

Abstract
Tumor necrosis factor-alpha (TNF-α) is a central mediator of inflammatory responses elicited by Toll-like receptor agonists, such as the Gram-negative bacterial outer membrane antigen lipopolysaccharide (LPS). TNF-α is responsible for altering vascular permeability and activating infiltrating inflammatory cells, such as monocytes and neutrophils. Interestingly, TNF-α has also demonstrated the ability to induce tolerance to subsequent challenges with TNF-α or LPS in monocyte and macrophage cell populations. Tolerance is characterized by the inability to mount a typical inflammatory response during subsequent challenges following the initial exposure to an inflammatory mediator such as LPS. The ability of TNF-α to induce a tolerant-like state with regard to LPS is most likely a regulatory mechanism to prevent excessive inflammation. We hypothesized that the induction of tolerance or the degree of tolerance is dependent upon the production of TNF-α during the primary response to LPS. To investigate TNF-α-dependent tolerance, human monocytic THP-1 cells were treated with TNF-α-neutralizing antibodies or antagonistic TNF-α receptor antibodies before primary LPS stimulation and then monitored for the production of TNF-α during the primary and challenge stimulation. During the primary stimulation, anti-TNF-α treatment effectively attenuated the production of TNF-α and interleukin-1β; however, this reduced production did not impact the induction of endotoxin tolerance. These results demonstrate that interfering with TNF-α signaling attenuates production of inflammatory cytokines without affecting the induction of tolerance.
Keywords: endotoxin tolerance, lipopolysaccharide, tumor necrosis factor-alpha, anti-tumor necrosis factor-alpha, THP-1 cells
Introduction
The endotoxin tolerance phenomenon is defined by an attenuated production of inflammatory mediators during subsequent challenges following the initial exposure to the lipopolysaccharide (LPS) of Gram-negative bacteria. Endotoxin tolerance is a suppressive immunological state that effectively regulates excessive production of inflammatory cytokines and mediators during a Gram-negative bacterial infection. The induction of tolerance has predominantly been observed and studied in monocytes and macrophages, which are the primary cells involved in microbial inflammatory responses.1,2 In vivo endotoxin tolerance can be induced by sublethal LPS administration that reduces morbidity and mortality during subsequent lethal challenges with endotoxin or Gram-negative bacteria.3,4 In addition, tolerant macrophages demonstrate increased phagocytic activity, which enhances the clearance of the microbial pathogen.5
Monocytes and macrophages are activated by LPS following recognition of the antigen by the germ-line encoded Toll-like receptor 4 (TLR4) which initiates an intracellular signaling cascade that activates the transcription factor nuclear factor-kappa B (NF-κB).6 NF-κB is responsible for the transcription of many of the inflammatory mediators involved in host resistance to Gram-negative bacterial infection.7,8 The potent cytokine tumor necrosis factor-alpha (TNF-α) is a well established mediator of inflammation capable of activating endothelial vascular permeability changes and promoting infiltration of inflammatory cells at the sites of infection.9,10 Similar to LPS, TNF-α signaling activates NF-κB11 to induce production of inflammatory cytokines.12,13 TNF-α signals through two receptors, TNFRI and TNFRII, and it has been demonstrated that this interaction is involved in cell survival, apoptosis, vascular permeability, and promoting the inflammatory response.10,14 Monitoring the production of TNF-α has been used extensively as an indicator of endotoxin tolerance, because the TNF-α response during subsequent challenges is attenuated.
During a Gram-negative bacterial infection, LPS recognized by TLR4 activates NF-κB;15 the activated transcription factor then translocates into the nucleus to promote the transcription of genes coding for a variety of inflammatory mediators, including TNF-α. TNF-α mRNA is translated and the secreted TNF-α protein signals back on the responding cell to stabilize and amplify NF-κB gene expression.16 If expression of the TNF-α receptors is knocked down by RNA interference during stimulation of LPS, production of TNF-α protein is suppressed.17 These reports indicate that TNF-α can amplify its own expression through an autocrine amplification loop.18 However, this TNF-α amplification system may be involved in excessive production of the inflammatory cytokine if unregulated. Interestingly, many of the LPS-induced pathophysiological consequences are associated with the damage caused by TNF-α.19,20 In severe sepsis or septic shock induced by Gram-negative bacteria, the most debilitating pathologies are directly related to the functions of TNF-α, such as hypotension, cell death, tissue damage, and multiple organ dysfunction syndrome.21–25 Due to the adverse effects of excessive TNF-α production during a systemic inflammatory response to bacterial infection, there have been numerous attempts to impede severe sepsis and septic shock with anti-TNF-α treatments.26–30 Unfortunately, this approach has not demonstrated efficacy in clinical trials, and the use of anti-TNF-α therapy in severe sepsis or septic shock has almost been abandoned.31–33 A major side effect of using anti-TNF-α treatments is the immunosuppressive state that leaves the host vulnerable to subsequent infections.34
In addition to amplifying the inflammatory response, anti-inflammatory effects have been associated with TNF-α.35,36 Probably the most important anti-inflammatory effect of TNF-α is the ability to induce a tolerant-like state in which the host is hyporesponsive to subsequent challenges with TNF-α or LPS.37–39 In 1998, Fraker et al demonstrated that sublethal TNF-α treatment could protect rats from lethal TNF-α administration, similar to the effects of sublethal LPS treatment and endotoxin tolerance.37 Fahmi and Chaby determined that LPS was not necessary to induce tolerance, but that culture supernatant from LPS-stimulated macrophages could induce a hyporesponsive state in naïve macrophages after the LPS was removed from the culture.38 In addition, demonstrating that TNF-α could induce tolerance to subsequent TNF-α stimulations, Ferlito et al designed an experimental system to investigate cross-tolerance between TNF-α and LPS.39 They demonstrated that human monocytic THP-1 cells treated with TNF-α were refractory to subsequent challenges with TNF-α or LPS, and that LPS induced tolerance to TNF-α or LPS challenges. In 2011, Park et al suggested a molecular mechanism for TNF-α-induced tolerance to subsequent LPS challenges.40 The fact that TNF-α production is critical to activating the inflammatory response to LPS, but can also induce a tolerant state, provided an opportunity to investigate the hypothesis that TNF-α production and autocrine signaling alone was responsible for induction of tolerance to LPS.
