

Abstract
CD4+CD25+/highFoxp3+ regulatory T cells (Tregs) are a subset of CD4+ T cells that play an essential role in maintaining peripheral immune tolerance. Several transcriptional co-factors have been recently identified, which form complexes with Tregs transcription factor Foxp3 and contribute in the suppressive function of Tregs. However, Foxp3 is still defined as a “master” (multiple pathway) regulator gene that controls the development and stability of Tregs. Due to its importance, the regulatory mechanisms underlying Foxp3 expression have been a focus of intensive investigation. Recent progress suggests that the epigenetics mechanisms responsible for regulating the Foxp3 gene expression are key components of Tregs suppressive activity. This review not only discusses the basic concepts of Tregs biology and epigenetic modifications. We also analyze the translational clinical aspect of Tregs epigenetic modifications, focusing on several ongoing clinical trials as well as FDA approved epigenetic based drugs. The new progress in identifying epigenetic enzymes functional in Treg cells is a new target for the development of novel therapeutic approaches for autoimmune and inflammatory diseases, graft-versus-host disease and cancers.
Keywords: regulatory T cells, immune suppression, epigenetic mechanisms, histone modifications, metabolic cardiovascular diseases
Introduction
CD4+CD25+/highFoxp3+ regulatory T cells are a subpopulation of CD4+ T cells specialized in the suppression of pathogenic responses from the host immune system against self or foreign antigens [1]. The suppressive function of Tregs in maintenance of self-tolerance and prevention of the development of autoimmune and chronic inflammatory diseases is mediated by different mechanisms such as cell-cell contact and/or secretion of anti-inflammatory cytokines such as like interleukin-10 (IL-10), IL-35 and transforming growth factor β (TGF-β) [2, 3].
One of the major milestones found in Tregs studies was the identification of Foxp3. Foxp3 is a member of the forkhead/winged-helix family of transcription factors, which acts as a “master” (multiple pathway) regulator gene for the development and suppressive function of Tregs [4-6]. The Foxp3 gene was identified by its significant mutations that cause fatal autoimmune diseases in early life, which is now termed Immunodysregulation, Polyendocrinopathy, Enteropathy, and X-linked (IPEX) syndrome in mice and humans. Since the discovery of the Foxp3 gene, its role and modification have been one of the potential topics in translational medicine field due to the essential function of Foxp3 in maintaining immune tolerance and homeostasis.
In inflammatory environments, the suppressive function of Tregs is perturbed and TGF-β-induced Tregs development is reduced by an epigenetic manner [7], suggesting that epigenetics regulation of Tregs function and development is pathophysiologically relevant. In correlation with this finding, Tregs suppression is also found to be disturbed in autoimmune type 1 diabetes, in which epigenetics is one of the pathological mechanisms involved [8]. Epigenetics is defined by heritable changes that occur in gene expression without modification in the DNA sequence of the genome. These epigenetic mechanisms, which include DNA methylation/demethylation, histones modifications and micro-RNAs (miRNA's) are the principal mechanisms involved in regulating chromosomal organization and gene expression via different and dynamic levels. More specifically, it has been demonstrated that epigenetic mechanisms play a significant role in regulating the expression of the Foxp3 gene and are leading to further regulations in Tregs functions [9-11]. Emerging epigenetic therapies are providing new therapeutic agents for the control of various diseases [12]. In this review we are focusing on understanding the mechanisms of epigenetic modifications in the Foxp3 gene in the development of autoimmune and inflammatory diseases, graft-versus-host disease (GVHD), cancer and therapeutic modalities in order to continue our long-term interest in identifying novel Tregs therapy-related targets [6, 13-19]. In addition, we will also analyze the progress in identifying epigenetic enzymes as potential therapeutic targets for novel Tregs-based therapy.
Regulatory T Cells
Originally termed suppressor T cells, the recognition of regulatory T cells as a cellular mechanism for immune tolerance resulted from experiments performed in the 1960s and 1970s by Gerson and Kondo, which described the induction of suppressor T cells capable of down-regulation of antigen-specific T-cell responses [20]. Due to the lack of known molecular markers, research on suppressor T cells ceased. However, in 1995, Sagakuchi et al. identified CD25 as a surface phenotypic marker for suppressive CD4 cells in mice [21]. Since then, suppressive T cells have been called regulatory T cells (Tregs). Later, the discovery of Foxp3 as a specific transcription factor and marker of natural occurring Tregs (nTregs) and adaptive/induced Tregs (iTregs) provided a molecular anchor to the population of Tregs [22]. The identification of these molecular markers led to an increase in research interest in regulatory T cells during the last decade, which has identified Tregs as a plausible therapeutic choice for several autoimmune diseases such as inflammatory bowel disease, systemic lupus erythematosus (SLE), rheumatoid arthritis (RA), type 1 diabetes mellitus (T1DM), and many other diseases.
Originally, the high expression of CD4 and CD25 surface markers was used to identify Tregs. However, because CD25+ has been found in other non-Tregs T cells such as activated T cells, the measurement of the intracellular expression of Foxp3 transcription factor allowed for a more specific analysis of Tregs. Since Foxp3 is also expressed in effector T cells, negative expression of CD127 is often used as an additional marker [23] due to its inverse correlation with FoxP3 expression and suppressive function of human CD4+ Tregs. Although the functional significance of the expression of these markers remains to be defined, several additional markers have been described such as cytotoxic T-lymphocyte associated molecule-4 (CTLA-4), glucocorticoid-induced tumor necrosis factor (TNF) receptor (GITR), CD39, and CD45RA.
Currently, several experimental systems are commercially available that simplify the identification, isolation, and characterization of Treg cells using fluorescent-conjugated antibodies for CD4, CD25, Foxp3 and CD127. Moreover, the isolation of mRNAs for cDNA synthesis is used to analyze Foxp3 expression in Tregs using a quantitative real-time PCR [24]. Tregs are characterized for the secretion of immunosuppressive/anti-inflammatory cytokines such as IL-10, IL-35 and TGF-β. Enzyme–linked immunoabsorbent assays (ELISA) and Western blots have been used for the detection of Tregs, while cytokines have been measured using a cytokine secretion assay [25]. In addition, only in Treg cells a certain region within the Foxp3 gene (Treg-specific-demethylated region, TSDR [26] or conserved noncoding sequence 2, CNS2 [27]) is found demethylated that allows for the monitoring of Treg cells through PCR reaction or other DNA-based analysis methods [26].
Tregs have indispensable functions in regards to maintaining immune homeostasis. They are essential in mediating peripheral immune tolerance, preventing autoimmune diseases, and suppressing inflammatory responses. Immune tolerance is defined as a function of the immune system which maintains immunological unresponsiveness to self-antigens and suppresses an exaggerated autoimmune response, which could ultimately lead to autoimmune diseases and atherosclerosis [28]. There are two types of immune tolerance known as central tolerance and peripheral tolerance; central tolerance is the elimination of self-reactive T cells within the thymus through a thymocyte developmental process termed negative selection, and peripheral tolerance is the elimination of self-reactive T cells outside of the thymus such as immunosuppressive activity of Tregs [29] and T cell anergy [30].
There are two classes of CD4+CD25high Tregs cells (nTregs), which comprise 5 - 10% of murine and human CD4+ cells, and iTregs that are cellular components of peripheral immune tolerance. The nTregs are matured within the thymus and express the Foxp3 transcription factor. Experimental evidence indicates that nTregs exist without peripheral antigenic stimulation [1, 28, 31]. On the other hand, iTregs are generated in the periphery from CD4+CD25− T cell population and are induced in response to the stimulations of particular antigens and cytokines.
Naturally Occurring Treg Cells (nTregs)
NTregs are developed in the thymus and are characterized by the expression of CD4, CD25 high and transcriptional factor Foxp3 [32]. Initially identified by their co-expression of CD4 and CD25 cell surface markers, subsequent reports have used other cell surface markers such as CD103, CD62L, lymphocyte activation gene 3 protein (LAG 3), C-C chemokine receptor type 5 (CCR5), neurophilin-1[33-35], the activation antigens GITR, and CTLA-4 (also known as CD152), as well as the lack of certain cell surface markers such as CD127 (the α chain of the IL-7 receptor) to identify nTregs [36]. They recognize specific self-antigens and prevent autoimmunity by the inhibition of pathogenic lymphocytes. The role of nTregs in experimental atherosclerosis was initially reported in 2006 by Ait-Oufella et al., showing an increase in atherosclerotic lesion size and vulnerability in ApoE−/− mice after depletion of peripheral Tregs [37].
NTregs express the transcription factor Foxp3. Fully-matured Foxp3+ nTregs exit the thymus and migrate to the secondary lymphoid organs where they suppress the proliferation of tissue-specific autoimmune T cells and their differentiation into type 1 T helper cells (Th1), Th2, and Th17 lineages in vivo [38]. NTregs inhibit polyclonal T cell activation and the function of antigen-presenting cells including B cells, macrophages, and dendritic cells (DCs) [38].
Adaptive Treg Cells (iTregs)
Adaptive, or inducible, Tregs (iTregs) are induced in the periphery from CD4+CD25− T cell precursors, which acquire the upregulation of CD25 (interleukin-2 receptor α chain; IL-2Rα). The inducible Tregs are developed from naïve CD4+ T cells in the lymphoid tissues in response to specific antigens in the presence of cytokines TGF-β1, IL-10, and IL-4, while in the absence of pro-inflammatory cytokines such as interferon- γ (IFN-γ), IL-1, IL-6, and IL-12. This antigen presentation in the absence of danger signals is referred to as tolerogenic, which is essential for the suppression of undesired immune reactivity against non-harmful materials such as airborne particles, commensal bacteria, and foods. In addition, iTregs depend on IL-2 for development and survival as previously reported [13-15, 17], which also explains why iTregs highly express CD25 and probably other IL-2 receptor components for survival which require required IL-2 signaling. Furthermore, iTregs may be able to redirect macrophage differentiation toward an anti-inflammatory cytokine-producing type 2 macrophage phenotype (M2) rather than pro-inflammatory type 1 macrophages (M1 phenotype) [21].
Different subsets of iTregs have been reported including T regulatory cell type 1 (Tr1) and T helper cell type 3 (Th3) [39]. Tr1 cells are CD25−FOXP3− characterized by the secretion of large amounts of IL-10, some IL-5 and IFN-γ with or without TGF-β, IL-2 or IL-4[40]. Tr1 can control the activation of naïve and memory T cells in vivo and in vitro, and also suppress the Th1 and Th2 immune responses to pathogens, tumors and alloantigen-expressed transplanted tissues [41]. The capacity of DCs to induce T cell proliferation is strongly reduced by the supernatant of activated Tr1 [42], suggesting that Tr1 suppression is mediated by secreted cytokines. It has been shown that Th3 cells produce high amounts of TGF-β when induced by oral tolerance in mucosal tissue in an antigen-specific manner [38]. In addition, CD4+LAP+ (latency associated peptide) Tregs have been identified recently as the third iTreg subtype whose suppression is mediated by TGF-β in immune diseases including experimental autoimmune encephalitis (EAE), T1DM, SLE, collagen-induced arthritis, type II diabetes, and atherosclerosis in mice [38, 43].
Foxp3 Gene Structure
As we discussed previously [6], Foxp3, which acts as a “master” regulator gene for the development and suppressive function of Tregs [12], is an X-chromosome encoded member of the forkhead TF family that controls differentiation and function of Treg cells [44]. The Foxp3 gene was first identified by Brunkow et al. in 2001 as a defective gene in the mouse strain scurfy, an X chromosome-linked recessive lymphoproliferative disease. The Scurfy mutation is lethal in hemizygous males, which exhibits hyperactivation of CD4+ T cells and overproduction of pro-inflammatory cytokines within a month after birth [45]. In humans, as discussed in a previous section, mutation of the Foxp3 gene leads to the development of IPEX syndrome, in which multiple organ autoimmune diseases, such as diabetes mellitus, allergy, and inflammatory bowel disease, are present in the patients [22]. The expression of Foxp3 is induced during thymic differentiation or upon activation of peripheral CD4+ T cells in response to the T cell antigen receptor (TCR) stimulation in combination with several other cytokine signals including IL-2 and TGF-β. Furthermore, forced expression of Foxp3 confers suppressive function in Treg precursor cells, and Foxp3 ablation in mature Tregs results in loss of lineage identity and immunosuppressive function [10].
The Foxp3 genes possess 11 coding and 3 noncoding exons [12, 45]. The 2 extreme 5’-noncoding exons (−2a and −2b) are spliced to a second common non-coding exon (−1) and are separated by 640 base pairs (bp). The −2b and −1 exons are separated by approximately 5,000 bp and possess several regulatory cis-elements. It has been demonstrated that a 2-bp insertion mutation in the amino acid sequence encoded by the exon 8 leads to scurfy mice [12, 45]. Sequencing analysis of a large cohort of IPEX patients shows that 60% of patients have a missense mutation mainly in the forkhead domain (exons 9, 10 and 11), and other mutations are distributed throughout the gene [46, 47]. The Foxp3 protein sequences of humans (NIH-NCBI protein database ID: NP_054728) and mouse (NIH-NCBI protein database ID: NP_ 473380) have 86% identity and 91% similarity in their amino acid sequence. Reports from Western blot analysis have showed that there are two isoforms of Foxp3 in human cells [12]. The upper band is similar to the mouse Foxp3 band, and the lower band is unique to humans because it lacks the exon 2-encoded sequence (amino acids 71-105) that is the part of the repressor domain in the Foxp3 protein. The expression of the exon 2 in Foxp3 sequence in human CD4+CD25−Foxp3− T cells leads to an increase in IL-2 secretion and proliferation in response to T cell antigen receptor (TCR) stimulation compared with the full-length of Foxp3.
