

Abstract
Human papillomavirus (HPV) DNA integrations may affect therapeutic responses in cancers through ATM network-related DNA damage response (DDR). We studied whether cisplatin-induced DDR was altered in human HK-2 renal tubular cells immortalized by HPV16 E6/E7 genes. Cytotoxicity assays utilized thiazolyl blue dye and DDR was identified by gene expression differences, double-strand DNA breaks, ATM promoter activity, and analysis of cell cycling and side population cells. After cisplatin, HK-2 cells showed greater ATM promoter activity indicating activation of this network, but DDR was muted, since little γH2AX was expressed, DNA strand breaks were absent and cells continued cycling. When HK-2 cells were treated with the MDM2 antagonist inducing p53, nutlin-3, or p53 transcriptional activator, tenovin-1, cell growth decreased but cisplatin toxicity was unaffected. By contrast, arsenic trioxide, which by inhibiting wild-type p53-induced phosphatase-1 that serves responses downstream of p53, and by depolymerizing tubulin, synergistically enhanced cisplatin cytotoxicity including loss of SP cells. Our findings demonstrated that HPV16 E6/E7 altered DDR through p53-mediated cell growth controls, which may be overcome by targeting of WIP1 and other processes, and thus should be relevant for treating renal cell carcinoma.
Keywords: Chemotherapy, Ataxia telangiectasia mutated, DNA damage response, Nephrotoxicity, Renal cell carcinoma
1. INTRODUCTION
Renal cell carcinoma (RCC) is a major worldwide problem with poor clinical outcomes (1). Typically, nonresectable RCC is resistant to conventional chemo- or radio-therapy, and highly effective molecular therapies are lacking, since oncogenetic events and mechanisms are less well understood in RCC (2). For cancer therapies in general, DNA damage/repair mechanisms related to ataxia telangiectasia mutated (ATM) gene network, are of considerable interest (3). The ATM network normally protects cells from DNA damage, such that impairment in network integrity could lead to organ failure (4). By contrast, dysregulation of ATM network may promote oncogenesis by helping protect cancer cells (5), which may serve nefarious goals. As DNA damage is a major mechanism in chemotherapy, e.g., as represented by cisplatin (Cis-P) and other commonly used drugs, avoiding bystander toxicity to healthy cells via greater cancer specificity by chemically modified drugs has gathered interest (6). While insights into ATM-mediated DNA damage response (DDR) will help characterize tumor biology and provide therapeutic directions in RCC (7), these areas need more work. In part, genetic differences in DNA damage/repair pathways with potential to alter DDR, may determine susceptibility of RCC to chemo- or radio-therapy (8). This possibility was supported by putative pathophysiological roles in mice with tyrosinemia type-1 and RCC and hepatocellular carcinoma (HCC) of molecules downstream of ATM in the network, e.g., cell cycle checkpoint controls regulated by p53 or p21 (9). Also, these types of mechanisms will be appropriate for assigning prognosis in people with RCC (10). Other genetic elements, e.g., oncogenes transmitted by cancer-associated viruses, may contribute in DDR and/or cell cycling (11,12). For instance, clinical studies of RCC recently identified presence of DNA from oncogenic human papillomavirus (HPV) 16 or 18 serotypes in 14%–30% of cases (13,14). This should be of interest because HPV oncoproteins may alter DDR and cell cycling (11,12).
We hypothesized that study of ATM-mediated DDR in suitable renal cells will help illuminate mechanisms of chemosusceptibility in RCC besides that of nephrotoxicity in chemotherapy recipients. This was examined with Cis-P as candidate drug in HK-2 human kidney cells, which were immortalized with a retroviral vector to express E6/E7 oncoproteins of HPV serotype 16 (15), and subsequently retained a proximal tubular epithelial phenotype (16). Despite their nonRCC origin, HK-2 cells offer parallels for understanding contributions of HPV genes in DDR related to RCC. Renal cell carcinoma cells with HPV DNA integrations are not available. Our studies included HuH-7 cells, which originated from an adult HCC (17), and displayed robust Cis-P-induced DDR (4). In these ways, we obtained information in respect to DDR in HK-2 cells, including cell growth regulation in the context of p53-related intracellular signaling, and the “side population (SP)” often associated with cancer stem cells (CSC) (18,19). This helped us to determine whether drugs could be identified with capacity for synergistically amplifying Cis-P toxicity in renal cells.
2. METHODS
2.1. Drugs and chemicals
The chemicals were from Sigma Chemical Co. (St. Louis, MO). Stocks were prepared as follows: 3.3 mM Cis-diammineplatinum (II) dichloride (Cis-P) (P4394, Sigma, St. Louis, MO) in normal saline; 676 μM tenovin-1 (13085, Cayman Chemical, Ann Arbor, MI) in dimethylsulfoxide (DMSO); 5 mM arsenic trioxide (ATO) (NG-I1690, Chem Service Inc., West Chester, PA) in water with pH to 6.5 with 1N NaOH; 1.7 mM nutlin-3 (222086, Santa Cruz Biotechnology, Santa Cruz, CA) in DMSO.
2.2. Cells
HuH-7 cells were originally from R. Stockert at Albert Einstein College of Medicine. HK-2 cells were from Dr. B. Goilav at Albert Einstein College of Medicine. Cells were maintained in Dulbecco's Modified Eagle Medium (DMEM, Sigma, St. Louis, MO) with 10% FBS and antibiotics in 5% CO2 atmosphere. For studies, 2.5×104 HK-2 or HuH-7 cells were plated per cm2 plastic in 24-well or larger dishes for 24 h followed by drugs in fresh medium for 12–16 h at 37°C. For 50% inhibitory concentrations (IC50), HK-2 and HuH-7 cells were cultured with 25–150 μM Cis-P in triplicate conditions, followed by cell viability analysis.
2.3. ATM promoter activity assay
We transduced HK-2 cells with lentiviral vector (LV) at 10 multiplicity of infection (MOI) to express tdTomato (tdT) fluorescent reporter under human ATM promoter, as described previously (4). For assays, cells were treated with drugs for 16–18 h, washed twice with phosphate buffered saline, pH 7.4 (PBS), and stained for 30 min with 6 μg/ml Hoechst 33258 (Sigma). After more PBS washes fluorescence at 520 nm (excitation) and 590 nm (emission) for Hoechst 33428 and at 355 nm (excitation) and 460 nm (emission) for tdT were measured by FLUOstar reader (BMG Lab Technologies, Life Technologies, Carlsbad, CA)
2.4. Cell viability assay
Cells were incubated for 2 h with 50 μg/mL thiazolyl blue (MTT) dye at 37°C. Intracellular formazan crystals were lysed in acid isopropanol and quantitated at 570 nm as previously described (20). Alternatively, cell nuclei were stained with Hoechst 33258 dye (Sigma) for fluorimetric DNA content measurements as previously described (4).
