

Abstract
HIV patients with severe periodontitis have high levels of residual virus in their saliva and plasma despite effective therapy (HAART). Multiple short chain fatty acids (SCFAs) from periodontal pathogens reactivate HIV-1 in both Jurkat and primary T-cell models of latency. SCFAs not only activate positive transcription elongation factor b (P-TEFb), which is an essential cellular cofactor for Tat, but can also reverse chromatin blocks by inducing histone modifications. SCFAs simultaneously increase histone acetylation by inhibiting class-1/2 histone deacetylases (HDACs) and decrease repressive histone tri-methylation at the proviral LTR by downregulating expression of the class-3 HDAC sirtuin-1 (SIRT1), and the histone methyltransferases enhancer of Zeste homolog 2 (EZH2) and suppressor of variegation 3–9 homolog 1 (SUV39H1). Our findings provide a mechanistic link between periodontal disease and enhanced HIV-1 replication, and suggest that treatment of periodontal disease, or blocking the activities of SCFAs, will have a therapeutic benefit for HIV patients.
Keywords: Short chain fatty acids, SIRT1, EZH2, P-TEFb, HIV-1 transactivation
Introduction
Periodontal pathogens and viruses, such as HIV, interact with each other in the oral arena to exacerbate disease progression (Mataftsi et al., 2011). Periodontal disease is initiated by infections with periodontal pathogens such as Porphyromonas gingivalis (Pg) and is associated with chronic inflammation (Holt et al., 1988; Pihlstrom et al., 2005). Larger numbers of bacteria responsible for periodontal disease have been found in the plaques of HIV-positive individuals than that of HIV-negative people (Chattin et al., 1999; Ramos et al., 2012; Zambon et al., 1990) and there is an increased prevalence and severity of chronic periodontitis in HIV-positive subjects (Mataftsi et al., 2011; Phiri et al., 2010; Tukutuku et al., 1990). Thus, HIV infection may predispose the patients to chronic periodontitis as a result of immune deficiency, while periodontal pathogens may enhance HIV infection and replication in the oral cavity, creating a vicious cycle of co-infections.
Anaerobic gram-negative bacteria such as Pg and Fusobacterium nucleatum (Fn) produce at least five different short chain fatty acids (SCFAs) as metabolic by-products (Niederman et al., 1996; Niederman et al., 1997). Millimolar levels of the different SCFAs are found in the gingival crevicular fluid of patients with severe periodontal disease while they are undetectable in the gingival crevicular fluid of healthy individuals (Niederman et al., 1996; Niederman et al., 1997). One of the SCFAs, butyric acid, was previously identified from the culture supernatants of Pg and Fn as the molecule responsible for stimulating HIV-1 transactivation (Imai and Ochiai, 2011; Imai et al., 2009; Imai et al., 2012a; Imai et al., 2012b; Imai et al., 2012c; Kantor et al., 2009). Butyric acid inhibits class-1/2 histone deacetylases (HDACs), leading to histone hyperacetylation and induction of viral gene expression and replication. Different than the previous reports, we recently demonstrated that the different SCFAs from periodontal pathogens dose-dependently and additively promote lytic replication of Kaposi’s sarcoma-associated herpesvirus (KSHV) in acutely infected oral epithelial cells and latently infected B lymphocytes (Yu et al., 2014). Therefore, it would be interesting to test if the different SCFAs have similar effects on HIV, which is a very different virus.
Since cytokines and bacterial metabolic products released during inflammation are known to stimulate HIV transcription and productive replication, the infected oral cavity can become a site of enhanced viral replication (Bafica et al., 2004; Mbopi-Keou et al., 2002). In a chronic inflammation milieu such as gingivitis, CD4+ T-cells that are latently infected by HIV are likely present along with uninfected T-cells and macrophages (Fenouillet et al., 1989; Le Naour et al., 1992; Mabondzo et al., 1991; Neuveut et al., 1991; von Briesen et al., 1990). We therefore postulated that when latently infected CD4+ T-cells are exposed to this environment of bacterial infection and chronic inflammation, the proviruses will become reactivated and this will lead to the release of infectious virus.
In the present study, we investigated whether the different SCFAs can induce latent HIV-1 proviral transcription in T-cells. Several features of the metabolism of resting CD4+ T-cells work in an interdependent manner to ensure that latent proviruses remain transcriptionally inactive. First, quiescent T-cells contain minimal levels of P-TEFb, a cellular elongation factor that is an essential cofactor for the HIV transactivator protein Tat and strictly required for efficient HIV transcription (Wei et al., 1998). In resting T-cells, CycT1 is expressed at minimal levels preventing P-TEFb assembly (Ghose et al., 2001). Second, epigenetic silencing due to recruitment of histone deacetylases (HDACs), histone methyltransferases (du Chene et al., 2007; Friedman et al., 2011; Keedy et al., 2009; Pearson et al., 2008) and DNA methylation (Blazkova et al., 2009; Kauder et al., 2009) greatly restrict transcription initiation during latency. Finally, the transcription initiation factors NF-κB and NFAT, which are used to reverse chromatin blocks on latent proviruses, are sequestered in the cytoplasm (Bosque and Planelles, 2008; Kinoshita et al., 1997; Nabel and Baltimore, 1987). Despite these multiple restrictions, stimulation of memory T-cells by cytokines or by T-cell receptor activation provides a powerful signal leading to the resumption of HIV transcription, replication and spread.
We found that all SCFAs, except for acetic acid, are able to potently stimulate latent HIV-1 transcription in both Jurkat-T cells and primary CD4+ T-cells in a dose-dependent and additive manner. Similar to our observations on the activation of KSHV in acutely infected oral epithelial cells and latently infected B lymphocytes (Yu et al., 2014), we found that SCFAs potently inhibit the activity of class-1/2 HDACs in T-cells. In addition, SCFAs downregulate expression of the class-3 HDAC SIRT1 (sirtuin-1, silent information regulator-1), which is a NAD+-dependent HDAC (Guarente, 2000). SCFAs also downregulate expression of EZH2 (enhancer of Zeste homolog2) and SUV39H1 (suppressor of variegation 3–9 homolog1), two histone lysine methyltransferases (HLMTs) that suppresses gene expression through histone-3 (H3) di- and tri-methylation at Lys27 and Lys9, respectively (Cao et al., 2002; Schotta et al., 2003; Sewalt et al., 2002; Shinkai and Tachibana, 2011). Thus, SCFAs simultaneously increase histone acetylation and decrease repressive histone methylation at the proviral promoter. These histone modifications in the promoter region have been previously associated with transactivation of the HIV provirus (Bernhard et al., 2011; Bouchat et al., 2012; du Chene et al., 2007; Friedman et al., 2011; Marban et al., 2007; Pearson et al., 2008; Tyagi et al., 2010; Van Lint et al., 1996; Verdin et al., 1993). Unexpectedly, we also found that SCFAs induce phosphorylation of CDK9 and upregulate expression of CyclinT1 to activate the positive transcription elongation factor b (P-TEFb), which is essential for active transcription elongation of the HIV-1 provirus (Karn, 2013; Massari et al., 2013; Mbonye et al., 2013; Novis et al., 2013). Because of their ability to upregulate P-TEFb, SCFAs are able to stimulate HIV production in primary resting memory T-cells. The novel mechanisms by which SCFAs transactivate latent HIV-1 proviruses provide new insights into how periodontal pathogens contribute to HIV-1 replication and suggest that treatment of periodontal disease, and/or blocking the activities of SCFAs, will have a therapeutic benefit for HIV patients.
Results
Culture supernatants of periodontal pathogens strongly induce HIV-1 transactivation
The culture supernatants of the periodontal pathogens Pg and Fn have been shown previously to induce HIV-1 transactivation (Imai et al., 2009; Imai et al., 2012c). Using a latently infected Jurkat T-cell line (2D10) carrying a fluorescent reporter in the provirus (Fig. 1A) (Friedman et al., 2011; Jadlowsky et al., 2014; Pearson et al., 2008), we observed similar effects of the bacterial supernatants on induction of latent HIV-1 proviruses. As shown in Fig. 1B, treatment with fresh medium (control) and supernatant of the non-oral bacteria Ec resulted in 3.46% and 3.65% of cells expressing the GFP marker respectively. In contrast, when the cells were stimulated with culture supernatants of Pg and Fn at a 1 to 25-fold dilution, 90.87% and 91.69% of the cells became GFP-positive, respectively. In addition, the amounts of GFP-positive cells increased in a dose-dependent manner (Fig. 1C) when 2D10 cells were treated with different dilutions of Pg and Fn supernatants.
Fig. 1.

Culture supernatants of periodontal pathogens Pg and Fn induce HIV-1 transactivation in HIV-1 latently infected Jurkat T-cells. (A) Diagram of the HIV-1 provirus, which is integrated in chromosome 16 at the methionine sulfoxide reductase B1 (MSRB1) locus in Jurkat T-cells, clone 2D10 (Jadlowsky et al., 2014). The provirus carries the fluorescent d2EGFP protein in place of the Nef gene as a marker for transcriptional activation. (B) Flow cytometry histograms showing differences in GFP expression in 2D10 cells treated with fresh medium (bacteria negative control), culture supernatant of Ec (non-oral bacteria and SCFAs-negative control), and culture supernatants of periodontal pathogens Pg and Fn at 1 (bacterial supernatant volume) to 25 (total cell culture medium volume) dilution for 24 hours, respectively. (C) Dose-dependent induction of GFP-expression in 2D10 cells by supernatants of Pg and Fn. Identical amounts of 2D10 cells were treated with different concentrations (2%, 4%, 6%, 8%, and 10% bacterial supernatant volume vs. total culture medium volume) of Pg and Fn supernatants for 24 hours. (D) Fraction of GFP-positive cells in 2D10 cells treated with fresh medium, supernatants of Pg and Fn, filtered Fn supernatant with other bacterial by-products removed while retaining SCFAs, and filtered and heated Fn supernatant with all bacterial by-products including SCFAs removed, respectively.
Since bacteria in the culture supernatants of Pg and Fn had been eliminated by passing through a 0.22 μM filter, the bacterial metabolic by-products in the supernatants were likely responsible for the potent induction of HIV-1 transactivation. Pg and Fn produce multiple metabolic by-products including high molecular weight lipopolysaccharide (LPS), fimbriae, proteinases, and low molecular weight short-chain fatty acids (SCFAs). We previously showed that SCFAs were responsible for reactivation of KSHV (Yu et al., 2014). To verify that SCFAs were responsible for induction of HIV-1 transactivation, we passed the supernatants of Pg and Fn through YM3 filters (Millipore), which only allows the low molecule weight (< 3 kDa) SCFAs through. We also heated the filtered SCFAs containing supernatants of Pg and Fn at 100 °C for 1 hour, which effectively removes the volatile SCFAs (Yu et al., 2014). No difference in the numbers of GFP-positive cells was seen between 2D10 cells treated with the filtered Fn supernatant and those treated with unfiltered Fn supernatants (Fig. 1D). In contrast, the heated Fn supernatant gave rise to substantially lower numbers of GFP-positive cells. Compared to treatment with fresh medium, the relatively higher number of GFP-positive cells from treatment with the heated Fn supernatant is likely due to residual levels of SCFAs in the supernatant after heating (Yu et al., 2014). Thus, SCFAs in the supernatants of Pg and Fn indeed were responsible for induction of HIV-1 transactivation in Jurkat T-cells.
