

Abstract
Sjögren’s syndrome is an autoimmune disease that targets exocrine glands, but often exhibits systemic manifestations. Infiltration of the salivary and lacrimal glands by lymphoid and myeloid cells orchestrates a perpetuating immune response leading to exocrine gland damage and dysfunction. Th1 and Th17 lymphocyte populations and their products recruit additional lymphocytes, including B cells, but also large numbers of macrophages, which accumulate with disease progression. In addition to cytokines, chemokines, chitinases, and lipid mediators, macrophages contribute to a proteolytic milieu, underlying tissue destruction, inappropriate repair, and compromised glandular functions. Among the proteases enhanced in this local environment are matrix metalloproteases (MMP) and plasmin, generated by plasminogen activation, dependent upon plasminogen activators, such as tissue plasminogen activator (tPA). Not previously associated with salivary gland pathology, our evidence implicates enhanced tPA in the context of inflamed salivary glands revolving around lymphocyte-mediated activation of macrophages. Tracking down the mechanism of macrophage plasmin activation, the cytokines IFNγ and to a lesser extent, IFNα, via Janus kinase (JAK) and signal transducer and activator of transcription (STAT) activation, were found to be pivotal for driving the plasmin cascade of proteolytic events culminating in perpetuation of the inflammation and tissue damage, and suggesting intervention strategies to blunt irreversible tissue destruction.
Keywords: tPA, macrophage, salivary gland, cytokine, plasminogen, MMP
1.0 INTRODUCTION
Sjögren’s syndrome (SS) is a complex autoimmune disease that not only targets lacrimal and salivary exocrine glands, but can also exhibit multi-organ systemic manifestations[1]. Although the underlying cause of SS remains ill defined [2], disease is characterized by mononuclear cell infiltration of the exocrine glands, tissue destruction, and chronic dysfunction, reflected clinically by xerostomia and keratoconjunctivitis sicca. Initial periductal infiltration of activated T cells leads to an accumulation of B cells, accompanied by antigen presenting macrophages and dendritic cells[3–5]. In addition to CD4+ T helper type 1 (Th1) lymphocytes expressing interferon gamma (IFNγ), CD4+ Th17 cells that secrete IL-17 dominate in the glandular lesions[6], and their collective products promote salivary gland pathology.
One of the substantive manifestations of the chronic immune response within the salivary glands stemming from the massive infiltration of immune cells is the destruction of the normal tissue architecture eventuating in compromised function of these key exocrine glands. While much emphasis has been placed on deciphering the immunological conflagration underlying this tissue pathology[3, 5, 6], less information has accumulated defining the mechanisms causing tissue destruction, acinar atrophy and the ensuing fibrotic attempt at tissue repair, which results in irreversible loss of secretory capacity. Based on evidence that macrophages and their products are emblematic of emerging and advanced disease[4, 5] and that macrophages generate a plethora of proteolytic enzymes that could contribute to tissue destruction, we explored salivary gland tissues for evidence that such mechanisms were contributory. Based on our original microarray profiling[5], we detected enhancement of multiple enzymes, including carboxypeptidase M, a membrane bound enzyme linked to macrophage differentiation[7], cathepsins, which have multiple functions in immune responses[8], matrix metalloproteases (MMP), and components of the plasminogen activation system.
Plasmin is a serine protease generated from its precursor, plasminogen, by plasminogen activators (PA), including urokinase-type PA (uPA) and tissue-type PA (tPA)[9, 10]. Most efficient plasmin generation occurs at the cell surface and is facilitated by plasminogen and PA binding to annexin A2, uPAR, and other docking sites that co-localize enzyme and substrate[11–13]. Once generated, plasmin has multiple substrates and activates matrix-degrading MMPs. In our recent studies, we demonstrated that in oral squamous cell carcinoma, heightened levels of plasmin occur in an environment of reduced expression of secretory leukocyte protease inhibitor (SLPI), which blunts plasminogen activation at the cell surface[14]. Moreover, SLPI directly inhibits macrophage MMP, possibly through inhibition of NFκB[15, 16], and plasmin activation through its interaction with annexin A2[14, 17] and the absence of SLPI is associated with enhanced proteolytic activity, delayed matrix accumulation, aberrant tissue repair and tumor invasion[14, 18–20]. In transcriptome analyses of minor salivary gland tissues from patients with SS, SLPI expression was reduced[5], suggesting that such pathways may be operational in salivary gland pathology. Moreover, in recent studies, most cell-free saliva samples tested and salivary gland tissues were found to contain tPA, but very few had uPA, and plasminogen activator inhibitor 2 (PAI-2), but not PAI-1 was detected[21], prompting us to focus on regulatory mechanisms by which the tissue plasminogen activation system might be operational in SS tissue pathology and dysfunction. Although to date, there is little evidence linking cytokines such as IFN and regulation of plasminolytic activity, we identify the IFN pathway as a crucial link in driving myeloid cell plasmin activity and setting off a cascade of proteolytic events that may contribute to pathogenesis and potentially serve as targets to interrupt irreversible exocrinopathy.
2.0 MATERIALS AND METHODS
2.1 MSG histology and immunohistochemistry
Minor SG (MSG) biopsies were obtained with informed consent from individuals undergoing diagnostic evaluation for sicca symptoms indicative of SS (n=16)[4, 5] and were diagnosed by American-European SS consensus criteria [22] as having primary Sjögren’s syndrome. The control group consisted of gender-matched individuals with subjective complaints of dry mouth or eyes but who did not fulfill SS criteria and lacked histopathological evidence of SS. None of the patients had evidence of lymphoma, sarcoidosis, essential mixed cryoglobulinaemia or infection by HIV, hepatitis B and C viruses at time of study. Biopsy specimens were fixed, embedded, sectioned(5μm), deparaffinized, rehydrated through graded alcohol and stained with H&E. All pSS patients presented a biopsy focus score (FS) ≥1–12(one focus=aggregate of ~50 inflammatory cells) per 4mm2 [23], whereas the control group had FS<1. Histopathologic lesions were categorized [5] as early SS[Tarpley score(TS) of 1; 1=1–2lymphocytic aggregates/lobule], intermediate(TS=2; 2=3 aggregates/lobule) and severe(TS=3–4; 3=diffuse infiltration through acini associated with partial destruction of acinar tissue; 4=diffuse infiltration associated with complete loss of tissue architecture). Histologic grading was also categorized based on inflammation as mild(1+), intermediate(2+) and advanced(3+/4+)[24]. Additional sections were stained with Masson’s Trichrome (collagen), Fraaser-Lendrum (fibrin), Verhoeff’s Elastica EVG (elastic) and Jone’s PAMS (basement membrane)(Histoserv, Inc., Germantown, MD).
For immunohistochemistry, sections were processed for antigen retrieval in a decloaking chamber(Biocare Medical, Concord, CA) with antigen unmasking solution (Vector Laboratories, Burlingame, CA) before blocking endogenous peroxidase with 3% H2O2 in 50% methanol 15min. Sections were incubated with blocking serum(goat or rabbit) for 30min, and incubated overnight at 4°C with primary antibody: IL-17A (rabbit polyclonal)(Santa Cruz Biotechnology, Santa Cruz, CA), IFNγ (clone 350B10G6, mouse IgG1)(Abcam, Cambridge, MA), tPA (rabbit polyclonal, Epitomics, Burlingame, CA), plasminogen (Santa Cruz Biotechnology) and MMP9 (rabbit polyclonal, Millipore/Chemicon, Billerica, MA). For control staining, primary antibodies were replaced with irrelevant isotype-matched antibodies(Jackson ImmunoResearch, West Grove, PA). After 30min with biotinylated secondary antibody, immunoreactive staining was developed using Avidin:Biotinylated enzyme Complex(Vectastain Elite Kit) or streptavidin peroxidase as described[5].