2.5. Expression of DDR genes
Total cellular RNA was extracted by TRIzol Reagent (Invitrogen, Carlsbad, CA). Complementary DNA was made from 1 μg RNAs using RT2 First Strand Kit (SA Biosciences, Frederick, MD). RT2 Profiler PCR Array with 84 Human DNA Damage Signaling Pathway genes (PAHS-029A-12; SA Biosciences, Frederick, MD) was used for quantitative real-time polymerase chain reactions (qRT-PCR) with SYBR Green PCR Master Mix (SA Biosciences, Frederick, MD) according to the manufacturer and as previously described (4). Data were analyzed by 2-ΔΔCt method. Gene expression was normalized against housekeeping genes in individual samples. Fold-change in gene expression was determined by log-normalized ratios of treated/untreated cells. Gene expression differences of ≥2-fold were considered significant.
2.6. Comet assays for DNA strand breaks
Cultured cells were detached by trypsin-EDTA, washed and embedded in 0.5% low melting point (LMP) agarose gel (50101, Cambrex Bio Science Rockland, Inc, Rockland, ME) on glass slides as described previously (4). Slides were incubated in lysis buffer with 10% laurylsarcosine, 5M NaCl, 0.5M EDTA pH 8, 1M Tris HCl pH 10 and 100% Triton X-100 for 1 h at 4°C, followed by alkaline hydrolysis in 10M NaOH, 0.5M EDTA pH 8 for 1 h at 4°C. Slides were transferred to tank with 1×Tris-acetate-RDTA (TAE) buffer and electrophoresed at 25 V for 1 h. After neutralization in 1M Tris HCl pH 7.5 for 5 min, washing in water for 5 min, and fixation in 70% ethanol for 5 min, slides were dried at 45°C, stained with 1 μg/ml ethidium bromide (Promega, Madison, WI) and observed under epifluorescence. Comets were counted in at least 50 nuclei per condition. Comet tail-length was analyzed by Image J (NCI, Bethesda, MD) with Comet Assay Plugin originally from H.M. Geller and added by R. Bagnell (Microscopy Services Laboratory, Chapel Hill, NC).
2.7. γH2AX immunostaining
Cells were fixed in 4% paraformaldehyde in PBS for 10 min, permeabilized in PBS with 0.5% Triton-X (PBS-T) for 10 min, and blocked in 5% goat serum in PBS-T for 1 h at room temperature. Cells were incubated with anti-human γH2AX IgG (1:300, Cell Signaling, Beverly, MA) for 1 h at room temperature, by rhodamine-conjugated anti-rabbit goat IgG (1:500, Sigma, St. Louis MO) for 1 h at room temperature, and DAPI counterstaining.
2.8. Western blot
Cells were lysed in buffer with protease inhibitor cocktail (05 892 970 001, Roche, Indianapolis, IN). After keeping on ice for 30 min, lysed cells were pelleted to collect supernatant. Proteins were quantified with Bradford reagent and analyzed by SDS-PAGE with 75 μg proteins in mini Protean TGX precast gels (456–1033, Biorad, Hercules, CA). Resolved proteins were transferred to PVDF membranes, which were blocked in 4% nonfat dry milk for 1 h, and incubated with primary antibodies overnight at 4°C: WIP1 (1:1000; Cell Signaling, Beverly, MA), and β-tubulin (1:1000; Sigma, St. Louis, MO). Blots were incubated for 1 h at room temperature with peroxidase-conjugated anti-mouse (1:1000, Sigma, St. Louis, MO) or anti-rabbit (1:1000, Sigma, St. Louis, MO) IgG. Blots were developed with Western Lightning Plus-ECL (Perkin Elmer, Waltham, MA). To re-probe blots were treated with Restore Plus Western Blot Stripping Buffer (46530, Thermo Scientific, Rockford, IL).
2.9. Cell cycle analysis
Cells were released by trypsin-EDTA, washed in PBS, and stored in ice-cold 70% ethanol at 4°C. Cells were washed with PBS and incubated with 40 μg/mL propidium iodide (PI) (81845, Sigma, St, Louis MO) plus 10 μg/mL RNAse A (Worthington Biochemicals, Lakewood, NJ) in PBS for 20 min on ice. Cells were analyzed by FACS Aria (Becton Dickinson) using FACSDiva Software (Becton Dickinson) to identify G0/G1, S and G2/M fractions.
2.10. Side population (SP)
Cultured cells were released by trypsin-EDTA, washed, resuspended in prewarmed DMEM, and incubated with 10 μg/mL Hoechst 33342 (4082S, Cell Signaling, Beverly, MA) for 90 min at 37°C. After pelleting, cells were stained with 2 μg/mL PI in ice-cold PBS for FACS at 450 and 650 nm. To identify SP, cells were incubated for 20 min with 50 μM verapamil (4629, Sigma, St. Louis, MO).
2.11. Statistical methods
Each condition was in triplicate. Data are shown as means ± SEM. The t-test or Chi-square test or ANOVA with posthoc Tukey tests were used for evaluating significance of differences. P values <0.05 were considered significant.
3. RESULTS
Cis-P was toxic for HuH-7 and HK-2 cells, with in each case overt cytotoxicity, including cell rounding, vacuolation and other morphological criteria such as cell membrane disruptions for necrosis (Fig. 1A). The IC50 doses for Cis-P in HuH-7 and HK-2 cells were 15 μM and 25 μM, respectively, as elucidated by well-established cell viability assays using reduction of MTT dye or nuclear DNA staining by Hoechst 33258 dye (4,20) (Fig. 1B and 1C). Therefore, we used these Cis-P concentrations in further HuH-7 and HK-2 cell studies.
Figure 1. Cis-P cytotoxicity in HuH-7 and HK-2 cells.
