] via B2 receptor which is accompanied by protrusion and volume changes.
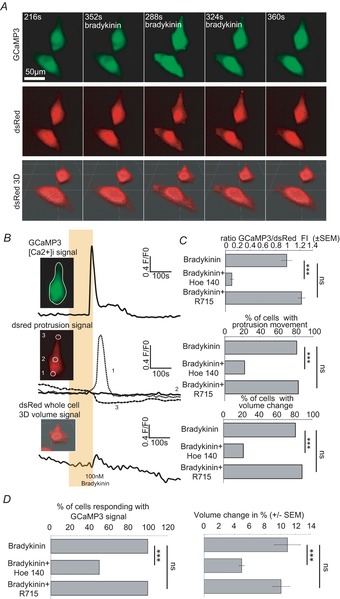
A, confocal time lapse series of D54 glioma cells in vitro before, during and after bradykinin application (100 nm, 120 s, 37°C). GCaMP3 fluorescence (upper series) and dsRed fluorescence (middle series) were recorded in parallel. GCaMP3 fluorescence was recorded in one confocal z-plane, whereas dsRed fluorescence was recorded in 14 confocal planes (1 μm intervals). We used dsRed fluorescence to determine protrusion movement by analysing z-projections. Furthermore we used dsRed fluorescence for calculating the cell's volume by 3D reconstruction for every time point (lowest series). B, quantification of the data shown in A. Upper trace shows GCaMP3 fluorescence ([Ca2+]i), middle trace dsRed fluorescence (protrusion movement) and lowest trace volume recordings during bradykinin application (indicated by the yellow box). White circles in the inserts indicate the regions of interest (ROI) that were analysed. C, summary bar graphs of GCaMP3 fluorescence intensity (FI), percentage of cells with protrusion movement and percentage of cells showing volume changes. D, summary bar graphs of percentage of cells responding with a GCaMP3 signal and percentage volume changes when treated with 100 nm bradykinin (n = 27), 100 nm bradykinin + 5 μm Hoe-140 (n = 18) or 100 nm bradykinin + 5 μm R715 (n = 18). One-way-ANOVA with a Bonferroni post hoc test was used to compare GCaMP3 FI/dsRed and volume changes (%). χ2 test was used to compare the numbers of cells.
Figure1B shows the individual traces of one cell from Fig.1A. On average, protrusions started to move 24.51 ± 16.93 s after the Ca2+ peak. Changes in cell volume were also detected. The cells changed their volume on average by 10.95 ± 1.7% (Fig.1D right). Volume changes were detected 30.93 ± 15.49 s after the Ca2+ peak. Ca2+ signals correlated well in time with the protrusion movements and volume changes (Fig.3A).
Figure 3. Ca2+ dependence of membrane blebbing and volume changes.
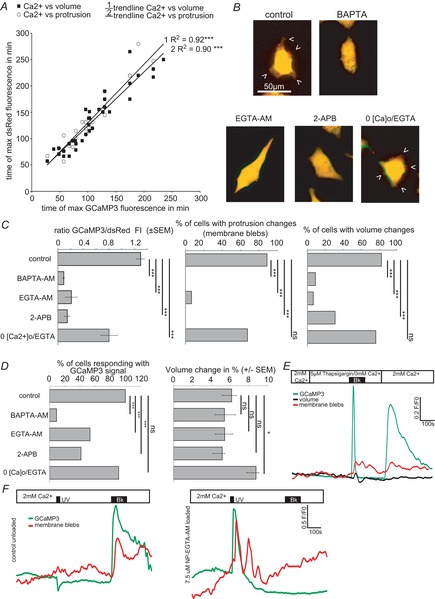
A, temporal Pearson-Correlation analysis of the maximal [Ca2+]i compared with the maximum of protrusion movement or maximum of volume change. B–D, D54 cells were incubated with blockers of the Ca2+ pathway before bradykinin application. Controls received 100 nm bradykinin for 2 min without pretreatment. B, pictures show dsRed fluorescence of example cells before (green) and during (red) 100 nm bradykinin application (overlay yellow). C, summary bar graphs of GCaMP3 fluorescence intensity (FI), percentage of cells with protrusion movement and percentage of cells showing volume changes. D, summary bar graphs of percentage of cells responding with a GCaMP3 signal and volume changes (%). Controls n = 23, 5 μm BAPTA-AM n = 22, 5 μm EGTA-AM n = 15, 100 μm 2-APB n = 29, 0[Ca2+]o/1 mm EGTA n = 34. One-way-ANOVA with a Bonferroni post hoc testwas used to compare GCaMP3 FI/dsRed and volume changes (%). χ2 test was used to compare the numbers of cells. E, Ca2+ from internal stores induces protrusion movement and volume changes. F, uncaging Ca2+ by UV light induces strong protrusion movements in NP-EGTA loaded cells. Left trace shows unloaded control cells (n = 11); right trace shows D54 cells loaded with 7.5 μm NP-EGTA. Ca2+ increased upon UV illumination for 20 s followed by protrusion movement (n = 16 of 26). Application of 100 nm bradykinin (Bk) or 20 s UV light is indicated by the black bars.
To analyse the receptor specificity of this response, we used blockers of the bradykinin receptors B1 and B2. Figure1C shows the comparison of the groups. We found that all cells responded with an increase in [Ca2+]i to bradykinin application. The rise in fluorescence intensity (FI) in bradykinin treated cells was 0.98 ± 0.08-fold. In the presence of 5 μm Hoe-140, a B2 receptor blocker, [Ca2+]i increased only 0.1 ± 0.33-fold when bradykinin was applied. The [Ca2+]i intensity was not significantly different from the application of bradykinin in the presence of 5 μm R 715, a B1 receptor antagonist. Protrusion changes have been found to occur in 81.45% of the bradykinin-treated control cells. Treatment with Hoe-140 reduced the number of cells that exhibit protrusions in response to bradykinin to 22.22%. Volume changes were observed in 81.45% of the control cells and in 22% of the Hoe-140-treated cells. R 715 did not affect either the bradykinin-induced protrusions or cell volume, suggesting that B1 receptor is not involved in this response. Figure1D shows the percentage of cells that responded with a Ca2+ change and summarizes the percentage volume changes observed under all conditions. Our results suggest that bradykinin increases [Ca2+]i, protrusion movement and volume changes via B2 receptor activation.