2.2 Isolation and culture of human blood monocytes
Peripheral blood mononuclear cells (PBMC) were obtained by leukapheresis of healthy volunteers (Department of Transfusion Medicine, NIH) and monocytes purified by counterflow centrifugal elutriation[17]. Monocytes were cultured in Dulbecco’s Modified Eagles Medium (DMEM, BioWhittaker, Walkersville, MD) supplemented with 2mM L-glutamine, 100μl/ml penicillin and 100μg/ml streptomycin (Sigma, St. Louis, MO). For monocyte-derived macrophages, monocytes were adhered and cultured in supplemented DMEM with 10% FBS for 7–10 days[14] before treatment with the cytokines, TNFα, IFNα (R&D Systems, Minneapolis, MN), IFNγ or IL-17 (Peprotech, Rocky Hill, NJ) or with lipopolysaccharide (LPS; Escherichia coli 055:B5, Sigma-Aldrich) at indicated concentrations.
2.3 Plasmin generation assays
Macrophages, pre-treated or not with IFNγ (10ng/ml) for 4hr or overnight were suspended in incubation buffer (Hepes-buffered saline containing 3mM CaCl2 and 1mM MgCl2; Cellgro-Mediatech, Inc., Herndon VA) (1x106 cells/ml, 100μl/tube) and incubated with 100nM N-terminal glutamic acid plasminogen (glu-plasminogen) (American Diagnostica, Greenwich CT) for 1hr at 4°C. The cells were washed with incubation buffer, tPA (12nM, Calbiochem, La Jolla, CA) and the fluorogenic plasmin substrate AFC-081 (166 μM, D-valine-leucine-lysine-7-amino-4-trifluoromethyl coumarin, Enzyme Systems Products, Aurora, OH) were added, and substrate hydrolysis measured at 5-min intervals at 400nm excitation and 505nm emission[14] in Wallac Victor(Perkin-Elmer, Boston MA).
2.4 RNA extraction and cDNA synthesis
MSG specimens were preserved in RNAlater (Ambion, Applied Biosystems) and stored at −80°C. Total RNA was extracted with RNeas y Mini Kit(Qiagen, Hilden, Germany). To eliminate genomic DNA contamination, samples were treated with RNase-free DNase (Qiagen). cDNA was prepared from 0.5μg RNA using oligo-dT primers (MWG Biotechnology, Ebersberg, Germany) and SuperScript-II reverse transcriptase(Invitrogen, Carlsbad, CA). Integrity of RNA was verified by amplification of β-actin mRNA. For myeloid cells, RNA was extracted and DNase digested using Qiagen RNeasy mini kit and RNase-Free DNase. RNA was reverse-transcribed using oligodeoxythymidylic acid primer (Invitrogen).
2.5 Semi-quantitative Real-time PCR
The resulting cDNA was amplified by PCR using ABI 7500 Sequence Detector (Applied Biosystems). Amplification was performed using the Taqman expression assays for IFNγ (Hs00989291_m1), IFNα (Hs00353738_s1), tPA (Hs 00263492_m1), uPA (Hs00170182_m1), annexin A2 (Hs00733393_m1), uPAR/CD87 (Hs00959822_m1), S100A10 (Hs00741221_m1) and COL1A1 (Hs00164004_m1), MMP2 (Hs00234422_m1), MMP7 (Hs01042796_m1), MMP9 (Hs00234579_m1), and GAPDH(Hs99999905_m1) as normalization control (Applied Biosystems). Data were examined using the 2-Δ Δ Ct method[5] and results expressed as fold increase.
2.6 Western blots
Human monocytes and monocyte-derived macrophages were treated or not with IFNα and IFNγ at 10ng/ml and harvested at the indicated time points after treatment. Cell lysates were prepared using a lysis buffer of 1% Nonidet P-40, 150mM NaCl, 20mM Tris-HCl(pH 7.5), 10mM NaF, 10mM NaPPi, 0.5mM EDTA, 1mM Na3VO4, 1mM phenylmethylsulfonyl fluoride, 5μg/ml Pepstatin, 5μg/ml leupeptin and 5μg/ml Aprotinin, set on ice for 20min, and cell debris removed by centrifugation (14,000 rpm, 20min, 4°C). Total protein concentration in lysates was determined using Bio-Rad DC Protein Assay (Bio-Rad) and samples analyzed by SDS-PAGE(10% Tris-Glycine gels, reducing conditions) followed by Western blot with the following antibodies: pSTAT1(Y701), pSTAT3(Y705), pSTAT5(Y694), pSTAT6(Y641) and pNFκB p65 (Cell Signaling). In indicated experiments, macrophages were pretreated with IFNγ receptor 2 (R2) or R1 antibody, CD118 antibody (R&D Systems), isotype-matched control antibody (eBiosciences), or JAK inhibitor I (Calbiochem) for 1hr prior to addition of IFN. Signal was analyzed by adding Alexa Fluor 680 goat anti–rabbit or Alexa Fluor 750 goat anti-mouse antibodies (LI-COR) secondary antibodies and the infrared fluorescence was detected with Odyssey infrared imaging system (LI-COR).
2.7 Statistics
Statistical analyses were performed by Mann-Whitney test for non-parametric biologic data sets (two-tailed) and by t test for in vitro samples (two-tailed). P values <0.05 were considered statistically significant. All analyses were performed using VassarStats (http://faculty.vassar.edu/lowry/VassarStats.html).
3.0 RESULTS
3.1 Salivary gland infiltrate and tissue damage
Primary Sjögren’s symptoms are accompanied by abnormal biopsy of salivary and lacrimal glands, and as pathology progresses, the periductal lymphoid infiltrate extends into the acini, and may destroy and replace acinar epithelial cells, although the ducts often appear to be spared (Fig. 1A,B). In our prior microarray analyses of severely diseased exocrine glands, enhanced expression of proteases (MMP, cathepsins, carboxypeptidases), coupled with macrophage infiltration, occurred, with a less apparent compensatory increase in protease inhibitors, such as plasminogen activation inhibitors (PAI) or tissue inhibitors of MMP (TIMPS) [5]. Based on these data, we considered that macrophage-derived proteases might mediate not only inflammatory cell invasion, but also tissue destruction. Since the macrophage plasminogen activation pathway is linked to invasive behavior[14], we examined heavily infiltrated MSG (Fig. 1B) for potential evidence of involvement of this pathway. As shown with anti-CD68 staining of macrophages, areas characterized by lymphocytic infiltration contain CD68+ cell populations (Fig. 1C). Moreover, these same regions demonstrate cell associated and/or cell free staining for tPA (Fig. 1D), while little or no staining for tPA was seen in control, noninflamed salivary gland tissues (Fig. 1E). As evident in stained tissue sections from multiple SS patients (Fig. 1C–K, FS=7–10), regions enriched in CD68+ macrophages were typically the areas with tPA staining, but it was noted that epithelial cells, lacking CD68, could also be tPA+. Although in non-diseased salivary glands, tPA has been identified in the ducts [21], this had not been explored in the context of tissue damage in Sjögren’s syndrome.
Figure 1. Salivary gland infiltration and plasminogen activators.
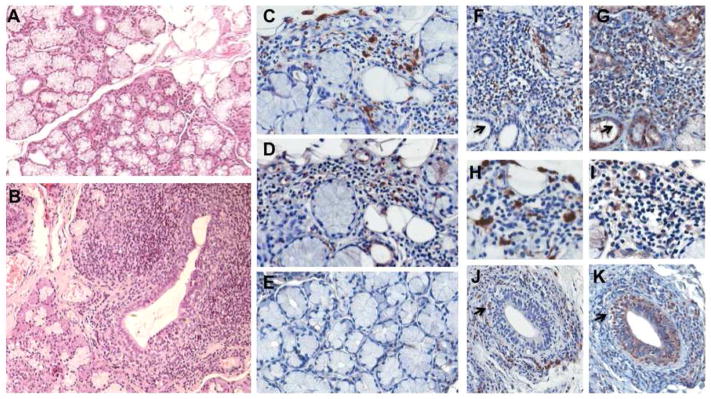
A) Control minor salivary gland illustrating acini, ducts and minimal periductal infiltration. H&E, original magnification 10X. B) Extensive mononuclear cell infiltration and tissue disruption in Sjögren’s syndrome MSG lead to compromised secretory function. Periductal lymphoid infiltrate extends into the acini and throughout parenchyma. Original magnification 10X. C) Immunohistochemical staining for CD68 positive macrophages in representative SS MSG indicates myeloid cells in the mononuclear cell infiltrate. 40X. D) Staining for a tissue component of the plasminogen activation system, tissue plasminogen activator (tPA), demonstrates positive staining in regions of inflammatory cell infiltrate and in some ductal cells. E) Limited evidence of tPA staining in noninflamed MSG tissues. F,G) Salivary gland tissue from SS patient with FS of 7 stained for CD68 (F) and tPA (G) showing staining in infiltrate and ducts. H,I) Higher magnification of CD68 (H) and tPA (I) staining from SS patient with FS = 9.5. J,K) SS (FS=10) MSG tissue stained for CD68 macrophages (J) (arrow denotes CD68+ macrophages appearing to invade duct) which are stained with tPA (K, arrow), but ductal cells are also tPA positive (K).