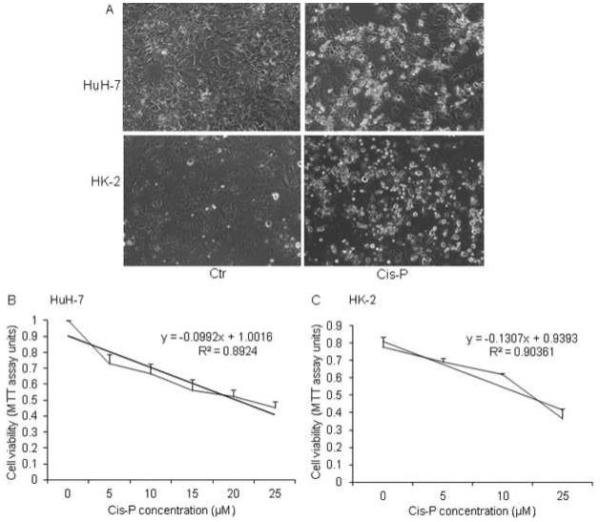
A. Phase contrast microscopy for morphological changes in Cis-P-treated HuH-7 and HK-2 cells. Shown are cells with or without IC50 doses of Cis-P. These were calculated with dose response analysis of Cis-P cytotoxicity by MTT cell viability assays and were 15 μM and 25 μM for HuH-7 and HK-2 cells, respectively (B).
3.1. DDR in HK-2 and HuH-7 cells
Despite similar IC50 doses of Cis-P in HuH-7 and HK-2 cells, DDR genes were expressed differently in these cell types. Based on the widely used parameter of ≥2-fold gene expression differences, 29 of 84 (35%) versus 45 of 84 (54%) genes were differently expressed in HuH-7 and HK-2 cells, respectively, p<0.05, Chi Square (Fig. 2A). Of these, in HuH-7 cells, cell cycle arrest and apoptosis genes were most prominently upregulated, whereas damaged DNA-binding genes were most prominently downregulated (Fig. 2B). This was different in HK-2 cells because more cell cycle arrest and apoptosis genes were downregulated instead of being upregulated, whereas damaged DNA-binding genes were only downregulated. In both HuH-7 and HK-2 cells ATM, ATR, CHEK1, and CHEK2 genes were downregulated after Cis-P. However, p53 was expressed more in HuH-7 cells and less in HK-2 cells, which suggested cell growth controls may have been altered (Table 1). After Cis-P, γH2AX was expressed in 81±1.8% of Cis-P-treated HuH-7 cells but in only 31±0.9% of HK-2 cells, which was 2.6-fold lower, p<0.05, Chi-square (Fig. 3A, 3B). This difference in DDR was borne out by ATM promoter activity, which increased after Cis-P by 5-fold in HuH-7 cells, p<0.05, t-test, and by only 1.3-fold in HK-2 cells, p=n.s., t-test (Fig. 3C). After Cis-P, we observed comets in 74.3±3.5% HuH-7 cells with relative comet tail-length of 9.04±1.35, whereas in HK-2 cells comets were noted less frequently after Cis-P, although this difference was not statistically significant, but might have suggested possible cell losses not involving DDR and double-strand DNA breaks (Fig. 3D, 3E).
Figure 2. Differences in expression of DNA damage-associated genes in Cis-P-treated HuH-7 and HK-2 cells.
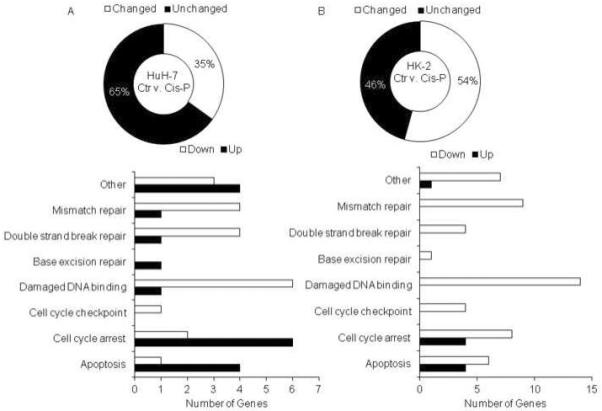
A. Overview showed significant divergence in ≥2 fold up- or down-regulated genes in HuH-7 cells versus HK-2 cells, p<0.05. B. Representation of DNA damage genes in various ontological classes indicated fewer DDR genes were upregulated in Cis-P-treated HK-2 cells compared to Cis-P-treated HuH-7 cells.
Table 1.
Gene expression in cisplatin-treated HuH-7 and HK-2 cells versus drug-untreated controls
| Gene Symbol | Gene Name | HuH-7 (Fold Up or Down) | HK-2 (Fold Up or Down) | Fold difference in HuH-7 and HK-2 cells |
|---|---|---|---|---|
| ABL1 | C-abl oncogene 1, receptor tyrosine kinase | −1.0 | −2.7 | 0.4 |
| ANKRD17 | Ankyrin repeat domain 17 | 1.8 | −5.3 | −0.3 |
| APEX1 | APEX nuclease (multifunctional DNA repair enzyme) 1 | 1.1 | −1.4 | −0.8 |
| ATM | Ataxia telangiectasia mutated | −2.9 | −5.5 | 0.5 |
| ATR | Ataxia telangiectasia and Rad3 related | −1.0 | −2.0 | 0.5 |
| ATRX | Alpha thalassemia/mental retardation syndrome X-linked | −7.3 | −24.0 | 0.3 |
| BRCA1 | Breast cancer 1, early onset | −1.5 | −5.4 | 0.3 |
| CCNH | Cyclin H | 1.4 | −1.1 | −1.3 |
| CDK7 | Cyclin-dependent kinase 7 | 2.2 | −5.2 | −0.4 |
| CHEK1 | CHK1 checkpoint homolog | 1.3 | −2.6 | −0.5 |
| CHEK2 | CHK2 checkpoint homolog | −1.7 | −5.9 | 0.3 |
| CIB1 | Calcium and integrin binding 1 | 1.8 | −1.0 | −1.8 |
| CRY1 | Cryptochrome 1 | 1.3 | −4.3 | −0.3 |
| DDB1 | Damage-specific DNA binding protein 1 | 1.1 | −1.6 | −0.7 |
| DDIT3 | DNA-damage-inducible transcript 3 | 3.6 | −1.8 | −2.0 |