To determine cell movement in response to bradykinin in detail we analysed z-projections of the dsRed channel more closely (Supporting information Video S1). On the left side of Fig.2A, an overlay of the same cell before (green) and during bradykinin application (red) is presented. Traces for the regions of interest (ROI) 1–10 are shown on the right side. The analysis reveals that the dsRed fluorescence in the cell center (ROI 5 and 6) does not change during bradykinin application. In adjacent areas (ROI 4 and 7), however, the dsRed fluorescence was reduced following the application, whereas in distant ROIs 1, 2, 3, 8 and 9 and 10 an increase in dsRed fluorescence was observed. We interpret this wave of dsRed fluorophore as cytoplasmic flow evoked by a contraction that occurs in the cell body (next to ROI 4 and 7). Finally when the cytoplasm reaches the plasma membrane blebs are formed in the periphery of the cell (ROI 1 and 10). Twenty-two of 27 cells responded with a comparable pattern. Individual cells varied in shape and size, and therefore cell length and position of the cell center varied. This made further quantifications challenging. Additional examples of cells moving in response to bradykinin are illustrated in Fig.2B.
Figure 2. Analysis of glioma cell movement during bradykinin application.
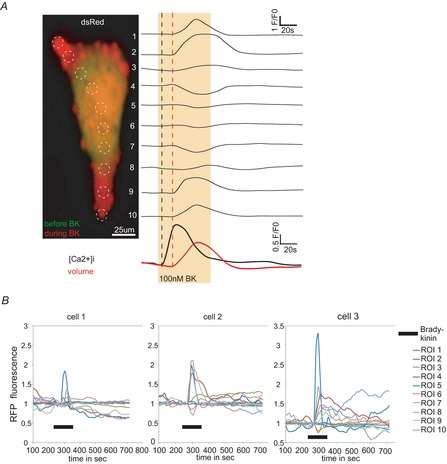
A, dsRed fluorescence of z-projections (see also Supporting information Video S1) for every time point was analysed within ROI 1–10 (indicated by dashed white circles). Picture shows cell before (green) and during a 2 min 100 nm bradykinin application (red). On the right side individual traces of every ROI are shown. Bradykinin application is indicated by a yellow box. For comparison traces of [Ca2+]i and cell volume are shown at the bottom. Notice that D54 cells show shape changes only in certain compartments of the cell. Membrane blebs occurred mainly in the periphery. B, dsRed fluorescence of 3 example glioma cells analysed like the cell in A. Traces have been normalized to mean baseline intensity (F0) Black bar indicates application of 100 nm bradykinin for 2 min.
Membrane blebs and cell volume changes are caused by a transient rise in intracellular Ca2+
Of the entire population studied, 67.4% of the cells responded to bradykinin application with both formation of membrane blebs and volume changes; 84.8% of the cells showed a Ca2+ response followed by bleb formation, and in 73.9% the Ca2+ change was followed by a change in cell volume. Temporal analysis of the peak Ca2+ level with the peak of bleb formation (R2 = 0.90, P < 0.0001) or volume changes (R2 = 0.93, P < 0.0001) was highly correlated, supporting the idea that the blebs and the volume changes are both evoked by changes in [Ca2+]i (Fig.3A). Bradykinin receptor 2 is a G-protein coupled receptor, which is linked to the formation of inositol-1,4,5-trisphosphate (IP3) and the subsequent release of Ca2+ from internal stores (Billups et al. 2006). To test the hypothesis that bleb formation and volume changes are dependent on changes of [Ca2+]i we undertook a series of experiments disrupting Ca2+ signalling during bradykinin application. Overlays of dsRed fluorescence (yellow) of representative cells before (green) and during (red) bradykinin applications are shown in Fig.3B for every treatment. Figures3C summarizes the recordings. We applied 100 nm bradykinin per bath application at 5 min for a duration of 2 min and recorded the cells every 15 s as described above. We loaded the cells with 5 μm BAPTA-AM, a fast Ca2+ chelator. Under this condition the increase in [Ca2+]i after bradykinin application was only 0.09 ± 0.01-fold in comparison to controls, which showed a 1.29 ± 0.06-fold rise in Ca2+. This resulted in an absence of bleb formation. The number of cells responding with volume changes was reduced to 9.09%. We loaded the cells with 5 μm EGTA-AM, a slower Ca2+ chelator than BAPTA-AM. This treatment also prevents Ca2+ propagation within the cell. The increase in [Ca2+]i was reduced to 0.21 ± 0.09-fold. In EGTA-AM-treated cells only 6.67% of the cells responded with membrane blebs and 6.67% with volume changes. To prevent the formation of IP3 we bath applied 100 μm 2-APB. This hindered [Ca2+]i to increase (0.14 ± 0.04-fold) and abolished bleb formation. Volume changes were detected in 31.01% of the cells. To test if protrusions and volume changes were dependent on Ca2+ influx via the plasma membrane, we omitted Ca2+ from the external solution and added the Ca2+ chelator EGTA (1 mm) to the external solution. The omission of extracellular Ca2+ decreased the Ca2+ signal 0.8 ± 0.13-fold. The number of cells responding with membrane blebs (67.65%) or volume changes (76.47%) did not significantly change in comparison to control cells. The percentage of cells responding with a CCaMP3 signal and percentage volume change are provided in Fig.3D.
Bradykinin-induced Ca2+ signals were complex, occurring mostly biphasically with an initial peak response followed by a plateau phase (Fig.2B). Typically the peak phase of such Ca2+ signals results from the release of Ca2+ from internal stores. Ca2+ release from stores opens store-operated Ca2+ channels (SOCs) in the plasma membrane, resulting in an additional Ca2+ entry into the cell, seen as a plateau phase. We dissected the bradykinin-induced Ca2+ signal by using Ca2+ free-bath solution (0[Ca2+]o; Fig.3E). Under this condition bradykinin evoked a short but intense Ca2+ signal that must result from internal stores, including potentially all organelles. The amount of Ca2+ released was sufficient to evoke membrane blebs and volume changes. To see if store-operated Ca2+ entry through the plasma membrane would additionally be able to evoke membrane blebs, we blocked Ca2+ reuptake into the stores with 5 μm thapsigargin, a blocker of the of the sarco-/endoplasmic reticulum Ca2+-ATPase (SERCA). By switching back to standard bath solution, Ca2+ was able to enter the cytoplasm only via channels in the plasma membrane. This Ca2+ was able to induce membrane blebs in some cells but did not evoke changes in cell volume. In some cells we found membrane blebs occurring at the same locations as previously from the ER signal.