3.2 Plasmin activation system in SS MSG
Further assessment of expression of these tissue constituents of the plasminogen activation system by RT-PCR revealed minimally enhanced tPA expression in MSG classified as early or intermediate disease relative to non-SS MSG, but further increased expression in tissues with severe disease (Fig. 2A). In addition, elevations were seen in membrane PA binding molecules, annexin A2 and S100A10(Fig. 2B,C), supporting a role for plasmin proteolytic pathways in exocrinopathy. While Fraaser-Lendrum staining revealed the presence of fibrin, a substrate for plasmin in SS MSG (Fig. 2D,E, fibrin is red; also stains some cytoplasmic granules)[25], a plethora of additional plasmin substrates are available within these tissues.
Figure 2. Enhanced plasmin activation system in inflamed salivary glands.

MSG tissues from control subjects (n=5), pSS subjects with early/intermediate disease (n=6) and those with severe SS (n=5) were tested by RT-PCR for tissue plasminogen activator and two cell binding partners. A) RT-PCR for tPA revealed increased plasminogen activator expression in individuals with severe SS. Data (expression normalized to GAPDH) represent fold difference compared to non-SS MSG. B) Increased expression of annexin A2 was modest, but significant in the diseased tissues, and S100A10 was also significantly elevated in early/intermediate and severe disease relative to control (non-SS) tissues (C), *p≤0.01; **p=0.06, Mann-Whitney, two-tailed. D,E) Fraaser-Lendrum staining for fibrin in which fibrin, keratin, and some cytoplasmic granules appear red, erythrocytes appear orange, and matrix is green in MSG from a SS patient (FS=10); original magnification 20X and inset 40X (D) and normal MSG (20X) (E).
3.3 Cytokine regulation of macrophage plasminogen activation system
Based on our prior evidence that the Th1 and Th17 cytokines, IFNγ and IL-17, products of CD3+ T cells, are highly expressed during SS tissue immunopathogenesis [3, 6] (Fig. 3A–E), we first examined whether these cytokines mediate expression of plasminogen activators and/or membrane docking stations that catalyze activation of plasminogen to plasmin. Addition of IFNγ to cultured monocytes resulted in striking increases in tPA expression as detected by RT-PCR (Fig. 3C). By comparison, IL-17 did not appear to substantively increase tPA (Fig. 3C). Both IFNγ and IL-17 increased annexin 2, recognized as a catalytic site for tPA and plasminogen interactions [11, 13]. Interestingly, concurrent addition of IFNγ and IL-17 to the monocyte cultures did not result in a synergistic enhancement of tPA expression at the optimal concentrations used (Fig. 3F). Emerging from these data, IFNγ appears to be an important driving force for the tPA pathway. Other cytokines linked to SS, such as TNFα and IFNα, were tested for their ability to upregulate tPA, as well as the uPA pathway. Compared to unstimulated macrophages (Ctrl), TNFα had no significant effect on tPA or uPA, whereas IFNγ and to a lesser extent, IFNα upregulated tPA, but not uPA (Fig. 3G). Relative to IFN signaling, which focused a tPA response, TLR-mediated signaling (LPS) appears to promote the uPA pathway (Fig. 3G inset), as seen in endotoxemia and sepsis[26]. To determine whether IFNα and IFNγ worked in concert to augment plasminogen activation, these two cytokines were added in a dose dependent manner (0–100ng/ml) independently and simultaneously to monocytes at the onset of culture and tPA expression monitored after 4 hours. As we have shown and evident in Fig. 4A, IFNα and IFNγ independently enhanced tPA expression (1–100ng/ml) and when added at the same time, there was typically further monocyte activation, but this depended on the concentrations of the two IFNs added.
Figure 3. Cytokine regulation of components of plasminogen activation.

A,B) Representative MSG tissues from patients with SS were stained for CD3 (A) and IFNγ (B) and demonstrate IFNγ+ cells in the inflamed tissues characterized by abundant infiltrating CD3+ cell populations. Original magnification 20X. C) Peripheral blood monocytes were treated with IFNγ (10ng/ml) or IL-17 (100ng/ml) and monitored for altered expression of components of the tPA plasminogen activation system by RT-PCR. IFNγ was an effective inducer of monocyte tPA (*p=0.004 compared to control/unstimulated cells) and IFNγ and IL-17 induced annexin A2 (**p<0.02); n=4. P values calculated using independent Vassar Stats, T test (two-tailed, equal variances). D,E) Representative MSG tissues from patients with SS were stained for CD3 (D) and IL-17 (E) and reveal IL-17+ cells in the inflamed tissues characterized by abundant infiltrating CD3+ cell populations. F) Monocytes were untreated (0), treated with IL-17 (10–300ng/ml), treated with IFNγ (1–100ng/ml) or treated with IFNγ (1–100ng/ml) in the presence of a fixed amount of IL-17 (10ng/ml) and monitored by RT-PCR for tPA expression. G) Comparison of cytokine mediation of tPA and uPA in monocyte-derived macrophages was assessed by RT-PCR. TNFα did not enhance plasminogen activators and IFNγ induced tPA to a greater extent than IFNα (*p=0.001, relative to control, unstimulated macrophages). Inset. LPS (50ng/ml) engagement of TLR triggers components of the uPA system, but not the tPA pathway in macrophages.
Figure 4. IFN enhancement of tPA in vitro and elevated expression in vivo.

A) Monocytes were untreated (0), treated with IFNγ only (1–100ng/ml), treated with IFNα only (1–100ng/ml) or treated with IFNγ (1–100ng/ml) in the presence of IFNα (1–100ng/ml) and monitored by RT-PCR for tPA expression. B,C) By RT-PCR, MSG tissues from non-SS MSG, SS patients with early/intermediate disease and severe disease exhibited variable IFNα levels with progressive disease (B), whereas significant increases in expression of IFNγ were evident in early/intermediate disease and in severe disease (*p≤0.01) as compared to control non-SS tissues (C). P values calculated using Vassar Stats, Mann Whitney (two-tailed).
Assessment of MSG relative expression levels of type I and type II IFNs in control, early/intermediate and advanced SS disease (Fig. 4B,C) revealed variable IFNα levels relative to non-SS tissues, whereas IFNγ expression escalated with increasing severity of disease (Fig. 4C, *p<0.01). The parallel increase in IFNγ and plasminogen activators in advanced pathogenesis, coupled with the ability of IFNγ to bolster macrophage tPA levels, point to an unrecognized ability of IFNγ to promote plasminolytic activity in vitro and also, in vivo. In this regard, addition of IFNγ to monocytes revealed that IFNγ augmented monocyte activation of plasmin (Fig. 5A), consistent with enhanced detection of plasmin in inflamed salivary glands (Fig. 5B–D).
Figure 5. Enhanced plasminogen activation by IFNγ in vitro and increased plasmin in vivo.

A) Monocytes were cultured in the presence or absence of IFNγ and tested for their ability to activate plasminogen resulting in plasmin formation as detected by proteolytic activity and cleavage of a colorimetric substrate (relative fluorescence units) over time, as indicated. B) Representative MSG tissues stained with an antibody to plasmin(ogen) reveal cell associated and intercellular positive staining within the inflammatory infiltrate, endothelial and ductal cells, relative to an isotype control antibody (C) and compared to control non-SS gland tissues (D). Original magnification 40X.