We treated human monocytic THP-1 cells with soluble TNF-α-neutralizing antibodies and/or antagonistic TNF-α receptor antibodies prior to stimulation with LPS and monitored the production of TNF-α and interleukin (IL)-1β. Treatment with the antibodies significantly attenuated production of the proinflammatory cytokines during the primary LPS response. However, when these same cells were challenged with another LPS stimulation, tolerance was induced even when TNF-α signaling was impaired. These results suggest that TNF-α production and autocrine signaling are not responsible for induction of LPS tolerance, and that LPS signaling activates the regulatory mechanisms involved in attenuating the inflammatory response to subsequent challenges.
Materials and methods
Cell culture
Human monocytic THP-1 cells were acquired from the American Tissue Culture Collection (Manassas, VA, USA) and grown in Roswell Park Memorial Institute medium with L-glutamine (HyClone, Thermo Fisher Scientific, Waltham, MA, USA; Cat#10-040-CM) containing 10% heat-inactivated fetal bovine serum (HyClone; Cat#SH30071.03), 100 U/mL penicillin, 100 μg/mL streptomycin (HyClone; Cat# SV30010), 25 mM HEPES (Thermo Fisher Scientific; Cat# BP299-500), and β-mercaptoethanol (1 mL per L medium, Gibco, Life Technologies, Carlsbad, CA, USA; Cat#21985-023).
TNF-α treatment
THP-1 cells (2.5×105) were stimulated with human recombinant TNF-α (BioLegend, San Diego, CA, USA; Cat#570109) for 8 hours at 37°C with 5.0% CO2. Following initial stimulation, the cells were washed twice with prewarmed sterile phosphate-buffered saline and resuspended with 100 ng/mL of purified LPS from Escherichia coli ([0111:B4], Sigma-Aldrich, St Louis, MO, USA; Cat#L4391) for 2 hours at 37°C. The supernatants were collected for cytokine analysis. Each experiment was repeated at least two times.
TNF-α or LPS-induced tolerance
THP-1 cells (2.5×105) were stimulated with 1 ng/mL of human recombinant TNF-α or 100 ng/mL of purified LPS from E. coli for 2 hours at 37°C with 5.0% CO2. Following initial stimulation, the cells were washed twice with prewarmed sterile phosphate-buffered saline and resuspended with fresh prewarmed complete medium. Cell culture supernatants were collected and the cells were resuspended in fresh prewarmed medium every 2 hours for 14 hours. At 16 hours, the THP-1 cells were challenged with 1 ng/mL human recombinant TNF-α or 100 ng/mL LPS for 2 hours. The supernatants were collected for cytokine analysis. Each experiment was repeated at least two times.
Anti-TNF-α or anti-TNF-α receptor treatment
THP-1 cells (2.5×105) were treated with 25 μg/mL soluble TNF-α-neutralizing antibody (BioLegend, Cat#502922) or 10 μg/mL of each anti-TNF-α receptor I (R&D Systems, Minneapolis, MN, USA; Cat#MAB625) and II (R&D Systems, Cat# MAB726) antibodies for one hour; the cells were then stimulated with 1 ng/mL human recombinant TNF-α or 100 ng/mL of purified LPS. The concentration of antibodies used to neutralize the soluble TNF-α or inhibit the receptors was determined by stimulating THP-1 cells with 1 ng/mL of recombinant TNF-α in the presence of varying concentrations of each antibody. In addition, a mouse IgG1 isotype control antibody (BioLegend, Cat#400124) was used as a negative control and resulted in identical results as the no antibody control. Following initial stimulation, the cells were washed twice with prewarmed sterile phosphate-buffered saline and resuspended with fresh prewarmed complete medium for 2 hours. The cell culture supernatant was removed and fresh pre-warmed complete media was added every 2 hours for the next 14 hours. At 16 hours, the THP-1 cells were challenged with a 100 ng/mL LPS dose for 2 hours. The supernatants were collected for cytokine analysis. Each experiment was repeated at least two times.
TNF-α and IL-1β ELISA
The THP-1 cell culture supernatants were harvested and stored at −30°C until analysis by enzyme-linked immunosorbent assay (ELISA). The supernatants were analyzed for TNF-α or IL-1β protein secretion using BioLegend ELISAMax kits (human TNF-α, Cat#78402; human IL-1β, Cat#437005). The analysis was carried out following the manufacturer’s instructions. Samples were analyzed in duplicate.
Statistical analysis
Data represented as the mean ± standard deviation of at least two assessments. The Student’s t-test (P<0.05) was used to test for statistical significance.