The constitutive expression of Foxp3 is essential for the suppressive function of Tregs. The nTregs and iTregs have different functional characteristics. Several studies suggest that nTregs are more stable than TGF-β-induced iTregs, which may correspond with epigenetic modifications to the Foxp3 gene. Several epigenetic modifications have been reported at the Foxp3 locus [48], such as histone acetylation, methylation, and cytosine residue methylation in CpG dinucleotides in Foxp3 DNA sequence which suggest epigenetic mechanisms are critical regulators for Treg differentiation, stability and suppressive functions.
In addition to the Foxp3 gene, recent reports indicate that Foxp3 is able to form complexes with a number of co-factors to execute cooperative effects during their interaction [49]. The largest group of Foxp3 co-factors is composed of as many as 11 sequence specific transcription factors including NFATc2, Runx1, Bcl11b, Foxp1, Foxp4, GATA-3, STAT3, Ikaros (Ikzf1), Aiolos (Ikzf3), Ets, and Cnot3. The majority of Foxp3 binding sites within the genome lack an identifiable forkhead-binding motif in Tregs, which suggests that a large number of Foxp3 co-factors facilitate the binding of Foxp3 to a given site [50-52]. These bindings could be possible either through direct recruitment of Foxp3-containing complexes or through facilitating interactions with Foxp3-bound sites containing the forkhead motif via loop formation. Moreover, the interaction of Foxp3 with other transcription factors is able to induce a common Tregs –type gene expression pattern that cannot be achieved just by Foxp3 [53].
Epigenetic Mechanisms
The term epigenetics was first introduced by Conrad Waddington in 1942, and was described as the causal interaction between genes and their products [54]. Previously introduced, epigenetics is defined as heritable changes in gene expression without changing the DNA sequence of the genome, which ultimately alters cell differentiation, phenotype and function. Epigenetics integrates organism genotypes by the influence and response of environmental stimuli on their phenotype. These gene modifications can take place in chromosomal DNA or in the proteins linked with the chromosomal DNA such as histones. In recent years, many epigenetic proteins have been experimentally and clinically investigated, while inhibitor development for modification enzymes has been the frontier for drug discovery. So far, epigenetic modifications have been grouped into four main categories: DNA methylation, histone modification, small and non-coding RNAs, as well as chromatin remodeling (Figure 1) [55, 56].
Figure 1. The schematic representation of our new working model about the role of epigenetics mechanisms in the Tregs biogenesis/suppressive function and the development of autoimmune and pro-inflammatory human diseases.
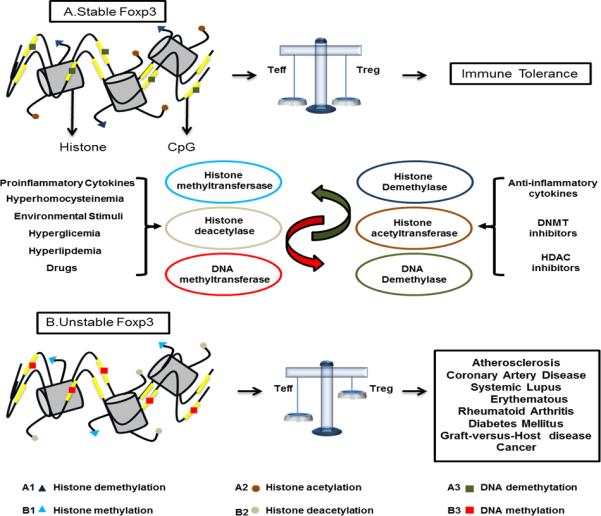
Several environmental factors influence the expression of Foxp3 gene via epigenetics mechanisms. The Foxp3 transcription factor determines the suppressive function and stability of Tregs. The epigenetics changes in modulating the Foxp3 expression are mediated by different mechanisms: 1. The histone acetylation/deacetylation of Foxp3 gene by histone acetyltransferase (HTA) and histone deacetylase (HDACs) complex. The increase in histone acetylation makes the Foxp3 gene more stable in enhancing the Tregs’ supressive function. 2. The DNA methylation/demethylation of Foxp3 gene by DNA methyltranferases (DNMTs) and DNA demethylases. The demethylation of CpG islands in the genomic DNA of Foxp3 gene increases the stability of Foxp3 gene expression and leads to the stabilized suppressive capacity of Tregs. 3. The histone demethylation/methylation of Foxp3 gene mediated by histone methyltransferases (HMT) and histone demethylases. Foxp3 histone demethylation increases the development and stable Tregs phenotype. The epigenetics modification by different stimuli like hyperlipidemia, hyperhomocysteinemia, hyperglycemia, inflammation-provoking drugs and pro-inflammatory cytokines, generates an unstable Foxp3 gene, generating an imbalance among conventional T cell (Tconv.) and Treg cells, promoting the development of autoimmune and inflammatory diseases, cancers and graft-versus-host disease (GVHD). Moreover, the anti-inflammatory drugs, DNMT inhibitors and histone deacetylase inhibitors (HDIs) have the capacity of increasing the stability of Foxp3 gene, increasing the suppressive capacity of Tregs, promoting an immune tolerance environment.
DNA Methylation
In mammals, DNA can be methylated on the fifth carbon (C5) in the cytosine base at CpG dinucleotides. The 5-methyl cytosine (Me5C) accounts for about 1% of the total DNA bases. It is estimated that the CpG dinucleotides with Me5C represent 70-80% of all the CpG dinucleotides in the genome [57]. The CpG dinucleotides are concentrated in dense “pockets” called CpG islands (CGIs). The methylation/demethylation process occurs in germ cells and pre-implantation embryos. The genome of mature sperms and eggs in mammals are highly methylated compared to somatic cells [58]. The CpGs in the CGI-containing promoter regions are demethylated during development as well as in normal tissues. When CpG dinucleotides are methylated, they often directly repress gene expression by blocking DNA recognition and the binding of transcription factors to their promoters and other distal regulatory elements. Consequently, methylated CpG dinucleotides suppress the recruitment of RNA polymerase II and indirectly associate with chromatin remodeling factors, such as methyl-DNA-binding proteins (MBDs), which results in the condensation of chromatin [59].
In the ENCODE project (Encyclopedia of DNA elements, www.encodeproject.org/Encode) an average of 1.2 million CpGs regions in each of 82 cell lines and tissues were assayed using reduced representation bisulphite sequencing (RRBS) in order to profile DNA methylation quantitatively. About 96% of CpGs exhibit differential methylation in at least one cell type or tissue. Rather than in promoters and upstream regulatory regions (terminal regions), the most variably methylated CpGs are found more often in gene bodies and intergenic regions. DNA methylation is mediated by the DNA methyltransferase enzymes (DNMTs) that catalyze the transfer of a methyl group from the methyl donor molecule S-adenosyl methionine (SAM) to a 5-position cytosine in certain CpG dinucleotides as we discussed previously [60]. Four DNA methyltransferases (Dnmt) enzymes have been identified including: Dnmt1, Dnmt3a, and Dnmt3b [61]. The Dnmt3a and Dnmt3b enzymes methylate germ cells and cells in early stages of development [62, 63]. The Dnmt1 enzyme binds preferentially to hemi-methylated DNA and re-establishes DNA methylation after DNA replication [61-63]. Furthermore, the process of DNA demethylation could be passive or active. The passive way can take place with no methylation during the synthesis of a new DNA strand. Active DNA demethylation can be achieved by the direct removal of a methyl group via a replication-independent process [64]. Ten-eleven translocation (TET) family enzymes can oxidize 5-methylcytosine and generate the 5-Hydroxymethylcytosine (5hmc), which is a pivotal nexus in demethylation. Consequently, 5hmc can either be passively depleted during DNA replication or actively undergo iterative oxidation and thymine DNA glycosylase (TDG)-mediated base excision repair to transform back to cytosine [65]. In contrast, demethylation of CpG motifs generates the relaxation of chromatin that is favorable for gene expression. Non-methylated CpG sequences recruit CXXC finger protein 1 (CFP1), which is associated with the histone 3 lysine 4 (H3K4) methyltransferase Setd1, and establish the methylated domains of H3K4me3. CFP1-Setd1 association thereby increases the accessibility of target sequences that promotes the binding of transcription factors [66]. Of note, the unmethylated CpG islands specifically bind a histone acetyltransferase P300, which is linked to an enhancer activity and further points out the inter-relationship between DNA demethylation and histone acetylation [67].
Histone Modification
Among the four groups of histones in the nucleosome, histone 3 (H3) and histone 4 (H4) are highly conserved and transmitted to progeny cells during the replication process. Histones are subject to a wide variety of posttranslational modifications including (1) lysine acetylation, (2) lysine and arginine methylation, (3) serine and threonine phosphorylation, (4) lysine ubiquitination, (5) lysine sumoylation and (6) ADP ribosylation. Each of these reactions has a specific function in transcriptional regulation, DNA repair, DNA replication, alternative splicing and chromosome condensation [55, 68, 69]. For example, histone acetylation/deacetylation has long been positively correlated with gene activation/repression, whereas histone lysine methylation has been associated with both gene activation and gene repression depending on the site and extent of methylation (Table 1) [70]. Histone modifications are proposed to affect chromatin remodeling through at least two distinct mechanisms. One proposed mechanism is the modification of histone protein tails’ electrostatic charge resulting in alteration of histone/DNA and/or nucleosome/nucleosome interactions [59]. The other mechanism suggests that these modifications can alter chromatin activity by creating binding sites for protein recognition molecules, such as proteins with bromodomains , chromodomains that recognize acetylated lysine or methylated lysine [71].
Table 1.
Histone modification and responsible enzymes
| Histone modification | Histone | “writer” | “eraser” | PUBMED ID Reference |
|---|---|---|---|---|
| Methylation | H3K4me1, H3K4me2 | SET 7 | LSD1 JARIDA-B | [19381457] |
| H3K36me3 | SET D2 | ? | [20920478] [20920475] | |
| H3K4 me2, H3K4me3 | MLL | LSD1 JARID1A-D | [19381457] [24011576] | |
| H3K79me2 | DOT1 | ? | [23754963] | |
| H3K9me3 | SUV39H1 | JMJD2A JHDM3A | [24011576], [2407543] [23739122] | |
| H4k20me4 | SUV420H2 | ? | [19381457] | |
| H3K27me2 H3K27me3 | Ezh2 | JMJD3 UTX | [24011576] [24141370] | |
| Acetylation | H4 (K5, K8, K12, K16) H3K14 | TIP60 | HDAC 7 HDAC9 | [19114310] [22124370] |
| H4K12 | P300 | HDAC1 HDAC2 | [14560007] | |
| H4k8 | CBP | HDAC1 HDAC2 | [14560007] | |
| H3k9, H3k14, H3k18 H3K56, H4K5, H4K8 | CBP / P300 | HDAC1 HDAC2 | [24075743] | |
| H3k9, H3K14, H3K18 | GCn5 PCAF | HDAC1 HDAC2 | [10365964] |
Histone methylation is catalyzed by a family of conserved proteins known as the histone methyltransferases (HMTs), which use S-adenosylmethionine (SAM) as the methyl donor. The enzymes responsible for histone methylation are grouped into three different classes. The first group is the lysine-specific SET domain-containing histone methyltranferases involved in the methylation of lysines at the amino acid positions 4, 9, 27, and 36 of H3 and lysine 20 of H4; the second group is non-SET domain-containing lysine methyltransferases involved in the methylation of arginine 79 of H3 and the third group is arginine methyltransferases involved in the methylation of arginines 2, 17, and 26 of H3 and arginine 3 of H4. The methylation of histones could be related to activation, elongation, or repression of gene expression [72, 73]. Modified histones in parental cells can also be divided and transmitted to the progeny cells. It has been proposed that the methylation of H3 and H4 acts as an epigenetic marker. H3 is more often methylated than histone H4, which makes it more stable than histone H4 [74, 75]. These methylated histones recruit methyl-binding proteins and other transcriptional repressors that maintain CpG DNA methylation and regulate gene transcription. The role of histone methylation is then less clear pertaining to the repair of damaged DNA.
The discovery of histone acetylation was in the 1960's when core histones were first discovered at the ∊ -amino group of specific lysine residues in the amino terminal tail [76, 77]. The acetylation of histone molecules is catalyzed by histone acetyltransferase (HTA). HATs acetylate the conserved ∊ -amino group of the lysine residue in the amino-terminus of the histone tail ultimately decreasing the overall positive charges that is the platform for the binding of transcription factors to the chromatin. Acetylated histone is a characteristic for open chromatin structure. Lysine acetylation is a reversible post-translational process, in which HATs and Histone deacetylases (HDACs) guide the dynamic equilibrium [78, 79]. HATs can be divided into three different groups: the Gcn5/PCAF family, which includes Gcn5, PCAF and Gcn5L, the p300/CBP family, which includes CBP and p300 and the MYST family, which includes Esal, MOF and TIP60 [80]. The hyperacetylation of histones is described as a hallmark of transcription process in active regions. Also, the acetylation of histones can affect DNA replication and repair.
The HDAC's are divided into 4 classes. Class I HDAC's (HDACs 1, 2, 3, and 8) are located in the nucleus, and are expressed ubiquitously in different cell lines and tissues. Class II HDACs (HDACs 4, 5, 6, 7, 9 and 10) are expressed in a tissue-specific manner and are located in between the nucleus and the cytoplasm. Class III HDACs includes the NAD+-dependent deacetylases including sirtuin 1 (SIRT1) to SIRT 7, which are not related to the class I and class II HDACs and require the NAD cofactor in order to be activated. Class IV consists of HDAC 11 and its classification is still under debate. HDACs are involved in several signaling pathways and are present in repressive chromatin complexes.
Protein phosphorylation represents an addition of a phosphate group (PO4) to a protein molecule. The most studied sites of histone phosphorylation is serine 10 of H3 (H3S10) and serine 139 of H2A variant. Phosphorylated H3 at positions serine 10 and serine 28 (H3S10 and H3S28) has been implicated with chromatin condensation during mitosis. Phosphorylated H3 is involved in transcriptional activation of genes resulting from stress or mitogen-stimuli, while mitogen stimuli kinase-1 (MSK1), a kinase activated by a growth factor and stress stimuli, has been demonstrated to be responsible for this serine 10 phosphorylation [81]. It was found that in chicken erythrocyte chromatin, the serine 28 phosphorylation is highly enriched in active/competent gene fractions. The H3 phosphorylated at serine 10 is present in all chromatin fractions, while H3K9me2 is correlated with the chromatin-containing repressed genes. In addition, it was proved that in H3 variant H3.3, the phosphorylated serine 28 is associated with the structure change of nucleosomes in active promoters.