| ERCC1 | Excision repair cross-complementing rodent repair deficiency, complementation group 1 | 1.3 | −1.5 | −0.9 |
| EXO1 | Exonuclease 1 | 1.9 | −1.5 | −1.3 |
| FANCG | Fanconi anemia, complementation group G | 2.0 | −1.1 | −1.8 |
| FEN1 | Flap structure-specific endonuclease 1 | 2.4 | −1.5 | −1.6 |
| GADD45A | Growth arrest and DNA-damage-inducible, alpha | 24.0 | 3.4 | 7.0 |
| GTF2H1 | General transcription factor IIH, polypeptide 1 | 1.6 | −3.4 | −0.5 |
| GTF2H2 | General transcription factor IIH, polypeptide 2 | 1.1 | −2.7 | −0.4 |
| GTSE1 | G-2 and S-phase expressed 1 | 1.0 | −2.4 | −0.4 |
| HUS1 | HUS1 checkpoint homolog | −1.2 | −5.6 | 0.2 |
| IGHMBP2 | Immunoglobulin mu binding protein 2 | 2.4 | −1.2 | −2.0 |
| IP6K3 | Inositol hexakisphosphate kinase 3 | −1.3 | −2.5 | 0.5 |
| XRCC6BP1 | XRCC6 binding protein 1 | 1.6 | −1.6 | −1.0 |
| MAPK12 | Mitogen-activated protein kinase 12 | 2.9 | 1.0 | 2.8 |
| MBD4 | Methyl-CpG binding domain protein 4 | 3.6 | −1.1 | −3.5 |
| MLH3 | MutL homolog 3 | 1.5 | −1.7 | −0.9 |
| MNAT1 | Menage a trois homolog 1, cyclin H assembly factor | −5.9 | −1.8 | 3.3 |
| MPG | N-methylpurine-DNA glycosylase | 1.7 | −1.3 | −1.3 |
| MSH3 | MutS homolog 3 | −8.1 | −3.1 | 2.7 |
| NBN | Nibrin | −2.1 | −9.2 | 0.2 |
| PCNA | Proliferating cell nuclear antigen | 2.2 | −1.1 | −1.9 |
| PMS1 | PMS1 postmeiotic segregation increased 1 | −1.5 | −4.0 | 0.4 |
| PMS2 | PMS2 postmeiotic segregation increased 2 | 1.8 | −2.4 | −0.8 |
| PMS2L3 | Postmeiotic segregation increased 2-like 3 | 1.5 | −2.8 | −0.5 |
| PNKP | Polynucleotide kinase 3'-phosphatase | 1.1 | −1.1 | −1.0 |
| PPP1R15A | Protein phosphatase 1, regulatory (inhibitor) subunit 15A | 11.0 | 2.5 | 4.3 |
| PRKDC | Protein kinase, DNA-activated, catalytic polypeptide | −4.7 | −1.7 | 2.7 |
| RAD17 | RAD17 homolog | 1.3 | −4.5 | −0.3 |
| RAD18 | RAD18 homolog | 1.0 | −1.7 | −0.6 |
| RAD21 | RAD21 homolog | 1.2 | −2.4 | −0.5 |
| RAD51 | RAD51 homolog | −1.2 | −3.1 | 0.4 |
| RAD51L1 | RAD51-like 1 | −6.0 | −2.9 | 2.0 |
| RAD9A | RAD9 homolog A | 2.0 | −1.0 | −2.0 |
| REV1 | REV1 homolog | −1.4 | −6.0 | 0.2 |
| SESN1 | Sestrin 1 | −1.5 | −21.2 | 0.1 |
| SUMO1 | SMT3 suppressor of mif two 3 homolog 1 | 1.3 | −1.3 | −1.0 |
| TP53 | Tumor protein p53 | 1.9 | −2.2 | −0.9 |
| TP73 | Tumor protein p73 | −3.3 | 1.7 | −1.9 |
| TREX1 | Three prime repair exonuclease 1 | 5.7 | 1.5 | 3.9 |
| XPA | Xeroderma pigmentosum, complementation group A | −1.1 | −3.7 | 0.3 |
| XPC | Xeroderma pigmentosum, complementation group C | 1.5 | −1.5 | −1.0 |
| XRCC1 | X-ray repair complementing defective repair 1 | 1.1 | −1.7 | −0.6 |
| XRCC2 | X-ray repair complementing defective repair 2 | −1.5 | −11.0 | 0.1 |
| XRCC3 | X-ray repair complementing defective repair 3 | 1.7 | −2.5 | −0.7 |
| B2M | Beta-2-microglobulin | 4.1 | 1.2 | 3.4 |
| HPRT1 | Hypoxanthine phosphoribosyltransferase 1 | −4.3 | −1.2 | 3.8 |
Figure 3. Evidences for ATM-related DDR in Cis-P-treated HuH-7 and HK-2 cells.
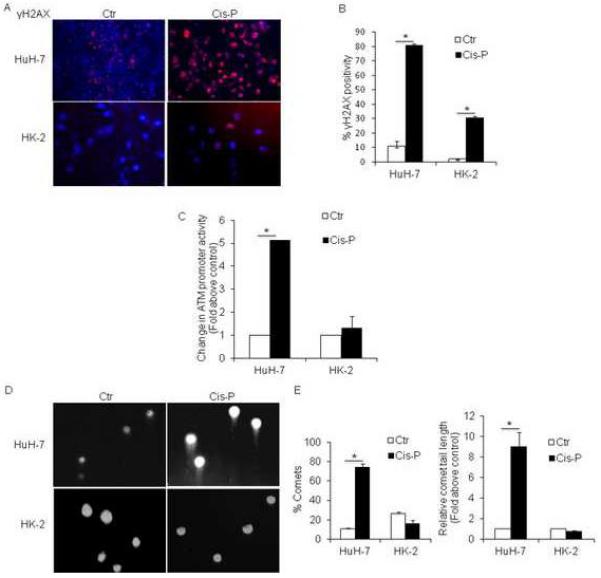
A. HuH-7 and HK-2 cells with or without Cis-P showed extensive γH2AX expression by immunostaining in the former, whereas γH2AX was expressed less frequently in the latter, as shown by quantitative analysis (B), 70–80% versus 20–40%, p<0.05 (asterisks). C. Analysis of ATM promoter activity with transgene reporter assay using tdT/Hoechst fluorescence with significant increase in Cis-P-treated HuH-7 cells but not in HK-2 cells, p<0.05 (asterisk). D. Photomicrographs showing formation of Comets in Cis-P-treated HuH-7 cells and not in HK-2 cells. E. Quantitation of Comets and Comet tail lengths in Cis-P-treated HuH-7 and HK-2 cells indicated this parameter of double-stranded DNA breaks was also significantly different in these cell lines, p<0.05 (asterisks).