Results
TNF-α treatment attenuates response to LPS in THP-1 cells
The inflammatory cytokine TNF-α has been demonstrated to induce a tolerant-like state in monocytes and macrophages on subsequent challenges with TNF-α or LPS.37,40 In our model, human monocytic THP-1 cells were treated with recombinant human TNF-α for 8 hours before being stimulated with 100 ng/mL LPS from E. coli for an additional 2 hours. Exogenous TNF-α stimulation caused a dose-dependent endogenous TNF-α response, and for most concentrations produced more than double the amount of TNF-α used for stimulation (Figure 1A). These results are in line with previous investigations demonstrating an autocrine TNF-α amplification loop.18 Following treatment with TNF-α, the THP-1 cells were washed and stimulated with LPS. An attenuation of TNF-α production was observed that correlated with the concentration of TNF-α used during treatment (Figure 1B). The reduced TNF-α response to LPS was significant for treatment with 250 pg/mL TNF-α, and at concentrations ≥500 pg/mL, the THP-1 cells were nonresponsive to LPS stimulation. These data demonstrate that a tolerant-like state is induced in THP-1 cells when they are treated with exogenous TNF-α.
Figure 1.

Pretreatment with exogenous TNF-α attenuates the endogenous TNF-α response to lipopolysaccharide.
Notes: exogenous TNF-α induces production of endogenous TNF-α in a dose-dependent manner (A). In cells pretreated with various concentrations of TNF-α, the subsequent production of TNF-α protein in response to lipopolysaccharide is attenuated in a dose-dependent manner (B). Data are representative of the mean ± standard deviation of two independent experiments analyzed in triplicate. Statistically significant compared with no TNF-α pretreatment control (*P<0.05, student’s t-test).
Abbreviation: TNF-α, tumor necrosis factor-alpha.
Because TNF-α induces production of TNF-α, we wanted to determine if treatment with exogenous TNF-α displays a kinetic profile similar to that of LPS in THP-1 cells. THP-1 cells were stimulated with 1 ng/mL human TNF-α or 100 ng/mL LPS for 2 hours. After the initial 2 hours of stimulation, the supernatants were collected, and the cells were washed and resuspended in fresh medium. Supernatants were collected and fresh medium was added every 2 hours up to 16 hours. Following collection at 16 hours, the stimulated THP-1 cells were challenged with either 1 ng/mL TNF-α or 100 ng/mL LPS for an additional 2 hours. Stimulation with LPS or TNF-α induced a transient production of TNF-α during the first 6 hours, and by 8 hours the THP-1 cells were no longer secreting TNF-α (Figure 2A and B). During subsequent challenge with either TNF-α or LPS following collection at 16 hours, both the LPS-treated and TNF-α-treated THP-1 cells displayed an attenuated response to either challenge, indicating that treatment with TNF-α induced tolerance to itself and cross-tolerance with LPS (Figure 2C).
Figure 2.
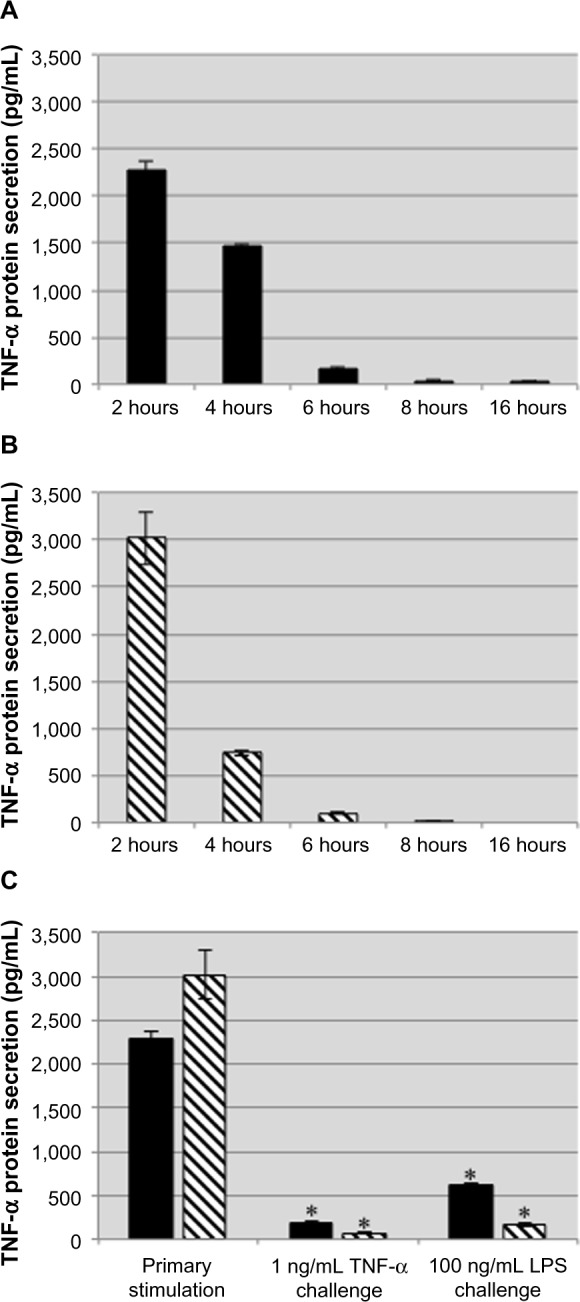
Exogenous TNF-α induces tolerance to subsequent TNF-α and LPS challenges.
Notes: THP-1 cells were stimulated with TNF-α (A, black bars) or LPS (B, striped bars) for 2 hours at which point they were washed and incubated with fresh medium for 14 hours before being challenged with TNF-α or LPS for 2 hours (C, black bars: primary stimulation TNF-α; striped bars: primary stimulation LPS). The nominal concentration of exogenous TNF-α was subtracted when preparing this graph. TNF-α or LPS induced tolerance to challenges from either stimulus. Data are representative of the mean ± standard deviation of two independent experiments analyzed in duplicate. Statistically significant compared with primary stimulations (*P<0.05, student’s t-test).