3.4 Disruption of IFN signal attenuates plasminogen activators
Building on the evidence that IFNγ appears to uniquely foster monocyte plasmin generation which may not only implicate myeloid cells as indispensable in creating a path through the endothelial barrier for mononuclear cells to traverse, but also contribute to tissue destruction, we focused our attention on IFNγ-dependent pathways underlying enhanced tPA expression. First, we exposed monocytes to antibodies that reportedly block IFNγ binding to its receptors. Whereas anti-IFNγR1 partially inhibited IFNγ signaling as monitored by signal transducer and activator of transcription (STAT) phosphorylation and tPA induction (Fig. 6A), anti-IFNγR2 more effectively blunted phosphorylation of STAT1/3/5 (Fig. 6A) and downstream IFNγ-mediated tPA expression (Fig. 6A; p<0.001). Similarly, an antibody to IFNAR (CD118) that blocks IFNα-induced signaling, evidenced by reduced phospho-STAT1/3/5, diminished IFNα-upregulated tPA expression (Fig. 6B).
Figure 6. Signaling pathways in macrophage plasminogen activation.

A) Macrophage cultures were stimulated or not with IFNγ in the presence or absence of antibodies to the IFNγ receptor (R1, R2) and tPA expression by RT-PCR and inset) p-STAT signaling monitored by Western blot. IFNγ significantly enhanced tPA relative to control, unstimulated macrophages (A, *p<0.001). As evident, phosphorylations of STAT1 and particularly, STAT3 and STAT5 were inhibited in the presence of antibody to R2 (inset), which resulted in significantly blunted expression of tPA relative to IFNγ-only treated macrophages. *p<0.001; **p<0.01. B. Macrophage cultures were stimulated or not with IFNα in the presence or absence of antibodies to IFNAR (CD118) and tPA levels monitored by RT-PCR (B) and p-STAT signaling monitored by Western blot (inset). Phosphorylations of STAT1, STAT3 and STAT5 were all inhibited in the presence of the antibody, which resulted in significant blockage of tPA expression relative to IFNα. *p<0.01. C,D) Using a Jak1/2 inhibitor, both IFNα and IFNγ signaling pathways were interrupted with near loss of STAT phosphorylation as determined by Western blot using STAT6 protein as loading control. No inhibition was seen on constitutive pNFκB. E) In the presence of the Jak inhibitor, IFNγ-induced tPA expression was significantly inhibited. *p=0.01 comparing IFNγ only with IFNγ + Jak Inh.
In subsequent experiments, the Jak1/Jak2 inhibitor, which interferes with IFN- induced Jak-dependent STAT phosphorylation (Fig. 6C–D) and downstream sequelae was found to suppress phosphorylation of STAT1, STAT3, STAT5, and/or STAT6 with significant inhibition of tPA expression (Fig. 6E, p=0.01), whether triggered by IFNγ or IFNα. These data confirm that IFN binding, particularly IFNγ, to its cognate receptors underlies induction of a signal that culminates in expression of multiple macrophage genes, including tPA.
3.5 Downstream substrates of plasmin
Among the downstream substrates of plasmin activity, besides fibrin, are the pro-MMPs, of which several are significantly overexpressed by transcriptome analysis in the severely infiltrated MSG tissues [5], including MMP9 (gelatinase B), MMP12, a potent macrophage elastase, and ADAM-like, decysin 1, a member of a disintegrin and metalloprotease (ADAM) superfamily of zinc binding metalloproteases produced by macrophages and DC[5, 27] (Fig. 7A inset), which may also also activate pro-MMPs[28]. By comparison, antagonists of MMPs, including tissue inhibitors of metalloproteases (TIMP) were not significantly elevated (TIMP1,3,4), with the exception of TIMP2, which was increased only 2–3 fold in severely diseased MSG (Fig. 7A inset), likely failing to provide inhibition of increased proteolytic activity. In addition to plasmin and ADAMDEC1, pro-MMP can also be activated by cathepsins[29], several of which, cathepsin S, C and B, we have shown to be upregulated from 2–7 fold in severely inflamed MSG[5]. Cathepsin B, a cysteine protease with both exo- and endopeptidase activity also represents an antigen-processing enzyme and occurs in pathways leading to apoptosis [30][31][32].
Figure 7. MMP expression in MSG.

A) By microarray, SS salivary glands with severe lesions exhibited enhanced MMP9, MMP12 and ADAMDEC1 expression relative to gland tissues from subjects without SS and minimally increased TIMP2 (inset). By RT-PCR, MSG tissues from additional populations (n=5–6/group) were analyzed for expression of MMP9. By comparison to non-SS MSG, those tissues with early/intermediate disease exhibited a significant elevation in MMP9 expression (*p=0.004), with a more dramatic elevation in the severely inflamed and damaged MSG (**p=0.006). B) Immunohistochemical staining for MMP9 in control MSG revealed staining in ductal cells and/or in the periductal regions. Original magnification 20X. C,D,E) Compared to control MSG, tissues obtained from patients with severe SS exhibited extensive staining for MMP in the infiltrate and around acini (C, original magnification 20X), consonant with basal lamina damage, detachment of acinar cells, loss of nuclear polarity and disrupted structural integrity (D, E). Original magnification 40X. F) Immunohistochemistry using Jone’s PAMS stain for basement membrane identifies areas of fragmented basement membrane, particularly evident around acinar structures (white arrows). Original magnification 40X. G) Monocytes were cultured in the presence or absence of IFNγ (10ng/ml) for 4hr and MMP2, MMP7 and MMP9 expression monitored by RT-PCR. * p<0.01, **p=0.05 compared to control unstimulated macrophages (no IFNγ).
By RT-PCR, MMP9 was significantly elevated not only in salivary gland tissues from patients with severe disease (Fig. 7A, p=0.006), but also in MSG with early/intermediate pathology (Fig. 7A, p<0.004). Relative to control MSG, the level of MMP9 staining was abundant in infiltrated tissues (Fig. 7B,C), localizing to infiltrates and around acini, consonant with loss of architectural integrity (Fig. 7D–F) and basal lamina damage (Fig. 7F, Jone’s PAM stain). Consistent with evidence that MMP9 has been linked to acinar damage and destruction of basal lamina surrounding acini and ducts [33], enabling infiltration by mononuclear cells, we found MMP9+ staining cells were often of myeloid appearance. Moreover, exposure of macrophages to IFNγ in culture directly induced MMP7, to a greater extent than MMP2 or MMP9 relative to IFNγ unstimulated control cells(Fig. 7G). Although IFNγ does not independently induce significant monocyte MMP1 or 9, it may alter their expression in the context of other microenvironmental stimuli, such as TNFα[34], abundantly found in inflamed MSG[3, 6]. Elevated MMP2 and MMP9 activity has been reported in human salivary gland cell lines treated with IFNγ[35] and cytokines, including TNFα and IL-1β, contribute to destruction of basal membrane of salivary acinar epithelial cells from SS patients, acting in part through generation and activation of MMP2 (gelatinase A)[36], which may involve chondroitin sulfate glycosaminoglycan[37].
Overexpression of MMP12 protein, which also colocalizes to macrophages, has been associated with local depletion of elastin fibers[28], as observed in SS MSG with extensive fragmentation, thinning and loss of elastic fibers. Thus, MMP12 likely directly contributes to MSG pathology, injury, remodeling and fibrosis, although a specific role in exocrine damage has not been identified. Expression levels of TIMP1, an important natural inhibitor of MMP12[38], were minimally affected (~2 fold) [5], further suggesting increased enzymatic actions of MMP12. TIMP2 was also increased by only 2–3 fold, consistent with earlier evidence of an imbalance in the ratio of MMP to TIMP[33].