To test if Ca2+ was sufficient to induce membrane blebs, we loaded the cells with 7.5 μm o-nitrophenyl EGTA acetoxymethyl ester (NP-EGTA-AM), a Ca2+ chelator that releases Ca2+ by stimulation with ultraviolet (UV) light (Fig.3F). To induce a ∼0.8-fold Ca2+ rise, comparable to the Ca2+ rise of the 0[Ca2+]o group in Fig.3C we used UV light flashes at 405 nm lasting 20 s. In unloaded control cells (left trace, Fig.3F), UV light induced a negative signal in the green channel, indicative for bleaching of GCaMP3 fluorescence by the UV flash. Subsequent bradykinin application evoked membrane blebs in these control cells. In NP-EGTA-AM loaded cells we recorded increases in [Ca2+]i upon UV activation (right trace, Fig.3F). These Ca2+ signals were accompanied by the formation of membrane blebs. Subsequent bradykinin application did not induce the formation of additional membrane blebs.
Taken together these experiments show that a transient rise in [Ca2+]i alone is sufficient to induce membrane blebs and cell volume changes by releasing Ca2+ from intracellular Ca2+ stores.
Bradykinin-induced membrane blebs and volume changes of D54 glioma cells are sensitive to blockers of cytoskeleton
Cell contraction and bleb formation has been described as being dependent on the cytoskeleton in germ cells and in macrophages (Blaser et al. 2006; Charras et al. 2006). To test if bradykinin-induced membrane blebbing and volume changes are dependent on the cytoskeleton in D54 glioma cells we applied 2.5 μm cytochalasin D, a blocker of actomyosin function. Cytochalasin D blocked bradykinin-induced membrane blebs and volume changes in all investigated cells (Fig.4A). Additionally we tested blebbistatin, a blocker of myosin II kinase activity. Myosin II kinase is required for retraction of membrane blebs (Charras et al. 2006). Blebbistatin (2.5 μm) increased the number of cells with membrane blebs significantly from 75% to 100% in our experiments. Interestingly, changes in volume were not affected by blebbistatin. The percentage of cells responding with a GCaMP3 signal and the percentage volume change are provided in Fig.4B. Figure4C shows glioma cells that have been fixed with 4% paraformaldehyde 25 s after bradykinin application at the maximum of bleb formation. The cells were stained for Alexa Fluor 488 Phalloidin, a marker for actin, with CellMask (Molecular Probes), a membrane marker, and 4′,6-diamidino-2-phenylindole (DAPI), a nuclear stain. We found membrane blebs in various regions of the cell.
Figure 4. Bradykinin-induced membrane blebs and volume changes are dependent on the cytoskeleton.
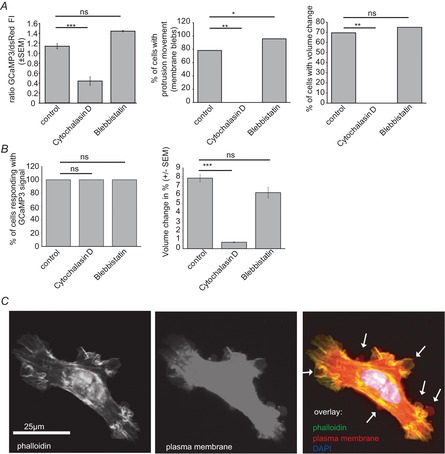
A and B, quantification of all groups treated with blockers of the cytoskeleton before bradykinin application. A, summary bar graphs of GCaMP3 fluorescence intensity (FI), percentage of cells with protrusion movement and percentage of cells showing volume changes. B, summary bar graphs of the percentage of cells responding with a GCaMP3 signal and volume changes. Control (no pretreatment) n = 24, 2.5 μm cytochalasin D n = 5, 2.5 μm blebbistatin n = 24. One-way-ANOVA with a Bonferroni post hoc test was used to compare GCaMP3 FI/dsRed and volume changes (%). χ2 test was used to compare the numbers of cells. C, D54 glioma were fixed during bradykinin application and stained for actin with Alexa Fluor 488 Phalloidin, the plasma membrane with CellMask (Molecular Probes) and the nucleus with DAPI. Arrows point to membrane blebs.
Bradykinin-induced membrane blebs and cell migration are both sensitive to blockers of Ca2+-dependent Cl− and K+ channels in D54 glioma cells
From previous studies we know that bradykinin opens Ca2+-dependent Cl− and K+ channels (Ransom & Sontheimer, 2001; Cuddapah et al. 2013). In the following experiments we tested if Ca2+-dependent Cl−and K+ channels play a role in bradykinin-induced membrane blebbing.
Figure5A shows overlays before and during bradykinin application of representative cells treated with different Cl− and K+ channel blockers. To test if Cl− channels are involved in bleb formation, we first replaced Cl− with 130 mm gluconate (Fig.5B). Gluconate reduced the percentage of cells with membrane blebs during application of 100 nm bradykinin from 79.17% in controls to 25%. Next we bathed the cells in 10 μm TMEM 16A inhibitor. TMEM 16A channels are Ca2+-operated Cl− channels and have been described as being overexpressed in cancer (Hartzell et al. 2009). Adding TMEM 16A inhibitor to the bath resulted, surprisingly, in bigger blebs (Fig.5A) and strongly increased membrane blebbing from 79.17% in control cells to 100% (Fig.5B). 4,4′-Diisothiocyanatostilbene-2,2′-disulfonic acid disodium salt (DIDS, 200 μm), a general blocker of Ca2+-activated chloride channels, decreased cells responding with membrane blebs to 15.38%. 5-Nitro-2-3-phenylpropylaminobenzoic acid (NPPB, 200 μm) reduced the population of cells with membrane blebs to 8%.
Figure 5. Regulation of bradykinin-induced membrane blebbing by Ca2+-dependent Cl− and K+ channels.
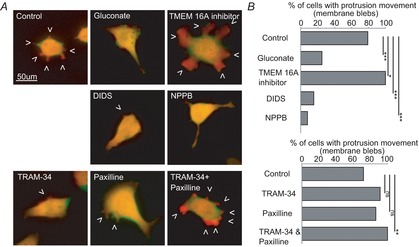
A, examples of dsRed fluorescence of D54 glioma cells before (green) and during application of 100 nm bradykinin (red) with different pretreatments (overlay yellow). B, summary of all cells. Upper graph: control (no pretreatment) n = 24, 130 mm gluconate n = 20, 10 μm TMEM 16Ainhibitor n = 28, 200 μm DIDS n = 26, 200 μm NPPB n = 25. Lower graph: control (no pretreatment) n = 25, 10 μm TRAM-34 n = 23, 2.5 μm paxiline n = 22, 10 μm TRAM-34 + 2.5 μm paxiline n = 25. χ2 test was used to compare protrusion movements.