Periodontal Pathogens produce multiple SCFAs that induce latent HIV-1 transactivation
Periodontal pathogens produce at least five different SCFAs including butyric acid, isobutyric acid, isovaleric acid, propionic acid, and acetic acid. The culture supernatants of Pg and Fn contain millimolar levels of the different SCFAs (Yu et al., 2014). Previous studies suggested that butyric acid was the sole SCFA that induced HIV-1 transcription (Imai et al., 2009; Imai et al., 2012c). In contrast to these reports, we found that the HIV-1 latently infected Jurkat T-cells (2D10) could respond to each of the SCFAs except acetic acid, in a dose-dependent manner (Fig. 2A and B). Among the four SCFAs that induced HIV-1 transactivation, butyric acid had the strongest effect while isobutyric acid had the weakest effect. When the cells were treated with different combinations of SCFAs at lower concentration (0.5 mM), an additive effect of each of these SCFAs was observed (Fig. 2C).
Fig. 2.

Different SCFAs induce HIV transactivation dose-dependently and additively. (A) Flow cytometry histograms showing changes in GFP-expression in 2D10 cells upon treatment with different SCFAs at 5 mM for 24 hours. (B) Fraction of GFP-positive cells in 2D10 cells that were treated for 24 hours with different concentrations of acetic acid, A; butyric acid, B; isobutyric acid, IB; isovaleric acid, IV; and propionic acid, P. (C) Fraction of GFP-positive cells in 2D10 cells that were treated with different combinations of SCFAs at 0.5 mM each for 24 hours.
To examine possible synergy between the different SCFAs, we conducted systematic synergy assays in 96-well plates by treating equal numbers of 2D10 cells in each well with two different SCFAs, each with one of the various concentrations (0, 0.1 mM, 0.25 mM, 0.5 mM, 1.0 mM, 1.5 mM, 2.0 mM, and 2.5 mM). Consistent with the results shown in Fig. 2C, additive effects were seen between butyric acid and any of the other SCFAs. A modest synergy was observed between isovaleric acid and propionic acid at 2.0 mM each (Fig. 3A and B). Together, these results suggest that all SCFAs but acetic acid contribute to HIV-1 transactivation, dose-dependently and additively.
Fig. 3.

Isovaleric acid and propionic acid induce HIV-1 transactivation synergistically. (A) Flow cytometry histograms showing changes in GFP expression in 2D10 cells that were treated with 2 mM isovaleric acid and 2 mM propionic acid, alone or in combination for 24 hours. Un-stimulated cells were used as a reference (control). (B) Fraction of GFP-positive cells in 2D10 cells from synergy assays that were treated with a combination of various concentrations of isovaleric acid and butyric acid (left panel) or propionic acid and isovaleric acid (right panel). Only additive effects were seen between isovaleric acid and butyric acid while a modest synergy was observed between propionic acid and isovaleric acid at 2 mM each.
SCFAs induce HIV-1 transcription in primary T-cell models of latency
To examine if SCFAs induce HIV-1 transactivation in primary CD4+ T-cells, we used a model for HIV latency in primary T-cells based on a modified protocol of Bosque and Planelles (Bosque et al., 2011; Bosque and Planelles, 2009; Kauder et al., 2009). Briefly, naïve helper T cells were stimulated using α-CD3/α-CD28 mAb and placed in defined cocktails of cytokines and neutralizing antibodies to polarize cells into a Th17 phenotype, the principle effector memory cell found in gut-associated lymphoid tissue. The polarized cells are then infected using a single round VSV pseudotyped reporter virus, CD8a-d2EGFP-IRES-Nef (Fig. 4A). This reporter carries the tat, rev, env and nef genes of HIV-1 clone NL4-3. In addition, the reporter carries a CD8-d2EGFP fusion protein, which permits cell surface expression of the N-terminal external domain of CD8 and allows for purification of infected cells with magnetic beads. The polarized cells are then expanded in the presence of IL-2 and/or IL-23 (for Th17 cells) and the infected cells are purified using magnetic activated cell sorting. After purification, the HIV-positive cells are then placed in a step down cytokine condition, which gradually reduces the cytokine levels and allows the cells to enter a quiescent cell phenotype. The infected cells were confirmed to be quiescent by flow cytometry analysis of expression of Ki67, CD25, CD69 and Cyclin D3 (data not shown), which are cell cycle arrest markers in these cells (Zack et al., 2013).
Fig. 4.

SCFAs activate P-TEFb and induce HIV-1 transactivation in a primary cell model for HIV-1 latency (Th-17 cells). (A) Schematic diagram of the recombinant HIV-1 virus used to infect Th-17 cells, which carries a “CD8a-d2EGFP-IRES-Nef” reporter in place of the Nef gene. The internal ribosome entry site (IRES) between d2EGFP and Nef ensures expression of both GFP and Nef proteins upon transcriptional activation. Infected cells were purified by antibody selection against CD8a and forced into quiescence by cytokine withdrawal. (B to I) Flow cytometry histograms showing changes in the numbers of cells expressing HIV-1 Nef protein (AF647 positive) and GFP (upper panels) or phosphorylated CDK9 (pS175-AF750) and Cyclin T1 (TRITC) (lower panels) upon stimulation. Unstimulated cells (B) were used as a reference. Fresh medium (E) was used as a bacteria-free control, and TCR activation with dynabeads conjugated to anti-CD3 and anti-CD28 antibodies (C) and treatment with SAHA (500 mM) (D) were used as positive controls for stimulation of HIV-1 reactivation. A mixture of five different SCFAs at 0.1 mM each (F) and Pg culture supernatant at 1:100 (G), 1:50 (H), and 1:25 (I) dilution (bacterial supernatant volume vs. total cell culture medium volume) were used to stimulate the cells. Percentages (%) of quiescent cells and activated cells from each treatment are indicated.
The cells were then stained with an AF647-conjugated antibody specific for HIV-1 Nef protein to monitor induction of the latent HIV provirus (Chang et al., 1998). Once the cells are in quiescence, the level of HIV Nef protein expression is reduced to almost undetectable levels (1.38%), which is indicative that the majority of cells carry latent HIV proviruses (Fig. 4B). GFP levels also decline compared to freshly infected cells but because of the high stability of the CD8a-GFP fusion protein a moderate level of GFP persists in the quiescent cells. Stimulation of the T-cell receptor (TCR) by Dynal beads coated with antibodies to CD28 and CD3 activated 95.48% of the cells which appeared in a unique region of the histogram as Nef+, GFPhi cells (Fig. 4C). Upon stimulation with the class 1/2 HDAC inhibitors, SAHA (500 nM, 24 hours), there was inefficient activation of the cells resulting in a minority of cells (18.04%) appearing in the Nef+, GFPhi region (Fig. 4D).
The quiescent cells were also treated with Pg supernatant and fresh medium (control) at different dilutions, as well as a mixture of five different SCFAs at 0.1 mM each. Treatment with the mixture of SCFAs resulted in 81.50% of Nef+, GFPhi (Fig. 4F). Treatment with Pg supernatant at 1/100, 1/50, and 1/25 dilutions gave rise to 64.10%, 79.26%, and 91.45% of cells that expressed Nef and d2EGFP proteins respectively (Fig. 4G, H, and I). These results clearly demonstrated that SCFAs strongly induce HIV-1 transactivation in latently infected primary T-cells in a dose-dependent manner.
SCFAs activate P-TEFb to permit proviral reactivation in primary cells
Recent studies show that there is a strict correlation between the induction of P-TEFb in primary T-cells and the induction of transcription of latent proviruses (Karn, 2013; Massari et al., 2013; Mbonye et al., 2013; Novis et al., 2013). In order to monitor P-TEFb activation (in the same samples evaluated for HIV-Nef induction), a TRITC-conjugated anti-Cyclin T1 (CycT1) antibody to monitor synthesis of CycT1 (which leads to assembly of P-TEFb) and an AF750-conjugated antibody specific for pSer175, a phosphorylated form of CDK9 which is associated with transcription and excluded from the 7SK RNP pool (Mbonye et al., 2013). As expected for quiescent T-cells, phosphorylation of CDK9 and expression of CycT1 are highly restricted in the unstimulated cells with only 0.18% of cells expressing phosphorylated CDK9 and CycT1 (Fig. 4B). Treatment of the cells with Pg supernatant resulted in a dose-dependent increase in the expression of both markers with 88.17% of the cells expressing P-TEFb and CycT1 at saturating concentration (Fig. 4I). Treatment with the mixture of five SCFAs (0.1 mM each) also resulted in high P-TEFb activation with 79.70% of the cells expressing both markers (Fig. 4F).
In contrast, the class-1/2 HDACs inhibitor SAHA only displayed a modest efficiency on P-TEFb activation, rendering 10.72% of the cells positive for both markers (Fig. 4D). As expected, there is a strong correlation between the number of CDK9-pS175+, CycT1+ cells and the number of Nef+ GFPhi cells, with the vast majority of cells showing co-expression of all four markers. Thus, SCFAs are unexpectedly able to potently activate P-TEFb to promote productive transcription of the latent proviruses in resting memory T-cells, which constitute the bulk of the HIV reservoir found in well-suppressed patients.
Because the protein kinase C (PKC) is typically used to upregulate P-TEFb activity in cells and PKC agonists promote reactivation of latent HIV-1 by increasing the level of P-TEFb (Bartholomeeusen et al., 2013; Bullen et al., 2014; Fujinaga et al., 2012; Mbonye and Karn, 2014), we reasoned that PKC activation might also play a role in the SCFA-dependent activation of P-TEFb in primary T-cells. To test this hypothesis, we treated the quiescent T-cells with fresh medium (control), supernatant of Pg, and a mixture of SCFAs at 0.1 mM each in the presence or absence of the PKC inhibitor Gö6983 (10 mM) for 16 hours. P-TEFb and NF-κB activation was followed by staining of the cells with anti-pS175 CDK9 and anti-pS529 NF-κB p65, a phosphoantibody specific for the nuclear form of NF-κB. In un-stimulated cells (Fig. 5A) and cells treated with fresh medium (Fig. 5C), we found no cells that were CDK9 pS175+, NF-κB pS529+ in the presence or absence of PKC inhibitor. Cells activated through TCR activation gave rise to 88.41% CDK9 pS175+, NF-κB pS529+ cells. The fraction of activated cells was reduced to 10.85% in the presence of PKC inhibitor (Fig. 5B). Cells treated with Pg supernatant at 1:100 and 1:25 dilutions gave rise to 45.55% and 59.88% of CDK9 pS175+, NF-κB pS529+ cells. The fractions of activated cells were reduced to 9.15% and 30.67% respectively in the presence of PKC inhibitor (Fig. 5D and E). It is important to note that although pS175 activation was almost completely abolished by Gö6983, NF-κB phosphorylation remained largely unaffected, suggesting that NF-κB was activated by a PKC-independent pathway induced by SCFAs. Cells treated with the mixture of SCFAs gave rise to 77.39% CDK9 pS175+, NF-κB pS529+ cells, while the proportion of activated cells was reduced to 19.98% in the presence of the PKC inhibitor (Fig. 5F). Thus, the bacterial by-products are able to activate P-TEFb through a PKC-dependent pathway, consistent with their ability to potently induce latent proviruses.
Fig. 5.

SCFAs activate P-TEFb via a PKC-dependent pathway. Resting memory T-cells were unstimulated (A) or treated with dynabeads conjugated to anti-CD3 and anti-CD28 antibodies for T-cell receptor activation (B), control media (C), supernatant of Pg at 1:100 and 1:25 dilutions (D and E), and a mixture of SCFAs at 0.1 mM each for 16 hours in the absence (control) or presence of PKC inhibitor Go 6983 (10 mM). The cells were then stained with antibodies to phosphorylated CDK9 (pS175) and NF-κB P65 (pS529), and analyzed by flow cytometry. Percentages (%) of quiescent and activated cells from each treatment are indicated.
Saliva from patients with severe periodontal disease contains high levels of SCFAs and can strongly induce latent HIV-1 proviral transcription
The gingival crevicular fluids from patients with severe periodontal disease contain millimolar levels of SCFAs (Niederman et al., 1996; Niederman et al., 1997). To demonstrate that SCFAs found in oral fluids have an impact on the host cells to promote HIV-1 transactivation, we collected saliva from seven patients with severe periodontal disease before treatment of the disease and seven individuals without noticeable periodontal disease as controls. As shown in Fig. 6A, significantly higher levels of butyric acid, isovaleric acid, propionic acid, and acetic acid were detected in the saliva of patients with severe periodontal disease.