3.6 Evidence of tissue fibrosis and adiposity
In diseased MSG, proteolytic degradation occurs and histological findings of fatty infiltration, fibrosis and mononuclear cell infiltration (Fig. 8) further compromise structural and functional integrity of the salivary glands in pSS. Beyond the extensive mononuclear cell infiltrates with germinal center formation, interstitial fibrosis and acinar atrophy are linked to irreversible destruction of the glandular tissue. It appears likely that the persistent attempt to heal the tissue damage mediated by excessive proteolytic activity results in matrix deposition, interacinar fibrosis and scar-like formation. Among the collagen molecules significantly upregulated in the diseased MSG by microarray are type IV collagen alpha chains, α3 (3.8 fold) and α4 (3 fold)[5], and type 1 collagen, confirmed by RT-PCR in late disease, but not early or intermediate stages, as represented by ColA1 (Fig. 8D, p<0.01), building blocks for basal lamina formation. Both ductal and acinar epithelial cells produce mRNA for alpha chains[39] and disorganization of the basal lamina has been linked to invasion of mononuclear cells[40], implicating a state of active remodeling.
Figure 8. Loss of salivary gland integrity and matrix accumulation.

A) In MSG from patients with advanced disease, the extensive lymphoid infiltrate with germinal centers leads to interstitial fibrosis (arrows) and acinar atrophy, as ECM and adipose cells impinge on structure and functionality, compared to noninflamed MSG (B) H&E, original magnification 10X. C) Trichrome stain delineating interstitial fibrosis, along with acinar loss and mononuclear infiltration. Original magnification 20X. D) By RT-PCR, a representative collagen molecule, Col1A1, expression was not increased in early/intermediate disease MSG, but was significantly enhanced in the MSG of subjects with chronic/severe SS relative to MSG of individuals without SS; p<0.01.
Collectively, the increased expression of proteolytic pathways linked to mononuclear cell invasion into the salivary glands, the ensuing protease-mediated tissue damage and the host response to repair this damage all contribute to acinar atrophy and replacement of the glandular structure with matrix molecules. These sequelae preclude restoration of the physiological secretory functional activities of the salivary glands and/or other exocrine glands.
4.0 DISCUSSION
The mononuclear infiltrate has been a main focus of efforts to decipher Sjögren’s exocrine pathogenesis[41], with lesser attention aimed at the ensuing tissue destructive events. From our studies aimed at understanding how tissue infiltrates lead to exocrine gland damage and architectural disruption underlying xerostomia, we find that IFNγ promotes production and release of components of the plasminogen activation system, and plasmin generation facilitates invasion of the glandular structure by inflammatory cells with resultant tissue damage. The finding that IFNγ promotes myeloid plasminolytic activity was unexpected because it has not previously been linked to enhanced tPA, a key mechanism for plasminogen activation to plasmin. IFNγ treated macrophages significantly augmented their tPA expression and this effect was abolished by either blocking IFNγ binding to IFN receptors or by JAK-STAT signal disruption. We focused on JAK-STAT-dependent signaling in large part because of evidence that STAT4 is a genetic risk factor for SS[42] and that the risk variant of STAT4 correlates with increased expression[43] and sensitivity to IFNα signaling, resulting in amplified downstream IFN-induced gene expression[44], propelling the underlying autoimmune process. Among their many context-dependent immunoregulatory functions, STATs guide IL-23-dependent Th17 expansion[45], also abundant in SS salivary glands[6]. Collectively, considerable evidence supports an IFN-STAT linked signature in PBMC and salivary glands of patients with SS, along with altered Ro52(TRIM21)[46] and our transcriptional profile lends credence to such a connection.3
IFNγ exposure also persistently triggered other genes known to exacerbate macrophage activation and release of harmful molecules capable of attacking the gland[5]. Consequently, blocking the IFNγ pathway could alter the damaging effects of the infiltrating cells on the acinar secretory system and may represent an effective immunotherapeutic alternative for treatment of a subset of patients, notably those with macrophage-rich infiltrates in their salivary glands.
The plasminogen activation system plays an integral role in the migration of macrophages in response to an inflammatory stimulus, and the binding of plasminogen to its cell-surface receptors can initiate this process. Our data suggest an important mechanism of inflammatory cell invasion mediated by increased tPA, together with cell surface annexin A2 and/or S100A10, platforms for plasmin generation[11, 13]. Plasmin not only creates gradients from the blood across the sinusoidal endothelium into the exocrine glands, but among the downstream substrates for plasmin are chemokines, cytokines, growth factors and other proteases[47], including MMP. In addition to macrophage recruitment, tPA promotes neutrophil degranulation and the release of MMP9[48].
MMPs represent endopeptidases that regulate extracellular matrix and its remodeling, and the type IV collagenases/gelatinases MMP2 and MMP9 have been linked to pathogenesis in SS[49], influencing deposition of ECM and as a consequence, destruction of basal lamina and acinar and ductal architecture[33]. An altered balance between MMPs and their inhibitors is associated with acinar damage[33] and since salivary gland acinar cells express both MMPs and TIMPs, these cells, in addition to the infiltrating populations, may play an important role in extracellular matrix destruction and in MSG pathophysiology. MMP9, evident in regions where acinar integrity was compromised, may contribute to detachment of acinar cells from basal lamina, mucus cells with abnormal nuclear polarity, ducts with dilated lumen that may contain cellular debris, and disrupted ducts surrounded by fibrotic tissue. Not only is type IV collagen a major component of the basal lamina targeted by MMP2 and/or MMP9, but its expression is enhanced in diseased MSG to further obstruct secretory functions. Since suppression of TNFα-induced MMP9 has been shown to prevent destruction of acinar tissue in the salivary glands of patients with SS[50], this further underscores the impact of these proteases on salivary gland pathogenesis.
Although MMP12 and ADAMDEC1 have been linked with pathogenesis in other conditions, including pulmonary sarcoidosis[28], we demonstrate the novel finding that MMP12 is also overexpressed within sites of salivary gland inflammation. Moreover, the expression levels of TIMP1, an important natural inhibitor of MMP12[38], were not correspondingly changed, and since elastin fibers were depleted, the enzymatic actions of MMP12 were likely increased. These findings suggest that MMP12 plays an important role in facilitating MSG remodeling, possibly, as reported, by recruiting inflammatory cells[51] and promoting fibrosis[38].
Among the more substantively augmented proteases in the advanced exocrine lesions, carboxypeptidase M (CPM; >16 fold in severe SS vs control MSG) [5] is a membrane glycoprotein which removes C-terminal basic residues from peptides and proteins[52, 53]. Although CPM is widely distributed, as a product of differentiated myeloid cells[54], it converts short-lived complement fragment C5a to long-lived C5adesArg by removing C-terminal arginine, consistent with elevated systemic levels of C5a and C5adesArg in patients with inflammatory disorders [55]. More mature myeloid cells express the C5a/C5adesArg receptor (CD88), which is significantly upregulated in severe MSG lesions [5], and myeloid cells migrate to and/or are activated by C5adesArg [56] to produce MMP9 [57, 58], CPM and MT1-MMP [59, 60] to degrade ECM macromolecules, cytokines, and chemokines, including SDF-1, activate pro-MMP2 and stimulate cell mobility and migration [61, 62]. Another carboxypeptidase, vitellogenic-like (CPVL) is enhanced 5–6 fold [5] and is a serine carboxypeptidase of unknown function that was first characterized in human macrophages [63] where it colocalizes with markers for endoplasmic reticulum, but is not found on the outer plasma membrane. CPLV has been linked to antigen processing, the secretory pathway and may play a role in lamellipodium formation.
Further contributing to the proteolytic milieu in the diseased MSG are the cystatins, a family of naturally occurring cysteine protease inhibitors, and cystatin F (leukocystatin), which we have previously shown to be upregulated 2–3 fold [5], is expressed selectively in immune cells and targets aminopeptidase cathepsin C, to regulate diverse immune cell effector functions[64]. Likewise, dipeptidase 2 (DPEP2), expressed >12 fold [5] in SS MSG, but not previously linked to Sjögren’s syndrome, can hydrolyze a variety of dipeptides, including leukotriene D4[65] and may be a product of M2 macrophages[66]. Thus, the multitude of proteases may have independent actions, but also intersect with each other to choreograph the cascade of events culminating in salivary gland distortion and dysfunction.