For investigation of K+ channels we used 10 μm TRAM-34, a blocker IK channels, and 2.5 μm paxiline, a blocker of BK channels. Only when we combined both blockers was membrane blebbing increased and found in 100% of the cells.
We investigated if the underlying ion channels were expressed on membrane blebs by immune staining (Fig.6A). D54 cells were treated with 100 nm bradykinin. After 25 s the cells were fixed with ice-cold 3% paraformaldehyde (PFA). To investigate membrane blebs we stained actin with phalloidin, nuclei with DAPI and ion channels with antibodies. Figure6B shows a magnification of the staining for the Ca2+-activated channels IK, BK, TMEM 16A and ClC3 channels on membrane blebs.
Figure 6. Regulation of bradykinin-induced glioma migration by Ca2+-dependent Cl− and K+ channels.
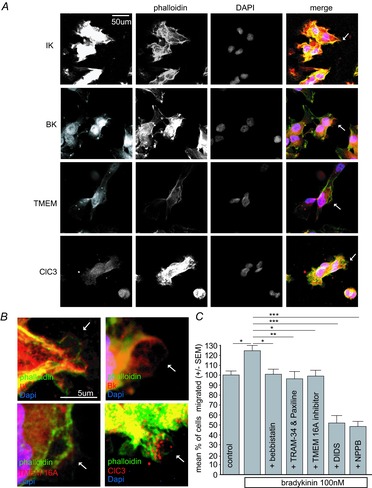
A, immunostainings of Ca2+-activated K+ and Cl− channels in blebbing D54 glioma cells. Confocal z-projections of receptor staining with antibodies against IK, BK, TMEM and ClC3 channels. Individual membrane blebs (arrows) are magnified in B. Receptors red, Alexa Fluor 488 Phalloidin stain green, DAPI stain blue. C, bradykinin-induced transwell migration was blocked by 2.5 μm blebbistatin, 10 μm TRAM-34 and 2.5 μm paxiline, 10 μm TMEM 16A inhibitor, 200 μm DIDS and 200 μm NPPB. N = 15 per group (3 × 5 repetitions).
We performed transwell migration assays to test the relevance of Ca2+-dependent ion channels in bradykinin-induced migration (Fig.6C). The transwells were coated with laminin. Basal migration of untreated D54 cells (control) was set to 100%. Bradykinin induced an increase in glioma migration to 124 ± 5.22%. Blebbistatin (100.99 ± 5.19%), TRAM-34 and paxiline in combination (96.48 ± 7.23%) and TMEM 16A inhibitor (99.13 ± 5.92%) reduced migration back to control levels. DIDS blocked transwell migration to 52 ± 7.4% and NPPB blocked migration to 48.4 ± 5.24%.
In summary, we conclude that Cl− depletion with gluconate and unspecific block of Cl− channels with DIDS and NPPB resulted in an absence of membrane blebbing, indicating that the underlying channels could be involved in cell contraction and bleb formation. Specific blocking of TEMEM 16A, IK and BK channels evoked increased bleb formation. We therefore suggest that these channels participate in bleb retraction. Interestingly, all blockers reduced transwell migration of glioma cells. With the help of immunohistochemistry we showed that K+ and Cl− channels are localized in membrane blebs.
Bradykinin induces protrusion movement of glioma cells in situ
To examine whether membrane blebs play a role in bradykinin-induced invasion we implanted the D45-GCaMP3/dsRed cells into the cortex of BALB/c scid mice. Acute brain slices were prepared 3–4.5 weeks after tumour implantation. Confocal stacks (z-interval 2 μm) of the GCaMP3/dsRed expressing cells were recorded every 15 s with a multiphoton confocal microscope (Fig.7). Figure7A shows an example cell in the periphery of its tumour mass. Following 100 nm bradykinin application for 2 min in the bath, [Ca2+] increased transiently and the cells moved (Fig.7B,; Supporting information Videos S2 and S3). By analysing the dsRed z-projections we found that bradykinin induced movement in the protrusions (Fig.7C, ROI 1–3), but not in the cell body (ROI 5, n = 6 of 9 cells in 3 recordings).
Figure 7. Bradykinin induces protrusion movement in situ.
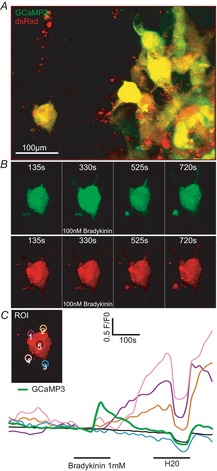
A, D54-GCaMP3/dsRed cells were implanted in the neocortex of mice. Cells in the periphery of the tumour were investigated. B, relevant time points before, during and after bradykinin application (100 nm, 2 min) are shown for the left cell in A (see also Supporting information videos S2 and S3). C, fluorescence traces of the 4 protrusions (ROI 1–4 indicated in the insert) and [Ca2+]i of the example cell. ROI 5 in the middle of the cell did not show dsRed fluorescence changes in response to bradykinin (n = 6 of 9 cells in 3 recordings).
Bradykinin facilitates tumour growth in situ by increasing migration speed of cells along blood vessels
We next implanted D54-EGFP cells into organotypic brain slices of BALB/c scid mice that have been cultured for 3 days. EGFP (enhanced green fluorescent protein) fluorescence was photographed 1 and 8 days after cell implantation (Fig.8A). Figure8B shows photographs of control, bradykinin- (100 nm) and bradykinin (100 nm) + Hoe-140 (5 μm)-treated brain slices at day 8. The tumour size was determined by normalizing the tumour area at day 8 to the tumour area at day 1. We found that the tumour size was significantly larger in cultured brain slices treated with 10 nm, 100 nm and 1 μm bradykinin than in non-treated control slices (Fig.8C). Treatment with 10 μm bradykinin did not show a significant difference between tumours of control slices and tumours of treated slices, probably due to additional bradykinin receptor activation of other cell types in the slice. Tumour growth in response to 100 nm bradykinin was blocked with the B2 receptor antagonist Hoe-140 (5 μm) (Fig.8C).