Fig. 6.

Saliva from patients with severe periodontal disease contains significantly higher levels of SCFAs and strongly induces HIV-1 transcription. (A) Levels of butyric acid, isobutyric acid, isovaleric acid, propionic acid, and acetic acid in saliva from 7 patients with severe periodontal disease and 7 healthy controls. (B) Fraction of GFP+ 2D10 cells treated with saliva from 7 patients with severe periodontal disease and 7 healthy controls at 1:5 dilution (saliva volume vs. total cell culture medium volume) for 24 hours. Unpaired student t test was performed for all comparisons. Difference with a P value < 0.05 is statistically significant.
We treated the HIV-1 latently infected Jurkat cells (2D10) with saliva from the two groups of participants at 1:5 dilution to assess their abilities to induce HIV-1 transcription. As shown in Fig. 6B, all seven saliva samples from patients with severe periodontal disease highly induced HIV-1 transcription and gave rise to 64.41 ± 8.51% of GFP-positive cells. In contrast, the seven saliva samples from healthy controls only gave rise to 7.12 ± 0.62% of GFP-positive cells. The reason that saliva samples from the control group also slightly increased the number of GFP-positive cells when compared to unstimulated cells (1.38% GFP-positive) is likely due to the fact that these samples also contained low levels of SCFAs, which may arise from bacterial infections that have not given rise to clinically significant gingivitis. These results suggest that the significantly higher levels of SCFAs in the saliva of patients with severe periodontal disease indeed promote HIV-1 emergence from latency.
SCFAs inhibit activity of class-1/2 HDACs
In addition to the restrictions imposed by limiting P-TEFb levels, latent HIV genomes are silenced through multiple epigenetic regulatory mechanisms including histone deacetylation and repressive histone methylation (Hakre et al., 2011; Mbonye and Karn, 2011; Van Lint et al., 2013). SCFAs are well known inhibitors of class-1/2 HDACs (Aoyama et al., 2010; Hinnebusch et al., 2002; Waldecker et al., 2008). Using a class-1/2 HDACs activity assay, we found that the supernatants of Pg and Fn, but not Ec, substantially inhibited class-1/2 HDACs activity in Jurkat T-cells (Fig. 7A), which is in full agreement with previous reports (Imai et al., 2009; Imai et al., 2012c).
Fig. 7.
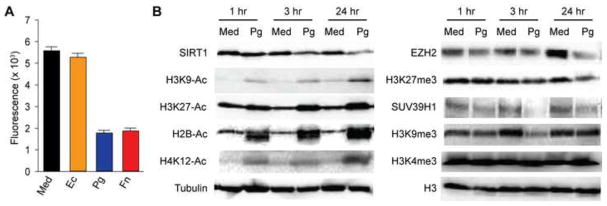
SCFAs inhibit class-1/2 HDACs and downregulate expression of class-3 HDAC SIRT1 and the histone lysine methyltransferases EZH2 and SUV39H1. (A) HDAC class 1/2 activity (fluorescence intensity) in Jurkat T-cells that were treated with fresh medium (Med), supernatant of Ec, and filtered supernatants of Pg and Fn at 1:10 dilution for 6 hours. Equal numbers (2.5×105) of cells were used for each treatment and subsequent measurement of the relative HDACs activity, using a fluorometric assay kit of Class I&II-specific HDACs activity from Sigma-Aldrich. (B and C) Western blot detection of SIRT1, EZH2, SUV39H1, acetylated histone marks H3K9-Ac, H3K27-Ac, H2B-Ac, H4K12-Ac, repressive histone tri-methylation marks H3K27me3 and H3K9me3, and activating histone tri-methylation mark H3K4me3. 2D10 cells were treated with medium (Med) or filtered Pg supernatant at 1:10 dilution for 1 hour, 3 hours, or 24 hours. The levels of β-tubulin and total histone-3 (H3) were used as loading controls and references.
SCFAs downregulate the class-3 HDAC SIRT1 and the histone methyltransferases EZH2 and SUV39H1
To test if SCFAs impact other epigenetic factors to enhance HIV replication as we demonstrated recently in KSHV infected cells (Yu et al., 2014), we examined their effects on the class-3 HDACs, histone methyltransferases and patterns of histone acetylation and methylation (Fig. 7B). Pg supernatants downregulated expression of the class-3 HDAC SIRT1 and the histone methyltransferases EZH2 and SUV39H1, which are responsible for the repressive histone tri-mehylation marks H3K27me3 and H3K9me3, respectively. As shown in Fig. 7B, the level of SIRT1 in 2D10 cells decreased as early as 1 hour post-treatment with Pg supernatant and progressively decreased after 3 hours and 24 hours exposure. In parallel, the levels of different histones acetylation marks including H3K9-Ac, H3K27-Ac, H2B-Ac, and H4K12-Ac increased significantly. Similarly, treatment with Pg supernatant also rapidly decreased the levels of EZH2 and SUV39H1. Consequently, the levels of the repressive histone tri-methylations marks H3K27me3 and H3K9me3 decreased. In contrast, treatment with Pg supernatant did not significantly increase the level of the activating histone methylation mark H3K4me3.
To investigate how SCFAs downregulate SIRT1 and EZH2, we first measured their mRNAs levels by real-time RT-PCR in 2D10 cells that were treated with medium (control) or Pg supernatant for 6 hours. As shown in Fig. 8A, the level of SIRT1 mRNA was 2.6 times lower in cells that were treated with Pg supernatant, suggesting that SCFAs suppress SIRT1 transcription. In contrast, the mRNA level of EZH2 was not significantly reduced. Because SCFA-mediated downregulation of SIRT1 and EZH2 occurred as early as 1 hour post-treatment (Fig. 7B), we suspected that SCFAs might also cause protein degradation in 2D10 cells. Indeed, consistent with our previous observations using KSHV-infected human oral epithelial cells (Yu et al., 2014), treatment of 2D10 cells with supernatant of Pg resulted in SIRT1 and EZH2 degradation (Fig. 8B). To confirm that SCFAs cause SIRT1 and EZH2 proteomic degradation, we treated 2D10 cells with Pg supernatant in the presence of proteasome inhibitor MG-132 and proteasome inhibitor II (P-inh-II), respectively. As shown in Fig. 8B, both proteasome inhibitors effectively blocked EZH2 degradation. In contrast, SIRT1 downregulation could not be completely inhibited by the two inhibitors, which is consistent with the fact that SCFAs suppress SIRT1 transcription. Together, these results confirm SCFAs downregulation of SIRT1 and EZH2 and suggest that these bacterial by-products exert much broader epigenetic impacts than simply acting as class-1/2 HDACs inhibitors.
Fig. 8.
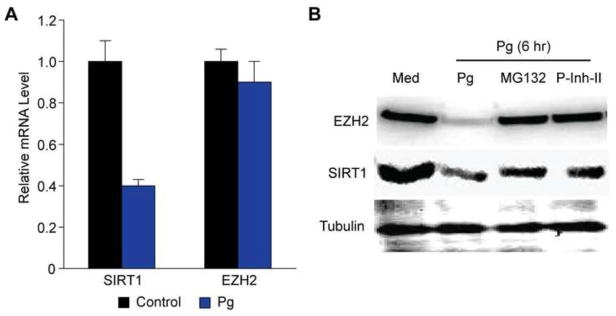
SCFAs inhibit SIRT1 transcription and induce SIRT1 and EZH2 degradation. (A) Relative levels of EZH2 and SIRT1 mRNAs from 2D10 cells that were treated with fresh medium (Med, control) or filtered Pg supernatant at 1:25 dilution for 6 hours. (B) Western blot detection of SIRT1 and EZH2 proteins from 2D10 cells that were treated with medium (Med) or Pg supernatant at 1:10 dilution in the absence or presence of 2 μM proteasome inhibitor MG-132 and proteasome inhibitor II (P-Inh-II) for 6 hours. Beta-tubulin was used as reference for loading control.
We next examined the effects of individual SCFAs on expression of SIRT1 and EZH2 and histone modifications. As shown in Fig. 9, all SCFAs with the exception of acetic acid increased the level of acetylated H3 (H3K27-Ac). The individual SCFAs also reduced expression of SIRT1 and EZH2 with a corresponding reduction in repressive histone mark H3K27me3, in a dose-dependent manner with butyric acid displaying the highest potency. The different SCFAs also induced expression of HIV-1 Tat protein in a dose-dependent manner. Notably, the global changes in histones modification marks correlated with Tat expression and the viral transactivation patterns shown previously in Fig. 2, further suggesting that SCFAs induce HIV-1 Tat-dependent transcription by simultaneously increasing histone acetylation and decreasing repressive histone tri-methylation.
Fig. 9.
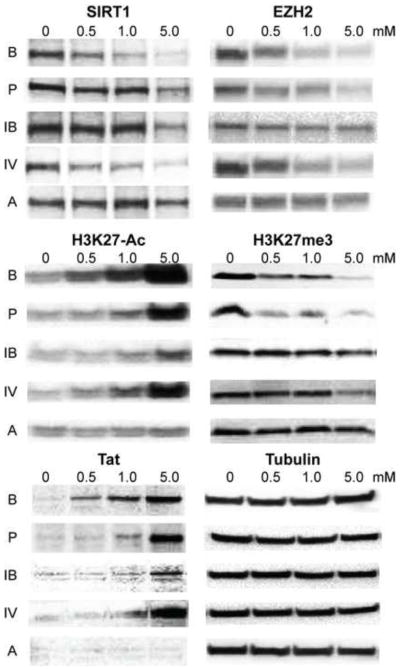
Western blot detection of SIRT1, EZH2, H3K27-Ac, H3K27me3, HIV-1 Tat protein, and β-tubulin in 2D10 cells that were treated with different concentrations (mM) of butyric acid, B; isobutyric acid, IB; propionic acid, P; isovaleric acid, IV and acetic acid, A for 24 hours.
SIRT1 and EZH2 are present as a complex at the HIV-1 LTR
We previously demonstrated that EZH2 is involved in epigenetic silencing of HIV-1 through H3K27 methylation and is present at the LTR of latent provirus (Friedman et al., 2011). Since both EZH2 and SIRT1 are simultaneously downregulated by SCFAs, we examined the possibility that these two proteins could physically associate with each other. In a co-IP experiment, we precipitated EZH2 from cell lysates of 2D10 cells that were either treated with medium (control) or supernatant of Pg for 6 hours using a rabbit anti-EZH2 antibody or control IgG. Data from Western blot analysis of the co-IP products indicated that the anti-EZH2 antibody pulled down not only EZH2 and another subunit of the PRC-2, SUZ12, but also SIRT1 (Fig. 10). In contrast, the control IgG could pull down neither protein. As expected, both EZH2 and SIRT1 were downregulated in cells that were treated with Pg supernatant. These results suggest that EZH2 and SIRT1 are both components of a larger Polycomb repressive complex assembled on the proviral genome, which includes both PRC2 (EZH2) and PRC4 (SIRT1) as previously suggested (Friedman et al., 2011; Kuzmichev et al., 2005).
Fig. 10.
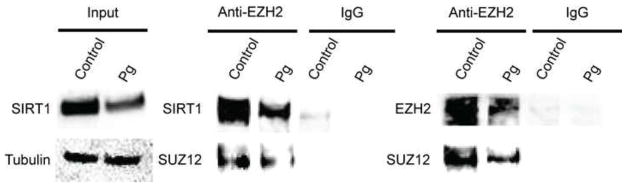
SIRT1 and EZH2 are present as a complex in HIV-1 latently infected Jurkat T-cells. Total proteins extracts from equal numbers (2×106) of HIV-1 latently infected Jurkat T-cells (2D10) that were treated with fresh medium (control) or filtered Pg supernatant at 1:25 dilution for 6 hours were immuno-precipitated (IP) with a rabbit anti-EZH2 polyclonal antibody or control rabbit IgG. The input protein samples and the resulting IP products were then subjected to SDS-PAGE and Western blot detection with antibodies to SIRT1, EZH2 or another PRC2 subunit, SUZ12.