Abbreviations: TNF-α, tumor necrosis factor-alpha; LPS, lipopolysaccharide.
Anti-TNF-α or anti-TNF-α receptor antibodies impair the primary LPS-induced inflammatory response
We have demonstrated that exogenous TNF-α stimulation can induce the production of endogenous TNF-α and generate a tolerant-like state upon subsequent challenges. To investigate the involvement of TNF-α signaling and an autocrine amplification loop in endotoxin tolerance, we treated THP-1 cells with antibodies that neutralized the soluble TNF-α cytokine and/or antagonize TNF-α receptors at the time when they were stimulated with 100 ng/mL LPS for 2 hours. The concentration of neutralizing TNF-α and TNF-α receptor antagonist antibodies needed to inhibit production of TNF-α was determined by titration of each antibody and stimulating with LPS (data not shown). In the presence of the soluble TNF-α neutralizing antibody, the levels of TNF-α detectable by ELISA were less than 95% of the TNF-α observed by the no antibody control following LPS stimulation. The neutralizing antibody was effective at reducing detectable levels of the LPS-induced TNF-α response; however, this experimental condition does not demonstrate how much of the overall TNF-α production is due to TNF-α autocrine signaling. To determine the production of TNF-α dependent on TNF-α autocrine signaling, THP-1 cells were treated with anti-TNF-α receptor antibodies prior to LPS stimulation, which would induce TNF-α production in response to LPS but would not allow TNF-α to signal through the receptors. In the presence of antagonist antibodies to TNF-α receptor 1 (p55) and TNF-α receptor 2 (p75), THP-1 cells generated approximately 30% of the TNF-α produced in the cells that were not treated with antibodies (Figure 3A, 3,014.43±157.90 pg/mL TNF-α [no antibody] versus 1,056.18±200.32 pg/mL TNF-α [anti-TNF receptor antibodies]). These results suggest that the TNF-α autocrine amplification loop accounts for more than 60% of the TNF-α produced in response to LPS. Both approaches used to impair TNF-α signaling were effective at attenuating overall production of TNF-α.
Figure 3.

Neutralizing soluble TNF-α and antagonistic TNF-α receptor antibodies impair the production of proinflammatory mediators in LPS-stimulated THP-1 cells.
Notes: The concentration of TNF-α detected from LPS-stimulated THP-1 treated with anti-TNF-α or TNF-α receptor antibodies was significantly attenuated (A). In addition, impairing TNF-α signaling attenuated the production of the proinflammatory cytokine, IL-1β (B). These data suggest that TNF-α signaling plays a prominent role in the induction of other proinflammatory mediators during LPS stimulation. Data representative of mean ± standard deviation of two independent experiments analyzed in duplicate. Statistically significant compared with no antibody controls (*P<0.05, student’s t-test).
Abbreviations: TNF-α, tumor necrosis factor-alpha; TNFR 1/II, tumor necrosis factor receptor I and II; LPS, lipopolysaccharide; IL-1β, interleukin-1β.
TNF-α signaling through NF-κB can induce production of other inflammatory cytokines, such as IL-1β and IL-6.13 Therefore, we wanted to determine if the impaired TNF-α response would affect the production of other proinflammatory mediators. We investigated IL-1β, as it is known to be induced by LPS stimulation,41,42 and has been implicated in chronic inflammatory disorders and as a therapeutic target for many noninfectious inflammatory conditions.43 Inhibition of TNF-α signaling by neutralizing and antagonist antibodies significantly attenuated production of IL-1β in THP-1 cells following stimulation with LPS (Figure 3B). These results suggest that impairing TNF-α production and autocrine signaling attenuates the production of other proinflammatory mediators. The impact of impairing TNF-α signaling on other proinflammatory mediators should be considered when administering anti-TNF-α treatment for acute and chronic inflammatory responses.44
Induction of tolerance during TNF-α signaling inhibition
We have demonstrated that TNF-α can induce a tolerant-like state in THP-1 cells (Figures 1 and 2). We hypothesized that limiting the production of TNF-α and inhibiting TNF-α autocrine signaling during the primary stimulation would reduce the degree of endotoxin tolerance induced by LPS. THP-1 cells were treated with anti-TNF-α or anti-TNF-α receptor antibodies and then stimulated with 100 ng/mL LPS or 1 ng/mL recombinant human TNF-α for 2 hours. The THP-1 cells were washed and fresh medium was added every 2 hours for 14 hours before the cells were challenged with the same stimulating dose of LPS. Production of TNF-α was measured after the subsequent challenge to determine if the THP-1 cells were still refractory even when the initial TNF-α response was inhibited by the soluble TNF-α-neutralizing antibody or anti-TNF-α receptor antibodies. Even in the presence of anti-TNF-α or anti-TNF-α receptor antibodies during the primary LPS stimulation, induction of tolerance to LPS challenge was observed (Figure 4A). To ensure that the anti-TNF-α or anti-TNF-α receptor antibodies that were added before the primary stimulation were still not inhibiting production of TNF-α, a set of THP-1 cells were treated with the antibodies but were not stimulated during the primary 0–2 hours of stimulation. Following the subsequent LPS challenge (Figure 4B, white bars), the antibody-treated control cells responded as expected to the LPS challenge, indicating that there were no lingering effects of anti-TNF-α or anti-TNF-α receptor antibodies from the primary stimulation. In addition, we stimulated THP-1 cells with recombinant TNF-α in the presence of anti-TNF-α and anti-TNF-α receptors and challenged with LPS to demonstrate that the neutralizing and antagonist antibodies were blocking TNF-α signaling. Under these experimental conditions, recombinant TNF-α did not induce tolerance to LPS in the presence of anti-TNF-α and anti-TNF-α receptor antibodies during the primary TNF-α stimulation (Figure 4B, black bars). These data suggest that LPS signaling alone activates the downregulatory mechanisms involved in controlling the inflammatory response, independent of TNF-α autocrine signaling. Induction of tolerance is desired during a systemic inflammatory response; however, these results provide insight into why anti-TNF-α treatment increases the risk of serious infection in chronic inflammatory conditions.34
Figure 4.