We fixed and stained slices with anti-laminin antibody, a marker of the basal membrane of blood vessels, and anti-GFP antibody for the detection of the EGFP expressing D54 cells (Fig.8D). A closer look at the implanted tumours revealed that single D54-EGFP cells left the tumour mass at the rim of the tumour. Many cells looked elongated and beaded. In chain migrating glioma cells were found to be attached to blood vessels. Interestingly, the laminin staining within the main tumour mass was defuse whereas in the periphery blood vessels looked well-structured and did not differ from the control staining of the contralateral hemisphere. Glioma cells have been described as invading the adjacent healthy brain tissue by perivascular migration (Farin et al. 2006; Zagzag et al. 2008). We recorded time-lapse movies of EGFP expressing D54 glioma cells at the edge of the D54-EGFP tumours in live cultured brain slices with different treatment (see Supporting information example videos: for control Video S4, for bradykinin Video S5, for bradykinin + Hoe-140 Video S6, for bradykinin + blebbistatin Video S7, for blebbistatin Video S8). In bradykinin-treated brain slices D54 glioma cells migrated longer distances within the same time (Fig.8E). This effect was blocked by Hoe-140. In slices treated with blebbistatin and blebbistatin combined with bradykinin, D54 cells migrated significantly less compared to the control level. Detailed statistics are listed in Table1. We additionally calculated the average migration speed of the cells and found significant differences in the averaged velocity of the cells treated with bradykinin (Fig.8F). In untreated control slices D54-EGFP cells migrated at 15.25±0.16 μm h−1. Bradykinin induced an elevation of the average migration speed to 19.85 ±0.17 μm h−1, which was reduced by Hoe-140 to 13.43 ±0.11 μm h−1 and by blebbistatin to 7.29 ±0.22 μm h−1. Basal migration speed of D54 cells in brain slices treated with blebbistatin alone was 10.23 ±0.29 μm h−1.
Table 1.
One-way analysis of variance (ANOVA) with Tukey–Kramer multiple comparisons test at time point 11.5 h
| Mean comparison | Difference (μm) | q | P value |
|---|---|---|---|
| Control vs. bradykinin | −56.515 | 5.044 | **P < 0.01 |
| Control vs. bradykinin + Hoe-140 | 2.625 | 0.2289 | n.s. P > 0.05 |
| Control vs. Hoe-140 | 27.302 | 1.897 | n.s. P > 0.05 |
| Control vs. bradykinin + blebbistatin | 86.910 | 5.201 | **P < 0.01 |
| Control vs. blebbistatin | 58.710 | 3.513 | n.s. P > 0.05 |
| Bradykinin vs. bradykinin + Hoe-140 | 59.140 | 5.157 | **P < 0.01 |
| Bradykinin vs. Hoe-140 | 83.817 | 5.824 | ***P < 0.001 |
| Bradykinin vs. bradykinin + blebbistatin | 143. 42 | 8.582 | ***P < 0.001 |
| Bradykinin vs. blebbistatin | 115.22 | 6.895 | ***P < 0.001 |
| Bradykinin + Hoe-140 vs. Hoe | 24.677 | 1.691 | n.s. P > 0.05 |
| Bradykinin + Hoe-140 vs. Bradykinin + blebbistatin | 84.285 | 4.990 | **P < 0.01 |
| Bradykinin + Hoe-140 vs. blebbistatin | 56.085 | 3.321 | n.s. P > 0.05 |
| Hoe vs. bradykinin + blebbistatin | 59.607 | 3.138 | n.s. P > 0.05 |
| Hoe vs. blebbistatin | 31.408 | 1.653 | n.s. P > 0.05 |
| Bradykinin + blebbistatin vs. blebbistatin | −28.200 | 1.355 | n.s. P > 0.05 |
n.s., not significant. P < 0.0001 (***) is considered extremely significant. If the value of q is greater than 4.071 then the P value is less than 0.05.
These results show that bradykinin enhances glioma expansion by facilitating migration of single glioma cells into the peritumoural tissue. Migration was reduced by the B2 receptor blocker Hoe-140 and by the myosin II kinase blocker blebbistatin, which suggests that bradykinin induces an amoeboid migration type in glioma cells.
Discussion
Previous studies showed that bradykinin induces chemotaxis of glioma cells in vitro (Montana & Sontheimer, 2011; Cuddapah et al. 2013), but the detailed mechanisms of bradykinin-driven migration have not been identified. This study provides insights into the mechanism of membrane bleb regulation and gives evidence for an important contribution of membrane blebs in bradykinin-induced amoeboid invasion of glioma cells.
Membrane blebs have been evoked via the activation of B2 receptors (Fig.9A). B2 receptor stimulation leads to the formation of IP3 and subsequent release of Ca2+ from intracellular stores (Fig.9B). Ca2+ is an important regulator of cell migration. Hence, by depletion of Ca2+ from the bath solution and Ca2+ uncaging experiments, we show that the formation of membrane blebs is dependent on Ca2+. We further found that transient Ca2+ release from intracellular stores is sufficient to cause membrane blebs in D54 glioma cells and that disturbing intracellular Ca2+ homeostasis prevents bleb formation.
Figure 9. Mechanism of bradykinin-induced amoeboid migration.
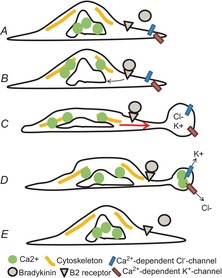
A, D54 cell before bradykinin stimulation. B, B2 receptor binding and release of Ca2+ from internal stores. C, contraction of the actomyosin cortex (yellow), cytoplasmic flow (red arrow) and bleb formation. D, activation of Ca2+-dependent K+ and Cl− channels at the bleb, bleb retraction and cell relaxation. E, cell after bradykinin stimulation.
We observed that bradykinin-induced Ca2+ release evoked a contraction within defined parts of the cell that leads to cytoplasmic flow resulting in hydrostatic pressure against the plasma membrane and subsequent formation of membrane blebs (Fig.9C). The mechanism of ligand-induced contraction of the actomyosin cortex with subsequent bleb formation was suggested first for primordial germ cells (Blaser et al. 2006; Paluch & Raz, 2013). A similar mechanism has subsequently been described in continuously blebbing human melanoma cells, where blebbing in response to serum stimulation was dependent on membrane tension and cytosolic pressure (Charras et al. 2008). In one of their experiments the authors microinjected polyethylene glycol-passivated quantum dots into the cytoplasm of blebbing cells (hydrodynamic diameter 25 nm). Dots were detected in forming membrane blebs, indicating that the blebs are filled with cytoplasm. Furthermore they observed the flow of lipids into the bleb through the bleb neck, which led to tearing of the membrane bilayer from the actomyosin cortex.