Ubiquitination (or ubiquitylation) is characterized by a post-translational modification consisting of a covalent attachment of one or more ubiquitin monomers of the ε-amino group of lysine residue. Ubiquitin is a 76 amino acid protein highly conserved in eukaryotes [82]. The polyubiquitination process marks a protein to be degraded by 26S proteasome, whereas mono-ubiquitination is in charge of modifying the protein function [83]. In considering nucleosome dynamics during transcription, histone H2B ubiquitylation (H2Bub1) specifically affects the chromatin structure by leading to H3-K4 and H3-K79 methylation. Along with increasing or decreasing H2Bub1 levels, the nucleosome stability is accordingly reduced or enhanced. When H2B ubiquitylation is abolished, it brings defects in cell growth, septation, and nuclear structure. But these phenotypes were not observed in cells lacking H3K4 methylation [84].
Similar to ubiquitylation, SUMO (small ubiquitin-related modifier) modification (SUMOylation) regulates gene expression and cell proliferation as coactivators and corepressors via altering activity and/or localization of related proteins. For example, H4 can be sumoylated by E1 (SUMO-activating enzyme) and E2 (SUMO-conjugating enzyme) resulting in gene silencing by recruiting HDAC1 [85].
Moreover, as a reversible post-translational protein modification, ADP-ribosylation has been detected in not only core histones (H2A, H2B, H3 and H4) but linker histone H1. ADP-ribosylation is a post-translational modification defined by the addition of an ADP-ribose moiety onto a protein using nicotinamide adenine dinucleotide (NAD) + as a substrate. Upon ADP-ribosylation of histones by diphtheria toxin-like ADP-ribosyltransferases, the chromatin relaxes, meanwhile, ADP-ribosylated H1 could promote chromatin unwinding [86]. According to the histone code hypothesis, post-translational modifications of chromatin alter the chromatin structure and regulate the transcription of genetic information encoded in DNA. Modifications such as ADP-ribosylation, acetylation, methylation, phosphorylation, ubiquitination and SUMOylation of histone tails constitute an epigenetic code for histones modifications.
Non-coding RNAs
Non-coding RNAs (ncRNAs) are a type of functional RNA molecule, which are not translated into proteins. More functional groups of ncRNAs have been categorized by the following: four short noncoding RNAs (17-31 bp) (microRNAs (miRNAs), Piwi-interacting RNAs (piRNAs), small interfering RNAs (siRNAs), transcription initiation RNAs), mid-size noncoding RNAs (<200 bp) (small nucleolar RNAs, promoter-associated small RNAs (PASRs), TSS-associated RNAs (TSSa-RNAs), promoter upstream transcripts (PROMPTs), long non-coding RNAs (lncRNAs, >200 bp) and its subgroups such as long intergenic noncoding RNAs (lincRNAs), enhancer RNAs (eRNAs), transcribed ultraconserved regions (T-UCRs) and other lncRNAs [87]. It has been widely shown that ncRNAs not only regulate gene expression at the transcriptional and post-transcriptional levels, but also play a role in the control of epigenetic pathways. For example, PASRs could complement the rDNA promoter and interact with TTF-I, the transcription factor located on the target site. Interestingly, this DNA: RNA triplex specifically mediates the recruitment of DNMT3b further revealing a new mechanism of RNA-dependent DNA methylation [88].
Role of Epigenetic Mechanisms in Regulating Treg Function
1. CpG DNA Methylation/Demethylation in Treg Development and Function
Epigenetics regulation by CpG methylation at specific gene sites in T cells controls the differentiation of T helper cells [12, 59, 89]. The stability of Foxp3 expression is correlated with DNA demethylation at Treg specific demethylated region (TSDR) in the Foxp3 gene. These conserved sequences within the Foxp3 locus are fully and selectively demethylated upon differentiation into [48]. It has been demonstrated that the TSDR region in Tregs is completely demethylated whereas the TDSR of conventional CD4+T cells and in vitro-induced Tregs is highly methylated [90]. Demethylation of the TDSR is required for the long-term Foxp3 maintenance.
The molecular characterization of the TSDR revealed that this region processes transcriptional enhancer activity that determines the stability of Foxp3 expression. For example, the methylation status of TSDR is important because it allows, or prevents, the binding of the methylation-sensitive transcription factor Ets-1 that controls the stability of Foxp3 expression in CD4+ T cells [91].
There are different regulatory cis-elements in the Foxp3 locus present upstream of the transcription site at TSDR. Zorn et al. have reported that the demethylation induced by the hypomethylating drug Decitabine (5-aza-2'-deoxycytidine, DAC) in human natural killer cells leads to Foxp3 expression [92]. Also, it has been reported that 10% to 45% of the CpG sites in the Foxp3 proximal promoter are methylated in CD4+CD25− T cells, whereas all of the CpG sites are demethylated in nTregs, and TGF-β induces the demethylation of CpG at these sites in CD4+CD25− T cells [93]. These studies demonstrate that the methylation of the proximal promoter is an important Foxp3 expression regulator.
Regulatory cis-elements present in TSDR between non-coding exons (−2b and −1) act as enhancers, specifically known as intronic enhancers [12]. The intronic enhancer of Foxp3 is responsible for the regulation of Foxp3 expression. The CpG residues in the intronic region from +4201 to +4500 are completely methylated in naïve CD4+CD25− cells and are fully demethylated in nTregs in mice and humans [93]. Studies have reported that this region has different levels of demethylation after TGF-β stimulation in mouse and human species [48, 94]. The first intronic CpG region (+4393 to +4506 bp, conserved noncoding sequence 3) has decreased CpG residue methylation after TGF-β signaling. After TCR signaling, the first intronic CpG region has an increase in the binding of cyclic-AMP binding response element-binding protein/activating transcription factor, which leads to an increase in Foxp3 expression [95].
Another CpG island upstream enhancer in the TDSR (−5786 to −5558 bp) is methylated in naïve peripheral CD4+CD25− T cells, activated CD4+ T cells, and TGF-β-induced Foxp3+ Tregs, but is demethylated in nTregs [61, 95, 96]. This CpG island region is bound by repressors DNMT1, DNMT3b, MeCP2, and MBD2 in naïve CD4+CD25− T cells, activated CD4+ T cells and TGF-β induced CD4+Foxp3+ iTregs. In nTregs this enhancer region has acetylated histone 3 binding indicating that this is a transcriptionally active site that interacts with the transcription factors Sp1 and TGF-β-induced early 1 product (TIEG1) [12].
Recent progress has identified DNA glycosylases as a DNA demethylase in plants and vertebrates. Zhen et al. have shown that the murine DNA base excision repair glycosylase Myh can act as a DNA demethylase involved in remodeling the IL-2 promoter for its transcription [97]. The enzyme is not expressed in naïve CD4+ T cells, but its expression can be transiently induced following T cell activation. The deficiency in Myh enzyme in T cells blunts the demethylation of the promoter and impairs IL-2 secretion but not IFN-γ secretion [97]. DNA glycosylases have long been implicated in active DNA demethylation, although the exact enzymes and their mechanisms of action have been controversial.
IL-6, which is a proinflammatory cytokine, suppresses the development and function of Tregs by enhancing the activity of DNMT1 and repressing Foxp3 expression [98-101]. IL-6 induces the signal transducer and activator of transcription-3 (STAT-3)-dependent methylation of the upstream Foxp3 enhancer by DNMT1 in nTregs, which causes the repression of Foxp3 gene [101].
These reports demonstrate that Foxp3 is regulated by epigenetic mechanisms, which involve extracellular signal-controlled transacting factors as well as chromatin remodeling through covalent modifications of CpG DNA. The DNA methylation/ demethylation process has an important role in the stabilizing Foxp3 gene.
2. HAT/HDACs Role in Treg Development and Function
2.1. HAT and Tregs
Protein acetylation is an important post-translational modification for the regulation of protein functions. The acetylation of Foxp3 is an important and required post-translational modification, which is regulated by components of if HAT/HDAC complex present in Tregs [102]. Acetylated Foxp3 appears to be more stable as HDAC inhibitors (HDACi) or HAT enzyme inhibits and limits Foxp3 degradation [103]. Foxp3 protein levels are controlled directly by acetylation, which is a process mediated by the inhibition of proteasomal degradation, that leads to resistance of ubiquitination and an increase of DNA binding [99, 103].
TIP60 is expressed in CD25+ Tregs and CD25− T cells, and is co-localized with Foxp3 in the nucleus of human cells, demonstrating the role of TIP60 in regulation of Foxp3 acetylation in physiological conditions. TregTIP60 acetylates the Foxp3 protein and enhances the repression ability on the IL-2 promoter [104, 105]. After the discovery of TIP60 acetylation in Tregs, evidence has shown that p300 promotes the repressive activity of Foxp3. P300 is localized in the nucleus with Foxp3 and increases its acetylation. Recent reports established that conditional deletion or pharmacologic inhibition of p300 (also known as Ep300 or KAT3B) enzyme in Foxp3+ Tregs increases T cell receptor–induced apoptosis in Tregs, impairs Treg suppressive function and peripheral Treg cell induction, and limits tumor growth in immunocompetent mice although not in immunodeficient mice [106]. This data demonstrated that p300 is important for Foxp3+ Treg function and homeostasis in vivo and in vitro, defining a mechanism by which appropriate small-molecule inhibitors can diminish Treg function without the impairment of T effector cell responses or inducing autoimmunity, suggesting a new approach for cancer immunotherapy. Different acetyltransferases may acetylate various sites of Foxp3 and lead to different consequences in its regulatory function. Xiao et al. have demonstrated that the combination of TIP60 and p300 promotes Foxp3 acetylation, but each alone results in a weak acetylation of Foxp3 [102]. Since HAT activity is required for the acetylation process, one possibility is that TIP60 and p300 cooperate with each other in the acetylation process of Foxp3 in order to make it more stable and increase the suppressive function of Tregs. Moreover, TIP60 promotes the acetylation of p300, which then acetylates Foxp3. The relationship between Tip60 and p300 is currently under investigation. It has been demonstrated that p300 and sirtuin-1 regulate the acetylation of Foxp3 and prevent its ubiquitination and turnover [103, 107].
2.2. HDACs and Tregs
The HAT/HDC complex plays a defining role in the regulation of Foxp3 activity due to the importance of increasing the suppressive function of Tregs and removal of the acetyl groups by histone deacetylases [108]. The function of Tregs can be regulated by the modulation of the HAT/HDC complex activity [12, 109]. Certain types of HDACs like HDAC6, HDAC7, HDAC9, and sirtuin1 have the capacity to decrease Tregs suppressive function capacity.
HDAC6 is primarily a cytosolic protein that regulates the acetylation of cytoskeletal proteins such as tubulin. Recently, it has been reported that HDAC6 is translocated to the nucleus of Tregs after the activation of the T cell co-stimulation receptor CD28 and CD3∊, a T cell antigen receptor (TCR) component [110]. Also, it was found that HDAC6−/− Tregs have an increased Foxp3 expression when compared to wild type Tregs, which is correlated well with a more acetylated Foxp3 in the absence of HDAC6 [110]. This data suggests that HDAC6 deacetylates Foxp3, and the loss of HDAC6 promotes Foxp3 acetylation that, in consequence, increases the stability of the suppressive function of Foxp3 and makes Foxp3+ Tregs resistant to proteasomal degradation.
It has been reported that HDAC7 and HDAC9 are associated with Foxp3 in a multimeric protein complex, which could also be in charge of Foxp3 deacetylation [105]. Real time quantitative PCR analysis demonstrated little difference between Tregs versus non-Tregs in murine expression of HDACs, whereas class II HDACs are mainly expressed by Tregs especially after TCR activation [109]. HDAC7 is recruited to the Foxp3 co-repressor complex where it deacetylates and inhibits the function of the Foxp3 protein [103]. Also, HDAC7 deacetylates histones in the Foxp3 promoter and represses the transcription process. It has been shown that HDAC7 undergoes phosphorylation and nuclear export after T cell activation therefore stimulating the depression of target genes [111, 112]. These studies suggest the importance of HDAC7 as a repressor of Foxp3 function in Tregs.
HDAC9 is found in the Foxp3 complex in resting human Tregs. With nuclear export of HDAC9 after T cell activation, TCR activation leads to a down-regulation of HDAC9 in non Tregs, whereas increases the regulation of HDAC9 by 30-fold in Tregs [109]. Suggesting that HDAC9 normally inhibits the function of Foxp3, the nuclear export of HDAC9 increases the suppressive function of Tregs, which is necessary for optimal Treg suppressive activity [113, 114]. HDAC9−/− mice have a moderate increase in Tregs numbers [109]. Research results showed that HDAC9−/− Tregs express more Foxp3 after activation and suppressive function then when it is compared with wild type Tregs [109]. These studies suggest that HDAC9 is a key repressor of Foxp3 function in Tregs.
It is known that Sirtuin-1 (Sirt-1) is negatively associated with T cell activation. The loss of Sirt-1 function results in an abnormal increase in T cell activation and break-down of CD4+ T cell tolerance [115]. In contrast, the upregulation of Sir-1 results in T-cell anergy: a peripheral immune tolerance mechanism when there are no co-stimulation signals [115]. Sirt1 deficient mice develop allergic encephalomyelitis and spontaneous autoimmunity. Recently, Van Loosdregt et al. showed that in nonimmune cells Sirt-1 is co-localized with Foxp3 and mediates its deacetylation and polyubiquitination [103]. However, other studies reported that loss of Sirt-1 activity in Tregs increases the expression of Foxp3 protein, which prolongs the allograft's overall survival [116]. Therefore, it is still unclear as to whether there is a unified pathway for Sirt-1 to regulate immune responses in allergy, autoimmune and allograft-related immune settings.