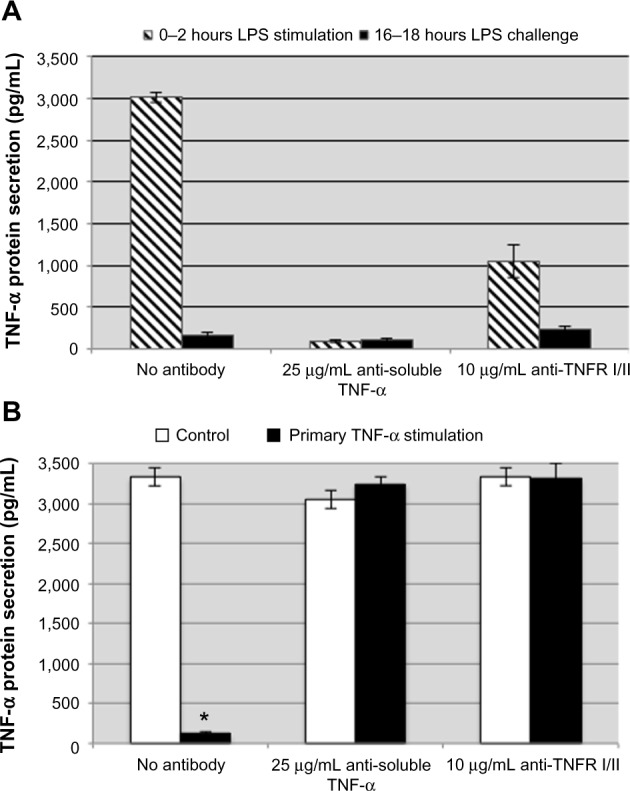
Neutralizing soluble TNF-α and antagonist TNF-α receptor antibodies do not attenuate the induction of LPS tolerance in THP-1 cells.
Notes: The concentration of TNF-α detected from LPS-stimulated THP-1 treated with anti-TNF-α or anti-TNF-α receptor antibodies was significantly attenuated during the primary LPS stimulation (A, striped bars). Impairing TNF-α signaling during the primary stimulation did not result in a reduced degree of tolerance (A, black bars). There were no lingering effects of the antibodies added during the primary stimulation as both control cells (B, white bars) and TNF-α-stimulated cells (B, black bars) did not display tolerance when challenged with LPS. Data are representative of the mean ± standard deviation of two independent experiments analyzed in duplicate. Statistically significant when compared with no antibody controls (*P<0.05, student’s t-test).
Abbreviations: TNF-α, tumor necrosis factor-alpha; TNFR 1/II, tumor necrosis factor receptor I and II; LPS, lipopolysaccharide.
Discussion
Elevated levels of proinflammatory cytokines such as TNF-α, IL-1β, and IL-6 are typically observed in patients with sepsis.19,45 Excessive production of these mediators has been associated with the immunopathogenesis of sepsis and other chronic inflammatory conditions. An extensive amount of research has focused on developing treatments to attenuate the production and functions of the inflammatory cytokine TNF-α. There have been several attempts to lower TNF-α concentrations and alleviate the detrimental effects of excessive production using anti-TNF-α treatment in a variety of inflammatory disorders.46–48 Unfortunately, anti-TNF-α therapy has not been successful when used for severe sepsis and septic shock.31,49 We believe these drugs are administered too late, and that the damage has already been done. Our data also demonstrate that the inflammatory response to LPS stimulation is attenuated in the presence of anti-TNF-α antibodies, but endotoxin tolerance is still induced. These events allow the pathogen to proliferate and increase the risk of septicemia because the immune cells are hyporesponsive to subsequent challenges.
The phenomenon of endotoxin tolerance is defined by a diminished production of inflammatory mediators during subsequent challenges following the initial exposure to endotoxin. Endotoxin tolerance is also a very important function during systemic infections, because this is the immune system’s attempt to regulate an excessive inflammatory response. Induction of tolerance can be extended to several TLR agonists50 as well as the inflammatory mediator TNF-α.40 TNF-α is the central inflammatory cytokine produced by most infections, and the recognition that it can induce tolerance made us consider the possibility that TNF-α is responsible for induction of tolerance.
Using the human monocytic THP-1 endotoxin tolerance model, we investigated the hypothesis that TNF-α is the critical mediator inducing tolerance following LPS stimulation. We treated THP-1 cells with anti-TNF-α or anti-TNF-α receptor antibodies prior to stimulating them with LPS. In the presence of these antibodies, the initial production of TNF-α was significantly attenuated. In addition, inhibition of the TNF-α signaling cascade impaired the production of IL-1β (Figure 4). After the initial LPS stimulation, the anti-TNF-α-treated cells were allowed to recover for 14 hours before being challenged with LPS to determine if blocking TNF-α production reduced the degree of tolerance. However, production of TNF-α was still attenuated during the challenge response, indicating that the downregulatory mechanisms involved in tolerance are expressed without the influence of TNF-α autocrine signaling. These results may seem contradictory to the conclusion reached by Park et al that the LPS tolerance induced by TNF-α is TNF-α-dependent via activation of glycogen synthase kinase-3.40 However, this study40 actually demonstrates that deubiquitinase A20 is critical to the suppressed response following treatment with LPS. Recently, Li et al demonstrated that LPS increases A20 levels by generation of reactive oxygen species and that increased expression is a component of the endotoxin tolerance.51 The study by Li et al did not stimulate macrophages in the presence of reagents capable of impairing TNF-α production and signaling, so it is possible that LPS-induced production of TNF-α ultimately elevated the A20 levels and attenuated the inflammatory response to subsequent challenges. Our results demonstrate that tolerance can still be induced even when TNF-α production and signaling is inhibited, indicating that LPS is capable of activating the regulatory mediators. It will be important to assess LPS or TNF-α expression patterns of the biochemical mediators involved in controlling the inflammatory response to subsequent challenges. The results of these experiments would provide information regarding which stimulus induces the expression of specific regulatory mediators.