To further test this hypothesis and examine how SCFAs stimulation affects the presence of the EZH2 and SIRT1 complex at the HIV-1 LTR, we next performed ChIP assays using chromatin isolated from 2D10 cells treated with medium (control) or Pg supernatant for 1 hour and 6 hours respectively. As described previously the protein distribution on the proviral LTR was measured by using three pairs of PCR primers specific for regions within or in the immediate vicinity of HIV-1 promoter (Friedman et al., 2011; Jadlowsky et al., 2014; Kim et al., 2011). As controls, one pair of primers was used to measure DNA outside of the LTR, and another pair of primers was used to examine the promoter region of the constitutively expressed housekeeping gene GAPDH.
As shown in Fig. 11, SIRT1 and EZH2 are present at the HIV-1 promoter and its vicinity in latently infected cells. Upon treatment with Pg supernatant, they are removed from the LTR within 1 hour. Consistent with these results, the levels of acetylated histone marks H4K12-Ac and H2B-Ac were increased in LTR upon stimulation with Pg supernatant while the level of suppressive histone marks H3K27me3 and H3K9me3 were reduced. These changes in histone marks were coupled with increased recruitment of RNA polymerase II (RNAP II). In contrast to antibodies specific for the different histone marks, ChIP assays performed with the control IgG pulled down 10-fold lower amounts of DNA and there were no obvious differences between cells with and without stimulation.
Fig. 11.

SCFAs simultaneously increase histone acetylation and decrease repressive histone tri-methylation in the HIV-1 promoter. Chromatin was isolated from equal numbers (6×107) of HIV-1 latently infected Jurkat T-cells (2D10) treated with fresh medium (control) or filtered Pg supernatant at 1:25 dilution for 1 hour or 6 hours. After sonication, DNA fragments were immuno-precipitated with antibodies to SIRT1, EZH2, H3, H4K12-Ac, H2B-Ac, H3K27me3 H3K9me3, RNA polymerase II (RNAP II) or control IgG. DNA was amplified by real-time PCR using primers specific for the Nuc-0 (−390 to −383), HIV-1 promoter (−116 to +4) and Nuc-1 (+30 to +134) regions. Primers specific for region outside of the HIV-1 LTR (+611 to +770) and primers for the host house-keeping gene GAPDH were used as controls. Data was expressed as a fraction of the sample input DNA. Error bars represent the standard deviations of three repeats of real-time PCR measurement.
Collectively, these results confirm that SCFAs modulate multiple epigenetic regulators to induce different histone modifications including increased levels of histone acetylation marks and decreased levels of repressive histone tri-methylation marks in the LTR to promote HIV-1 transactivation.
Knock-down or inhibition of SIRT1 induces HIV-1 transactivation
The role of SIRT1 in controlling HIV-1 silencing and transactivation has been controversial (Pagans et al., 2005; Pinzone et al., 2013; Zhang and Wu, 2009). To verify that SCFAs downregulation of SIRT1 contributes to HIV-1 induction, we infected 2D10 cells with lentiviruses expressing five different SIRT1-pecific shRNAs and a control shRNA. The fraction of GFP-positive cells increased significantly in all the cells transduced with the individual SIRT1-specific shRNAs compared to cells trasduced with the control shRNA (Fig. 12A and B), suggesting that knock-down of SIRT1 expression is sufficient to induce HIV-1 transactivation. As expected all five SIRT1-specific shRNAs effectively knocked down expression in 2D10 cells (Fig. 12C).
Fig. 12.

Knockdown of SIRT1 induces HIV-1 transcription. (A) Flow cytometry showing changes in GFP-expression in 2D10 cells that were infected with lentiviruses expressing five SIRT1-specific shRNAs or control shRNA. (B) Mean percentages (%) of GFP-positive cells from 2D10 cells that were transduced with lentiviruses expressing five different SIRT1-specific shRNAs or control shRNA. (C) Western blot detection of SIRT1 and beta-tubulin from 2D10 cells that were transduced with lentiviruses expressing five different SIRT1-specific shRNAs or control shRNA.
To further confirm the involvement of SIRT1 in HIV-1 latency, we next treated 2D10 cells with SIRT1 inhibitor Sirtinol (10 μM) in the presence or absence of the class-1/2 HDACs inhibitor SAHA (0.5 μM). Both Sirtinol and SAHA increased the numbers of GFP-positive cells (Fig. 13A and B), and the combination of SAHA and Sirtinol further increased the numbers of GFP-positive cells in an additive manner. Thus, SIRT1 appears to contribute directly to HIV latency and suppression of SIRT1 induces HIV-1 transactivation.
Fig. 13.

Inhibition of SIRT1 activity induces HIV-1 transactivation. (A) Flow cytometry histograms showing changes in GFP-expression in 2D10 cells that were treated with SIRT1 inhibitor Sirtinol (10 μM) and the class-1/2 HDACs inhibitor SAHA (0.5 μM), alone or in combination for 24 hours, in comparison to cells treated with placebo (DMSO, control). (B) Fraction of GFP-positive cells in 2D10 cells that were treated with Sirtinol and SAHA, alone or in combination.
Discussion
Induction of HIV transcription by bacterial SCFAs
Although HARRT can effectively inhibit HIV replication and limit the viral load to nearly undetectable levels in HIV patients, sporadic reactivation of the latent virus can still occur and many patients show “blips” of viremia during the course of their treatment. This is particularly true in HIV patients who have severe periodontal disease, in which the milieu of pathogenic bacterial infection and chronic inflammation appears to enhance HIV replication. Not only are more periodontal pathogens such as Pg found in HIV-positive individuals than in healthy controls (Chattin et al., 1999; Scully et al., 1999), but there was a significant correlation between the clinical stage of periodontitis and HIV-1 proviral DNA load in the gingival crevicular fluid and HIV-1 viral RNA load in the plasma and saliva (Maticic et al., 2000; Shugars et al., 2000). The interactions between periodontal pathogens and HIV are therefore likely to create a vicious cycle of enhanced viral induction and bacterial replication in the oral cavity of HIV patients.
Understanding the molecular mechanisms of this microbial interaction may hold the key to developing effective treatment for both periodontal disease and limiting viral infection in the oral cavity of HIV patients. In this study, we demonstrated that SCFAs from anaerobic gram-negative bacteria such as Pg and Fn very strongly induced HIV-1 transactivation in latently infected T-cells. Importantly, similar activities were found in both Jurkat T-cell models of latency and in primary T-cells. One of the SCFAs, butyric acid, has been previously reported to promote HIV transactivation by inhibiting class-1/2 histone deacetylases (HDACs) (Imai et al., 2012a; Imai et al., 2009; Imai et al., 2012c; Kantor et al., 2009), leading to histone hyperacetylation and induction of viral gene expression and replication. However, in contrast to the previous reports, we found that all SCFAs, with the exception of acetic acid, induced HIV-1 transactivation in a dose-dependent manner.
Most of the different SCFAs exhibited additive effects while isovaleric acid and propionic acid displayed a modest synergy. In a highly inflammatory microenvironment such as periodontal disease, excessive amounts of infiltrating inflammatory cells and lymphocytes, including HIV-latently infected CD4+ T-cells (Fenouillet et al., 1989; Le Naour et al., 1992; Mabondzo et al., 1991; Neuveut et al., 1991; von Briesen et al., 1990), could be present in the oral arena of HIV patients. Because millimolar levels of the different SCFAs are simultaneously present in the gingival crevicular fluid and saliva of patients with severe periodontal disease, these bacterial by-products may act together to constantly stimulate the HIV-latently infected CD4+ T-cells to induce HIV transactivation and promote productive viral replication. Our finding that saliva from patients with severe periodontal disease very strongly induced HIV transactivation in HIV-1 latently infected Jurkat T-cells in vitro further supports this notion. The oral cavity of HIV patients could therefore be an important viral source for recurrent HIV induction. It is, therefore, necessary to take the interaction between periodontal pathogens and HIV into consideration when it comes to finding strategies for improving anti-viral therapy and eradication of the latent HIV reservoir.
SCFAs activate P-TEFb: a novel mechanism for HIV-1 transactivation in primary T-cells
In primary resting T-cells, RNA polymerase II transcription elongation and Tat activity is highly restricted due to very low levels of P-TEFb expression in the quiescent cells (Chiang et al., 2012; Herrmann et al., 1998; Ramakrishnan et al., 2009; Ramakrishnan and Rice, 2011). P-TEFb is a heterodimer of the CDK9 serine/threonine kinase and a C-type regulatory cyclin, Cyclin T1 (CycT1). Human Cyclin T1 (hCycT1) binds directly to Tat and enhances the co-operative binding of P-TEFb/Tat to TAR RNA by binding to its apical loop (Bieniasz et al., 1998; Fujinaga et al., 1999; Wei et al., 1998). P-TEFb is then able to stimulate HIV transcription elongation by phosphorylating a variety of positive and negative factors. For positive regulation, hyperphosphorylation of serine residues of the heptad repeats at the CTD of RNAP II by Tat-stimulated P-TEFb enhances its processivity (Isel and Karn, 1999; Kim et al., 2002; Marciniak and Sharp, 1991; Parada and Roeder, 1996). Similarly, phosphorylation of the C-terminal region of the SPT5 subunit of DSIF by P-TEFb transforms it into a positive elongation factor (Bourgeois et al., 2002; Yamada et al., 2006). Removal of transcriptional blocks is due to the phosphorylation of the E-subunit of the negative elongation factor NELF by P-TEFb which forces its dissociation from paused RNAP II complexes and allows resumption of productive elongation (Fujinaga et al., 2004; Jadlowsky et al., 2014; Natarajan et al., 2013). It has also been recently discovered that Tat mediates the recruitment of a large “super elongation complex” containing numerous additional elongation factors (He et al., 2011; He et al., 2010; Liu et al., 2012; Sobhian et al., 2010).
Using a primary CD4+ T-cell model of HIV-1 latency, we found unexpectedly that SCFAs are capable of inducing phosphorylation of CDK9 at Ser175, a modification that we have previously shown is associated with the transcriptionally active form of P-TEFb (Mbonye et al., 2013b). In addition, SCFAs also strongly induce CycT1 expression, further suggesting that these bacterial by-products activate P-TEFb to promote transcription elongation of the integrated provirus. Importantly, we found that the PKC inhibitor Gö6983 strongly inhibits SFCA-dependent activation of P-TEFb, without blocking NF-κB activation, indicating that PKC mediates SCFAs activation of P-TEFb. While the roles of PKC in activation of P-TEFb and HIV-1 transactivation is extremely well documented (Bartholomeeusen et al., 2013; Fujinaga et al., 2012; Mbonye and Karn, 2014), how SCFAs impact the PKC signaling pathway remains to be determined. One possibility is that SCFAs bind to their cellular receptors GPR41 and/or GPR43, which are G-protein coupled receptors (Eberle et al., 2014; Kimura et al., 2014) that are known to activate PKC upon stimulation with GPCR agonists (Naor et al., 2000; Rozengurt, 2007). This type of mechanism may explain why SCFAs activate P-TEFb much more efficiently than SAHA (Fig. 4), a HDACs inhibitor that is just as potent as SCFAs in inducing histone modifications. In summary, our results highlight an additional unexpected mechanism of SCFAs for the induction of HIV-1 transactivation involving the induction of P-TEFb in resting primary T-cells.