Along with the histological findings of inflammatory disease, fatty infiltration, acinar loss and interstitial fibrosis become prominent which may be driven in part, by enhanced TGF-β and/or other growth factors[6]. With chronicity, the tissue develops fibrotic changes, and ECM and adipose cells further impinge on structure and functionality. Moreover, adiponectin is constitutively produced by activated salivary gland epithelial cells from patients with primary SS and may play a role[67]. The exocrine glands become functionally bereft due to compromised secretory activity. With the persistent inflammatory foci, fibrosis, atrophy and fatty changes that underscore lymphocytic adenitis and degeneration of the exocrine glands, it is important to decipher these pathways[68], since once fibrosis sets in, response to therapy becomes limited. The effect of therapy on salivary secretion is reported to be more marked in patients having moderate lymphocytic infiltration and moderate or less severe acinar atrophy and intralobular fibrosis at baseline [69]. If the pathways of tissue destruction leading to fibrosis are defined, intervention strategies may be revealed, albeit earlier intervention remains preferable.
Highlights.
Cell infiltrates orchestrate a perpetuating response underlying gland dysfunction
Th1 and Th17 cell products recruit additional lymphocytes, but also macrophages
Macrophages promote proteolysis, underlying tissue damage and inappropriate repair
Among the proteases is plasmin, generated by plasminogen activation through tPA
IFN drives tPA and plasmin cascade of proteolytic events in macrophages in vitro
Acknowledgments
The authors are grateful to Calley Grace for editorial assistance and Alfredo Molinolo, NIDCR, NIH for expertise in immunohistochemistry and scanning. This research was supported in part by the Intramural Research Program of the NIH, National Institute of Dental and Craniofacial Research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
6.0 REFERENCES
- 1.Voulgarelis M, Dafni UG, Isenberg DA, Moutsopoulos HM. Malignant lymphoma in primary Sjogren’s syndrome: a multicenter, retrospective, clinical study by the European Concerted Action on Sjogren’s Syndrome. Arthritis Rheum. 1999;42:1765–72. doi: 10.1002/1529-0131(199908)42:8<1765::AID-ANR28>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 2.Ice JA, Li H, Adrianto I, Lin PC, Kelly JA, Montgomery CG, et al. Genetics of Sjögren’s syndrome in the genome-wide association era. J Autoimmun. 2012;39:57–63. doi: 10.1016/j.jaut.2012.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Katsifis GE, Moutsopoulos NM, Wahl SM. T lymphocytes in Sjogren’s syndrome: contributors to and regulators of pathophysiology. Clin Rev Allergy Immunol. 2007;32:252–64. doi: 10.1007/s12016-007-8011-8. [DOI] [PubMed] [Google Scholar]
- 4.Manoussakis MN, Boiu S, Korkolopoulou P, Kapsogeorgou EK, Kavantzas N, Ziakas P, et al. Rates of infiltration by macrophages and dendritic cells and expression of interleukin-18 and interleukin-12 in the chronic inflammatory lesions of Sjogren’s syndrome: correlation with certain features of immune hyperactivity and factors associated with high risk of lymphoma development. Arthritis Rheum. 2007;56:3977–88. doi: 10.1002/art.23073. [DOI] [PubMed] [Google Scholar]
- 5.Greenwell-Wild T, Moutsopoulos NM, Gliozzi M, Kapsogeorgou E, Rangel Z, Munson PJ, et al. Chitinases in the salivary glands and circulation of patients with Sjogren’s syndrome: Macrophage harbingers of disease severity. Arthritis Rheum. 2011;63:3103–15. doi: 10.1002/art.30465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Katsifis GE, Rekka S, Moutsopoulos NM, Pillemer S, Wahl SM. Systemic and local interleukin-17 and linked cytokines associated with Sjogren’s syndrome immunopathogenesis. Am J Pathol. 2009;175:1167–77. doi: 10.2353/ajpath.2009.090319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Deiteren K, Hendriks D, Scharpe S, Lambeir AM. Carboxypeptidase M: Multiple alliances and unknown partners. Clin Chim Acta. 2009;399:24–39. doi: 10.1016/j.cca.2008.10.003. [DOI] [PubMed] [Google Scholar]
- 8.Conus S, Simon HU. Cathepsins and their involvement in immune responses. Swiss Med Wkly. 2010;140:w13042. doi: 10.4414/smw.2010.13042. [DOI] [PubMed] [Google Scholar]
- 9.Duffy MJ. The urokinase plasminogen activator system: role in malignancy. Curr Pharm Des. 2004;10:39–49. doi: 10.2174/1381612043453559. [DOI] [PubMed] [Google Scholar]
- 10.Sharma MC, Sharma M. The role of annexin II in angiogenesis and tumor progression: a potential therapeutic target. Curr Pharm Des. 2007;13:3568–75. doi: 10.2174/138161207782794167. [DOI] [PubMed] [Google Scholar]
- 11.Brownstein C, Deora AB, Jacovina AT, Weintraub R, Gertler M, Khan KM, et al. Annexin II mediates plasminogen-dependent matrix invasion by human monocytes: enhanced expression by macrophages. Blood. 2004;103:317–24. doi: 10.1182/blood-2003-04-1304. [DOI] [PubMed] [Google Scholar]
- 12.Shi Z, Stack MS. Urinary-type plasminogen activator (uPA) and its receptor (uPAR) in squamous cell carcinoma of the oral cavity. Biochem J. 2007;407:153–9. doi: 10.1042/BJ20071037. [DOI] [PubMed] [Google Scholar]
- 13.O’Connell PA, Surette AP, Liwski RS, Svenningsson P, Waisman DM. S100A10 regulates plasminogen-dependent macrophage invasion. Blood. 2010;116:1136–46. doi: 10.1182/blood-2010-01-264754. [DOI] [PubMed] [Google Scholar]
- 14.Wen J, Nikitakis NG, Chaisuparat R, Greenwell-Wild T, Gliozzi M, Jin W, et al. Secretory Leukocyte Protease Inhibitor (SLPI) Expression and Tumor Invasion in Oral Squamous Cell Carcinoma. Am J Pathol. 2011;178:2866–78. doi: 10.1016/j.ajpath.2011.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang Y, DeWitt DL, McNeely TB, Wahl SM, Wahl LM. Secretory leukocyte protease inhibitor suppresses the production of monocyte prostaglandin H synthase-2, prostaglandin E2, and matrix metalloproteinases. J Clin Invest. 1997;99:894–900. doi: 10.1172/JCI119254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Taggart CC, Cryan SA, Weldon S, Gibbons A, Greene CM, Kelly E, et al. Secretory leucoprotease inhibitor binds to NF-kappaB binding sites in monocytes and inhibits p65 binding. J Exp Med. 2005;202:1659–68. doi: 10.1084/jem.20050768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ma G, Greenwell-Wild T, Lei K, Jin W, Swisher J, Hardegen N, et al. Secretory leukocyte protease inhibitor binds to annexin II, a cofactor for macrophage HIV-1 infection. J Exp Med. 2004;200:1337–46. doi: 10.1084/jem.20041115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ashcroft GS, Lei K, Jin W, Longenecker G, Kulkarni AB, Greenwell-Wild T, et al. Secretory leukocyte protease inhibitor mediates non-redundant functions necessary for normal wound healing. Nat Med. 2000;6:1147–53. doi: 10.1038/80489. [DOI] [PubMed] [Google Scholar]