Conclusion
In this study we have demonstrated that although TNF-α autocrine signaling enhances its own production, such signaling is not required for LPS-induced tolerance. Survival in Gram-negative bacterial infections requires the production of inflammatory mediators like TNF-α. However, excessive production can lead to septic shock and death, while insufficient production can lead to overwhelming bacteremia and death. A better understanding of the regulation of endotoxin tolerance will allow therapeutic interventions that balance the production of inflammatory mediators like TNF-α and their necessary control.
Acknowledgments
This work was supported in part by a grant from Nutritional Science Corporation (Liberty, TX, USA).
Footnotes
Author contributions
CL designed and performed the experiments concerning cell culture, anti-TNF-α treatment, LPS stimulation, and ELISA, assisted with interpretation of the results, and helped draft the manuscript. KH assisted with interpretation of the results and helped draft the manuscript. Both authors read and approved the final manuscript.
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.West MA, Heagy W. Endotoxin tolerance: a review. Crit Care Med. 2002;30(Suppl 1):S64–S73. [PubMed] [Google Scholar]
- 2.Biswas SK, Lopez-Collazo E. Endotoxin tolerance: new mechanisms, molecules and clinical significance. Trends Immunol. 2009;30(10):475–487. doi: 10.1016/j.it.2009.07.009. [DOI] [PubMed] [Google Scholar]
- 3.Murphey ED, Fang G, Sherwood ER. Endotoxin pretreatment improves bacterial clearance and decreases mortality in mice challenged with Staphylococcus aureus. Shock. 2008;29(4):512–518. doi: 10.1097/shk.0b013e318150776f. [DOI] [PubMed] [Google Scholar]
- 4.Shi DW, Zhang J, Jiang HN, et al. LPS pretreatment ameliorates multiple organ injuries and improves survival in a murine model of polymicrobial sepsis. Inflamm Res. 2011;60(9):841–849. doi: 10.1007/s00011-011-0342-5. [DOI] [PubMed] [Google Scholar]
- 5.Wheeler DS, Lahni PM, Denenberg AG, et al. Induction of endotoxin tolerance enhances bacterial clearance and survival in murine polymicrobial sepsis. Shock. 2008;30(3):267–273. doi: 10.1097/shk.0b013e318162c190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chow JC, Young DW, Golenbock DT, Christ WJ, Gusovsky F. Toll-like receptor-4 mediates lipopolysaccharide-induced signal transduction. J Biol Chem. 1999;274(16):10689–10692. doi: 10.1074/jbc.274.16.10689. [DOI] [PubMed] [Google Scholar]
- 7.Baeuerle PA, Henkel T. Function and activation of NF-kappa B in the immune system. Annu Rev Immunol. 1994;12:141–179. doi: 10.1146/annurev.iy.12.040194.001041. [DOI] [PubMed] [Google Scholar]
- 8.Lawrence T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb Perspect Biol. 2009;1(6):a001651. doi: 10.1101/cshperspect.a001651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Royall JA, Berkow RL, Beckman JS, Cunningham MK, Matalon S, Freeman BA. Tumor necrosis factor and interleukin 1 alpha increase vascular endothelial permeability. Am J Physiol. 1989;257(6 Pt 1):L399–L410. doi: 10.1152/ajplung.1989.257.6.L399. [DOI] [PubMed] [Google Scholar]
- 10.Hofmann S, Grasberger H, Jung P, et al. The tumour necrosis factor-alpha induced vascular permeability is associated with a reduction of VE-cadherin expression. Eur J Med Res. 2002;7(4):171–176. [PubMed] [Google Scholar]
- 11.Schütze S, Wiegmann K, Machleidt T, Krönke M. TNF-induced activation of NF-kappa B. Immunobiology. 1995;193(2–4):193–203. doi: 10.1016/s0171-2985(11)80543-7. [DOI] [PubMed] [Google Scholar]
- 12.Zhang YH, Lin JX, Vilcek J. Interleukin-6 induction by tumor necrosis factor and interleukin-1 in human fibroblasts involves activation of a nuclear factor binding to a kappa B-like sequence. Mol Cell Biol. 1990;10(7):3818–3823. doi: 10.1128/mcb.10.7.3818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Turner NA, Mughal RS, Warburton P, O’Regan DJ, Ball SG, Porter KE. Mechanism of TNFalpha-induced IL-1alpha, IL-1beta and IL-6 expression in human cardiac fibroblasts: effects of statins and thiazolidinediones. Cardiovasc Res. 2007;76(1):81–90. doi: 10.1016/j.cardiores.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 14.Wajant H, Pfizenmaier K, Scheurich P. Tumor necrosis factor signaling. Cell Death Differ. 2003;10(1):45–65. doi: 10.1038/sj.cdd.4401189. [DOI] [PubMed] [Google Scholar]
- 15.Hofmann MA, Drury S, Fu C, et al. RAGE mediates a novel proinflammatory axis: a central cell surface receptor for S100/calgranulin polypeptides. Cell. 1999;97(7):889–901. doi: 10.1016/s0092-8674(00)80801-6. [DOI] [PubMed] [Google Scholar]