3. Role of Histone Methylation in Treg Function
The histone demethylase LSD1 was discovered in 2004, and since then, several histone demethylases have been identified that play an important role in the regulation of gene expression, cellular differentiation and development [117]. The histone methylation process has been correlated with the expression of genes associated with proliferation, differentiation and survival of antigen-activated T cells [118]. Chromatin immunoprecipitation experiments (ChIP) demonstrated that CD4+CD25+ Tregs have more modified histones compared with conventional T cells [48]. The major differences between two subsets are found in the acetylation and trimethylation of H3, whereas the minor differences between two subsets are found in acetylation of H4. This data depicted that, within the conventional CD4+CD25− T cells, the Foxp3 locus is packed in a more condensed and inaccessible chromatin structure compared to an open euchromatin in CD4+CD25+ Tregs.
There are genome-wide H3K4me1 (monomethylation in lysine 4 of histone 3) and H3K4me3 (trimethylation in lysine 4 of histone 3) modification regions in Tregs and Tconv cells. These enhancers are probably important in driving Treg cell-type specific patterns of gene expression. The majority of the H3K4me1 regions differing between Treg and aTconv cells are located at distal-promoter region. In other hand the modifications of H3K4me3 are located in the proximal-promoter regions, which are nearly identical in both Treg and Tconv cells, with the exception of a few promoters of genes, such as FOXP3 and CCR7, which are uniquely expressed in Treg cells. The Tregs- and Tconv- specific H3K4me1 and H3K43 patterns may have the functions as significant mediators of differentiation events, lineage commitment and cell type-specific gene expression [119].
The reprogramming of Treg cells is associated with differential histone modifications. The decrease of H3K4me3 within the downregulated Treg's genes like Foxp3, CTLA4 and LRRC32 correlates with an increase of H3K4me3 in the Th-2 associated locus such as IL-4 and IL-5. These results concluded that that the Foxp3-losing Treg cells are reprogrammed into cells with a gene expression signature dominated by Th2 lineage-associated genes and that histone methylation may contribute to this reprogramming [120].
In a recent study using a murine T cell transfer model of colitis, the investigators found that T cell–intrinsic expression of the histone lysine methyltransferase G9A is required for the development of pathogenic T cells and intestinal inflammation [121]. The methyltransferrase G9A mediates dimethylation of histone H3 lysine 9 (H3K9me2) restricted Th17 and Tregs differentiation in vitro and in vivo. H3K9me2 is found at high levels in naive Th cells and is lost following Th cell activation. The loss of G9A in naive T cells is associated with increased chromatin accessibility and increased sensitivity to TGF-β1. The inhibition of G9A methyltransferase activity in WT T cells promotes the differentiation of Th17 and Treg cells. This data indicates that G9A-dependent H3K9me2 is an epigenetic modification that regulates Th17, Tregs, TGF-β1 responses by limiting chromatin accessibility. This suggests that the G9A enzyme is a therapeutic target for treating intestinal inflammation.
The methyl-binding domain (Mbd) proteins recruit histone-modifying and chromatin-remodeling complexes to methylated sites. A recent study showed that the Mbd2 target promotes demethylation of Foxp3 and Tregs numbers or Tregs function [122]. They used chromatin immunoprecipitation (ChIP) analysis and showed the binding of the Mbd2 proteins with the Foxp3-associated TSDR site in Treg cells. Mbd2 targeting by homologous recombination, or small interfering RNA (siRNA), decreases Tregs numbers and impairs Tregs-suppressive function in vitro and in vivo. Moreover, a complete TSDR demethylation is found in wild-type (WT) Treg cells but >75% methylation in Mbd2−/− Treg cells, whereas reintroduction of Mbd2 into Mbd2-null Treg cells restored TSDR demethylation, Foxp3 gene expression, and Tregs-suppressive function was found. Lastly, thymic Treg cells from Mbd2−/−_ mice have normal TSDR demethylation, but compared to WT Treg cells, peripheral Mbd2−/− Treg cells have a marked impairment of Tet2 binding, the DNA demethylase enzyme, at the TSDR site. This data established that Mbd2 plays a key role in promoting TSDR demethylation, Foxp3 expression, and Tregs-suppressive function.
4. MiRNA-155 in Tregs Function
Much evidence demonstrates the necessary role of microRNAs (miRNAs) in the immune system. RNA enzyme III (Dicer), which is important for the generation of mature miRNAs, results in impaired thymic development and a decrease in helper T cell differentiation with the conditional deletion of T lymphocytes [123, 124]. This correlation has been demonstrated in nTreg cell-specific Dicer knock-out mice and it was found that miRNAs seem to be important for T cell development, homeostasis and activation. Also, miRNAs seem to play a role in the suppressive function of n Tregs [124-127]. The miR-155 is encoded by a small region of the proto-oncogen BIC, which was originally described as a common site of a viral DNA integration in virally-induced lymphomas in chicken [127, 128].
The role of miR-155 in the immune system was proven using miR-155−/− knock-out mice in 2007 [129, 130]. These mice show a severe autoimmune phenotype of the lung characterized by leukocyte invasion in bronco-alveolar lavage (BAL) and increased airway remodeling. These results suggest that miR-155 has an impact in the immune system response to self-antigens. To determine the role of Foxp3 in the regulation of BIC/miR-155, Stahl et al. used resting and activated CD4+ T cells from Foxp3- mutant scurfy mice and compared it with WT mice in order to analyze the expression of miR-155 [131]. Since CD4+ T cells from scurfy and wild type mice have the same expression level of miR-155 after induction, miR-155 is not necessarily regulated by Foxp3. Using murine and human cells, they demonstrated that an increase in miR-155 expression in nTregs did not influence their abilities to suppress CD4+ effector Th cells, but instead showed that the overexpression of miR-155 decreases the susceptibility of CD4+ effector Th cells to nTregs suppression. Recently, it was found that miR-155 is required for the development of Tregs but is dispensable for the function of Tregs in regulating conventional T cells [132, 133]. In 2012, Liao et al. showed that miR-155 has a critical role in driving Treg/Th17 cell differentiation and enhancing Th17 cell function by the inhibition of a negative regulator of Janus Kinase (JAK)/signal transducers and activators of transcription (STAT) signaling pathway, suppressor of cytokine signaling-1 (SOCS1) [134]. The specific deletion of SOCS1 results in an increase in the proportion and absolute number of Tregs in the thymus [133, 135]. The imbalance of Treg/Th17 cells induced by miR-155 might contribute to the imbalance of the JAK/STAT and TGF-β/SMAD5 signaling pathways. The inhibition of the SOCS1 expression in activated CD4+ T cells by miR-155 contributes to the activation of IL-2/STAT, which is essential for the maintenance and homeostasis of Tregs and suppression of Th17 differentiation [136, 137]. In addition, inhibition of SOCS1 expression in T cells by miR-155 contributes to the activation of IL6/STAT3, which is indispensable for Th17 cell differentiation and Treg inhibition [136]. The TGF-β/SMAD5 signaling pathway contains a critical, non-positively regulated by miR-155, role in Treg function. In summary, although the production of IL-10 and TGF-β is not increased, the production of IL-17 and differentiation of Treg and Th17 cells is increased when the expression of SOCS1 is inhibited by miR-155 [138-140].
The miR-155 is considered a proinflammatory miRNA. The exposure of ox-LDL induces miR-155 expression in human THP-1 macrophages [141]. Although miR-155 is considered a proinflammatory miRNA, in vitro studies reported anti-inflammatory effects in lipid-loaded cells [142]. These in vitro studies were done using bone marrow transplantation from miR-155 deficient mice, or wild type mice, to hyperlipidemic mice in order to analyze the development of atherosclerosis. The hematopoietic deficiency of miR-155 enhances the development of atherosclerosis and decreases plaque stability. These inflammatory stages in miR-155 KO mice show a decrease in circulating CD4+CD25+/highFoxp3+ Tregs, an increase in inflammatory monocyte subset (CD11b+LY6G−LY6Chigh) and a reduction in resident monocytes (CD11+Ly6G−Ly6Clow). The role of the miR-155 as a multifunctional miRNA in the development of immune and inflammatory diseases is under intensive investigation, which could offer a variety of resources for the development of new therapeutic approaches in the clinical field. It is essential that more studies are completed to further determine the specific role of the miR-155 in the development of human diseases.
Epigenetic Tregs Modifications, Human Diseases and Therapeutic Approaches
The role of environmental factors in the pathogenesis of autoimmune diseases has been extensively discussed [143-147]. In recent years, the role of epigenetic modifications has been demonstrated through DNA methylation and histone modification process in the etiology of autoimmune and inflammatory diseases [148, 149]. As an important component of the immune system, miR-155 promotes autoimmunity and is also oncogenic under certain conditions [150, 151]. It has been demonstrated that there is an epigenetic mechanism that plays a role in the activity of the Foxp3 gene leading to regulation of the Tregs function [9, 10]. Tregs driven by the Foxp3 transcription factor are responsible for limiting autoimmunity and chronic inflammation.
Epigenetic therapy is emerging in the pharmacology field, which could help discover novel therapeutic agents and increase our control on various diseases [12]. Currently, epigenetic drugs can be divided into two groups: DNMT inhibitors and HDAC inhibitors. DNMT inhibitors induce strong Foxp3 expression but are associated with cell toxicity and induction of Th1 and Th2 cytokines that limit their use. The HAT/HDAC complex is important in the stability and function of the Foxp3 gene. Experimental studies have demonstrated that HDAC inhibitors have an immunomodulatory activity in in vivo and in vitro models of inflammation, autoimmunity and transplantation. Also, HDACs are involved in oncogenic transformation.
DNA methylation is a well-established epigenetic mechanism that ranges from parenteral imprinting to X-chromosomal inactivation [152]. Recent studies have shown that a DNA methylation process regulates the expression of Foxp3 in Tregs, and Foxp3+ Tregs can be used as a possible treatment for graft-versus-host disease (GVHD), diabetes, and other autoimmune diseases [153, 154]. Decitabine (5-aza-2’-deoxycytidine, DAC) and 5-azacitidine (Azac) are hypomethylating agents approved by the FDA for the treatment of myelodysplatic syndrome and other leukemias [155]. The Dnmt1 inhibitor, DAC increases the expression of Foxp3 in WT conventional T cells and promotes their conversion into iTregs [5]. Dnmt1 interacts with Dmnt3a, Dnmt3b and additional silencing proteins like HDAC1 and HDAC2. The stability of Dnmt1 is regulated by several posttranslational modifications, such as phosphorylation, acetylation, ubiquitination, methylation and sumoylation [156, 157]. The deletion of DNMT1 in conventional T cells increases their Foxp3 expression after stimulation with TCR ligation. These results have clearly demonstrated that DNMT1 limits the capacity of CD4+ T cells to express Foxp3 and become functional Tregs. In other studies, Liquing et al. reported the role of DNMT1 in Tregs [61]. They deleted the DNMT1 enzyme in mice Tregs, which showed a decrease in number and function of Tregs as well as a decrease in the conversion of Foxp3+ iTregs from conventional T cells. They also reported that mice with a conditional deletion of DNMT1 in their Tregs died of autoimmunity by 3-4 weeks of age. The conditional deletion of DNMT1 does not affect the methylation of CpG sites in the Foxp3 gene, but decreases the global methylation of DNAs and alters the expression of pro-inflammatory and other genes in Tregs. Taken together, these results demonstrated that DNMT1 inhibitors need to be used with caution when present in the development of Treg-based cellular therapies. In other hand the histone methylation process has been correlated with the expression of genes. The use of a histone methylation inhibitor 3-Deazaneplanocin A (DZNep), arrests the ongoing GVHD by the induction of apoptosis in alloreactive T cells [158].
Animal experimental studies have shown that the induction of Foxp3 in Tregs stabilizes atherosclerotic plaque [37, 159, 160]. Oxidized low density lipoprotein (OxLDL) is a risk factor for the development of atherosclerosis, reduction of the demethylation rate in Foxp3 gene, decreased suppressive function of Tregs and increased destabilization of atherosclerotic plaque [161]. In addition, hyperhomocysteinemia has a pathogenic role in vascular diseases that is correlated with DNA hypomethylation [60, 162, 163]. A decrease in Treg number and impairment of its function has been reported in patients with acute coronary syndrome (ACS) [164, 165]. Other studies report that Foxp3 is overexpressed in coronary artery disease (CAD) patients with no correlation to the severity of coronary atherosclerosis [166-168]. The role of Tregs in CAD is still controversial, mainly due to the lack of Treg-specific markers. The unmethylation of the CpG-enriched element in Foxp3 intron 1 (Foxp3 i 1) is specific in Tregs and can be used to identify the role of Tregs in clinical diseases. A recent study analyzed the demethylated status of Foxp3 i 1 in circulating Tregs in healthy and ACS patients [169]. They found a decrease in the numbers of Tregs in ACS patient groups using a Foxp3 i 1 demethylation assay, but did not see any differences when they used flow cytometric analysis, suggesting that intron demethylation assays are more sensitive than flow cytometry in detecting Foxp3+ Tregs. This result demonstrated a quantitative defect in Tregs of ACS patients. Moreover, they used the DNA hypomethylation agent DAC to treat the CD4+CD25+/high T cells from ACS patients, which resulted in an increase in the demethylation of Foxp3i 1 in a dose-dependent manner. Also, the use of DAC increases the production of anti-inflammatory cytokine IL-10 and decreases the expression of pro-inflammatory cytokine IFN-γ in ACS patients. These results demonstrated the increase of Foxp3 Tregs from T conv cells of ACS patients after the induction with the hypomethylating agent DAC, suggesting the use of epigenetic-based therapy in patients with ACS.