- 19.Zhu J, Nathan C, Jin W, Sim D, Ashcroft GS, Wahl SM, et al. Conversion of proepithelin to epithelins: roles of SLPI and elastase in host defense and wound repair. Cell. 2002;111:867–78. doi: 10.1016/s0092-8674(02)01141-8. [DOI] [PubMed] [Google Scholar]
- 20.Angelov N, Moutsopoulos N, Jeong MJ, Nares S, Ashcroft G, Wahl SM. Aberrant mucosal wound repair in the absence of secretory leukocyte protease inhibitor. Thromb Haemost. 2004;92:288–97. doi: 10.1160/TH03-07-0446. [DOI] [PubMed] [Google Scholar]
- 21.Virtanen OJ, Siren V, Multanen J, Farkkila M, Leivo I, Vaheri A, et al. Plasminogen activators and their inhibitors in human saliva and salivary gland tissue. Eur J Oral Sci. 2006;114:22–6. doi: 10.1111/j.1600-0722.2006.00264.x. [DOI] [PubMed] [Google Scholar]
- 22.Vitali C, Bombardieri S, Jonsson R, Moutsopoulos HM, Alexander EL, Carsons SE, et al. Classification criteria for Sjogren’s syndrome: a revised version of the European criteria proposed by the American-European Consensus Group. Ann Rheum Dis. 2002;61:554–8. doi: 10.1136/ard.61.6.554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Daniels TE, Whitcher JP. Association of patterns of labial salivary gland inflammation with keratoconjunctivitis sicca. Analysis of 618 patients with suspected Sjogren’s syndrome. Arthritis Rheum. 1994;37:869–77. doi: 10.1002/art.1780370615. [DOI] [PubMed] [Google Scholar]
- 24.Tarpley TM, Jr, Anderson LG, White CL. Minor salivary gland involvement in Sjogren’s syndrome. Oral Surg Oral Med Oral Pathol. 1974;37:64–74. doi: 10.1016/0030-4220(74)90160-1. [DOI] [PubMed] [Google Scholar]
- 25.Zeher M, Szegedi G, Csiki Z, Cseh A, Adany R. Fibrinolysis-resistant fibrin deposits in minor labial salivary glands of patients with Sjogren’s syndrome. Clin Immunol Immunopathol. 1994;71:149–55. doi: 10.1006/clin.1994.1065. [DOI] [PubMed] [Google Scholar]
- 26.Abraham E, Gyetko MR, Kuhn K, Arcaroli J, Strassheim D, Park JS, et al. Urokinase-type plasminogen activator potentiates lipopolysaccharide-induced neutrophil activation. J Immunol. 2003;170:5644–51. doi: 10.4049/jimmunol.170.11.5644. [DOI] [PubMed] [Google Scholar]
- 27.Bates EE, Fridman WH, Mueller CG. The ADAMDEC1 (decysin) gene structure: evolution by duplication in a metalloprotease gene cluster on chromosome 8p12. Immunogenetics. 2002;54:96–105. doi: 10.1007/s00251-002-0430-3. [DOI] [PubMed] [Google Scholar]
- 28.Crouser ED, Culver DA, Knox KS, Julian MW, Shao G, Abraham S, et al. Gene expression profiling identifies MMP-12 and ADAMDEC1 as potential pathogenic mediators of pulmonary sarcoidosis. Am J Respir Crit Care Med. 2009;179:929–38. doi: 10.1164/rccm.200803-490OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mort JS, Buttle DJ. Cathepsin B. Int J Biochem Cell Biol. 1997;29:715–20. doi: 10.1016/s1357-2725(96)00152-5. [DOI] [PubMed] [Google Scholar]
- 30.Steinfeld S, Maho A, Chaboteaux C, Daelemans P, Pochet R, Appelboom T, et al. Prolactin up-regulates cathepsin B and D expression in minor salivary glands of patients with Sjogren’s syndrome. Lab Invest. 2000;80:1711–20. doi: 10.1038/labinvest.3780181. [DOI] [PubMed] [Google Scholar]
- 31.Mizuochi T, Yee ST, Kasai M, Kakiuchi T, Muno D, Kominami E. Both cathepsin B and cathepsin D are necessary for processing of ovalbumin as well as for degradation of class II MHC invariant chain. Immunol Lett. 1994;43:189–93. doi: 10.1016/0165-2478(94)90221-6. [DOI] [PubMed] [Google Scholar]
- 32.Deiss LP, Galinka H, Berissi H, Cohen O, Kimchi A. Cathepsin D protease mediates programmed cell death induced by interferon-gamma, Fas/APO-1 and TNF-alpha. EMBO J. 1996;15:3861–70. [PMC free article] [PubMed] [Google Scholar]
- 33.Perez P, Kwon YJ, Alliende C, Leyton L, Aguilera S, Molina C, et al. Increased acinar damage of salivary glands of patients with Sjogren’s syndrome is paralleled by simultaneous imbalance of matrix metalloproteinase 3/tissue inhibitor of metalloproteinases 1 and matrix metalloproteinase 9/tissue inhibitor of metalloproteinases 1 ratios. Arthritis Rheum. 2005;52:2751–60. doi: 10.1002/art.21265. [DOI] [PubMed] [Google Scholar]
- 34.Zhou M, Zhang Y, Ardans JA, Wahl LM. Interferon-gamma differentially regulates monocyte matrix metalloproteinase-1 and-9 through tumor necrosis factor-alpha and caspase 8. J Biol Chem. 2003;278:45406–13. doi: 10.1074/jbc.M309075200. [DOI] [PubMed] [Google Scholar]
- 35.Wu AJ, Lafrenie RM, Park C, Apinhasmit W, Chen ZJ, Birkedal-Hansen H, et al. Modulation of MMP-2 (gelatinase A) and MMP-9 (gelatinase B) by interferon-gamma in a human salivary gland cell line. J Cell Physiol. 1997;171:117–24. doi: 10.1002/(SICI)1097-4652(199705)171:2<117::AID-JCP1>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 36.Azuma M, Motegi K, Aota K, Hayashi Y, Sato M. Role of cytokines in the destruction of acinar structure in Sjogren’s syndrome salivary glands. Lab Invest. 1997;77:269–80. [PubMed] [Google Scholar]
- 37.Iida J, Wilhelmson KL, Ng J, Lee P, Morrison C, Tam E, et al. Cell surface chondroitin sulfate glycosaminoglycan in melanoma: role in the activation of pro-MMP-2 (pro-gelatinase A) Biochem J. 2007;403:553–63. doi: 10.1042/BJ20061176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Matute-Bello G, Wurfel MM, Lee JS, Park DR, Frevert CW, Madtes DK, et al. Essential role of MMP-12 in Fas-induced lung fibrosis. Am J Respir Cell Mol Biol. 2007;37:210–21. doi: 10.1165/rcmb.2006-0471OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Poduval P, Sillat T, Virtanen I, Porola P, Konttinen YT. Abnormal basement membrane type IV collagen alpha-chain composition in labial salivary glands in Sjogren’s syndrome. Arthritis Rheum. 2009;60:938–45. doi: 10.1002/art.24388. [DOI] [PubMed] [Google Scholar]
- 40.Molina C, Alliende C, Aguilera S, Kwon YJ, Leyton L, Martinez B, et al. Basal lamina disorganisation of the acini and ducts of labial salivary glands from patients with Sjogren’s syndrome: association with mononuclear cell infiltration. Ann Rheum Dis. 2006;65:178–83. doi: 10.1136/ard.2004.033837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tzioufas AG, Kapsogeorgou EK, Moutsopoulos HM. Pathogenesis of Sjögren’s syndrome: What we know and what we should learn. J Autoimmun. 2012;39:4–8. doi: 10.1016/j.jaut.2012.01.002. [DOI] [PubMed] [Google Scholar]
- 42.Gestermann N, Mekinian A, Comets E, Loiseau P, Puechal X, Hachulla E, et al. STAT4 is a confirmed genetic risk factor for Sjogren’s syndrome and could be involved in type 1 interferon pathway signaling. Genes Immun. 2010;11:432–8. doi: 10.1038/gene.2010.29. [DOI] [PubMed] [Google Scholar]
- 43.Sigurdsson S, Nordmark G, Garnier S, Grundberg E, Kwan T, Nilsson O, et al. A risk haplotype of STAT4 for systemic lupus erythematosus is over-expressed, correlates with anti-dsDNA and shows additive effects with two risk alleles of IRF5. Hum Mol Genet. 2008;17:2868–76. doi: 10.1093/hmg/ddn184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kariuki SN, Kirou KA, MacDermott EJ, Barillas-Arias L, Crow MK, Niewold TB. Cutting edge: autoimmune disease risk variant of STAT4 confers increased sensitivity to IFN-alpha in lupus patients in vivo. Journal of immunology. 2009;182:34–8. doi: 10.4049/jimmunol.182.1.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mathur AN, Chang HC, Zisoulis DG, Stritesky GL, Yu Q, O’Malley JT, et al. Stat3 and Stat4 direct development of IL-17-secreting Th cells. Journal of immunology. 2007;178:4901–7. doi: 10.4049/jimmunol.178.8.4901. [DOI] [PubMed] [Google Scholar]