To confirm that bradykinin-induced contraction in glioma cells was dependent on proteins of the actomyosin cortex, we blocked membrane blebbing by using cytocalasin D, a blocker of actin polymerization. Cytocalasin D inhibited cell contraction and membrane blebbing completely. Secondly, we used blebbistatin, a blocker of myosin II kinase. In contrast to cytochalasin D, membrane blebbing was increased, suggesting that bleb retraction rather than bleb formation was impaired. At first glance this result is in contrast to the observations made on continuously blebbing cells, where blebbistatin blocks membrane blebbing (Charras et al. 2005, 2006; Kapustina et al. 2013). Continuous blebbing is based on oscillating fluctuations of Ca2+ that trigger rhythmic actomyosin cortex contractions followed by bleb formation in the plasma membrane and subsequent bleb retraction (Charras et al. 2006). Since bleb formation involves disruption of the actomyosin cortex itself, blocking myosin II kinase also blocks actomyosin cortex regeneration and hinders contraction of the actomyosin cortex in the next phase. Consequently the formation of new membrane blebs is prevented. In accordance with our results, the authors indeed report that myosin II kinase function was necessary for bleb retraction after uncoupling contractility of the actomyosin cortex from bleb retraction. Our results suggest that bradykinin-induced bleb formation is myosin II kinase independent and we agree with the hypothesis that the protein is involved in the retraction of membrane blebs, for instance by the formation of a new actomyosin cortex underneath the retracted bleb.
We found that bradykinin-induced membrane blebs are accompanied by small cell volume changes of approximately 10%. A recent paper shows that glioma cells change their volume between 10 and 30% when the cells invade peritumoural tissue (Watkins & Sontheimer, 2011). A previous study also found that blocking transient Ca2+ increase or Ca2+-dependent ion channels blocked bradykinin-induced glioma migration (Cuddapah et al. 2013). In Walker carcinosarcoma cells K+ accumulation has been found in the vicinity of forming blebs (Vanhecke et al. 2008). Since membrane blebs present local volume changes we tested if Ca2+-dependent Cl− and K+ channels are involved in the regulation of membrane blebs. We show that Ca2+-activated Cl− channels regulate membrane blebbing by using DIDS and NPPB, unspecific blockers of Ca2+-dependent Cl− channels. These substances blocked membrane blebbing completely. TMEM 16A inhibitor, a blocker of TMEM channels, increased blebbing in response to bradykinin. The different channels might be involved in different phases of the bradykinin-induced cell response. DIDS- and NPPB-sensitive Cl− channels could be involved, for instance, in the contraction of the cytoskeleton via ClC3 channels and could therefore block bleb initiation, suggesting that these unspecific blockers interfere with cell movement in general. This would also explain why DIDS and NPPB block transwell migration of D54 glioma cells below control levels. In comparison TMEM 16A inhibitor blocked bleb retraction and bradykinin-induced transwell migration like blebbistatin. We also found that IK and BK channels are involved in the retraction of membrane blebs, since blocking these channels with TRAM-34 and paxiline in combination evoked, like TMEM 16A inhibitor, an increase in membrane blebbing. IK and BK channels might serve redundant roles, since increased membrane blebbing is only achieved by blocking both channels. In transwell migration assays, treatment with TMEM 16A, TRAM-34 + paxiline or blebbistatin blocked bradykinin-induced transwell migration. Interestingly, in our previous study TRAM-34 alone was able to block transwell migration of D54 gliomas, if the transwells were coated with vitronectin instead of laminin (Cuddapah et al. 2013). Since TMEM 16A, IK and BK channels blocked bleb retraction we suggest that the channels are involved in moving Cl− and K+ out of the cell with the consequence of a reduction in bleb volume by osmotic water release (Fig.9D and E). Nevertheless, since subtype-specific channel blockers are missing, it remains to be clarified which specific channels are involved, which may be accomplished through knockout and overexpression experiments.
Many of the glioma cells in culture appeared bipolar with a leading and lagging side. Membrane blebs occurred preferentially in either of these polarized sides. The distribution of osmolytes in certain subcompartments such as lamellipodia or protrusions could lead to local increase in volume and fast changes in cell shape. Specified microdomains might have the necessary machinery already assembled in lipid rafts probably including cytoskeletal elements and Ca2+ sensitive ion channels before Ca2+ activates these proteins.
It has been reported that B1 receptors also mediate glioma cell migration by acting on the phosphoinositide 3-kinase and AKT proteins (Lu et al. 2010). In our experiments we found no evidence that B1 receptors are involved in the glioma response to bradykinin.
Other ligands have been also found to induce membrane blebbing in a Ca2+-dependent way. Substance P-induced membrane blebbing in HEK cells and U373MG astrocytoma cells is accompanied by a rise in intracellular Ca2+ and leads to activation of Rho-associated protein kinase (ROCK), a mediator of myosin function (Meshki et al. 2009, 2011). Interestingly, stromal cell-derived factor 1 (SDF-1), another ligand which induces membrane blebbing in glioma cells, was found to stimulate migration by acting on its G-protein coupled receptor CXCR4. SDF-1-induced migration was also Ca2+ dependent and required the activation of Ca2+-dependent K+ channels (Sciaccaluga et al. 2010). In zebra fish primordial germ cells, SDF-1-activated membrane blebbing was dependent on Ca2+ accumulation within the formed bleb (Blaser et al. 2006). In light of these results it needs to be clarified whether bradykinin, substance P and SDF-1 act through the same pathway.
Since membrane blebbing is not described in intact tissue, we tested the response of glioma cells to bradykinin application in situ. We found that bradykinin also results in protrusion movement in acute brain slices. The density of the tissue might favour extension of the cells processes instead the formation of blebs. By investigating D54 glioma cells in brain slice cultures we found that bradykinin treatment leads to faster migration in the periphery of the tumour, resulting in increased tumour growth. Blebbistatin blocked migration in transwell migration assays and in situ migration assays, suggesting a role for amoeboid type migration during invasion.
Bradykinin is most likely released by cells of the vasculature. B2 receptors might be also associated with vascular regulation in astrocytes (Abbott, 2000). Nevertheless the exact release site has not been identified. The fact that glioma cells seeded on top of brain slices migrate to blood vessels in response to bradykinin treatment (Montana & Sontheimer, 2011) agrees well with our results that bradykinin increases the speed of glioma migration. Based on these arguments we favour the idea that bradykinin-induced membrane blebbing is one of the major mechanisms involved in glioma cell migration. Whether bradykinin participates in vascular coupling of glioma cells remains unknown. Microscopic techniques with advanced spatial and temporal resolution are necessary to investigate the protrusion movements in situ in more detail.