3.2. Effects on cell cycling
Cell cycle profiles showed G2/M decreased and G0/G1 increased but S was unaffected in Cis-P-treated HuH-7 cells (Fig. 4A). After Cis-P in HK-2 cells, G2/M decreased and G0/G1 increased similarly, p<0.05, Chi-square. However, S also decreased in Cis-P-treated HK-2 cells, suggesting replicative stress in these cells after Cis-P, which involves mechanisms additional to DDR. Nonetheless, approximately 12% of HK-2 cells were in S and, therefore, these were still cycling. Next, we examined susceptibility of nonreplicative cell subpopulations to Cis-P, e.g., CSC, which commonly included SP cells, as defined by MDR transporter expression and efflux of Hoechst dyes. We found HuH-7 cells and HK-2 cells contained similar fractions of SP with verapamil-sensitive Hoechst 33342 dye efflux, 1.1% and 1.8%, respectively, p=n.s., Chi-square (Fig. 4B). After Cis-P, SP fractions declined in both HuH-7 and HK-2 cells, to 0.3% and 0.6% of total. Therefore, Cis-P toxicity extended to noncycling CSC and was likely mediated by mechanisms besides replicative stress.
Figure 4. Cell cycle and side population analyses.
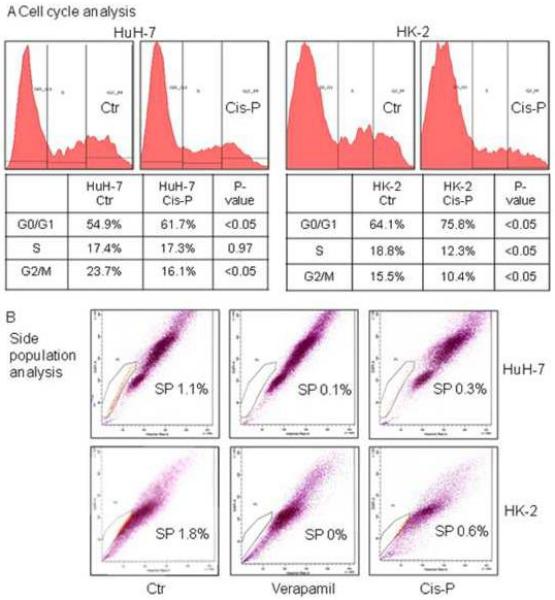
A. Shows cell cycle profiles in HuH-7 and HK-2 cells with or without Cis-P. The fractions in G0/G1, S or G2/M are given and indicated interference with cycling in both cell lines although the fraction in S was relatively more depleted in HK-2 cells. B. Both HuH-7 and HK-2 cells contained SP cell population as indicated by loss of these cells after treatment with verapamil, which characterizes Hoechst dye efflux in this cell population. After Cis-P, SP cells declined in HuH-7 as well as HK-2 cells, which indicated cytotoxicity in this relatively slower or noncycling cell population.
3.3. Drug regulation of p53 signaling
As p53 is a major mediator of ATM kinase-regulated DDR, and after DNA damage, may activate cell cycle restriction or apoptosis (21), we determined whether cell growth alterations involved this mechanism. To dissect limbs of p53 signaling, we used well-characterized small molecule drugs (22–24). Control of p53 expression through upstream transcriptional regulator, MDM2, is of major interest for cancer therapies, including RCC, when p53 is not mutated and retains normal function (24). The small molecule, nutlin-3, was developed as an MDM2 antagonist and exerts differential cytotoxic effects in cells depending upon presence or absence of wild-type p53 (23,24). When HuH-7 and HK-2 cells were cultured with nutlin-3, we observed trends for cytostatic effects with MTT assays but these differences did not reach statistical significance (Fig. 5A). Moreover, after nutlin-3 was combined with Cis-P, we did not observe further cytotoxicity. Similarly, small molecule p53 activators, such as tenovin-1, may decrease cell cycling by transcriptional upregulation of wild-type p53 expression without such effects on mutant p53 in cultured cells (22). We found tenovin-1 alone in the highest dose of 10 μM exerted some cytostatic effects in HuH-7 and HK-2 cells but these differences from corresponding untreated control cells did not reach statistical significance (Fig. 5B). Although mRNA expression analysis had shown 1.9-fold greater p53 expression in Cis-P-treated HuH-7 cells with significant decreases in cell viability compared with untreated controls consistent with appropriate inhibitory cell cycling effects of p53, treatment with tenovin-1 plus Cis-P did not further affect HuH-7 cell viability. Similarly, we did not observe additional cytotoxicity after treatment with Cis-P plus tenovin-1 in HK-2 cells, which showed 2.2-fold lower p53 mRNA expression after Cis-P. These results indicated that although HuH-7 and HK-2 cells were somewhat responsive to tenovin-1-regulatable cell cycle controls, other downstream mechanisms were likely involved in cell survival since after Cis-P p53 increased in the former but decreased in the latter cell type, and tenovin-1 did not synergistically amplify Cis-P cytotoxicity in either case.
Figure 5. Effects on Cis-P cytotoxicity with regulation of p53 activity by nutlin-3 or tenovin-1.
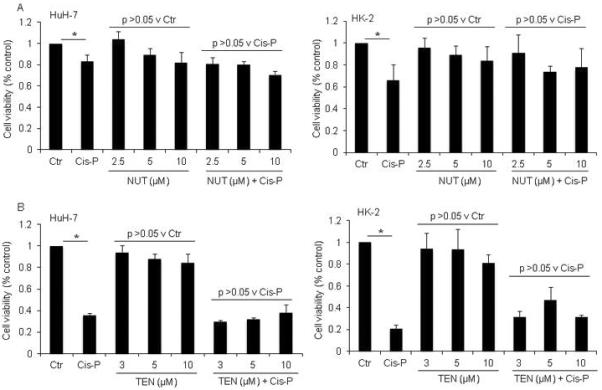
A. MTT cell viability assays in HuH-7 or HK-2 cells with limited dose-dependent cytostatic effects of nutlin-3, which was consistent with MDM2 antagonism-based activation of p53, as well as of tenovin-1, which indicated possible further transcriptional regulation of p53 activity, although these differences did not reach statistical significance by ANOVA. However, neither nutlin-3 nor tenovin-1 had synergistic effects upon Cis-P cytotoxicity. Asterisks indicate p<0.05.