- 16.Covert MW, Leung TH, Gaston JE, Baltimore D. Achieving stability of lipopolysaccharide-induced NF-kappaB activation. Science. 2005;309(5742):1854–1857. doi: 10.1126/science.1112304. [DOI] [PubMed] [Google Scholar]
- 17.Verma N, Chaudhury I, Kumar D, Das RH. Silencing of TNF-alpha receptors coordinately suppresses TNF-alpha expression through NF-kappaB activation blockade in THP-1 macrophage. FEBS Lett. 2009;583(17):2968–2974. doi: 10.1016/j.febslet.2009.08.007. [DOI] [PubMed] [Google Scholar]
- 18.Neels JG, Pandey M, Hotamisligil GS, Samad F. Autoamplification of tumor necrosis factor-alpha: a potential mechanism for the maintenance of elevated tumor necrosis factor-alpha in male but not female obese mice. Am J Pathol. 2006;168(2):435–444. doi: 10.2353/ajpath.2006.050699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dinarello CA. Proinflammatory and anti-inflammatory cytokines as mediators in the pathogenesis of septic shock. Chest. 1997;112(Suppl 6):321S–329S. doi: 10.1378/chest.112.6_supplement.321s. [DOI] [PubMed] [Google Scholar]
- 20.Cohen J. The immunopathogenesis of sepsis. Nature. 2002;420(6917):885–891. doi: 10.1038/nature01326. [DOI] [PubMed] [Google Scholar]
- 21.Jansen MJ, Hendriks T, Vogels MT, van der Meer JW, Goris RJ. Inflammatory cytokines in an experimental model for the multiple organ dysfunction syndrome. Crit Care Med. 1996;24(7):1196–1202. doi: 10.1097/00003246-199607000-00022. [DOI] [PubMed] [Google Scholar]
- 22.Burdon D, Tiedje T, Pfeffer K, Vollmer E, Zabel P. The role of tumor necrosis factor in the development of multiple organ failure in a murine model. Crit Care Med. 2000;28(6):1962–1967. doi: 10.1097/00003246-200006000-00045. [DOI] [PubMed] [Google Scholar]
- 23.Kilbourn RG, Gross SS, Jubran A, et al. NG-methyl-L-arginine inhibits tumor necrosis factor-induced hypotension: implications for the involvement of nitric oxide. Proc Natl Acad Sci U S A. 1990;87(9):3629–3632. doi: 10.1073/pnas.87.9.3629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xaus J, Comalada M, Valledor AF, et al. LPS induces apoptosis in macrophages mostly through the autocrine production of TNF-alpha. Blood. 2000;95(12):3823–3831. [PubMed] [Google Scholar]
- 25.Yang FL, Li CH, Hsu BG, et al. The reduction of tumor necrosis factor-alpha release and tissue damage by pentobarbital in the experimental endotoxemia model. Shock. 2007;28(3):309–316. doi: 10.1097/SHK.0b013e31803dd04d. [DOI] [PubMed] [Google Scholar]
- 26.Vincent JL, Bakker J, Marécaux G, Schandene L, Kahn RJ, Dupont E. Administration of anti-TNF antibody improves left ventricular function in septic shock patients. Results of a pilot study. Chest. 1992;101(3):810–815. doi: 10.1378/chest.101.3.810. [DOI] [PubMed] [Google Scholar]
- 27.Fisher CJ, Opal SM, Dhainaut JF, et al. Influence of an anti-tumor necrosis factor monoclonal antibody on cytokine levels in patients with sepsis. The CB0006 Sepsis Syndrome Study Group. Crit Care Med. 1993;21(3):318–327. doi: 10.1097/00003246-199303000-00006. [DOI] [PubMed] [Google Scholar]
- 28.Boillot A, Capellier G, Racadot E, Wijdenes J, Herve P, Barale F. Pilot clinical trial of an anti-TNF alpha monoclonal antibody for the treatment of septic shock. Clin Intensive Care. 1995;6(2):52–56. [PubMed] [Google Scholar]
- 29.Gallagher J, Fisher C, Sherman B, et al. A multicenter, open-label, prospective, randomized, dose-ranging pharmacokinetic study of the anti-TNF-alpha antibody afelimomab in patients with sepsis syndrome. Intensive Care Med. 2001;27(7):1169–1178. doi: 10.1007/s001340100973. [DOI] [PubMed] [Google Scholar]
- 30.Panacek EA, Marshall JC, Albertson TE, et al. Efficacy and safety of the monoclonal anti-tumor necrosis factor antibody F(ab’)2 fragment afelimomab in patients with severe sepsis and elevated interleukin-6 levels. Crit Care Med. 2004;32(11):2173–2182. doi: 10.1097/01.ccm.0000145229.59014.6c. [DOI] [PubMed] [Google Scholar]
- 31.Reinhart K, Karzai W. Anti-tumor necrosis factor therapy in sepsis: update on clinical trials and lessons learned. Crit Care Med. 2001;29(Suppl 7):S121–S125. doi: 10.1097/00003246-200107001-00037. [DOI] [PubMed] [Google Scholar]
- 32.Gawad KA, Schneider C, Brinken B, et al. Anti-TNF antibody treatment has no positive effect on survival in a model of pneumococcal sepsis in pigs. J Invest Surg. 2001;14(5):291–297. doi: 10.1080/089419301753170075. [DOI] [PubMed] [Google Scholar]
- 33.Lv S, Han M, Yi R, Kwon S, Dai C, Wang R. Anti-TNF-α therapy for patients with sepsis: a systematic meta-analysis. Int J Clin Pract. 2014;68(4):520–528. doi: 10.1111/ijcp.12382. [DOI] [PubMed] [Google Scholar]