This study adds to a growing appreciation that targeting glioma bradykinin 2 receptors might be beneficial for glioma therapy. We found that Hoe-140 blocked bradykinin-induced bleb formation and in situ migration of D54 glioma cells. Hoe-140, the B2 receptor blocker which we used, is already approved for the treatment of hereditary angioedema under the name icatibant (Cicardi et al. 2010), and hence the path to clinical use for gliomas could be accelerated.
Acknowledgments
Thanks to Dr Vishnu Cuddapah for discussion and Dr Stephanie Robert for help with Western Blots.
Glossary
- 0[Ca2+]o
without extracellular Ca2+
- 2-APB
2-aminoethyl diphenylborinate
- ANOVA
analysis of variance
- ACSF
artificial cerebrospinal fluid
- B1R
bradykinin receptor 1
- B2R
bradykinin receptor 2
- BAPTA-AM
1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetrakis acetoxymethyl ester
- BK
big conductance K+ channels
- [Ca2+]i
intracellular Ca2+ concentration
- DIDS
4,4′-diisothiocyanatostilbene-2,2′-disulfonic acid disodium salt
- dsRed
red fluorescent protein of Discosoma sp.
- EGTA-AM
ethylene glycol-bis (2-aminoethylether)- N,N,N′,N′-tetraacetic acid tetrakis acetoxymethyl ester
- EGFP
enhanced green fluorescent protein
- ER
endoplasmatic reticulum
- FI
fluorescence intensity
- GCaMP3
EGFP-based Ca2+ probe
- IK
intermediate conductance K+ channels
- IP3
inositol-1,4,5-trisphosphate
- NPPB
5-nitro-2-(3-phenylpropylamino)benzoic acid
- PFA
paraformaldehyde
- ROI
region of interest
- SERCA
sarco-/endoplasmic reticulum Ca2+ ATPase
- SOCE
store-operated Ca2+ entry
- TRPC
canonical transient receptor potential
- NP-EGTA-AM
o-nitrophenyl EGTA acetoxymethyl ester
- UV
ultraviolet
Additional information
Competing interests
The authors have no competing interests to disclose.
Author contributions
Dr S. Seifert is the first author and was responsible for study conception, surgical procedures, animal care, all data collection, data analyses and interpretation, and drafting of the manuscript. Prof. Dr H. Sontheimer is the senior author and was responsible for study conception, data interpretation, drafting of the manuscript, and final approval of the manuscript.
Funding
This work was supported by grant 2RO1 NS-036692 from the National Institutes of Health.
Key points
Glioma multiforme is a fast expanding and aggressive type of brain tumour that invades healthy brain tissue by migration of single glioma cells along blood vessels.
We show that bradykinin, a neuropeptide of the vasculature, induces the formation of small bleb-like protrusions at the plasma membrane of glioma cells in vitro, which are regulated by intracellular Ca2+, resulting in contraction of the cytoskeleton, cytoplasmatic flow and activation of Ca2+-dependent K+ and Cl− channels.
From our in vitro experiments we conclude that bradykinin facilitates glioma invasion by stimulating an amoeboid phenotype of migration.
In situ experiments confirmed that bradykinin increases the speed of glioma cell migration, which we have been able to block with blebbistatin, an inhibitor of membrane blebbing, and with the B2 receptor agonist Hoe-140.
The study reveals novel mechanisms of bradykinin-induced glioma migration and suggests pharmacological targets to reduce glioma invasion.
Supporting Information
The following supporting information is available in the online version of this article.
In vitro D54 cell response to 100nM bradykinin (3D)
In situ D54 cell response to 100nM bradykinin (GCaMP3)
In situ D54 cell response to 100nM bradykinin (GCaMP3)
In situ migration assay (control)
In situ migration assay (bradykinin)
In situ migration assay (bradykinin+Hoe-140)
In situ migration assay (bradykinin+blebbistatin)
In situ migration assay (blebbistatin)
References
- Abbott NJ. Inflammatory mediators and modulation of blood–brain barrier permeability. Cell Mol Neurobiol. 2000;20:131–147. doi: 10.1023/A:1007074420772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billups D, Billups B, Challiss RAJ. Nahorski SR. Modulation of Gq-protein-coupled inositol trisphosphate and Ca2+ signalling by the membrane potential. J Neurosci. 2006;26:9983–9995. doi: 10.1523/JNEUROSCI.2773-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaser H, Reichman-Fried M, Castanon I, Dumstrei K, Marlow FL, Kawakami K, Solnica-Krezel L, Heisenberg C-P. Raz E. Migration of zebrafish primordial germ cells: a role for myosin contraction and cytoplasmic flow. Dev Cell. 2006;11:613–627. doi: 10.1016/j.devcel.2006.09.023. [DOI] [PubMed] [Google Scholar]
- Charras GT, Coughlin M, Mitchison TJ. Mahadevan L. Life and times of a cellular bleb. Biophys J. 2008;94:1836–1853. doi: 10.1529/biophysj.107.113605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charras GT, Hu C-K, Coughlin M. Mitchison TJ. Reassembly of contractile actin cortex in cell blebs. J Cell Biol. 2006;175:477–490. doi: 10.1083/jcb.200602085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charras GT, Yarrow JC, Horton MA, Mahadevan L. Mitchison TJ. Non-equilibration of hydrostatic pressure in blebbing cells. Nature. 2005;435:365–369. doi: 10.1038/nature03550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cicardi M, Banerji A, Bracho F, Malbrán A, Rosenkranz B, Riedl M, Bork K, Lumry W, Aberer W, Bier H, et al. Icatibant, a new bradykinin-receptor antagonist, in hereditary angioedema. N Engl J Med. 2010;363:532–541. doi: 10.1056/NEJMoa0906393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuddapah VA, Turner KL, Seifert S. Sontheimer H. Bradykinin-induced chemotaxis of human gliomas requires the activation of KCa3.1 and ClC-3. J Neurosci. 2013;33:1427–1440. doi: 10.1523/JNEUROSCI.3980-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fackler OT. Grosse R. Cell motility through plasma membrane blebbing. J Cell Biol. 2008;181:879–884. doi: 10.1083/jcb.200802081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farin A, Suzuki SO, Weiker M, Goldman JE, Bruce JN. Canoll P. Transplanted glioma cells migrate and proliferate on host brain vasculature: a dynamic analysis. Glia. 2006;53:799–808. doi: 10.1002/glia.20334. [DOI] [PubMed] [Google Scholar]