Next, we focused on downstream p53 signaling by studying ATO, which inhibits wild-type p53-induced phosphatase (WIP)-1 activity, including in DDR (22). In cell culture assays with ATO alone, we observed cytostatic effects, although cytostatic effects required less ATO in HK-2 cells versus HuH-7 cells, 5 μM versus 10 μM (Fig. 6A). We noted improvements in cell viability after culture with 2.5 μM to 5 μM ATO – this presumably represented a paradoxical effect of ATO that may on occasion be observed with various regulators of cell growth controls. However, in greater amounts, ATO showed additional toxicity with Cis-P in HK-2 cells but not in HuH-7 cells. HK-2 cells showed less WIP1 protein after ATO with or without Cis-P (Fig. 6B). Also, ATO decreased β-tubulin content, which was in agreement with its well-known effects on tubulin depolymerization (25). To determine whether these effects of ATO included SP fractions, we profiled ATO-treated HuH-7 and HK-2 cells. The SP cells in HK-2 cells decreased after ATO alone and were undetectable after Cis-P plus ATO versus HuH-7 cells, p<0.05 (Fig. 6C). This was in agreement with synergistic amplification in HK-2 cells of Cis-P toxicity after WIP1 inhibition downstream of p53 gene.
Figure 6. Regulation in HuH-7 and HK-2 cells of WIP1 by ATO and effects on Cis-P cytotoxicity.
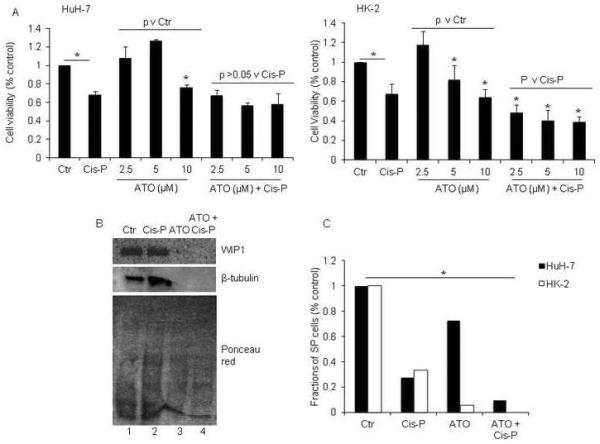
A. MTT cell viability assays in HuH-7 and HK-2 cells showed ATO was cytotoxic in both cell lines in 10 μM or 5 μM and 10 μM concentrations, respectively, p<0.05, asterisks, Chi-square tests. Moreover, ATO increased Cis-P cytotoxicity in HK-2 cells, asterisks, p<0.05, whereas that was not the case in HuH-7 cells. B. Western blot showing inhibition of WIP1 compared with control cells (top panel, lanes 1, 2) in ATO-treated HK-2 cells either with or without Cis-P (top panel, lanes 3, 4). Also, reprobing of the blot showed ATO decreased β-tubulin in these cells (middle panel, lanes 3, 4). Ponceau red staining after completion of westerns confirmed proteins were present in all samples (bottom panel). C. Analysis of SP cells after ATO and Cis-P in HuH-7 and HK-2 cells indicated these were depleted in both cases, asterisk, p<0.05. In HK-2 cells, SP fractions were no longer detectable.
4. DISCUSSION
These studies established differences in DDR following Cis-P toxicity in HK-2 and HuH-7 cells with or without HPV genes, respectively. Although equivalent cytotoxicity required more Cis-P in HK-2 cells relative to HuH-7 cells, this by itself was not surprising, because cell type-specific mechanisms, such as those determining drug uptake and removal, could explain such differences. For instance, as Cis-P shares copper-related import and export mechanisms in cells (26), in case of HuH-7 cells, Cis-P should have been excreted by the apical metal transporter, ATP7B, without contributions from the basolateral metal transporter, ATP7A, since the latter is not expressed in hepatocytes (27). By contrast, renal tubular cells express ATP7B as well as ATP7A (28), and both of these should have contributed in transporting Cis-P from HK-2 cells. Nonetheless, despite substantial IC50-dependent Cis-P toxicity, expression patterns of DDR genes in case of HK-2 cells were different, including for p53 expression. The extent of γH2AX expression, ATM promoter activity, and DNA strand breaks, as elicited by Comet assays confirmed that in HK-2 cells, Cis-P-induced DDR was less robust. Previously, in studies of oxidative DNA damage induced by ochratoxin A in HK-2 cells (16), DNA strand breaks were also limited, which was in agreement with our findings. Remarkably, signaling pathways both upstream and downstream of p53, which is a major transducer of cell survival and cell death decisions in the ATM network, no longer were active in imparting Cis-P toxicity in HuH-7 or HK-2 cells. Taken together, we concluded that HK-2 cells had acquired autonomy from p53-mediated controls that are typically present after DDR in normal cells. On the other hand, our results showing that Cis-P toxicity may be synergistically enhanced by ATO offers encouragement that some of the existing drugs may well be useful for people with RCC.
Presence in HK-2 cells of E6/E7 HPV 16 serotype genes (15) was significant because HPV and other DNA viruses may alter or even subvert DDR in cells for their own benefits (11,12). The interwoven relationship between this virus and ATM network is exemplified by the dependency of HPV replication in differentiated cells on ATM and CHK2 (11). High-risk HPV serotypes, e.g., 16 or 18, contribute multiple viral proteins, including E1, E2, E6 and E7, with ability to interfere with DDR involving ATM, ATR or DNA-dependent protein kinase (11,12). The E1 and E2 proteins of HPV 16 may colocalize at sites of viral DNA replication with phosphorylated H2AX, ATM and p53, including selective decreases in gene expression (29), as was the case here for γH2AX in HK-2 cells. Also, besides multiple alterations during HPV-induced cell transformation, it should be noteworthy that after DNA damage HPV E7 protein may help override G2/M checkpoint for continued cell proliferation (30). Similarly, HPV E6 and E7 proteins may facilitate proteasome-dependent p53 and RB degradation, which may facilitate bypassing of G1 checkpoint and entry into S (31), thereby further dysregulation of DDR after chemotherapy. Therefore, HK-2 cell transformation by HPV E6/E7 was likely responsible for incomplete ATM network-related DDR and autonomy from p53-mediated cell growth control.
This possibility regarding p53 was borne out by our studies with the MDM2 antagonist, nutlin-3 (22). MDM2 negatively controls p53 transcription and is a major drug target for cancers. Similarly, tenovin-1 is thought to increase p53 transcription, and thereby decreases cell proliferation (23). Our studies with nutlin-3 and tenovin-1 showed these did not substantially affect HK-2 cell proliferation and neither was synergistic for Cis-P-induced cytotoxicity. Therefore, the conclusion should be reasonable that in HK-2 cells p53 was no longer under control of MDM2 or regulatory elements amenable to drug classes represented by nutlin-3 and tenovin-1. Such was not so for nutlin-3 when p53 was regulated in a wild-type manner in RCC (24).