It has been proved that HDAC inhibitors (HDACi) have an effective therapeutic function in several murine models. In murine models of allogeneic bone marrow transplantation, the HDACi vorinostat (SAHA), reduced acute graft-versus-host disease by the suppression of pro-inflammatory cytokines such as TNF-α, IL-1, and IFN-γ [170]. Vorinostat was approved by the Food and Drugs administration (FDA) in 2006 for the treatment of patients with cutaneous T cell lymphoma (CTCL) [171-174]. Givinostat is an inhibitor for class I and class II HDACs, which was approved in 2010 as an orphan drug for the treatment of juvenile idiopathic arthritis in Europe [175].
The use of HDAC inhibitor increases the Foxp3 expression in Tregs in murine models of collagen-induced arthritis, allograft rejection and colitis [102]. Also, these models showed that the HDAC inhibitor enhances the number and suppressive function of Tregs, which leads to the prevention and improvement of autoimmune diseases. The histone deacetylase inhibitor trichostatin A (TSA) is a small molecule compound that inhibits class I, II, and IV HDAC enzyme families. Previous studies in mouse models have shown that the administration of TSA in vivo promotes the generation and function of Tregs, which depicts its beneficial effects in cardiac allograft transplant, inflammatory bowel disease and SLE [176-178]. Cristian et al. have demonstrated that TSA increases the generation of CD4+Foxp3+ Tregs from naive T cells in vitro, and the increase in Tregs is correlated with the hyperacetylation of histone H3 [177]. This evidence suggests that TSA could promote hyperacetylation of histone H3 in the Foxp3 promoter. Moreover, it has been reported that Foxp3 becomes acetylated and stabilized from proteasomal degradation. Indeed, it could be possible that TSA fulfills its function via a combined pathway as follows: firstly, an increase of Foxp3 gene expression by the hyperacetylation of histone H3 on the Foxp3 promoter and secondly an increase in the Foxp3 protein half-life. TSA, as the HDACi, provides useful and valuable tools for enhancing Tregs production and suppressive function, thus it is a novel therapeutic modality for the treatment of autoimmune diseases. Grabiec et al. studied the effect of TSA and NIC (Class III, sirtuin HDACi) on the function of peripheral and local macrophages in healthy and RA patients [179]. TSA and NIC reduce IL-6 production by RA synovial fluid macrophages and RA monocyte-derived macrophages from healthy subjects and RA patients. Pauley et al. reported that the expression of miR-155 is correlated with the activity of the disease and also overexpressed in polymorphonuclears (PMN) leukocytes from RA patients versus osteoarthritis (OA) patients [180].
The roles of Tregs have been identified in tumor immune tolerance and were also found to maintain peripheral immune tolerance for self-antigens and prevent autoimmune responses [181, 182]. Tregs are predominant in various cancers such as prostate cancer [183]. Tregs promotion and expansion occurs after immunotherapy in cancer patients [183-186]. High doses of Tregs-survival cytokine IL-2 are an FDA-approved treatment for selected cases of metastatic clear cell renal cell carcinoma but have a reported limited efficacy [187, 188]. A vaccine for prostate cancer (Sipuleucel T) was approved in 2010 by the FDA, in which, however, Tregs play an important role for the low efficacy of the vaccine therapy. Other clinical research studies report that the depletion of Tregs may enhance the antitumor response in cancer patients [189, 190]. The HDACs are involved in oncogenic transformation by mediating the transcriptional regulation of genes involved in cell cycle progression, proliferation and apoptosis [191]. HDAC inhibitors have demonstrated antitumor activity in different tumors. Entinostat, a synthetic benzamide, is a selective HDAC class I inhibitor that decreases the expression of Foxp3 in Tregs and inhibits their suppressive function [190]. Li Shen et al. reported that Etinostat inhibits Tregs and enhances the antitumor activity of IL-2 cytokine and the peptide vaccine treatment SurVaxM (modified survivin peptide vaccine) in a castration resistant prostate cancer model [190].
DNA Methylation Inhibitors and Histone Deacetylase inhibitors - Clinical Trials
We mentioned that the FDA has approved some epigenetic drugs for the treatment of several diseases. Moreover, there are several ongoing clinical trials that are testing these epigenetic drugs in human diseases, which are divided into the DNMT inhibitors group (Table 2) and HDAC inhibitors group (Table 3).
Table 2.
DNMT inhibitors ongoing clinical trials
| Medication | Condition | Phase | Clinical Trial | Other drugs |
|---|---|---|---|---|
| Azacitidine | Acute Myeloid Leukemia Acute Myelogenous Leukemia |
Phase 2 | NCT01358734 | Lenalidomide |
| Leukemia AML MDS |
Phase 3 | NCT00887068 | ||
| Prostate Cancer | Phase I/II | NCT00503984 | Docetaxel Prednisone |
|
| Acute Myeloid Leukemia | Phase II | NCT00492401 | ||
| Decitabine | Metastatic Papillary Thyroid Cancer Follicular Thyroid Cancer |
Phase II | NCT00085293 | |
| Acute Myeloid Leukemia | Phase II | NCT00492401 | ||
| Sickle Cell Disease | Phase 1 | NCT01685515 | THU | |
| Colon cancer | Interventional | NCT01882660 | ||
| Myelodysplastic Syndromes Acute Myeloid Leukemia |
Phase 4 | NCT01806116 |
Table 3.
HDAC inhibitors ongoing clinical trials
| Medication | Condition | Phase | Clinical Trial | Other drugs |
|---|---|---|---|---|
| Etinostat | Non-Small Cell Lung Cancer | Phase I/II Study | NCT00387465 | 5-Azacytidine |
| HER2-Positive Metastatic Breast Cancer | Phase I Phase II |
NCT01434303 | Lapatinib Trastuzumab |
|
| Acute Lymphoblastic Leukemia Bilineage Biphenotipic leukemia |
Phase I Study | NCT01132573 | Clofarabine | |
| Hodgkin's Lymphoma | Phase 2 | NCT00866333 | ||
| Metastatic Renal Cell Carcinoma | Phase I/II Study | NCT01038778 | Interleukin 2 Aldesleukin |
|
| Breast Cancer | Phase 2 | NCT02115594 | Fulvestrant | |
| Non-Small Lung Cancer, Epigenetic Therapy | Phase 2 | NCT01928576 | Azacitidine CC-486 |
|
| Vorinostat | Neimann-Pick Disease | Phase 1 Phase 2 |
NCT02124083 | |
| Non-Small-Cell Lung Carcinoma | Phase 1 | NCT02151721 | Gefitinib | |
| Von Hippel-Lindau Disease | Phase 1 | NCT02108002 | ||
| Cutaneous T-Cell Lymphoma | Phase 3 | NCT01728805 | Biological: KW-0761 | |
| Gastric Cancer | Phase 1 Phase 2 |
NCT01045538 | capecitabine cisplatin |
|
| Locally Advanced Non-small Cell Lung Cancer | Phase 1 | NCT01059552 | ||
| Sickle Cell Disease Sickle Cell Anemia |
Phase 2 | NCT01000155 | ||
| Panobinostat (LBH589) | Graft-Versus-Host Disease | Phase 1 Phase 2 |
NCT01111526 | |
| Melanoma Skin Cancer |
Phase 1 | NCT02032810 | Ipilimumab | |
| Sickle Cell Disease | Phase 1 | NCT01245179 | ||
| Prostate Cancer Prostatic Neoplasms |
Phase 1 | NCT00878436 | Bicalutamide | |
| Diffuse Large B Cell Lymphoma | Phase 2 | NCT01282476 | Rituximab | |
| Romidepsin Istodax | Lymphoma T-Cell Lymphoma Cutaneous Lymphoma |
Phase 1 | NCT01902225 | Doxil |
| Givinostat | Chronic Myeloproliferative Neoplasms (cMPN) | Phase 2 | NCT01761968 | |
| (Belinostat) (PDX101) | Small Cell Lung Carcinoma Malignant Epithelial Neoplasms |
Phase 1 | NCT00926640 | |
| Neoplasms Lymphomas |
Phase 1 | NCT01273155 | ||
| Pracinostat | Myelodysplastic Syndrome | Phase 2 | NCT01873703 | Azacitidine |
| Valproic Acid | Cancer | Phase 1 | NCT01007695 | |
| High Grade Sarcoma | Phase 1 | NCT01010958 |
Many ongoing clinical trials are recruiting patients to test the therapeutic efficacy, as well as secondary effects, of DNMT inhibitors on patients with several diseases and especially cancers. Also, an additional clinical trial is in process to determine whether Decitabine can increase fetal hemoglobin levels and improve the symptoms of sickle cell disease. The overall purpose is to develop disease modifying treatment for sickle cell disease that is less cytotoxic than the current standard of care, and which can directly and more efficiently reactivate fetal hemoglobin levels (ClinicalTrials.gov Identifier NCT01685515). Because colon cancer is the second leading cause of cancer-related death worldwide, the Clinical Trial NCT01882660 has the primary objective to determine whether short-course pre-operative treatment with decitabine can increase Wnt target gene expression as measured in resected tumors compared to pretreatment biopsies. The secondary objective of this study is to assess patients with primary colon cancer whether or not short-course pre-operative treatment with decitabine can revert CpG methylation and induce more favorable tumor characteristics as measured in resected tumors compared to pretreatment biopsies. The tertiary objective is to compare changes in Wnt target gene expression, CpG methylation and tumor characteristics for Wnt methylated and nonmethylated tumors as measured in resected tumors compared to pretreatment biopsies as well as identify new stratification markers.
Similar to DNMT inhibitor clinical trials, there are several ongoing HDAC inhibitor clinical trials working to determine the efficacy of these types of epigenetic drugs for the treatment of human diseases. A phase I/II trial NCT01038778 is studying the best dosage of entinostat, when given with aldesleukin, along with side effects to determine how well this works in treating patients with metastatic kidney cancer. Entinostat may stop the growth of tumor cells by blocking some of the enzymes that are necessary for cell growth. Together with aldesleukin, entinostat may kill more tumor cells. Von Hippel-Lindau (VHL) disease is a genetic disease. Patients with VHL often have a brain tumor called hemangioblastoma that is often treated with risky surgery. Some patients with VHL have mutations, which make abnormal proteins that break quickly and forms tumors.The goal of clinical trial NC T02108002 is to determine if the drug Vorinostat will slow the growth of hemangioblastomas in patients with VHL by preventing the breakdown of the mutant protein.
Conclusion
The role of Tregs in the immune system is well established. The Foxp3 gene has been described as a transcription factor that regulates the development, stability and suppressive function of Tregs as part of the immune system. It has been clearly demonstrated that Tregs have indispensable functions in maintaining immune homeostasis, mediating peripheral tolerance, preventing autoimmune diseases, and suppressing inflammatory responses as a suppressor. Epigenetic is defined as heritable changes in gene expression without changing the DNA sequence of the genome by different mechanisms. The epigenetic mechanism and enzymes involved with the Foxp3 gene stability and the suppressive capacity of Tregs has been studied, which ultimately created a new translational aspect in the epigenetic Tregs modifications biology as a target for new therapeutic modalities in human diseases. The ongoing clinical trials and the FDA approval of hypomethylating agents for the treatment of myelodysplastic syndromes and the HDACi for the treatment of CTCL confirm that the epigenetic mechanism on Tregs is an important tool for the development of new therapeutic approaches. Further investigations are needed to determine what other epigenetic mechanisms are involved with Foxp3 and its cofactors, which will lead to the development of new, specific and safe therapeutic targets for the development of new drugs.
Acknowledgements
We are very grateful to Dr. B. Ashby for critical reading of this manuscript. This work is partially supported by NIH grants to Drs. XF. Yang and H. Wang.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The authors have read the journal's authorship agreement and journal's policy on conflicts of interest. The authors declare no competing financial interests.