Regulation of SIRT1, EZH2 and SUV39H1 by SCFAs
SCFAs are well-known for their ability to inhibit class-1/2 HDACs, which results in histone hyperacetylation and promote gene expression (Chen et al., 2003; Kuzmichev et al., 2005; Santini et al., 2007). Previous studies have associated the ability of SCFAs to inhibit class-1/2 with the induction of HIV-1 transcription (Imai et al., 2009; Imai et al., 2012c; Kantor et al., 2009). However, complete silencing of HIV genome involves multiple epigenetic silencing mechanisms including DNA methylation and specific patterns of histone methylation (Hakre et al., 2011; Mbonye and Karn, 2011; Van Lint et al., 2013). In order to assess the broader epigenetic effects of SCFAs, we investigated whether these molecules also impact the class-3 HDAC SIRT1 and the histone methyltransferases EZH2 and SUV39H1. We found surprisingly that SCFAs not only inhibit the activity of class-1/2 HDACs but also downregulate expression of SIRT1, EZH2, and SUV39H1, leading to concomitant increases in the levels of histone acetylation and decreases in the levels of the repressive histone marks H3K27me3 and H3K9me3. SCFAs both directly inhibit SIRT1 transcription and cause proteomic degradation of the SIRT1 and EZH2 proteins.
Importantly, data from co-IP and ChIP experiments indicate that SIRT1 and EZH2 interact with each other and are present as part of a large complex recruited to the LTR of HIV-1 proviral genomes during latent infections. Treatment of cells with SCFAs reduces expression of both proteins and rapidly lowers their presence in the viral LTR. In consequence, the levels of acetylated histones such as H4K12-Ac and H2B-Ac increase while the level of repressive histone mark H3K27me3 decrease in the viral promoter region following SCFA exposure. Thus, SCFAs not only suppress HDACs to increase histone acetylation but also downregulate EZH2 to decrease repressive histone methylation at specific viral chromatins to induce HIV-1 transactivation.
These data confirm and extends our previous finding that transcription silencing complex PRC2 plays an important role in silencing HIV-1 genome (Friedman et al., 2011). The association of EZH2 and SIRT1 in a complex at the HIV-1 promoter region is unexpected but may reflect interactions between PRC2 and the SIRT1-containing PRC4 complex suggested previously (Kuzmichev et al., 2005). Based on these new findings, we conclude that the mechanism of SCFAs induction of viral replication is far more complicated than simply acting as HDACs inhibitors as previously claimed. Instead, these bacterial by-products modulate multiple components of the host epigenetic regulatory machinery to orchestrate viral transactivation by inducing multiple histone modifications including increased histone acetylation and decreased repressive histone methylation.
The role of SIRT1 in HIV transcription is only poorly understood. Multiple studies have demonstrated that HIV-1 Tat protein is a substrate of SIRT1 for deacetylation and functional inhibition or knockdown of SIRT1 is known to enhance Tat’s ability to transactivate HIV-1 (Pinzone et al., 2013; Zhang et al., 2010; Zhang and Wu, 2009). By contrast, in SIRT1-null mouse cells, where there is no SIRT1 expression, Tat transactivation is defective but can be rescued by expression of SIRT1 (Pagans et al., 2005). SIRT1 is also known to deacetylate lysine 310 in the p65 subunit of nuclear factor-κB (NF-κB), and inhibition of SIRT1 results in acetylation of p65 and activation of the NF-κB pathway and enhancement of HIV transcription (Kwon et al., 2008).
Our results are consistent with a dual role for SIRT1. Functional inhibition by SCFAs or Sirtinol, or knockdown of SIRT1 expression by shRNA, results in the induction of latent HIV proviral transcription, suggesting that SIRT1 contributes to HIV-1 latency. For example, reduced levels of SIRT1 may enhance Tat activity through the accumulation of acetylated Tat protein. However, we hypothesize that residual SIRT1 is required in these experiments in order to support HIV transcription. Consistent with this idea, the ChIP assays showed an increased level of SIRT1 at the HIV-1 LTR at 6 hours post-treatment with Pg supernatant compared to 1 hour post-treatment. It seems likely that this increase is due to the recycling of a SIRT1-Tat complex used during HIV-1 transactivation.
Conclusion
We have demonstrated that multiple SCFAs from periodontal bacteria can act as potent activators of latent HIV-1 transactivation in both cell models and latently infected primary cells. Our study reveals novel mechanisms that allow these bacterial metabolic by-products to create a permissive environment of HIV transcription by upregulating P-TEFb and downregulating the epigenetic silencing machinery. These new findings provide further insights into the epidemiological links between chronic periodontitis and increased HIV replication in the oral cavity and highlight the importance of treating periodontal disease in HIV-infected patients.
Materials and Methods
Reagents
Molecular grade SCFAs butyric acid, isobutyric acid, isovaleric acid, propionic acid, and acetic acid, proteomic inhibitor MG132, SIRT1 inhibitors Sirtinol, class-1/2 HDACs inhibitor suberoyanilide hydroxamic acid (SAHA), and EZH2 inhibitor UNC were purchased from Sigma-Aldrich. Antibodies against EZH2, SIRT1, SUV39H1, RNA polymerase II, histone-3 (H3), acetylated histones including H3K27-Ac, H3K9-Ac, H4K12-Ac, and H2B-Ac, repressive histone marks H3K27me3 and H3K9me3, and activating histone methylation mark H3K4me3 were purchased from Millipore.
Cell culture
2D10 cells are a clone of Jurkat T-cells that is latently infected with a defective recombinant HIV-1 carrying a d2EGFP reporter protein to monitor viral transactivation (Pearson et al., 2008). 2D10 cells were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS), penicillin (100 IU/ml), streptomycin (100 μg/ml) and 25mM HEPES in 5% CO2 at 37 °C.
To test the efficiency of SCFAs on HIV-1 latency in primary T-cells, a polarized model of HIV-1 latency modified from a previously described method was generated based on the methods of Bosque and Planelles (Bosque et al., 2011; Bosque and Planelles, 2009; Kauder et al., 2009). Briefly, naïve primary CD4+ T-cells from healthy donors were isolated from leukapheresis packs, activated by anti-CD3/CD28 stimulation of the TCR and polarized into a Th-17 cell phenotype. During the exponential growth phase, the cells were infected by a recombinant HIV-1 carrying a CD8-EGFP fusion protein using spinoculation. The infected cells were purified on magnetic beads and then allowed to enter quiescence by placing the cells in a restrictive cytokine environment. Cells were demonstrated to be quiescent prior to exposure to SCFAs by monitoring CD25, CD69 and CyclinD3 levels using flow cytometry.
Bacterial strains and culture conditions
Periodontopathgens Pg (ATCC 33277) and Fn (ATCC 25586) were maintained in blood agar plates (Fisher) and grown in Enriched Trypticase Doy Broth (ETDB) as described previously (Syed, 1980) in an anaerobic system (5% CO2, 10% H2, and 85% N2 at 37 °C) to a late log phase. The supernatants were collected by centrifugation at 10,000 x g for 10 minutes at 4 °C to remove bacteria, followed by sterilization through a 0.22 μm pore size membrane filter. Escherichia coli (Ec) were cultured in LB medium under aerobic condition (37 °C with shaking). Under such condition, no SCFA was detected in the culture supernatant of Ec.
Assessment of periodontal disease and collection of saliva
We used a protocol that is very similar to what has been reported (Vernon et al., 2009) to define and assess the severity of periodontal disease of all recruited participants, which includes: 1) probing depth (PD) and clinical attachment level (CAL) at six sites per tooth; 2) gingival recession; and 3) percentage of sites with bleeding on probing (BOP). The study was performed by fully following an IRB protocol (IRB-2012-253, Epigenetic regulation of viral replication by periodontal bacteria) approved by the Ethics Committee of Case Western Reserve University and with the written consent of all participants. Participants having CAL ≥ 6 mm with at least 5 mm PD in 30% of the sites were considered as patients with severe periodontal disease. Participants having CAL ≤ 2 mm with PD ≤ 3 mm on all teeth were selected as healthy controls. About 5 ml saliva was collected from each participant. After centrifugation to remove cells and debris, the supernatant of the collected saliva was sterilized by passing through a 0.22 μm filter, which was either used immediately for experiment or stored at −80 °C.
Quantification of SCFAs
Gas phase chromatography was used to determine the concentrations of butyric acid, isobutyric acid, isovaleric acid, propionic acid, and acetic acid in the collected saliva, as described previously (Niederman et al., 1996; Niederman et al., 1997). A series of dilutions of each of the pure SCFAs were first run to establish a standard curve reflecting the correlation between signal pick area and the concentration of the analyte, and the concentration of the analyte in the sample was determined by comparing its pick area with the standard curve. The concentration of the SCFA in question for a given sample was calculated as the average value from triplicate measurement.
Co-immunoprecipitation (co-IP) and Western blot analysis
HIV-1 latently infected Jurkat T-cells (2×106 cell/ml) were treated with Pg supernatants or fresh medium (control) at 1 (supernatant volume) to 25 (total cell culture medium volume) dilution for 6 hours. The cells were harvested and re-suspended in ice cold lysis buffer containing 400 mM NaCl, 1 mM EDTA, 1 mM DTT, 100 mM Tris-HCl (pH 7.4), 0.5% Triton-X, 0.2% NP-40, and proteinase inhibitors cocktail, and incubated on ice for 30 minutes. After centrifugation (10,000 g, 10 minutes), the supernatants were collected and incubated with a rabbit polyclonal anti-EZH2 antibody or control rabbit IgG at 4°C for overnight with shaking, followed by adding Protein-A/Protein-G agarose beads and incubation at room temperature for 2 hours. After two rounds of washing with the lysis buffer and two rounds of washing with phosphate saline, the protein-antibody complexes were eluted in lysis buffer plus 2% SDS and subjected to SDS-PAGE analysis and Western blot analysis.
Measurement of HIV-1 transactivation by flow cytometry
To determine the efficiency of SCFAs in inducing HIV-1 transactivation, HIV-1 latently infected Jurkat T-cell line 2D10 was stimulated with various doses of SCFAs or bacterial supernatants at different dilutions. The cells were then washed in Auto-MACs running buffer and the numbers of cells with d2EGFP expression were determined by flow cytomerty (BD LSRFortessa cell analyzer) analysis.
To measure HIV-1 transactivation in primary T-cells that were latently infected with HIV-1, following stimulation with bacterial supernatants or SCFAs, the cells were stained for HIV-1 Nef protein expression by using an AF647 conjugated antibody to Nef protein (AIDS Reagents, Item#709), an AF750 conjugated antibody to phosphorylated pTEF-b (pS175) (Mbonye et al., 2013), and a TRITC-conjugated antibody to Cyclin T1 (Santa Cruz Biotechnology, Inc), followed by flow cytometry quantification of Nef-positive, pS175-positive, and Cyclin T1-positive cells.
HDAC activity assay
Measurement of the relative activity of class-1/2 HDACs was conducted by using a fluorometric histone deacetylase assay kit from Sigma-Aldrich following the manufacturer’s instruction. Equal numbers (2.5×105) of Jurkat T-cells were treated with fresh medium, supernatant of non-oral bacteria Ec, and supernatants of Pg and Fn at 1 (bacterial supernatant volume) to 25 (total cell culture medium volume) dilution for 6 hours. The cells were harvested and re-suspended in the assay buffer, then loaded in 96-well plate. Upon incubation with the substrate, the plate was read under a fluorimeter at excitation wavelength of 380 nm and emission wavelength of 480 nm. The relative HDACs activity of the sample is represented as the mean value of fluorescence intensity from three repeats.