Testing of the role of a downstream p53 mediator, WIP1, established that HK-2 cells did respond to cell growth control when WIP1 was downregulated by ATO, although β-tubulin content was additionally altered. This should be of considerable interest because WIP1 may normally have roles in restoring cells to a healthy state after DNA damage and completion of DNA repair, including through dephosphorylation and possibly inactivation of major ATM signaling transducers, i.e., CHK1 and p53, as well as ATM kinase itself (32). On the other hand, this sequence of events will naturally be advantageous for replication or survival of HPV and other such viruses. Therefore, the synergistic effects of ATO in Cis-P toxicity via WIP1 inhibition will be in agreement with the otherwise restorative effects of the latter. The combination of Cis-P and ATO also depleted CSC, thereby offering yet other mechanisms for targeting RCC. The SP population in RCC may often be in G0/G1 although this may need more characterization (18,19).
As ATO has been widely given to people for acute promyelocytic leukemia based on its effects upon cellular targets, such as retinoic acid receptor-α, mitogen-activated protein kinases, and p53 (33), its combination with Cis-P for RCC should be feasible. Based on our results, the effects of ATO on β-tubulin may suggest that drugs affecting microtubules, e.g., paclitaxel, etc. (34), could also be useful. We did not study potential benefits of combined therapy with Cis-P and ATO in animal models of human RCC. These types of animal studies could offer more information although the biology of various renal tumor cell lines might be substantially different from primary renal tumors in people, particularly those with HPV in vivo. Since such questions may arise about the availability of effective animal models for HPV-associated RCC, whether clinical studies could be developed based on existing experience with ATO and other drugs in people, may need consideration for expedited trials. Therefore, our findings offer further therapeutic possibilities in HPV-associated RCC. On the other hand, whether certain drug combinations might produce DNA damage in healthy renal cells to increase risk for nephrotoxicity will simultaneously need to be kept in mind, and should require more study.
Highlights.
Presence of human papillomavirus DNA sequences in cancers may alter DNA damage responses
This study examined the integrity of ATM gene network-related DNA damage response after exposure to cisplatinum of cells with or without human papillomavirus genes
In renal tubular cells with human papillomavirus genes DNA damage responses related to ATM gene network were muted
Cytotoxicity of cisplatinum was synergistically amplified by arsenic trioxide via signaling downstream of p53 and tubulin depolymerization to provide further avenues for treating renal cell carcinoma
Acknowledgments
Sources of funding: Supported in part by NIH grants T32 DK007110, R01 DK071111, R01 DK 088561, P30 DK41296 and P30 CA013330
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
5. DISCLOSURE The authors declare no conflicts of interest exist.
6. REFERENCES
- 1.Wahlgren T, Harmenberg U, Sandström P, Lundstam S, Kowalski J, Jakobsson M, Sandin R, Ljungberg B. Treatment and overall survival in renal cell carcinoma: a Swedish population-based study (2000–2008) Br J Cancer. 2013;108:1541–9. doi: 10.1038/bjc.2013.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mattei J, da Silva RD, Sehrt D, Molina WR, Kim FJ. Targeted therapy in metastatic renal carcinoma. Cancer Lett. 2014;343:156–60. doi: 10.1016/j.canlet.2013.09.038. [DOI] [PubMed] [Google Scholar]
- 3.Tian K, Rajendran R, Doddananjaiah M, Krstic-Demonacos M, Schwartz JM. Dynamics of DNA damage induced pathways to cancer. PLoS One. 2013;8:e72303. doi: 10.1371/journal.pone.0072303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bandi S, Viswanathan P, Gupta S. Evaluation of cytotoxicity and DNA damage response with analysis of intracellular ATM signaling pathways. Assay Drug Dev Technol. 2014;12:272–81. doi: 10.1089/adt.2014.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang X, Nie Y, Li X, Wu G, Huang Q, Cao J, Du Y, Li J, Deng R, Huang D, Chen B, Li S, Wei B. Pathol Oncol Res. Feb 16, 2014. MicroRNA-181a functions as an oncomir in gastric cancer by targeting the tumour suppressor gene ATM. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 6.Jones AR, Bell-Horwath TR, Li G, Rollmann SM, Merino EJ. Novel oxidatively activated agents modify DNA and are enhanced by ercc1 silencing. Chem Res Toxicol. 2012;25:2542–52. doi: 10.1021/tx300337j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Selvarajah J, Nathawat K, Moumen A, Ashcroft M, Carroll VA. Chemotherapy-mediated p53-dependent DNA damage response in clear cell renal cell carcinoma: role of the mTORC1/2 and hypoxia-inducible factor pathways. Cell Death Dis. 2013;4:e865. doi: 10.1038/cddis.2013.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bachmann HS, Rübben H, Schmid KW, Siffert W, Riemann K. DNA repair gene XRCC1 polymorphisms and outcome of renal cell carcinoma in Caucasian patients. Anticancer Res. 2009;29:5131–5. [PubMed] [Google Scholar]
- 9.Willenbring H, Sharma AD, Vogel A, Lee AY, Rothfuss A, Wang Z, Finegold M, Grompe M. Loss of p21 permits carcinogenesis from chronically damaged liver and kidney epithelial cells despite unchecked apoptosis. Cancer Cell. 2008;14:59–67. doi: 10.1016/j.ccr.2008.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weiss RH, Borowsky AD, Seligson D, Lin PY, Dillard-Telm L, Belldegrun AS, Figlin RA, Pantuck AD. p21 is a prognostic marker for renal cell carcinoma: implications for novel therapeutic approaches. J Urol. 2007;177:63–8. doi: 10.1016/j.juro.2006.08.073. [DOI] [PubMed] [Google Scholar]