References
- 1.Sakaguchi S, et al. Naturally arising Foxp3-expressing CD25+CD4+ regulatory T cells in self-tolerance and autoimmune disease. Curr Top Microbiol Immunol. 2006;305:51–66. doi: 10.1007/3-540-29714-6_3. [DOI] [PubMed] [Google Scholar]
- 2.Collison LW, et al. The composition and signaling of the IL-35 receptor are unconventional. Nat Immunol. 2012;13(3):290–9. doi: 10.1038/ni.2227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li X, et al. IL-35 is a novel responsive anti-inflammatory cytokine--a new system of categorizing anti-inflammatory cytokines. PLoS ONE. 2012;7(3):e33628. doi: 10.1371/journal.pone.0033628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Katoh H, Zheng P, Liu Y. FOXP3: genetic and epigenetic implications for autoimmunity. J Autoimmun. 2013;41:72–8. doi: 10.1016/j.jaut.2012.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lal G, et al. Epigenetic regulation of Foxp3 expression in regulatory T cells by DNA methylation. J Immunol. 2009;182(1):259–73. doi: 10.4049/jimmunol.182.1.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang XF, et al. The FOX transcription factors regulate vascular pathology, diabetes and Tregs. Front Biosci (Schol Ed) 2009;1:420–36. doi: 10.2741/s35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bettini ML, et al. Loss of epigenetic modification driven by the Foxp3 transcription factor leads to regulatory T cell insufficiency. Immunity. 2012;36(5):717–30. doi: 10.1016/j.immuni.2012.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jeker LT, Bour-Jordan H, Bluestone JA. Breakdown in peripheral tolerance in type 1 diabetes in mice and humans. Cold Spring Harb Perspect Med. 2012;2(3):a007807. doi: 10.1101/cshperspect.a007807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003;299(5609):1057–61. doi: 10.1126/science.1079490. [DOI] [PubMed] [Google Scholar]
- 10.Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. 2003;4(4):330–6. doi: 10.1038/ni904. [DOI] [PubMed] [Google Scholar]
- 11.Vahedi G, et al. Helper T-cell identity and evolution of differential transcriptomes and epigenomes. Immunol Rev. 2013;252(1):24–40. doi: 10.1111/imr.12037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lal G, Bromberg JS. Epigenetic mechanisms of regulation of Foxp3 expression. Blood. 2009;114(18):3727–35. doi: 10.1182/blood-2009-05-219584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xiong Z, et al. Higher expression of Bax in regulatory T cells increases vascular inflammation. Front Biosci. 2008;13:7143–55. doi: 10.2741/3217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xiong Z, et al. Expression of TCTP antisense in CD25(high) regulatory T cells aggravates cuff-injured vascular inflammation. Atherosclerosis. 2009;203(2):401–8. doi: 10.1016/j.atherosclerosis.2008.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yan Y, Xiong Z, Zhang S, Song J, Huang Y, Thornton AM, Wang H, Yang X-F. CD25high T cells with a prolonged survival inhibit development of diabetes. International Journal of Immunopathology and Pharmacology. 2008;21(4):767–780. doi: 10.1177/039463200802100401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mai J, Wang H, Yang XF. Th 17 cells interplay with Foxp3+ Tregs in regulation of inflammation and autoimmunity. Front Biosci (Landmark Ed) 2010;15:986–1006. doi: 10.2741/3657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang XF. Factors regulating apoptosis and homeostasis of CD4+CD25highFOXP3+ regulatory T cells are new therapeutic targets. Front Biosci. 2008:1472–99. doi: 10.2741/2775. [DOI] [PubMed] [Google Scholar]
- 18.Yang XF, et al. The forkhead transcription factors play important roles in vascular pathology and immunology. Adv Exp Med Biol. 2009;665:90–105. doi: 10.1007/978-1-4419-1599-3_7. [DOI] [PubMed] [Google Scholar]
- 19.Pastrana JL, et al. Regulatory T cells and Atherosclerosis. J Clin Exp Cardiolog. 2012;2012(Suppl 12):2. doi: 10.4172/2155-9880.S12-002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gershon RK, Kondo K. Cell interactions in the induction of tolerance: the role of thymic lymphocytes. Immunology. 1970;18(5):723–37. [PMC free article] [PubMed] [Google Scholar]
- 21.Sakaguchi S, et al. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol. 1995;155(3):1151–64. [PubMed] [Google Scholar]
- 22.Bennett CL, et al. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet. 2001;27(1):20–1. doi: 10.1038/83713. [DOI] [PubMed] [Google Scholar]
- 23.Simonetta F, et al. Increased CD127 expression on activated FOXP3+CD4+ regulatory T cells. Eur J Immunol. 2010;40(9):2528–38. doi: 10.1002/eji.201040531. [DOI] [PubMed] [Google Scholar]
- 24.Liu J, et al. T regulatory cells in cord blood--FOXP3 demethylation as reliable quantitative marker. PLoS One. 5(10):e13267. doi: 10.1371/journal.pone.0013267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen J, et al. Pygo2 associates with MLL2 histone methyltransferase and GCN5 histone acetyltransferase complexes to augment Wnt target gene expression and breast cancer stem-like cell expansion. Mol Cell Biol. 2010;30(24):5621–35. doi: 10.1128/MCB.00465-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wieczorek G, et al. Quantitative DNA methylation analysis of FOXP3 as a new method for counting regulatory T cells in peripheral blood and solid tissue. Cancer Res. 2009;69(2):599–608. doi: 10.1158/0008-5472.CAN-08-2361. [DOI] [PubMed] [Google Scholar]
- 27.Kitagawa Y, Ohkura N, Sakaguchi S. Molecular determinants of regulatory T cell development: the essential roles of epigenetic changes. Front Immunol. 2013;4:106. doi: 10.3389/fimmu.2013.00106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sakaguchi S, et al. Regulatory T cells and immune tolerance. Cell. 2008;133(5):775–87. doi: 10.1016/j.cell.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 29.Ketelhuth DF, Hansson GK. Cellular immunity, low-density lipoprotein and atherosclerosis: break of tolerance in the artery wall. Thromb Haemost. 2011;106(5):779–86. doi: 10.1160/TH11-05-0321. [DOI] [PubMed] [Google Scholar]
- 30.Fathman CG, Lineberry NB. Molecular mechanisms of CD4+ T-cell anergy. Nat Rev Immunol. 2007;7(8):599–609. doi: 10.1038/nri2131. [DOI] [PubMed] [Google Scholar]
- 31.Sakaguchi S, et al. Foxp3+ CD25+ CD4+ natural regulatory T cells in dominant self-tolerance and autoimmune disease. Immunol Rev. 2006;212:8–27. doi: 10.1111/j.0105-2896.2006.00427.x. [DOI] [PubMed] [Google Scholar]
- 32.Stephens GL, Shevach EM. Foxp3+ regulatory T cells: selfishness under scrutiny. Immunity. 2007;27(3):417–9. doi: 10.1016/j.immuni.2007.08.008. [DOI] [PubMed] [Google Scholar]
- 33.Tordjman R, et al. A neuronal receptor, neuropilin-1, is essential for the initiation of the primary immune response. Nat Immunol. 2002;3(5):477–82. doi: 10.1038/ni789. [DOI] [PubMed] [Google Scholar]
- 34.Yadav M, et al. Neuropilin-1 distinguishes natural and inducible regulatory T cells among regulatory T cell subsets in vivo. J Exp Med. 2012;209(10):1713–22. S1–19. doi: 10.1084/jem.20120822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Weiss JM, et al. Neuropilin 1 is expressed on thymus-derived natural regulatory T cells, but not mucosa-generated induced Foxp3+ T reg cells. J Exp Med. 2012;209(10):1723–42. S1. doi: 10.1084/jem.20120914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sakaguchi S. Naturally arising CD4+ regulatory t cells for immunologic self-tolerance and negative control of immune responses. Annu Rev Immunol. 2004;22:531–62. doi: 10.1146/annurev.immunol.21.120601.141122. [DOI] [PubMed] [Google Scholar]
- 37.Ait-Oufella H, et al. Natural regulatory T cells control the development of atherosclerosis in mice. Nat Med. 2006;12(2):178–80. doi: 10.1038/nm1343. [DOI] [PubMed] [Google Scholar]
- 38.Sasaki N, et al. Regulatory T cells in atherogenesis. J Atheroscler Thromb. 19(6):503–15. doi: 10.5551/jat.10934. [DOI] [PubMed] [Google Scholar]
- 39.Jonuleit H, Schmitt E. The regulatory T cell family: distinct subsets and their interrelations. J Immunol. 2003;171(12):6323–7. doi: 10.4049/jimmunol.171.12.6323. [DOI] [PubMed] [Google Scholar]
- 40.Hippen KL, et al. Clinical perspectives for regulatory T cells in transplantation tolerance. Semin Immunol. 2011;23(6):462–8. doi: 10.1016/j.smim.2011.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Groux H. Type 1 T-regulatory cells: their role in the control of immune responses. Transplantation. 2003;75(9 Suppl):8S–12S. doi: 10.1097/01.TP.0000067944.90241.BD. [DOI] [PubMed] [Google Scholar]
- 42.Roncarolo MG, Levings MK, Traversari C. Differentiation of T regulatory cells by immature dendritic cells. J Exp Med. 2001;193(2):F5–9. doi: 10.1084/jem.193.2.f5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen Y, et al. The autocrine regulatory effect of vasoactive intestinal peptide on the growth of human pancreatic carcinoma cells. Chin Med Sci J. 1994;9(4):215–9. [PubMed] [Google Scholar]
- 44.Littman DR, Rudensky AY. Th17 and regulatory T cells in mediating and restraining inflammation. Cell. 2010;140(6):845–58. doi: 10.1016/j.cell.2010.02.021. [DOI] [PubMed] [Google Scholar]
- 45.Brunkow ME, et al. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat Genet. 2001;27(1):68–73. doi: 10.1038/83784. [DOI] [PubMed] [Google Scholar]
- 46.Ziegler SF. FOXP3: of mice and men. Annu Rev Immunol. 2006;24:209–26. doi: 10.1146/annurev.immunol.24.021605.090547. [DOI] [PubMed] [Google Scholar]
- 47.Wu Y, et al. FOXP3 controls regulatory T cell function through cooperation with NFAT. Cell. 2006;126(2):375–87. doi: 10.1016/j.cell.2006.05.042. [DOI] [PubMed] [Google Scholar]
- 48.Floess S, et al. Epigenetic control of the foxp3 locus in regulatory T cells. PLoS Biol. 2007;5(2):e38. doi: 10.1371/journal.pbio.0050038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rudra D, et al. Transcription factor Foxp3 and its protein partners form a complex regulatory network. Nat Immunol. 2012;13(10):1010–9. doi: 10.1038/ni.2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Marson A, et al. Foxp3 occupancy and regulation of key target genes during T-cell stimulation. Nature. 2007;445(7130):931–5. doi: 10.1038/nature05478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zheng Y, et al. Genome-wide analysis of Foxp3 target genes in developing and mature regulatory T cells. Nature. 2007;445(7130):936–40. doi: 10.1038/nature05563. [DOI] [PubMed] [Google Scholar]
- 52.Samstein RM, et al. Foxp3 exploits a pre-existent enhancer landscape for regulatory T cell lineage specification. Cell. 2012;151(1):153–66. doi: 10.1016/j.cell.2012.06.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fu W, et al. A multiply redundant genetic switch 'locks in' the transcriptional signature of regulatory T cells. Nat Immunol. 2012;13(10):972–80. doi: 10.1038/ni.2420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dvir A, Conaway JW, Conaway RC. Mechanism of transcription initiation and promoter escape by RNA polymerase II. Curr Opin Genet Dev. 2001;11(2):209–14. doi: 10.1016/s0959-437x(00)00181-7. [DOI] [PubMed] [Google Scholar]
- 55.Portela A, Esteller M. Epigenetic modifications and human disease. Nat Biotechnol. 2010;28(10):1057–68. doi: 10.1038/nbt.1685. [DOI] [PubMed] [Google Scholar]
- 56.Arrowsmith CH, et al. Epigenetic protein families: a new frontier for drug discovery. Nat Rev Drug Discov. 2012;11(5):384–400. doi: 10.1038/nrd3674. [DOI] [PubMed] [Google Scholar]
- 57.Ehrlich M, et al. Amount and distribution of 5-methylcytosine in human DNA from different types of tissues of cells. Nucleic Acids Res. 1982;10(8):2709–21. doi: 10.1093/nar/10.8.2709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16(1):6–21. doi: 10.1101/gad.947102. [DOI] [PubMed] [Google Scholar]
- 59.Wilson CB, Rowell E, Sekimata M. Epigenetic control of T-helper-cell differentiation. Nat Rev Immunol. 2009;9(2):91–105. doi: 10.1038/nri2487. [DOI] [PubMed] [Google Scholar]
- 60.Jamaluddin MS, Yang X, Wang H. Hyperhomocysteinemia, DNA methylation and vascular disease. Clin Chem Lab Med. 2007;45(12):1660–6. doi: 10.1515/CCLM.2007.350. [DOI] [PubMed] [Google Scholar]
- 61.Wang L, et al. Foxp3+ T-regulatory cells require DNA methyltransferase 1 expression to prevent development of lethal autoimmunity. Blood. 2013;121(18):3631–9. doi: 10.1182/blood-2012-08-451765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lan J, et al. DNA methyltransferases and methyl-binding proteins of mammals. Acta Biochim Biophys Sin (Shanghai) 2010;42(4):243–52. doi: 10.1093/abbs/gmq015. [DOI] [PubMed] [Google Scholar]
- 63.He XJ, Chen T, Zhu JK. Regulation and function of DNA methylation in plants and animals. Cell Res. 2011;21(3):442–65. doi: 10.1038/cr.2011.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wu SC, Zhang Y. Active DNA demethylation: many roads lead to Rome. Nat Rev Mol Cell Biol. 2010;11(9):607–20. doi: 10.1038/nrm2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kohli RM, Zhang Y. TET enzymes, TDG and the dynamics of DNA demethylation. Nature. 2013;502(7472):472–9. doi: 10.1038/nature12750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Thomson JP, et al. CpG islands influence chromatin structure via the CpG-binding protein Cfp1. Nature. 2010;464(7291):1082–6. doi: 10.1038/nature08924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dobosy JR, Selker EU. Emerging connections between DNA methylation and histone acetylation. Cell Mol Life Sci. 2001;58(5-6):721–7. doi: 10.1007/PL00000895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128(4):693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 69.Rando OJ, Chang HY. Genome-wide views of chromatin structure. Annu Rev Biochem. 2009;78:245–71. doi: 10.1146/annurev.biochem.78.071107.134639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Latham JA, Dent SY. Cross-regulation of histone modifications. Nat Struct Mol Biol. 2007;14(11):1017–24. doi: 10.1038/nsmb1307. [DOI] [PubMed] [Google Scholar]