Isolation of total RNAs and quantification of mRNA by qRT-PCR
Total RNAs from HIV-1 latently infected Jurkat T-cells (2D10) were isolated using a RNA purification kit from QIAGEN, which includes a step to remove residual genomic DNA prior to RNA purification. Reverse transcription (RT) of total RNA was performed by using Superscript Transcriptase II (Invitrogen, Carlsbad, CA). Quantitative RT-PCR was performed to measure the relative levels of SIRT1 and EZH2 mRNAs using the following pairs of primers: 5′CCTGACTTCAGATCAAGAGACGGTA3′ (SIRT1 forward) and 5′CTGATTAAAAATGTCTCCACGAACAG3′ (SIRT1 reverse); and 2) 5′GTGGAGAGATTATTTCTCAAGATG3′ (EZH2 forward) and 5′CCGACATACTTCAGGGCATCAGCC3′ (EZH2 reverse). The mRNA level of the house-keeping gene β-actin was used as reference for normalization and determined by using the primers 5′ATTGCCGACAGGATGCAGA3′ (actin forward) and 5′GAGTACTTGCGCTCAGGAGGA3′ (actin reverse). All qRT-PCR reactions consist of three repeats.
Chromatin immuno-precipitation (ChIP) assay
Equal numbers of HIV-1 latently infected Jurkat T-cells 2D10 (6×107) were treated with filtered culture supernatant of Pg or fresh medium (control) at 1 (supernatant volume) to 25 (total cell culture medium volume) dilution for 1 hour and 6 hours respectively, followed by fixation with 0.5% formaldehyde for 15 minutes. Preparation of chromatin suspensions from these cells and ChIP were conducted by using a ChIP assay kit from Invitrogen and following instruction from the manufacture. Antibodies against EZH2, SIRT1, RNA polymerase II (RNAP II), total histone-3 (H3), acetylated histones (H4K12-Ac and H2B-Ac), and methylated histone-3 (H3K27me3 and H3K9me3), all from Millipore, were used for the procedure. DNA from input and the end ChIP products were isolated by using DNA purification kit from QIAGEN. The purified DNA were re-suspended in 100 μl sterile water and used for real-time PCR quantification of DNA fragments within or in the vicinity of the HIV promoter region in LRT. Primers and their genomic locations were as described previously (Friedman et al., 2011).
Lentiviruses expressing SIRT1 specific shRNA
For efficient knock-down of SIRT1 protein expression in latently infected 2D10 cells, Puromycin selection marker containing pLKO.1 vector harboring specific shRNA sequence against SIRT1 (MISSION shRNA constructs from SIGMA-Aldrich) were used. Vesicular stomatitis viruses G-pseudotyped were produced with shRNAs sequences that specifically target SIRT1 mRNA to knockdown their expression, in HEK 293T cell line using Lipofectamine as described previously (Kim et al., 2006). The sequences of the five SIRT1 specific shRNAs were the followings:
5′GTACCGGCATGAAGTGCCTCAGATATTACTCGAGTAATATCTGAGGCACTT CA TGTTTTTTG3′ (shRNA34);
5′CCGGGCAAAGCCTTTCTGAATCTATCTCGAGATAGATTCAGAAAGGCTTTG C TTTTT3′ (shRNA79);
5′CCGGCCTCGAACAATTCTTAAAGATCTCGAGATCTTTAAGAATTGTTCGAG GTTTTT3′ (shRNA80);
5′CCGGGCGGGAATCCAAAGGATAATTCTCGAGAATTATCCTTTGGATTCCCG C TTTTT3′ (shRNA81); and
5′CCGGGCGGCTTGATGGTAATCAGTACTCGAGTACTGATTACCATCAAG CCGCTTTTT3′ (shRNA83). A control shRNA lentivirus was made with the following sequence: 5′ACCGGGCGCGATAGCGCTAATAATTTCTCGAGAAATTATTAGCGCTATCGC GCTTTTT3′.
The virus titers were determined by infecting 1×106 Jurkat T-cell with a serial dilution of concentrated and purified virus stocks from harvested culture supernatant by ultra-centrifugation. The virus MOI was determined by using Lentivirus qPCR Titer kit from Applied Biological Materials Inc., following the manufacturer’s instruction. To knock-down the expression of SIRT1 protein, latently infected 2D10 cells were infected with 3.6×106 IU MOI of shRNA containing virus for 16 hours. The transduced cells were then washed in fresh RPMI 1640 complete medium and re-suspended in 2μg/ml Puromycin containing medium for selection and maintenance of shRNA transduced cells. Expression of SIRT1 after shRNA knock-down was verified by Western blot analysis with a SIRT1-specific antibody.
Research highlights.
Multiple short chain fatty acids (SCFAs) transactivate latent HIV-1;
The different SCFAs transactivate latent HIV-1 dose dependently and additively;
SCFAs activate P-TEFb to promote HIV-1 transcription;
SCFAs downregulate SIRT1 to increase histone acetylation at HIV-1 promoter;
SCFAs also downregulate EZH2 to decrease repressive histone tri-methylation.
Acknowledgments
This study was supported by the grant R56DE023912 from National Institute of Dental & Craniofacial Research of the National Institutes of Health to Fengchun Ye and Jonathan Karn. Additional supports came from a developmental grant from the CWRU/UH Center for AIDS Research (CFAR) to Fengchun Ye (P30-AI036219), and grants DPI DA028869 and R01-AI067093 to Jonathan Karn. We also thank the CWRU/UH CFAR for provision of flow cytometry services and Alicia N. Holbert for technical assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aoyama M, Kotani J, Usami M. Butyrate and propionate induced activated or non-activated neutrophil apoptosis via HDAC inhibitor activity but without activating GPR-41/GPR-43 pathways. Nutrition. 2010;26:653–661. doi: 10.1016/j.nut.2009.07.006. [DOI] [PubMed] [Google Scholar]
- Bafica A, Scanga CA, Schito M, Chaussabel D, Sher A. Influence of coinfecting pathogens on HIV expression: evidence for a role of Toll-like receptors. J Immunol. 2004;172:7229–7234. doi: 10.4049/jimmunol.172.12.7229. [DOI] [PubMed] [Google Scholar]
- Bartholomeeusen K, Fujinaga K, Xiang Y, Peterlin BM. Histone deacetylase inhibitors (HDACis) that release the positive transcription elongation factor b (P-TEFb) from its inhibitory complex also activate HIV transcription. The Journal of biological chemistry. 2013;288:14400–14407. doi: 10.1074/jbc.M113.464834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernhard W, Barreto K, Saunders A, Dahabieh MS, Johnson P, Sadowski I. The Suv39H1 methyltransferase inhibitor chaetocin causes induction of integrated HIV-1 without producing a T cell response. FEBS Lett. 2011;585:3549–3554. doi: 10.1016/j.febslet.2011.10.018. [DOI] [PubMed] [Google Scholar]
- Bieniasz PD, Grdina TA, Bogerd HP, Cullen BR. Recruitment of a protein complex containing Tat and cyclin T1 to TAR governs the species specificity of HIV-1 Tat. EMBO J. 1998;17:7056–7065. doi: 10.1093/emboj/17.23.7056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blazkova J, Trejbalova K, Gondois-Rey F, Halfon P, Philibert P, Guiguen A, Verdin E, Olive D, Van Lint C, Hejnar J, Hirsch I. CpG methylation controls reactivation of HIV from latency. PLoS Pathog. 2009;5:e1000554. doi: 10.1371/journal.ppat.1000554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosque A, Famiglietti M, Weyrich AS, Goulston C, Planelles V. Homeostatic proliferation fails to efficiently reactivate HIV-1 latently infected central memory CD4+ T cells. PLoS pathogens. 2011;7:e1002288. doi: 10.1371/journal.ppat.1002288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosque A, Planelles V. Induction of HIV-1 latency and reactivation in primary memory CD4+ T cells. Blood. 2008;113:58–65. doi: 10.1182/blood-2008-07-168393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchat S, Gatot JS, Kabeya K, Cardona C, Colin L, Herbein G, de Wit S, Clumeck N, Lambotte O, Rouzioux C, Rohr O, van Lint C. Histone methyltransferase inhibitors induce HIV-1 recovery in resting CD4+ T cells from HIV-1+ HAART-treated patients. AIDS. 2012 doi: 10.1097/QAD.0b013e32835535f5. [DOI] [PubMed] [Google Scholar]
- Bourgeois CF, Kim YK, Churcher MJ, West MJ, Karn J. Spt5 cooperates with Tat by preventing premature RNA release at terminator sequences. Mol Cell Biol. 2002;22:1079–1093. doi: 10.1128/MCB.22.4.1079-1093.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullen CK, Laird GM, Durand CM, Siliciano JD, Siliciano RF. New ex vivo approaches distinguish effective and ineffective single agents for reversing HIV-1 latency in vivo. Nat Med. 2014;20:425–429. doi: 10.1038/nm.3489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao R, Wang L, Wang H, Xia L, Erdjument-Bromage H, Tempst P, Jones RS, Zhang Y. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science. 2002;298:1039–1043. doi: 10.1126/science.1076997. [DOI] [PubMed] [Google Scholar]
- Chang AH, Hoxie JA, Cassol S, O’Shaughnessy M, Jirik F. Construction of single-chain antibodies that bind an overlapping epitope of HIV-1 Nef. FEBS letters. 1998;441:307–312. doi: 10.1016/s0014-5793(98)01569-5. [DOI] [PubMed] [Google Scholar]
- Chattin BR, Ishihara K, Okuda K, Hirai Y, Ishikawa T. Specific microbial colonizations in the periodontal sites of HIV-infected subjects. Microbiology and immunology. 1999;43:847–852. doi: 10.1111/j.1348-0421.1999.tb01219.x. [DOI] [PubMed] [Google Scholar]
- Chen JS, Faller DV, Spanjaard RA. Short-chain fatty acid inhibitors of histone deacetylases: promising anticancer therapeutics? Current cancer drug targets. 2003;3:219–236. doi: 10.2174/1568009033481994. [DOI] [PubMed] [Google Scholar]
- Chiang K, Sung TL, Rice AP. Regulation of Cyclin T1 and HIV-1 Replication by MicroRNAs in Resting CD4+ T Lymphocytes. J Virol. 2012;86:3244–3252. doi: 10.1128/JVI.05065-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- du Chene I, Basyuk E, Lin YL, Triboulet R, Knezevich A, Chable-Bessia C, Mettling C, Baillat V, Reynes J, Corbeau P, Bertrand E, Marcello A, Emiliani S, Kiernan R, Benkirane M. Suv39H1 and HP1gamma are responsible for chromatin-mediated HIV-1 transcriptional silencing and post-integration latency. EMBO J. 2007;26:424–435. doi: 10.1038/sj.emboj.7601517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberle JA, Widmayer P, Breer H. Receptors for short-chain fatty acids in brush cells at the “gastric groove”. Frontiers in physiology. 2014;5:152. doi: 10.3389/fphys.2014.00152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenouillet E, Clerget-Raslain B, Gluckman JC, Guetard D, Montagnier L, Bahraoui E. Role of N-linked glycans in the interaction between the envelope glycoprotein of human immunodeficiency virus and its CD4 cellular receptor. Structural enzymatic analysis. The Journal of experimental medicine. 1989;169:807–822. doi: 10.1084/jem.169.3.807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman J, Cho WK, Chu CK, Keedy KS, Archin NM, Margolis DM, Karn J. Epigenetic silencing of HIV-1 by the Histone H3 lysine 27 Methyltransferase Enhancer of Zeste 2 (EZH2) J Virol. 2011;85:9078–9089. doi: 10.1128/JVI.00836-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujinaga K, Barboric M, Li Q, Luo Z, Price DH, Peterlin BM. PKC phosphorylates HEXIM1 and regulates P-TEFb activity. Nucleic acids research. 2012;40:9160–9170. doi: 10.1093/nar/gks682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujinaga K, Irwin D, Huang Y, Taube R, Kurosu T, Peterlin BM. Dynamics of human immunodeficiency virus transcription: P-TEFb phosphorylates RD and dissociates negative effectors from the transactivation response element. Mol Cell Biol. 2004;24:787–795. doi: 10.1128/MCB.24.2.787-795.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujinaga K, Taube R, Wimmer J, Cujec TP, Peterlin BM. Interactions between human cyclin T, Tat, and the transactivation response element (TAR) are disrupted by a cysteine to tyrosine substitution found in mouse cyclin T. Proc Natl Acad Sci U S A. 1999;96:1285–1290. doi: 10.1073/pnas.96.4.1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghose R, Liou LY, Herrmann CH, Rice AP. Induction of TAK (cyclin T1/P-TEFb) in purified resting CD4(+) T lymphocytes by combination of cytokines. J Virol. 2001;75:11336–11343. doi: 10.1128/JVI.75.23.11336-11343.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guarente L. Sir2 links chromatin silencing, metabolism, and aging. Genes & development. 2000;14:1021–1026. [PubMed] [Google Scholar]