- 46.Oke V, Wahren-Herlenius M. The immunobiology of Ro52(TRIM21) in autoimmunity: A critical review. J Autoimmun. 2012;39:77–82. doi: 10.1016/j.jaut.2012.01.014. [DOI] [PubMed] [Google Scholar]
- 47.Myohanen H, Vaheri A. Regulation and interactions in the activation of cell-associated plasminogen. Cell Mol Life Sci. 2004;61:2840–58. doi: 10.1007/s00018-004-4230-9. [DOI] [PubMed] [Google Scholar]
- 48.Cuadrado E, Ortega L, Hernandez-Guillamon M, Penalba A, Fernandez-Cadenas I, Rosell A, et al. Tissue plasminogen activator (t-PA) promotes neutrophil degranulation and MMP-9 release. J Leukoc Biol. 2008;84:207–14. doi: 10.1189/jlb.0907606. [DOI] [PubMed] [Google Scholar]
- 49.Konttinen YT, Halinen S, Hanemaaijer R, Sorsa T, Hietanen J, Ceponis A, et al. Matrix metalloproteinase (MMP)-9 type IV collagenase/gelatinase implicated in the pathogenesis of Sjogren’s syndrome. Matrix Biol. 1998;17:335–47. doi: 10.1016/s0945-053x(98)90086-5. [DOI] [PubMed] [Google Scholar]
- 50.Azuma M, Aota K, Tamatani T, Motegi K, Yamashita T, Ashida Y, et al. Suppression of tumor necrosis factor alpha-induced matrix metalloproteinase 9 production in human salivary gland acinar cells by cepharanthine occurs via down-regulation of nuclear factor kappaB: a possible therapeutic agent for preventing the destruction of the acinar structure in the salivary glands of Sjogren’s syndrome patients. Arthritis Rheum. 2002;46:1585–94. doi: 10.1002/art.10315. [DOI] [PubMed] [Google Scholar]
- 51.Houghton AM, Quintero PA, Perkins DL, Kobayashi DK, Kelley DG, Marconcini LA, et al. Elastin fragments drive disease progression in a murine model of emphysema. J Clin Invest. 2006;116:753–9. doi: 10.1172/JCI25617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Reverter D, Maskos K, Tan F, Skidgel RA, Bode W. Crystal structure of human carboxypeptidase M, a membrane-bound enzyme that regulates peptide hormone activity. J Mol Biol. 2004;338:257–69. doi: 10.1016/j.jmb.2004.02.058. [DOI] [PubMed] [Google Scholar]
- 53.Deiteren K, Surpateanu G, Gilany K, Willemse JL, Hendriks DF, Augustyns K, et al. The role of the S1 binding site of carboxypeptidase M in substrate specificity and turn-over. Biochim Biophys Acta. 2007;1774:267–77. doi: 10.1016/j.bbapap.2006.11.017. [DOI] [PubMed] [Google Scholar]
- 54.Rehli M, Krause SW, Andreesen R. The membrane-bound ectopeptidase CPM as a marker of macrophage maturation in vitro and in vivo. Adv Exp Med Biol. 2000;477:205–16. doi: 10.1007/0-306-46826-3_23. [DOI] [PubMed] [Google Scholar]
- 55.Burger R, Zilow G. Complement-derived anaphylatoxins in natural immunity. In: Sim E, editor. The Natural Immune System: Humoral Factors. New York: Oxford; 1993. p. 209. [Google Scholar]
- 56.Levesque JP, Winkler IG. Mobilization of hematopoietic stem cells: state of the art. Curr Opin Organ Transplant. 2008;13:53–8. doi: 10.1097/MOT.0b013e3282f42473. [DOI] [PubMed] [Google Scholar]
- 57.Jalili A, Shirvaikar N, Marquez-Curtis L, Qiu Y, Korol C, Lee H, et al. Fifth complement cascade protein (C5) cleavage fragments disrupt the SDF-1/CXCR4 axis: further evidence that innate immunity orchestrates the mobilization of hematopoietic stem/progenitor cells. Exp Hematol. 2010;38:321–32. doi: 10.1016/j.exphem.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.DiScipio RG, Schraufstatter IU, Sikora L, Zuraw BL, Sriramarao P. C5a mediates secretion and activation of matrix metalloproteinase 9 from human eosinophils and neutrophils. Int Immunopharmacol. 2006;6:1109–18. doi: 10.1016/j.intimp.2006.02.006. [DOI] [PubMed] [Google Scholar]
- 59.McQuibban GA, Butler GS, Gong JH, Bendall L, Power C, Clark-Lewis I, et al. Matrix metalloproteinase activity inactivates the CXC chemokine stromal cell-derived factor-1. J Biol Chem. 2001;276:43503–8. doi: 10.1074/jbc.M107736200. [DOI] [PubMed] [Google Scholar]
- 60.Marquez-Curtis L, Jalili A, Deiteren K, Shirvaikar N, Lambeir AM, Janowska-Wieczorek A. Carboxypeptidase M expressed by human bone marrow cells cleaves the C-terminal lysine of stromal cell-derived factor-1alpha: another player in hematopoietic stem/progenitor cell mobilization? Stem Cells. 2008;26:1211–20. doi: 10.1634/stemcells.2007-0725. [DOI] [PubMed] [Google Scholar]
- 61.Barbolina MV, Stack MS. Membrane type 1-matrix metalloproteinase: substrate diversity in pericellular proteolysis. Semin Cell Dev Biol. 2008;19:24–33. doi: 10.1016/j.semcdb.2007.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Matias-Roman S, Galvez BG, Genis L, Yanez-Mo M, de la Rosa G, Sanchez-Mateos P, et al. Membrane type 1-matrix metalloproteinase is involved in migration of human monocytes and is regulated through their interaction with fibronectin or endothelium. Blood. 2005;105:3956–64. doi: 10.1182/blood-2004-06-2382. [DOI] [PubMed] [Google Scholar]
- 63.Harris J, Schwinn N, Mahoney JA, Lin HH, Shaw M, Howard CJ, et al. A vitellogenic-like carboxypeptidase expressed by human macrophages is localized in endoplasmic reticulum and membrane ruffles. Int J Exp Pathol. 2006;87:29–39. doi: 10.1111/j.0959-9673.2006.00450.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hamilton G, Colbert JD, Schuettelkopf AW, Watts C. Cystatin F is a cathepsin C-directed protease inhibitor regulated by proteolysis. EMBO J. 2008;27:499–508. doi: 10.1038/sj.emboj.7601979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Habib GM, Shi ZZ, Cuevas AA, Lieberman MW. Identification of two additional members of the membrane-bound dipeptidase family. FASEB J. 2003;17:1313–5. doi: 10.1096/fj.02-0899fje. [DOI] [PubMed] [Google Scholar]
- 66.Oliveira LJ, McClellan S, Hansen PJ. Differentiation of the endometrial macrophage during pregnancy in the cow. PLoS One. 2010;5:e13213. doi: 10.1371/journal.pone.0013213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Katsiougiannis S, Kapsogeorgou EK, Manoussakis MN, Skopouli FN. Salivary gland epithelial cells: a new source of the immunoregulatory hormone adiponectin. Arthritis Rheum. 2006;54:2295–9. doi: 10.1002/art.21944. [DOI] [PubMed] [Google Scholar]
- 68.Seror R, Bootsma H, JVS, Dömer T, Gottenberg JE, Mariette X, et al. Outcome measures for primary Sjögren’s syndrome. J Autoimmun. 2012;39:97–102. doi: 10.1016/j.jaut.2012.01.013. [DOI] [PubMed] [Google Scholar]
- 69.Nakayamada S, Fujimoto T, Nonomura A, Saito K, Nakamura S, Tanaka Y. Usefulness of initial histological features for stratifying Sjogren’s syndrome responders to mizoribine therapy. Rheumatology (Oxford) 2009;48:1279–82. doi: 10.1093/rheumatology/kep228. [DOI] [PubMed] [Google Scholar]