- 71.Cosgrove MS, Boeke JD, Wolberger C. Regulated nucleosome mobility and the histone code. Nat Struct Mol Biol. 2004;11(11):1037–43. doi: 10.1038/nsmb851. [DOI] [PubMed] [Google Scholar]
- 72.Krogan NJ, et al. Methylation of histone H3 by Set2 in Saccharomyces cerevisiae is linked to transcriptional elongation by RNA polymerase II. Mol Cell Biol. 2003;23(12):4207–18. doi: 10.1128/MCB.23.12.4207-4218.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ng HH, et al. Targeted recruitment of Set1 histone methylase by elongating Pol II provides a localized mark and memory of recent transcriptional activity. Mol Cell. 2003;11(3):709–19. doi: 10.1016/s1097-2765(03)00092-3. [DOI] [PubMed] [Google Scholar]
- 74.Santos-Rosa H, et al. Active genes are tri-methylated at K4 of histone H3. Nature. 2002;419(6905):407–11. doi: 10.1038/nature01080. [DOI] [PubMed] [Google Scholar]
- 75.Bernstein BE, et al. Methylation of histone H3 Lys 4 in coding regions of active genes. Proc Natl Acad Sci U S A. 2002;99(13):8695–700. doi: 10.1073/pnas.082249499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Vidali G, Gershey EL, Allfrey VG. Chemical studies of histone acetylation. The distribution of epsilon-N-acetyllysine in calf thymus histones. J Biol Chem. 1968;243(24):6361–6. [PubMed] [Google Scholar]
- 77.Allfrey VG, Faulkner R, Mirsky AE. Acetylation and Methylation of Histones and Their Possible Role in the Regulation of Rna Synthesis. Proc Natl Acad Sci U S A. 1964;51:786–94. doi: 10.1073/pnas.51.5.786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Turner BM. Cellular memory and the histone code. Cell. 2002;111(3):285–91. doi: 10.1016/s0092-8674(02)01080-2. [DOI] [PubMed] [Google Scholar]
- 79.Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403(6765):41–5. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 80.Yang XJ. The diverse superfamily of lysine acetyltransferases and their roles in leukemia and other diseases. Nucleic Acids Res. 2004;32(3):959–76. doi: 10.1093/nar/gkh252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sun JM, et al. Phosphorylated serine 28 of histone H3 is associated with destabilized nucleosomes in transcribed chromatin. Nucleic Acids Res. 2007;35(19):6640–7. doi: 10.1093/nar/gkm737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ciechanover A. Intracellular protein degradation: from a vague idea thru the lysosome and the ubiquitin-proteasome system and onto human diseases and drug targeting. Cell Death Differ. 2005;12(9):1178–90. doi: 10.1038/sj.cdd.4401692. [DOI] [PubMed] [Google Scholar]
- 83.Voorhees PM, Orlowski RZ. The proteasome and proteasome inhibitors in cancer therapy. Annu Rev Pharmacol Toxicol. 2006;46:189–213. doi: 10.1146/annurev.pharmtox.46.120604.141300. [DOI] [PubMed] [Google Scholar]
- 84.Tanny JC, et al. Ubiquitylation of histone H2B controls RNA polymerase II transcription elongation independently of histone H3 methylation. Genes Dev. 2007;21(7):835–47. doi: 10.1101/gad.1516207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Shiio Y, Eisenman RN. Histone sumoylation is associated with transcriptional repression. Proc Natl Acad Sci U S A. 2003;100(23):13225–30. doi: 10.1073/pnas.1735528100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Messner S, Hottiger MO. Histone ADP-ribosylation in DNA repair, replication and transcription. Trends Cell Biol. 2011;21(9):534–42. doi: 10.1016/j.tcb.2011.06.001. [DOI] [PubMed] [Google Scholar]
- 87.Esteller M. Non-coding RNAs in human disease. Nat Rev Genet. 2011;12(12):861–74. doi: 10.1038/nrg3074. [DOI] [PubMed] [Google Scholar]
- 88.Schmitz KM, et al. Interaction of noncoding RNA with the rDNA promoter mediates recruitment of DNMT3b and silencing of rRNA genes. Genes Dev. 2010;24(20):2264–9. doi: 10.1101/gad.590910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lee PP, et al. A critical role for Dnmt1 and DNA methylation in T cell development, function, and survival. Immunity. 2001;15(5):763–74. doi: 10.1016/s1074-7613(01)00227-8. [DOI] [PubMed] [Google Scholar]
- 90.Toker A, et al. Active demethylation of the Foxp3 locus leads to the generation of stable regulatory T cells within the thymus. J Immunol. 2013;190(7):3180–8. doi: 10.4049/jimmunol.1203473. [DOI] [PubMed] [Google Scholar]
- 91.Polansky JK, et al. Methylation matters: binding of Ets-1 to the demethylated Foxp3 gene contributes to the stabilization of Foxp3 expression in regulatory T cells. J Mol Med (Berl) 2010;88(10):1029–40. doi: 10.1007/s00109-010-0642-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zorn E, et al. IL-2 regulates FOXP3 expression in human CD4+CD25+ regulatory T cells through a STAT-dependent mechanism and induces the expansion of these cells in vivo. Blood. 2006;108(5):1571–9. doi: 10.1182/blood-2006-02-004747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Baron U, et al. DNA demethylation in the human FOXP3 locus discriminates regulatory T cells from activated FOXP3(+) conventional T cells. Eur J Immunol. 2007;37(9):2378–89. doi: 10.1002/eji.200737594. [DOI] [PubMed] [Google Scholar]
- 94.Polansky JK, et al. DNA methylation controls Foxp3 gene expression. Eur J Immunol. 2008;38(6):1654–63. doi: 10.1002/eji.200838105. [DOI] [PubMed] [Google Scholar]
- 95.Kim HP, Leonard WJ. CREB/ATF-dependent T cell receptor-induced FoxP3 gene expression: a role for DNA methylation. J Exp Med. 2007;204(7):1543–51. doi: 10.1084/jem.20070109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zheng Y, et al. Role of conserved non-coding DNA elements in the Foxp3 gene in regulatory T-cell fate. Nature. 2010;463(7282):808–12. doi: 10.1038/nature08750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wu L, Zheng Q. Active demethylation of the IL-2 Promoter in CD4+ T cells is mediated by an inducible DNA glycosylase, Myh. Mol Immunol. 2014;58(1):38–49. doi: 10.1016/j.molimm.2013.10.016. [DOI] [PubMed] [Google Scholar]
- 98.Dominitzki S, et al. Cutting edge: trans-signaling via the soluble IL-6R abrogates the induction of FoxP3 in naive CD4+CD25 T cells. J Immunol. 2007;179(4):2041–5. doi: 10.4049/jimmunol.179.4.2041. [DOI] [PubMed] [Google Scholar]
- 99.Samanta A, et al. TGF-beta and IL-6 signals modulate chromatin binding and promoter occupancy by acetylated FOXP3. Proc Natl Acad Sci U S A. 2008;105(37):14023–7. doi: 10.1073/pnas.0806726105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Doganci A, et al. Pathological role of IL-6 in the experimental allergic bronchial asthma in mice. Clin Rev Allergy Immunol. 2005;28(3):257–70. doi: 10.1385/CRIAI:28:3:257. [DOI] [PubMed] [Google Scholar]
- 101.Tang M, et al. [Stat 3 signal transduction pathway correlates with proliferation of vascular smooth muscle cells of rats]. Zhonghua Xin Xue Guan Bing Za Zhi. 2005;33(9):832–5. [PubMed] [Google Scholar]
- 102.Xiao Y, et al. Histone acetyltransferase mediated regulation of FOXP3 acetylation and Treg function. Curr Opin Immunol. 2010;22(5):583–91. doi: 10.1016/j.coi.2010.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.van Loosdregt J, et al. Regulation of Treg functionality by acetylation-mediated Foxp3 protein stabilization. Blood. 2010;115(5):965–74. doi: 10.1182/blood-2009-02-207118. [DOI] [PubMed] [Google Scholar]
- 104.Li B, Greene MI. FOXP3 actively represses transcription by recruiting the HAT/HDAC complex. Cell Cycle. 2007;6(12):1432–6. [PubMed] [Google Scholar]
- 105.Li B, et al. FOXP3 interactions with histone acetyltransferase and class II histone deacetylases are required for repression. Proc Natl Acad Sci U S A. 2007;104(11):4571–6. doi: 10.1073/pnas.0700298104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Liu Y, et al. Inhibition of p300 impairs Foxp3(+) T regulatory cell function and promotes antitumor immunity. Nat Med. 2013;19(9):1173–7. doi: 10.1038/nm.3286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.van Loosdregt J, et al. Rapid temporal control of Foxp3 protein degradation by sirtuin- 1. PLoS One. 2011;6(4):e19047. doi: 10.1371/journal.pone.0019047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Akimova T, et al. Histone/protein deacetylase inhibitors increase suppressive functions of human FOXP3+ Tregs. Clin Immunol. 2010;136(3):348–63. doi: 10.1016/j.clim.2010.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Tao R, et al. Histone deacetylase inhibitors and transplantation. Curr Opin Immunol. 2007;19(5):589–95. doi: 10.1016/j.coi.2007.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Beier UH, et al. Histone deacetylases 6 and 9 and sirtuin-1 control Foxp3+ regulatory T cell function through shared and isoform-specific mechanisms. Sci Signal. 2012;5(229):ra45. doi: 10.1126/scisignal.2002873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Parra M, et al. Protein kinase D1 phosphorylates HDAC7 and induces its nuclear export after T-cell receptor activation. J Biol Chem. 2005;280(14):13762–70. doi: 10.1074/jbc.M413396200. [DOI] [PubMed] [Google Scholar]
- 112.Dequiedt F, et al. Phosphorylation of histone deacetylase 7 by protein kinase D mediates T cell receptor-induced Nur77 expression and apoptosis. J Exp Med. 2005;201(5):793–804. doi: 10.1084/jem.20042034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zhang CL, McKinsey TA, Olson EN. Association of class II histone deacetylases with heterochromatin protein 1: potential role for histone methylation in control of muscle differentiation. Mol Cell Biol. 2002;22(20):7302–12. doi: 10.1128/MCB.22.20.7302-7312.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Zhang CL, et al. Class II histone deacetylases act as signal-responsive repressors of cardiac hypertrophy. Cell. 2002;110(4):479–88. doi: 10.1016/s0092-8674(02)00861-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Zhang J, et al. The type III histone deacetylase Sirt1 is essential for maintenance of T cell tolerance in mice. J Clin Invest. 2009;119(10):3048–58. doi: 10.1172/JCI38902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Beier UH, et al. Sirtuin-1 targeting promotes Foxp3+ T-regulatory cell function and prolongs allograft survival. Mol Cell Biol. 2011;31(5):1022–9. doi: 10.1128/MCB.01206-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Lan F, Nottke AC, Shi Y. Mechanisms involved in the regulation of histone lysine demethylases. Curr Opin Cell Biol. 2008;20(3):316–25. doi: 10.1016/j.ceb.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.He S, et al. The histone methyltransferase Ezh2 is a crucial epigenetic regulator of allogeneic T-cell responses mediating graft-versus-host disease. Blood. 2013;122(25):4119–28. doi: 10.1182/blood-2013-05-505180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Tian Y, et al. Global mapping of H3K4me1 and H3K4me3 reveals the chromatin state-based cell type-specific gene regulation in human Treg cells. PLoS One. 2011;6(11):e27770. doi: 10.1371/journal.pone.0027770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.He H, et al. Histone methylation mediates plasticity of human FOXP3(+) regulatory T cells by modulating signature gene expressions. Immunology. 2014;141(3):362–76. doi: 10.1111/imm.12198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Antignano F, et al. Methyltransferase G9A regulates T cell differentiation during murine intestinal inflammation. J Clin Invest. 2014;124(5):1945–55. doi: 10.1172/JCI69592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Wang L, et al. Mbd2 promotes foxp3 demethylation and T-regulatory-cell function. Mol Cell Biol. 2013;33(20):4106–15. doi: 10.1128/MCB.00144-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Cobb BS, et al. T cell lineage choice and differentiation in the absence of the RNase III enzyme Dicer. J Exp Med. 2005;201(9):1367–73. doi: 10.1084/jem.20050572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Muljo SA, et al. Aberrant T cell differentiation in the absence of Dicer. J Exp Med. 2005;202(2):261–9. doi: 10.1084/jem.20050678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Liston A, et al. Dicer-dependent microRNA pathway safeguards regulatory T cell function. J Exp Med. 2008;205(9):1993–2004. doi: 10.1084/jem.20081062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Zhou X, et al. Selective miRNA disruption in T reg cells leads to uncontrolled autoimmunity. J Exp Med. 2008;205(9):1983–91. doi: 10.1084/jem.20080707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Clurman BE, Hayward WS. Multiple proto-oncogene activations in avian leukosis virus-induced lymphomas: evidence for stage-specific events. Mol Cell Biol. 1989;9(6):2657–64. doi: 10.1128/mcb.9.6.2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Eis PS, et al. Accumulation of miR-155 and BIC RNA in human B cell lymphomas. Proc Natl Acad Sci U S A. 2005;102(10):3627–32. doi: 10.1073/pnas.0500613102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Rodriguez A, et al. Requirement of bic/microRNA-155 for normal immune function. Science. 2007;316(5824):608–11. doi: 10.1126/science.1139253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Thai TH, et al. Regulation of the germinal center response by microRNA-155. Science. 2007;316(5824):604–8. doi: 10.1126/science.1141229. [DOI] [PubMed] [Google Scholar]
- 131.Stahl HF, et al. miR-155 inhibition sensitizes CD4+ Th cells for TREG mediated suppression. PLoS One. 2009;4(9):e7158. doi: 10.1371/journal.pone.0007158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Kohlhaas S, et al. Cutting edge: the Foxp3 target miR-155 contributes to the development of regulatory T cells. J Immunol. 2009;182(5):2578–82. doi: 10.4049/jimmunol.0803162. [DOI] [PubMed] [Google Scholar]