- Hagmann J, Burger MM. Dagan D. Regulation of plasma membrane blebbing by the cytoskeleton. J Cell Biochem. 1999;73:488–499. [PubMed] [Google Scholar]
- Hartzell HC, Yu K, Xiao Q, Chien L-T. Qu Z. Anoctamin/TMEM16 family members are Ca2+-activated Cl– channels. J Physiol. 2009;587:2127–2139. doi: 10.1113/jphysiol.2008.163709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ifuku M, Färber K, Okuno Y, Yamakawa Y, Miyamoto T, Nolte C, Merrino VF, Kita S, Iwamoto T, Komuro I, et al. Bradykinin-induced microglial migration mediated by B1-bradykinin receptors depends on Ca2+ influx via reverse-mode activity of the Na+/Ca2+ exchanger. J Neurosci. 2007;27:13065–13073. doi: 10.1523/JNEUROSCI.3467-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapustina M, Elston TC. Jacobson K. Compression and dilation of the membrane-cortex layer generates rapid changes in cell shape. J Cell Biol. 2013;200:95–108. doi: 10.1083/jcb.201204157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu D-Y, Leung Y-M, Huang S-M. Wong K-L. Bradykinin-induced cell migration and COX-2 production mediated by the bradykinin B1 receptor in glioma cells. J Cell Biochem. 2010;110:141–150. doi: 10.1002/jcb.22520. [DOI] [PubMed] [Google Scholar]
- Markovic DS, Vinnakota K, Chirasani S, Synowitz M, Raguet H, Stock K, Sliwa M, Lehmann S, Kälin R, van Rooijen N, et al. Gliomas induce and exploit microglial MT1-MMP expression for tumor expansion. Proc Natl Acad Sci U S A. 2009;106:12530–12535. doi: 10.1073/pnas.0804273106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meshki J, Douglas SD, Hu M, Leeman SE. Tuluc F. Substance P induces rapid and transient membrane blebbing in U373MG cells in a p21-activated kinase-dependent manner. PLoS One. 2011;6:e25332. doi: 10.1371/journal.pone.0025332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meshki J, Douglas SD, Lai J-P, Schwartz L, Kilpatrick LE. Tuluc F. Neurokinin 1 receptor mediates membrane blebbing in HEK293 cells through a Rho/Rho-associated coiled-coil kinase-dependent mechanism. J Biol Chem. 2009;284:9280–9289. doi: 10.1074/jbc.M808825200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montana V. Sontheimer H. Bradykinin promotes the chemotactic invasion of primary brain tumors. J Neurosci. 2011;31:4858–4867. doi: 10.1523/JNEUROSCI.3825-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paluch EK. Raz E. The role and regulation of blebs in cell migration. Curr Opin Cell Biol. 2013;25:582–590. doi: 10.1016/j.ceb.2013.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ransom CB. Sontheimer H. BK channels in human glioma cells. J Neurophysiol. 2001;85:790–803. doi: 10.1152/jn.2001.85.2.790. [DOI] [PubMed] [Google Scholar]
- Reetz G. Reiser G. [Ca2+]i oscillations induced by bradykinin in rat glioma cells associated with Ca2+ store-dependent Ca2+ influx are controlled by cell volume and by membrane potential. Cell Calcium. 1996;19:143–156. doi: 10.1016/s0143-4160(96)90083-4. [DOI] [PubMed] [Google Scholar]
- Reiser G, Binmöller FJ. Hamprecht B. The regulatory influence of bradykinin and inositol-1,4,5-trisphosphate on the membrane potential in neural cell lines. Biomed Biochim Acta. 1987;46:S682–687. [PubMed] [Google Scholar]
- Schulze-Topphoff U, Prat A, Prozorovski T, Siffrin V, Paterka M, Herz J, Bendix I, Ifergan I, Schadock I, Mori MA, Van Horssen J, et al. Activation of kinin receptor B1 limits encephalitogenic T lymphocyte recruitment to the central nervous system. Nat Med. 2009;15:788–793. doi: 10.1038/nm.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sciaccaluga M, Fioretti B, Catacuzzeno L, Pagani F, Bertollini C, Rosito M, Catalano M, D'Alessandro G, Santoro A, Cantore G, et al. CXCL12-induced glioblastoma cell migration requires intermediate conductance Ca2+-activated K+ channel activity. Am J Physiol, Cell Physiol. 2010;299:C175–C184. doi: 10.1152/ajpcell.00344.2009. [DOI] [PubMed] [Google Scholar]
- Vanhecke D, Bellmann R, Baum O, Graber W, Eggli P, Keller H. Studer D. Pseudovacuoles – immobilized by high-pressure freezing – are associated with blebbing in walker carcinosarcoma cells. J Microsc. 2008;230:253–262. doi: 10.1111/j.1365-2818.2008.01982.x. [DOI] [PubMed] [Google Scholar]
- Watkins S. Sontheimer H. Hydrodynamic cellular volume changes enable glioma cell invasion. J Neurosci. 2011;31:17250–17259. doi: 10.1523/JNEUROSCI.3938-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf K, Mazo I, Leung H, Engelke K, von Andrian UH, Deryugina EI, Strongin AY, Bröcker E-B. Friedl P. Compensation mechanism in tumor cell migration: mesenchymal–amoeboid transition after blocking of pericellular proteolysis. J Cell Biol. 2003;160:267–277. doi: 10.1083/jcb.200209006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zagzag D, Esencay M, Mendez O, Yee H, Smirnova I, Huang Y, Chiriboga L, Lukyanov E, Liu M. Newcomb EW. Hypoxia- and vascular endothelial growth factor-induced stromal cell-derived factor-1α/CXCR4 expression in glioblastomas: one plausible explanation of Scherer's structures. Am J Pathol. 2008;173:545–560. doi: 10.2353/ajpath.2008.071197. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
In vitro D54 cell response to 100nM bradykinin (3D)
In situ D54 cell response to 100nM bradykinin (GCaMP3)
In situ D54 cell response to 100nM bradykinin (GCaMP3)
In situ migration assay (control)
In situ migration assay (bradykinin)
In situ migration assay (bradykinin+Hoe-140)
In situ migration assay (bradykinin+blebbistatin)
In situ migration assay (blebbistatin)