- Hakre S, Chavez L, Shirakawa K, Verdin E. Epigenetic regulation of HIV latency. Curr Opin HIV AIDS. 2011;6:19–24. doi: 10.1097/COH.0b013e3283412384. [DOI] [PubMed] [Google Scholar]
- He N, Chan CK, Sobhian B, Chou S, Xue Y, Liu M, Alber T, Benkirane M, Zhou Q. Human Polymerase-Associated Factor complex (PAFc) connects the Super Elongation Complex (SEC) to RNA polymerase II on chromatin. Proc Natl Acad Sci U S A. 2011;108:E636–645. doi: 10.1073/pnas.1107107108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He N, Liu M, Hsu J, Xue Y, Chou S, Burlingame A, Krogan NJ, Alber T, Zhou Q. HIV-1 Tat and host AFF4 recruit two transcription elongation factors into a bifunctional complex for coordinated activation of HIV-1 transcription. Mol Cell. 2010;38:428–438. doi: 10.1016/j.molcel.2010.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann CH, Carroll RG, Wei P, Jones KA, Rice AP. Tat-associated kinase, TAK, activity is regulated by distinct mechanisms in peripheral blood lymphocytes and promonocytic cell lines. J Virol. 1998;72:9881–9888. doi: 10.1128/jvi.72.12.9881-9888.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinnebusch BF, Meng S, Wu JT, Archer SY, Hodin RA. The effects of short-chain fatty acids on human colon cancer cell phenotype are associated with histone hyperacetylation. The Journal of nutrition. 2002;132:1012–1017. doi: 10.1093/jn/132.5.1012. [DOI] [PubMed] [Google Scholar]
- Holt SC, Ebersole J, Felton J, Brunsvold M, Kornman KS. Implantation of Bacteroides gingivalis in nonhuman primates initiates progression of periodontitis. Science. 1988;239:55–57. doi: 10.1126/science.3336774. [DOI] [PubMed] [Google Scholar]
- Imai K, Inoue H, Tamura M, Cueno ME, Takeichi O, Kusama K, Saito I, Ochiai K. The periodontal pathogen Porphyromonas gingivalis induces the Epstein-Barr virus lytic switch transactivator ZEBRA by histone modification. Biochimie. 2012a;94:839–846. doi: 10.1016/j.biochi.2011.12.001. [DOI] [PubMed] [Google Scholar]
- Imai K, Ochiai K. Role of histone modification on transcriptional regulation and HIV-1 gene expression: possible mechanisms of periodontal diseases in AIDS progression. Journal of oral science. 2011;53:1–13. doi: 10.2334/josnusd.53.1. [DOI] [PubMed] [Google Scholar]
- Imai K, Ochiai K, Okamoto T. Reactivation of Latent HIV-1 Infection by the Periodontopathic Bacterium Porphyromonas gingivalis Involves Histone Modification. Journal of Immunology. 2009;182:3688–3695. doi: 10.4049/jimmunol.0802906. [DOI] [PubMed] [Google Scholar]
- Imai K, Victoriano AFB, Ochiai K, Okamoto T. Microbial Interaction of Periodontopathic Bacterium Porphyromonas gingivalis and HIV-Possible Causal Link of Periodontal Diseases to AIDS Progression- Current HIV research. 2012b;10:238–244. doi: 10.2174/157016212800618183. [DOI] [PubMed] [Google Scholar]
- Imai K, Yamada K, Tamura M, Ochiai K, Okamoto T. Reactivation of latent HIV-1 by a wide variety of butyric acid-producing bacteria. Cellular and molecular life sciences: CMLS. 2012c doi: 10.1007/s00018-012-0936-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isel C, Karn J. Direct evidence that HIV-1 Tat activates the Tat-associated kinase (TAK) during transcriptional elongation. J Mol Biol. 1999;290:929–941. doi: 10.1006/jmbi.1999.2933. [DOI] [PubMed] [Google Scholar]
- Jadlowsky JK, Wong JY, Graham AC, Dobrowolski C, Devor RL, Adams MD, Fujinaga K, Karn J. The negative elongation factor (NELF) is required for the maintenance of proviral latency but does not induce promoter proximal pausing of RNAP II on the HIV LTR. Mol Cell Biol. 2014;34:1911–1928. doi: 10.1128/MCB.01013-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantor B, Ma H, Webster-Cyriaque J, Monahan PE, Kafri T. Epigenetic activation of unintegrated HIV-1 genomes by gut-associated short chain fatty acids and its implications for HIV infection. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:18786–18791. doi: 10.1073/pnas.0905859106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karn J. A new BET on the control of HIV latency. Cell Cycle. 2013;12:545–546. doi: 10.4161/cc.23679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauder SE, Bosque A, Lindqvist A, Planelles V, Verdin E. Epigenetic regulation of HIV-1 latency by cytosine methylation. PLoS pathogens. 2009;5:e1000495. doi: 10.1371/journal.ppat.1000495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keedy KS, Archin NM, Gates AT, Espeseth A, Hazuda DJ, Margolis DM. A limited group of class I histone deacetylases act to repress human immunodeficiency virus type-1 expression. J Virol. 2009;83:4749–4756. doi: 10.1128/JVI.02585-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YK, Bourgeois CF, Isel C, Churcher MJ, Karn J. Phosphorylation of the RNA polymerase II carboxyl-terminal domain by CDK9 is directly responsible for human immunodeficiency virus type 1 Tat-activated transcriptional elongation. Mol Cell Biol. 2002;22:4622–4637. doi: 10.1128/MCB.22.13.4622-4637.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YK, Bourgeois CF, Pearson R, Tyagi M, West MJ, Wong J, Wu SY, Chiang CM, Karn J. Recruitment of TFIIH to the HIV LTR is a rate-limiting step in the emergence of HIV from latency. The EMBO journal. 2006;25:3596–3604. doi: 10.1038/sj.emboj.7601248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YK, Mbonye U, Hokello J, Karn J. T-Cell Receptor Signaling Enhances Transcriptional Elongation from Latent HIV Proviruses by Activating P-TEFb through an ERK-Dependent Pathway. Journal of molecular biology. 2011;410:896–916. doi: 10.1016/j.jmb.2011.03.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura I, Inoue D, Hirano K, Tsujimoto G. The SCFA Receptor GPR43 and Energy Metabolism. Frontiers in endocrinology. 2014;5:85. doi: 10.3389/fendo.2014.00085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita S, Su L, Amano M, Timmerman LA, Kaneshima H, Nolan GP. The T cell activation factor NF-ATc positively regulates HIV-1 replication and gene expression in T cells. Immunity. 1997;6:235–244. doi: 10.1016/s1074-7613(00)80326-x. [DOI] [PubMed] [Google Scholar]
- Kuzmichev A, Margueron R, Vaquero A, Preissner TS, Scher M, Kirmizis A, Ouyang X, Brockdorff N, Abate-Shen C, Farnham P, Reinberg D. Composition and histone substrates of polycomb repressive group complexes change during cellular differentiation. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:1859–1864. doi: 10.1073/pnas.0409875102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon HS, Brent MM, Getachew R, Jayakumar P, Chen LF, Schnolzer M, McBurney MW, Marmorstein R, Greene WC, Ott M. Human immunodeficiency virus type 1 Tat protein inhibits the SIRT1 deacetylase and induces T cell hyperactivation. Cell Host Microbe. 2008;3:158–167. doi: 10.1016/j.chom.2008.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Naour R, Raoul H, Mabondzo A, Ripoll L, Bartholeyns J, Romet-Lemonne JL, Dormont D. Functional consequences of monocyte/macrophage infection by HIV1. Research in immunology. 1992;143:49–56. doi: 10.1016/0923-2494(92)80079-z. [DOI] [PubMed] [Google Scholar]
- Liu M, Hsu J, Chan C, Li Z, Zhou Q. The Ubiquitin Ligase Siah1 Controls ELL2 Stability and Formation of Super Elongation Complexes to Modulate Gene Transcription. Mol Cell. 2012;46:325–334. doi: 10.1016/j.molcel.2012.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mabondzo A, Le Naour R, Raoul H, Clayette P, Lafuma C, Barre-Sinoussi FC, Cayre Y, Dormont D. In vitro infection of macrophages by HIV: correlation with cellular activation, synthesis of tumour necrosis factor alpha and proteolytic activity. Research in virology. 1991;142:205–212. doi: 10.1016/0923-2516(91)90058-b. [DOI] [PubMed] [Google Scholar]
- Marban C, Suzanne S, Dequiedt F, de Walque S, Redel L, Van Lint C, Aunis D, Rohr O. Recruitment of chromatin-modifying enzymes by CTIP2 promotes HIV-1 transcriptional silencing. EMBO J. 2007;26:412–423. doi: 10.1038/sj.emboj.7601516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marciniak RA, Sharp PA. HIV-1 Tat protein promotes formation of more-processive elongation complexes. EMBO J. 1991;10:4189–4196. doi: 10.1002/j.1460-2075.1991.tb04997.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massari S, Sabatini S, Tabarrini O. Blocking HIV-1 replication by targeting the Tat-hijacked transcriptional machinery. Current pharmaceutical design. 2013;19:1860–1879. doi: 10.2174/1381612811319100010. [DOI] [PubMed] [Google Scholar]
- Mataftsi M, Skoura L, Sakellari D. HIV infection and periodontal diseases: an overview of the post-HAART era. Oral diseases. 2011;17:13–25. doi: 10.1111/j.1601-0825.2010.01727.x. [DOI] [PubMed] [Google Scholar]
- Maticic M, Poljak M, Kramar B, Tomazic J, Vidmar L, Zakotnik B, Skaleric U. Proviral HIV-1 DNA in gingival crevicular fluid of HIV-1-infected patients in various stages of HIV disease. Journal of dental research. 2000;79:1496–1501. doi: 10.1177/00220345000790071101. [DOI] [PubMed] [Google Scholar]
- Mbonye U, Karn J. Control of HIV latency by epigenetic and non-epigenetic mechanisms. Current HIV research. 2011;9:554–567. doi: 10.2174/157016211798998736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mbonye U, Karn J. Transcriptional control of HIV latency: Cellular signaling pathways, epigenetics, happenstance and the hope for a cure. Virology. 2014;454–455C:328–339. doi: 10.1016/j.virol.2014.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mbonye UR, Gokulrangan G, Datt M, Dobrowolski C, Cooper M, Chance MR, Karn J. Phosphorylation of CDK9 at Ser175 enhances HIV transcription and is a marker of activated P-TEFb in CD4(+) T lymphocytes. PLoS Pathog. 2013;9:e1003338. doi: 10.1371/journal.ppat.1003338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mbopi-Keou FX, Belec L, Teo CG, Scully C, Porter SR. Synergism between HIV and other viruses in the mouth. The Lancet infectious diseases. 2002;2:416–424. doi: 10.1016/s1473-3099(02)00317-1. [DOI] [PubMed] [Google Scholar]