- 133.Lu LF, et al. Foxp3-dependent microRNA155 confers competitive fitness to regulatory T cells by targeting SOCS1 protein. Immunity. 2009;30(1):80–91. doi: 10.1016/j.immuni.2008.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Yao R, et al. MicroRNA-155 modulates Treg and Th17 cells differentiation and Th17 cell function by targeting SOCS1. PLoS One. 2012;7(10):e46082. doi: 10.1371/journal.pone.0046082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Zhan Y, et al. SOCS1 negatively regulates the production of Foxp3+ CD4+ T cells in the thymus. Immunol Cell Biol. 2009;87(6):473–80. doi: 10.1038/icb.2009.23. [DOI] [PubMed] [Google Scholar]
- 136.Wei L, Laurence A, O'Shea JJ. New insights into the roles of Stat5a/b and Stat3 in T cell development and differentiation. Semin Cell Dev Biol. 2008;19(4):394–400. doi: 10.1016/j.semcdb.2008.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Turka LA, Walsh PT. IL-2 signaling and CD4+ CD25+ Foxp3+ regulatory T cells. Front Biosci. 2008;13:1440–6. doi: 10.2741/2773. [DOI] [PubMed] [Google Scholar]
- 138.Flowers LO, Subramaniam PS, Johnson HM. A SOCS-1 peptide mimetic inhibits both constitutive and IL-6 induced activation of STAT3 in prostate cancer cells. Oncogene. 2005;24(12):2114–20. doi: 10.1038/sj.onc.1208437. [DOI] [PubMed] [Google Scholar]
- 139.Jager LD, et al. The kinase inhibitory region of SOCS-1 is sufficient to inhibit T-helper 17 and other immune functions in experimental allergic encephalomyelitis. J Neuroimmunol. 2011;232(1-2):108–18. doi: 10.1016/j.jneuroim.2010.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Yu CR, et al. Suppressor of cytokine signaling-1 (SOCS1) inhibits lymphocyte recruitment into the retina and protects SOCS1 transgenic rats and mice from ocular inflammation. Invest Ophthalmol Vis Sci. 2011;52(9):6978–86. doi: 10.1167/iovs.11-7688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Chen T, et al. MicroRNA-125a-5p partly regulates the inflammatory response, lipid uptake, and ORP9 expression in oxLDL-stimulated monocyte/macrophages. Cardiovasc Res. 2009;83(1):131–9. doi: 10.1093/cvr/cvp121. [DOI] [PubMed] [Google Scholar]
- 142.Donners MM, et al. Hematopoietic miR155 deficiency enhances atherosclerosis and decreases plaque stability in hyperlipidemic mice. PLoS One. 2012;7(4):e35877. doi: 10.1371/journal.pone.0035877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Miller FW, et al. Epidemiology of environmental exposures and human autoimmune diseases: findings from a National Institute of Environmental Health Sciences Expert Panel Workshop. J Autoimmun. 2012;39(4):259–71. doi: 10.1016/j.jaut.2012.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Miller FW, et al. Criteria for environmentally associated autoimmune diseases. J Autoimmun. 2012;39(4):253–8. doi: 10.1016/j.jaut.2012.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Ngalamika O, et al. Epigenetics, autoimmunity and hematologic malignancies: a comprehensive review. J Autoimmun. 2012;39(4):451–65. doi: 10.1016/j.jaut.2012.09.002. [DOI] [PubMed] [Google Scholar]
- 146.Selmi C, et al. Mechanisms of environmental influence on human autoimmunity: a National Institute of Environmental Health Sciences expert panel workshop. J Autoimmun. 2012;39(4):272–84. doi: 10.1016/j.jaut.2012.05.007. [DOI] [PubMed] [Google Scholar]
- 147.Selmi C, Lu Q, Humble MC. Heritability versus the role of the environment in autoimmunity. J Autoimmun. 2012;39(4):249–52. doi: 10.1016/j.jaut.2012.07.011. [DOI] [PubMed] [Google Scholar]
- 148.Javierre BM, Hernando H, Ballestar E. Environmental triggers and epigenetic deregulation in autoimmune disease. Discov Med. 2011;12(67):535–45. [PubMed] [Google Scholar]
- 149.Jenke AC, Zilbauer M. Epigenetics in inflammatory bowel disease. Curr Opin Gastroenterol. 2012;28(6):577–84. doi: 10.1097/MOG.0b013e328357336b. [DOI] [PubMed] [Google Scholar]
- 150.O'Connell RM, et al. MicroRNA-155 promotes autoimmune inflammation by enhancing inflammatory T cell development. Immunity. 2010;33(4):607–19. doi: 10.1016/j.immuni.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Costinean S, et al. Pre-B cell proliferation and lymphoblastic leukemia/high-grade lymphoma in E(mu)-miR155 transgenic mice. Proc Natl Acad Sci U S A. 2006;103(18):7024–9. doi: 10.1073/pnas.0602266103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Chen ZX, Riggs AD. DNA methylation and demethylation in mammals. J Biol Chem. 2011;286(21):18347–53. doi: 10.1074/jbc.R110.205286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Zheng Q, et al. Induction of Foxp3 demethylation increases regulatory CD4+CD25+ T cells and prevents the occurrence of diabetes in mice. J Mol Med (Berl) 2009;87(12):1191–205. doi: 10.1007/s00109-009-0530-8. [DOI] [PubMed] [Google Scholar]
- 154.Choi J, et al. In vivo administration of hypomethylating agents mitigate graft-versus-host disease without sacrificing graft-versus-leukemia. Blood. 2010;116(1):129–39. doi: 10.1182/blood-2009-12-257253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Issa JP. Optimizing therapy with methylation inhibitors in myelodysplastic syndromes: dose, duration, and patient selection. Nat Clin Pract Oncol. 2005;2(Suppl 1):S24–9. doi: 10.1038/ncponc0355. [DOI] [PubMed] [Google Scholar]
- 156.Bronner C. Control of DNMT1 abundance in epigenetic inheritance by acetylation, ubiquitylation, and the histone code. Sci Signal. 2011;4(157):pe3. doi: 10.1126/scisignal.2001764. [DOI] [PubMed] [Google Scholar]
- 157.Esteve PO, et al. A methylation and phosphorylation switch between an adjacent lysine and serine determines human DNMT1 stability. Nat Struct Mol Biol. 2011;18(1):42–8. doi: 10.1038/nsmb.1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.He S, et al. Inhibition of histone methylation arrests ongoing graft-versus-host disease in mice by selectively inducing apoptosis of alloreactive effector T cells. Blood. 2012;119(5):1274–82. doi: 10.1182/blood-2011-06-364422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Mallat Z, et al. Induction of a regulatory T cell type 1 response reduces the development of atherosclerosis in apolipoprotein E-knockout mice. Circulation. 2003;108(10):1232–7. doi: 10.1161/01.CIR.0000089083.61317.A1. [DOI] [PubMed] [Google Scholar]
- 160.Mor A, et al. Role of naturally occurring CD4+ CD25+ regulatory T cells in experimental atherosclerosis. Arterioscler Thromb Vasc Biol. 2007;27(4):893–900. doi: 10.1161/01.ATV.0000259365.31469.89. [DOI] [PubMed] [Google Scholar]
- 161.Jia L, et al. Methylation of FOXP3 in regulatory T cells is related to the severity of coronary artery disease. Atherosclerosis. 2013;228(2):346–52. doi: 10.1016/j.atherosclerosis.2013.01.027. [DOI] [PubMed] [Google Scholar]
- 162.Castro R, et al. Increased homocysteine and S-adenosylhomocysteine concentrations and DNA hypomethylation in vascular disease. Clin Chem. 2003;49(8):1292–6. doi: 10.1373/49.8.1292. [DOI] [PubMed] [Google Scholar]
- 163.Jamaluddin MD, et al. Homocysteine inhibits endothelial cell growth via DNA hypomethylation of the cyclin Agene. Blood. 2007;110(10):3648–55. doi: 10.1182/blood-2007-06-096701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Mor A, et al. Altered status of CD4(+)CD25(+) regulatory T cells in patients with acute coronary syndromes. Eur Heart J. 2006;27(21):2530–7. doi: 10.1093/eurheartj/ehl222. [DOI] [PubMed] [Google Scholar]
- 165.Zhao Z, et al. Activation of Th17/Th1 and Th1, but not Th17, is associated with the acute cardiac event in patients with acute coronary syndrome. Atherosclerosis. 2011;217(2):518–24. doi: 10.1016/j.atherosclerosis.2011.03.043. [DOI] [PubMed] [Google Scholar]
- 166.de Boer OJ, et al. Low numbers of FOXP3 positive regulatory T cells are present in all developmental stages of human atherosclerotic lesions. PLoS One. 2007;2(8):e779. doi: 10.1371/journal.pone.0000779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Patel S, et al. The “atheroprotective” mediators apolipoprotein A-I and Foxp3 are over-abundant in unstable carotid plaques. Int J Cardiol. 2010;145(2):183–7. doi: 10.1016/j.ijcard.2009.05.024. [DOI] [PubMed] [Google Scholar]
- 168.Ammirati E, et al. Circulating CD4+CD25hiCD127lo regulatory T-Cell levels do not reflect the extent or severity of carotid and coronary atherosclerosis. Arterioscler Thromb Vasc Biol. 2010;30(9):1832–41. doi: 10.1161/ATVBAHA.110.206813. [DOI] [PubMed] [Google Scholar]
- 169.Lu CX, et al. FOXP3 demethylation as a means of identifying quantitative defects in regulatory T cells in acute coronary syndrome. Atherosclerosis. 2013;229(1):263–70. doi: 10.1016/j.atherosclerosis.2013.05.007. [DOI] [PubMed] [Google Scholar]
- 170.Reddy P, et al. Histone deacetylase inhibitor suberoylanilide hydroxamic acid reduces acute graft-versus-host disease and preserves graft-versus-leukemia effect. Proc Natl Acad Sci U S A. 2004;101(11):3921–6. doi: 10.1073/pnas.0400380101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Kepp O, Galluzzi L, Kroemer G. Immune effectors required for the therapeutic activity of vorinostat. Oncoimmunology. 2013;2(11):e27157. doi: 10.4161/onci.27157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Mann BS, et al. FDA approval summary: vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist. 2007;12(10):1247–52. doi: 10.1634/theoncologist.12-10-1247. [DOI] [PubMed] [Google Scholar]
- 173.Straus DJ, et al. Results of a phase II trial of oral bexarotene (Targretin) combined with interferon alfa-2b (Intron-A) for patients with cutaneous T-cell lymphoma. Cancer. 2007;109(9):1799–803. doi: 10.1002/cncr.22596. [DOI] [PubMed] [Google Scholar]
- 174.Duvic M, et al. A phase II open-label study of recombinant human interleukin-12 in patients with stage IA, IB, or IIA mycosis fungoides. J Am Acad Dermatol. 2006;55(5):807–13. doi: 10.1016/j.jaad.2006.06.038. [DOI] [PubMed] [Google Scholar]
- 175.Vojinovic J, et al. Safety and efficacy of an oral histone deacetylase inhibitor in systemic-onset juvenile idiopathic arthritis. Arthritis Rheum. 2011;63(5):1452–8. doi: 10.1002/art.30238. [DOI] [PubMed] [Google Scholar]
- 176.Tao R, et al. Deacetylase inhibition promotes the generation and function of regulatory T cells. Nat Med. 2007;13(11):1299–307. doi: 10.1038/nm1652. [DOI] [PubMed] [Google Scholar]
- 177.Donas C, et al. Trichostatin A promotes the generation and suppressive functions of regulatory T cells. Clin Dev Immunol. 2013:679804. doi: 10.1155/2013/679804. 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Mishra N, et al. Histone deacetylase inhibitors modulate renal disease in the MRL-lpr/lpr mouse. J Clin Invest. 2003;111(4):539–52. doi: 10.1172/JCI16153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Grabiec AM, et al. Histone deacetylase inhibitors suppress rheumatoid arthritis fibroblast-like synoviocyte and macrophage IL-6 production by accelerating mRNA decay. Ann Rheum Dis. 2012;71(3):424–31. doi: 10.1136/ard.2011.154211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Pauley KM, et al. Upregulated miR-146a expression in peripheral blood mononuclear cells from rheumatoid arthritis patients. Arthritis Res Ther. 2008;10(4):R101. doi: 10.1186/ar2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Jonuleit H, et al. Identification and functional characterization of human CD4(+)CD25(+) T cells with regulatory properties isolated from peripheral blood. J Exp Med. 2001;193(11):1285–94. doi: 10.1084/jem.193.11.1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Read S, Powrie F. CD4(+) regulatory T cells. Curr Opin Immunol. 2001;13(6):644–9. doi: 10.1016/s0952-7915(01)00273-4. [DOI] [PubMed] [Google Scholar]
- 183.Yokokawa J, et al. Enhanced functionality of CD4+CD25(high)FoxP3+ regulatory T cells in the peripheral blood of patients with prostate cancer. Clin Cancer Res. 2008;14(4):1032–40. doi: 10.1158/1078-0432.CCR-07-2056. [DOI] [PubMed] [Google Scholar]
- 184.Liyanage UK, et al. Prevalence of regulatory T cells is increased in peripheral blood and tumor microenvironment of patients with pancreas or breast adenocarcinoma. J Immunol. 2002;169(5):2756–61. doi: 10.4049/jimmunol.169.5.2756. [DOI] [PubMed] [Google Scholar]
- 185.Dannull J, et al. Enhancement of vaccine-mediated antitumor immunity in cancer patients after depletion of regulatory T cells. J Clin Invest. 2005;115(12):3623–33. doi: 10.1172/JCI25947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Zhang H, et al. Lymphopenia and interleukin-2 therapy alter homeostasis of CD4+CD25+ regulatory T cells. Nat Med. 2005;11(11):1238–43. doi: 10.1038/nm1312. [DOI] [PubMed] [Google Scholar]
- 187.Abrams JS, et al. High-dose recombinant interleukin-2 alone: a regimen with limited activity in the treatment of advanced renal cell carcinoma. J Natl Cancer Inst. 1990;82(14):1202–6. doi: 10.1093/jnci/82.14.1202. [DOI] [PubMed] [Google Scholar]
- 188.Negrier S, et al. Recombinant human interleukin-2, recombinant human interferon alfa-2a, or both in metastatic renal-cell carcinoma. Groupe Francais d'Immunotherapie. N Engl J Med. 1998;338(18):1272–8. doi: 10.1056/NEJM199804303381805. [DOI] [PubMed] [Google Scholar]
- 189.Barnett B, et al. Regulatory T cells in ovarian cancer: biology and therapeutic potential. Am J Reprod Immunol. 2005;54(6):369–77. doi: 10.1111/j.1600-0897.2005.00330.x. [DOI] [PubMed] [Google Scholar]
- 190.Shen L, et al. Class I histone deacetylase inhibitor entinostat suppresses regulatory T cells and enhances immunotherapies in renal and prostate cancer models. PLoS One. 2012;7(1):e30815. doi: 10.1371/journal.pone.0030815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Shen L, Pili R. Class I histone deacetylase inhibition is a novel mechanism to target regulatory T cells in immunotherapy. Oncoimmunology. 2012;1(6):948–950. doi: 10.4161/onci.20306. [DOI] [PMC free article] [PubMed] [Google Scholar]