- 34.Galloway JB, Hyrich KL, Mercer LK, et al. Anti-TNF therapy is associated with an increased risk of serious infections in patients with rheumatoid arthritis especially in the first 6 months of treatment: updated results from the British Society for Rheumatology Biologics Register with special emphasis on risks in the elderly. Rheumatology (Oxford) 2011;50(1):124–131. doi: 10.1093/rheumatology/keq242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zakharova M, Ziegler HK. Paradoxical anti-inflammatory actions of TNF-alpha: inhibition of IL-12 and IL-23 via TNF receptor 1 in macrophages and dendritic cells. J Immunol. 2005;175(8):5024–5033. doi: 10.4049/jimmunol.175.8.5024. [DOI] [PubMed] [Google Scholar]
- 36.Blüml S, Binder NB, Niederreiter B, et al. Antiinflammatory effects of tumor necrosis factor on hematopoietic cells in a murine model of erosive arthritis. Arthritis Rheum. 2010;62(6):1608–1619. doi: 10.1002/art.27399. [DOI] [PubMed] [Google Scholar]
- 37.Fraker DL, Stovroff MC, Merino MJ, Norton JA. Tolerance to tumor necrosis factor in rats and the relationship to endotoxin tolerance and toxicity. J Exp Med. 1988;168(1):95–105. doi: 10.1084/jem.168.1.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fahmi H, Chaby R. Selective refractoriness of macrophages to endotoxin-induced production of tumor necrosis factor, elicited by an autocrine mechanism. J Leukoc Biol. 1993;53(1):45–52. doi: 10.1002/jlb.53.1.45. [DOI] [PubMed] [Google Scholar]
- 39.Ferlito M, Romanenko OG, Ashton S, Squadrito F, Halushka PV, Cook JA. Effect of cross-tolerance between endotoxin and TNF-alpha or IL-1beta on cellular signaling and mediator production. J Leukoc Biol. 2001;70(5):821–829. [PubMed] [Google Scholar]
- 40.Park SH, Park-Min KH, Chen J, Hu X, Ivashkiv LB. Tumor necrosis factor induces GSK3 kinase-mediated cross-tolerance to endotoxin in macrophages. Nat Immunol. 2011;12(7):607–615. doi: 10.1038/ni.2043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sacco RE, Nibbelink SK, Baarsch MJ, Murtaugh MP, Wannemuehler MJ. Induction of interleukin (IL)-1beta and IL-8 mRNA expression in porcine macrophages by lipopolysaccharide from Serpulina hyodysenteriae. Infect Immun. 1996;64(10):4369–4372. doi: 10.1128/iai.64.10.4369-4372.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lang CH, Silvis C, Deshpande N, Nystrom G, Frost RA. Endotoxin stimulates in vivo expression of inflammatory cytokines tumor necrosis factor alpha, interleukin-1beta, -6, and high-mobility-group protein-1 in skeletal muscle. Shock. 2003;19(6):538–546. doi: 10.1097/01.shk.0000055237.25446.80. [DOI] [PubMed] [Google Scholar]
- 43.Dinarello CA. Interleukin-1 in the pathogenesis and treatment of inflammatory diseases. Blood. 2011;117(14):3720–3732. doi: 10.1182/blood-2010-07-273417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Connor V. Anti-TNF therapies: a comprehensive analysis of adverse effects associated with immunosuppression. Rheumatol Int. 2011;31(3):327–337. doi: 10.1007/s00296-009-1292-x. [DOI] [PubMed] [Google Scholar]
- 45.Blackwell TS, Christman JW. Sepsis and cytokines: current status. Br J Anaesth. 1996;77(1):110–117. doi: 10.1093/bja/77.1.110. [DOI] [PubMed] [Google Scholar]
- 46.Geiler J, Buch M, McDermott MF. Anti-TNF treatment in rheumatoid arthritis. Curr Pharm Des. 2011;17(29):3141–3154. doi: 10.2174/138161211798157658. [DOI] [PubMed] [Google Scholar]
- 47.D’Haens G. Anti-TNF-alpha treatment strategies: results and clinical perspectives. Gastroenterol Clin Biol. 2009;33(Suppl 3):S209–S216. doi: 10.1016/S0399-8320(09)73156-2. [DOI] [PubMed] [Google Scholar]
- 48.Kircik LH, Del Rosso JQ. Anti-TNF agents for the treatment of psoriasis. J Drugs Dermatol. 2009;8(6):546–559. [PubMed] [Google Scholar]
- 49.Qiu P, Cui X, Barochia A, Li Y, Natanson C, Eichacker PQ. The evolving experience with therapeutic TNF inhibition in sepsis: considering the potential influence of risk of death. Expert Opin Investig Drugs. 2011;20(11):1555–1564. doi: 10.1517/13543784.2011.623125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dalpke AH, Lehner MD, Hartung T, Heeg K. Differential effects of CpG-DNA in Toll-like receptor-2/-4/-9 tolerance and cross-tolerance. Immunology. 2005;116(2):203–212. doi: 10.1111/j.1365-2567.2005.02211.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li Y, Zhang P, Wang C, et al. Immune responsive gene 1 (IRG1) promotes endotoxin tolerance by increasing A20 expression in macrophages through reactive oxygen species. J Biol Chem. 2013;288(23):16225–16234. doi: 10.1074/jbc.M113.454538. [DOI] [PMC free article] [PubMed] [Google Scholar]