- Nabel G, Baltimore DA. An inducible transcription factor activates expression of human immunodeficiency virus in T cells. Nature. 1987;326:711–713. doi: 10.1038/326711a0. [DOI] [PubMed] [Google Scholar]
- Naor Z, Benard O, Seger R. Activation of MAPK cascades by G-protein-coupled receptors: the case of gonadotropin-releasing hormone receptor. Trends in endocrinology and metabolism: TEM. 2000;11:91–99. doi: 10.1016/s1043-2760(99)00232-5. [DOI] [PubMed] [Google Scholar]
- Natarajan M, Schiralli-Lester GM, Lee C, Missra A, Wasserman GA, Steffen M, Gilmour DS, Henderson AJ. NELF coordinates RNA polymerase II pausing, premature termination and chromatin remodeling to regulate HIV transcription. J Biol Chem. 2013 doi: 10.1074/jbc.M113.496489. E print ahead of publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuveut C, Suzan M, Querat G, Sire J. HIV1 infection of human monocytes and macrophages promotes induction or translocation of NF-KB-related factors. Research in virology. 1991;142:227–231. doi: 10.1016/0923-2516(91)90061-7. [DOI] [PubMed] [Google Scholar]
- Niederman R, Buyle-Bodin Y, Lu BY, Naleway C, Robinson P, Kent R. The relationship of gingival crevicular fluid short chain carboxylic acid concentration to gingival inflammation. Journal of clinical periodontology. 1996;23:743–749. doi: 10.1111/j.1600-051x.1996.tb00604.x. [DOI] [PubMed] [Google Scholar]
- Niederman R, Buyle-Bodin Y, Lu BY, Robinson P, Naleway C. Short-chain carboxylic acid concentration in human gingival crevicular fluid. Journal of dental research. 1997;76:575–579. doi: 10.1177/00220345970760010801. [DOI] [PubMed] [Google Scholar]
- Novis CL, Archin NM, Buzon MJ, Verdin E, Round JL, Lichterfeld M, Margolis DM, Planelles V, Bosque A. Reactivation of latent HIV-1 in central memory CD4(+) T cells through TLR-1/2 stimulation. Retrovirology. 2013;10:119. doi: 10.1186/1742-4690-10-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagans S, Pedal A, North BJ, Kaehlcke K, Marshall BL, Dorr A, Hetzer-Egger C, Henklein P, Frye R, McBurney MW, Hruby H, Jung M, Verdin E, Ott M. SIRT1 regulates HIV transcription via Tat deacetylation. PLoS biology. 2005;3:e41. doi: 10.1371/journal.pbio.0030041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parada CA, Roeder RG. Enhanced processivity of RNA polymerase II triggered by Tat-induced phosphorylation of its carboxy-terminal domain. Nature. 1996;384:375–378. doi: 10.1038/384375a0. [DOI] [PubMed] [Google Scholar]
- Pearson R, Kim YK, Hokello J, Lassen K, Friedman J, Tyagi M, Karn J. Epigenetic silencing of human immunodeficiency virus (HIV) transcription by formation of restrictive chromatin structures at the viral long terminal repeat drives the progressive entry of HIV into latency. J Virol. 2008;82:12291–12303. doi: 10.1128/JVI.01383-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phiri R, Feller L, Blignaut E. The severity, extent and recurrence of necrotizing periodontal disease in relation to HIV status and CD4+ T cell count. Journal of the International Academy of Periodontology. 2010;12:98–103. [PubMed] [Google Scholar]
- Pihlstrom BL, Michalowicz BS, Johnson NW. Periodontal diseases. Lancet. 2005;366:1809–1820. doi: 10.1016/S0140-6736(05)67728-8. [DOI] [PubMed] [Google Scholar]
- Pinzone MR, Cacopardo B, Condorelli F, Di Rosa M, Nunnari G. Sirtuin-1 and HIV-1: an overview. Current drug targets. 2013;14:648–652. doi: 10.2174/1389450111314060005. [DOI] [PubMed] [Google Scholar]
- Ramakrishnan R, Dow EC, Rice AP. Characterization of Cdk9 T-loop phosphorylation in resting and activated CD4(+) T lymphocytes. J Leukoc Biol. 2009;86:1345–1350. doi: 10.1189/jlb.0509309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramakrishnan R, Rice AP. Cdk9 T-loop phosphorylation is regulated by the calcium signaling pathway. J Cell Physiol. 2011;227:609–617. doi: 10.1002/jcp.22760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos MP, Ferreira SM, Silva-Boghossian CM, Souto R, Colombo AP, Noce CW, Goncalves LS. Necrotizing periodontal diseases in HIV-infected Brazilian patients: a clinical and microbiologic descriptive study. Quintessence Int. 2012;43:71–82. [PubMed] [Google Scholar]
- Rozengurt E. Mitogenic signaling pathways induced by G protein-coupled receptors. Journal of cellular physiology. 2007;213:589–602. doi: 10.1002/jcp.21246. [DOI] [PubMed] [Google Scholar]
- Santini V, Gozzini A, Ferrari G. Histone deacetylase inhibitors: molecular and biological activity as a premise to clinical application. Current drug metabolism. 2007;8:383–393. doi: 10.2174/138920007780655397. [DOI] [PubMed] [Google Scholar]
- Schotta G, Ebert A, Reuter G. SU(VAR)3–9 is a conserved key function in heterochromatic gene silencing. Genetica. 2003;117:149–158. doi: 10.1023/a:1022923508198. [DOI] [PubMed] [Google Scholar]
- Scully C, Porter SR, Mutlu S, Epstein JB, Glover S, Kumar N. Periodontopathic bacteria in English HIV-seropositive persons. AIDS patient care and STDs. 1999;13:369–374. doi: 10.1089/apc.1999.13.369. [DOI] [PubMed] [Google Scholar]
- Sewalt RG, Lachner M, Vargas M, Hamer KM, den Blaauwen JL, Hendrix T, Melcher M, Schweizer D, Jenuwein T, Otte AP. Selective interactions between vertebrate polycomb homologs and the SUV39H1 histone lysine methyltransferase suggest that histone H3-K9 methylation contributes to chromosomal targeting of Polycomb group proteins. Molecular and cellular biology. 2002;22:5539–5553. doi: 10.1128/MCB.22.15.5539-5553.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinkai Y, Tachibana M. H3K9 methyltransferase G9a and the related molecule GLP. Genes & development. 2011;25:781–788. doi: 10.1101/gad.2027411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shugars DC, Slade GD, Patton LL, Fiscus SA. Oral and systemic factors associated with increased levels of human immunodeficiency virus type 1 RNA in saliva. Oral surgery, oral medicine, oral pathology, oral radiology, and endodontics. 2000;89:432–440. doi: 10.1016/s1079-2104(00)70124-7. [DOI] [PubMed] [Google Scholar]
- Sobhian B, Laguette N, Yatim A, Nakamura M, Levy Y, Kiernan R, Benkirane M. HIV-1 Tat assembles a multifunctional transcription elongation complex and stably associates with the 7SK snRNP. Mol Cell. 2010;38:439–451. doi: 10.1016/j.molcel.2010.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syed SA. Characteristics of Bacteroides asaccharolyticus from dental plaques of beagle dogs. Journal of clinical microbiology. 1980;11:522–526. doi: 10.1128/jcm.11.5.522-526.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tukutuku K, Muyembe-Tamfum L, Kayembe K, Mavuemba T, Sangua N, Sekele I. Prevalence of dental caries, gingivitis, and oral hygiene in hospitalized AIDS cases in Kinshasa, Zaire. Journal of oral pathology & medicine: official publication of the International Association of Oral Pathologists and the American Academy of Oral Pathology. 1990;19:271–272. doi: 10.1111/j.1600-0714.1990.tb00840.x. [DOI] [PubMed] [Google Scholar]
- Tyagi M, Pearson RJ, Karn J. Establishment of HIV latency in primary CD4+ cells is due to epigenetic transcriptional silencing and P-TEFb restriction. J Virol. 2010;84:6425–6437. doi: 10.1128/JVI.01519-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Lint C, Bouchat S, Marcello A. HIV-1 transcription and latency: an update. Retrovirology. 2013;10:67. doi: 10.1186/1742-4690-10-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Lint C, Emiliani S, Ott M, Verdin E. Transcriptional activation and chromatin remodeling of the HIV-1 promoter in response to histone acetylation. EMBO J. 1996;15:1112–1120. [PMC free article] [PubMed] [Google Scholar]
- Verdin E, Paras P, Jr, Van Lint C. Chromatin disruption in the promoter of human immunodeficiency virus type 1 during transcriptional activation. Embo J. 1993;12:3249–3259. doi: 10.1002/j.1460-2075.1993.tb05994.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernon LT, Demko CA, Whalen CC, Lederman MM, Toossi Z, Wu M, Han YW, Weinberg A. Characterizing traditionally defined periodontal disease in HIV+ adults. Community dentistry and oral epidemiology. 2009;37:427–437. doi: 10.1111/j.1600-0528.2009.00485.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Briesen H, Andreesen R, Esser R, Brugger W, Meichsner C, Becker K, Rubsamen-Waigmann H. Infection of monocytes/macrophages by HIV in vitro. Research in virology. 1990;141:225–231. doi: 10.1016/0923-2516(90)90025-e. [DOI] [PubMed] [Google Scholar]
- Waldecker M, Kautenburger T, Daumann H, Busch C, Schrenk D. Inhibition of histone-deacetylase activity by short-chain fatty acids and some polyphenol metabolites formed in the colon. The Journal of nutritional biochemistry. 2008;19:587–593. doi: 10.1016/j.jnutbio.2007.08.002. [DOI] [PubMed] [Google Scholar]
- Wei P, Garber ME, Fang SM, Fischer WH, Jones KA. A novel cdk9-associated c-type cyclin interacts directly with HIV-1 Tat and mediates its high-affinity, loop specific binding to TAR RNA. Cell. 1998;92:451–462. doi: 10.1016/s0092-8674(00)80939-3. [DOI] [PubMed] [Google Scholar]
- Yamada T, Yamaguchi Y, Inukai N, Okamoto S, Mura T, Handa H. P-TEFb-mediated phosphorylation of hSpt5 C-terminal repeats is critical for processive transcription elongation. Mol Cell. 2006;21:227–237. doi: 10.1016/j.molcel.2005.11.024. [DOI] [PubMed] [Google Scholar]
- Yu X, Shahir AM, Sha J, Feng Z, Eapen B, Nithianantham S, Das B, Karn J, Weinberg A, Bissada NF, Ye F. Short Chain Fatty Acids From Periodontal Pathogens Suppress HDACs, EZH2, and SUV39H1 to Promote Kaposi’s Sarcoma-Associated Herpesvirus Replication. Journal of virology. 2014 doi: 10.1128/JVI.03326-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zack JA, Kim SG, Vatakis DN. HIV restriction in quiescent CD4(+) T cells. Retrovirology. 2013;10:37. doi: 10.1186/1742-4690-10-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zambon JJ, Reynolds HS, Genco RJ. Studies of the subgingival microflora in patients with acquired immunodeficiency syndrome. Journal of periodontology. 1990;61:699–704. doi: 10.1902/jop.1990.61.11.699. [DOI] [PubMed] [Google Scholar]
- Zhang HS, Sang WW, Wang YO, Liu W. Nicotinamide phosphoribosyltransferase/sirtuin 1 pathway is involved in human immunodeficiency virus type 1 Tat-mediated long terminal repeat transactivation. Journal of cellular biochemistry. 2010;110:1464–1470. doi: 10.1002/jcb.22704. [DOI] [PubMed] [Google Scholar]
- Zhang HS, Wu MR. SIRT1 regulates Tat-induced HIV-1 transactivation through activating AMP-activated protein kinase. Virus research. 2009;146:51–57. doi: 10.1016/j.virusres.2009.08.005. [DOI] [PubMed] [Google Scholar]