- 11.Moody CA, Laimins LA. Human papillomaviruses activate the ATM DNA damage pathway for viral genome amplification upon differentiation. PLoS Pathog. 2009;5:e1000605. doi: 10.1371/journal.ppat.1000605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Turnell AS, Grand RJ. DNA viruses and the cellular DNA-damage response. J Gen Virol. 2012;93:2076–2097. doi: 10.1099/vir.0.044412-0. [DOI] [PubMed] [Google Scholar]
- 13.Salehipoor M, Khezri A, Behzad-Behbahani A, Geramizadeh B, Rahsaz M, Aghdaei M, Afrasiabi MA. Role of viruses in renal cell carcinoma. Saudi J Kidney Dis Transpl. 2012;23:53–7. [PubMed] [Google Scholar]
- 14.Farhadi A, Behzad-Behbahani A, Geramizadeh B, Sekawi Z, Rahsaz M, Sharifzadeh S. High-risk human papillomavirus infection in different histological subtypes of renal cell carcinoma. J Med Virol. 2014;86:1134–44. doi: 10.1002/jmv.23945. [DOI] [PubMed] [Google Scholar]
- 15.Ryan MJ, Johnson G, Kiiu J, Fuerstenberg SM, Zager RA, Torok-Storb B. HK-2: An immortalized proximal tubule epithelial cell line from normal adult human kidney. Kidney Internat. 1994;45:48–57. doi: 10.1038/ki.1994.6. [DOI] [PubMed] [Google Scholar]
- 16.Arbillaga L, Azqueta A, Ezpeleta O, Lo'pez de Cerain A. Oxidative DNA damage induced by ochratoxin A in the HK-2 human kidney cell line: evidence of the relationship with cytotoxicity. Mutagenesis. 2007;22:35–42. doi: 10.1093/mutage/gel049. [DOI] [PubMed] [Google Scholar]
- 17.Nakabayashi H, Taketa K, Miyano K, Yamane T, Sato J. Growth of human hepatoma cells lines with differentiated functions in chemically defined medium. Cancer Res. 1982;42:3858–63. [PubMed] [Google Scholar]
- 18.Addla SK, Brown MD, Hart CA, Ramani VA, Clarke NW. Characterization of the Hoechst 33342 side population from normal and malignant human renal epithelial cells. Am J Physiol Renal Physiol. 2008;295:F680–7. doi: 10.1152/ajprenal.90286.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang B, Huang YJ, Yao ZJ, Chen X, Guo SJ, Mao XP, Wang DH, Chen JX, Qiu SP. Cancer stem cell-like side population cells in clear cell renal cell carcinoma cell line 769P. PLoS One. 2013;8:e68293. doi: 10.1371/journal.pone.0068293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gagandeep S, Novikoff PM, Ott M, Gupta S. Paclitaxel shows cytotoxic activity in human hepatocellular carcinoma cell lines. Cancer Lett. 1999;136:109–18. doi: 10.1016/s0304-3835(98)00388-7. [DOI] [PubMed] [Google Scholar]
- 21.Imreh G, Norberg HV, Imreh S, Zhivotovsky B. Chromosomal breaks during mitotic catastrophe trigger γH2AX-ATM-p53-mediated apoptosis. J Cell Sci. 2011;124:2951–63. doi: 10.1242/jcs.081612. [DOI] [PubMed] [Google Scholar]
- 22.Wang L, Mosel AJ, Oakley GG, Peng A. Deficient DNA damage signaling leads to chemoresistance to cisplatin in oral cancer. Mol Cancer Ther. 2012;11:2401–9. doi: 10.1158/1535-7163.MCT-12-0448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.van Leeuwen IMM, Rao B, Sachweh MCC, Laín S. An evaluation of small-molecule p53 activators as chemoprotectants ameliorating adverse effects of anticancer drugs in normal cells. Cell Cycle. 2012;11:1851–1861. doi: 10.4161/cc.20254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Polański R, Noon AP, Blaydes J, Philips A, Rubbi CP, Parsons K, Vlatković N, Boyd MT. Senescence induction in renal carcinoma cells by Nutlin-3: a potential therapeutic strategy based on MDM2 antagonism. Cancer Lett. 2014;S0304–3835(14):00371–1. doi: 10.1016/j.canlet.2014.07.024. [DOI] [PubMed] [Google Scholar]
- 25.Ling YH, Jiang JD, Holland JF, Perez-Soler R. Arsenic trioxide produces polymerization of microtubules and mitotic arrest before apoptosis in human tumor cell lines. Mol Pharmacol. 2002;62:529–38. doi: 10.1124/mol.62.3.529. [DOI] [PubMed] [Google Scholar]
- 26.Safaei R. Role of copper transporters in the uptake and efflux of platinum containing drugs. Cancer Lett. 2006;234:34–9. doi: 10.1016/j.canlet.2005.07.046. [DOI] [PubMed] [Google Scholar]
- 27.Lutsenko S, Barnes NL, Bartee MY, Dmitriev OY. Function and regulation of human copper-transporting ATPases. Physiol Rev. 2007;87:1011–46. doi: 10.1152/physrev.00004.2006. [DOI] [PubMed] [Google Scholar]
- 28.Moore SD, Cox DW. Expression in mouse kidney of membrane copper transporters Atp7a and Atp7b. Nephron. 2002;92:629–34. doi: 10.1159/000064075. [DOI] [PubMed] [Google Scholar]
- 29.Sakakibara N, Mitra R, McBride AA. The papillomavirus E1 helicase activates a cellular DNA damage response in viral replication foci. J Virol. 2011;85:8981–8995. doi: 10.1128/JVI.00541-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Spardy N, Covella K, Cha E, Hoskins EE, Wells SI, Duensing A, Duensing S. Human papillomavirus 16 E7 oncoprotein attenuates DNA damage checkpoint control by increasing the proteolytic turnover of claspin. Cancer Res. 2009;69:7022–7029. doi: 10.1158/0008-5472.CAN-09-0925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moody CA, Laimins LA. Human papillomavirus oncoproteins: pathways to transformation. Nat Rev Cancer. 2010;10:550–560. doi: 10.1038/nrc2886. [DOI] [PubMed] [Google Scholar]
- 32.Lu X, Nannenga B, Donehower LA. PPM1D dephosphorylates Chk1 and p53 and abrogates cell cycle checkpoints. Genes Dev. 2005;19:1162–74. doi: 10.1101/gad.1291305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dos Santos GA, Kats L, Pandolfi PP. Synergy against PML-RARa: targeting transcription, proteolysis, differentiation, and self-renewal inacute promyelocytic leukemia. J Exp Med. 2013;210:2793–802. doi: 10.1084/jem.20131121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Duan XF, Wu YL, Xu HZ, Zhao M, Zhuang HY, Wang XD, Yan H, Chen GQ. Synergistic mitosis-arresting effects of arsenic trioxide and paclitaxel on human malignant lymphocytes. Chem Biol Interact. 2010;183:222–30. doi: 10.1016/j.cbi.2009.09.012. [DOI] [PubMed] [Google Scholar]