

Abstract
Morquio A syndrome is an autosomal recessive disorder, one of 50 lysosomal storage diseases (LSDs), and is caused by the deficiency of N-acetylgalactosamine-6-sulfate sulfatase (GALNS). Deficiency of this enzyme causes specific glycosaminoglycan (GAG) accumulation: keratan sulfate (KS) and chondroitin-6-sulfate (C6S). The majority of KS is produced in the cartilage, therefore, the undegraded substrates accumulate mainly in cartilage and in its extracelluar matrix (ECM), causing direct leads to direct impact on cartilage and bone development and leading to the resultant systemic skeletal spondyloepiphyseal dysplasia. Chondrogenesis, the earliest phase of skeletal formation that leads to cartilage and bone formation is controlled by cellular interactions with the ECM, growth and differentiation factors and other molecules that affect signaling pathways and transcription factors in a temporal-spatial manner. In Morquio A patients, in early childhood or even at birth, the cartilage is disrupted presumably as a result of abnormal chondrogenesis and/or endochondral ossification. The unique clinical features are characterized by a marked short stature, odontoid hypoplasia, protrusion of the chest, kyphoscoliosis, platyspondyly, coxa valga, abnormal gait, and laxity of joints.
In spite of many descriptions of the unique clinical manifestations, diagnosis delay still occurs. The pathogenesis of systemic skeletal dysplasia in Morquio A syndrome remains an enigmatic challenge. In this review article, screening, diagnosis, pathogenesis and current and future therapies of Morquio A are discussed.
Keywords: mucopolysaccharidosis IVA, enzyme assay, keratan sulfate, tandem mass spectrometry, GALNS, enzyme replacement therapy, bone marrow transplantation, pathogenesis, Morquio tissue repository bank
Introduction
Morquio A syndrome (Mucopolysaccharidosis type IVA, MPA IVA) is an autosomal recessive lysosomal storage disorder (LSD) caused by deficiency of N-acetylgalactosamine-6-sulfate sulfatase (GALNS). This enzyme deficiency leads to progressive accumulation of excessive glycosaminoglycans (GAGs), keratan sulfate (KS) and chondroitin-6-sulfate (C6S) primarily in the lysosomes of bone, cartilage, and ligaments and in the extracellular matrix (ECM) of these tissues,(1-4), since KS is produced mainly in cartilage tissue. The excessive storage of GAGs causes systemic skeletal spondyloepiphyseal dysplasia seen as striking short trunk stature, cervical spinal cord compression, pectus carinatum, kyphoscoliosis, knock-knee, hypermobile joints, and an abnormal gait with an increased tendency to fall.(5-7) (Figure 1) Many patients become wheelchair-dependent in their second decade and undergo multiple surgeries to alleviate serious medical complications. The respiratory failure from obstructive and restrictive lung and spinal cord injury results in significant mortality. Patients often do not survive beyond their twenties.(5-7)
Figure 1.
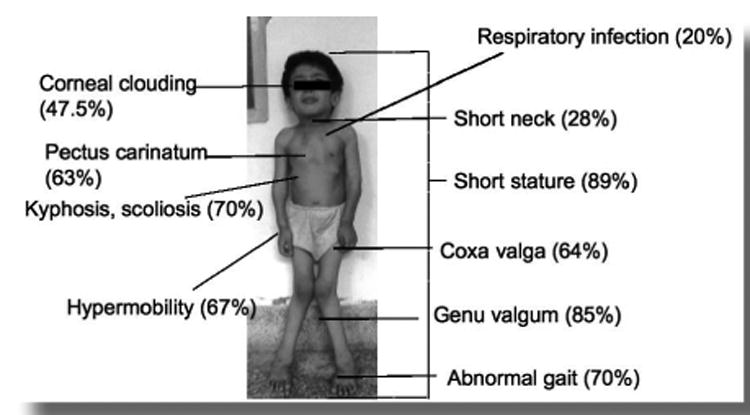
Clinical manifestations of Morquio A disease. Percentage of present symptoms based upon Morquio A database (photo; permitted by Morquio family).
Patients with Morquio A generally appear healthy at birth, but abnormal radiographs of the spine are observed even at newborn prior to other clinical manifestations.(8) However, diagnosis of Morquio A patients are often not made until two - three years of age with more prominent skeletal dysplasia since total urine GAG level is within a normal limit. Meanwhile, we have developed KS assay system by tandem mass spectrometry and have shown importance of measurements of KS levels to screen this disorder and assess the clinical status. (6-15)
Therapies for MPS include enzyme replacement therapy (ERT), gene therapy, hematopoietic stem cell transplantation (HSCT), and substrate reduction therapy (SRT), all of which lead to the partial improvement of clinical phenotypes.
Supportive measures are often provided. For joint pain, patients may receive non-steroidal anti-inflammatory drugs, and antibiotics are prescribed for otolaryngology infections. Surgical procedures are generally needed throughout life, including adenoidectomy, tonsillectomy, ear placement, cervical decompression/fusion, corrective knee surgery, and hip correction surgery.
Morquio A Diagnosis
Blood and urine KS: Urinary analysis of GAGs is useful as a preliminary investigative test for MPS, however, substantial overlapping in GAG levels between Morquio A patients and the age-matched controls was reported,(9-14) leading to delay of diagnosis or misdiagnosis.(9) Therefore, a more accurate screening biomarker for Morquio A is required. Deficiency of GALNS activity results in the build-up of C6S and KS in lysosomes leading to progressive skeletal dysplasia. Consequently, excessive undegraded KS mainly synthesized in cartilage cells and responsible for skeletal dysplasia is released into circulation and is thus an important biomarker for screening and assessing Morquio A.
A tandem mass spectrometry (MS/MS) method has been developed, which is highly specific and sensitive to measure KS.(10-14) In healthy individuals, blood KS levels rise progressively during the first 4 years of life and remain elevated until 12 years of age. At that time, KS levels decline markedly and after 15 years of age the levels continue to fall gradually until they stabilize around age 20.(11,13,14) The decline of KS levels after 13 years of age is consistent with the fact that the growth rate in normal children decreases after age 13. In Morquio A patients, blood KS levels peak between 5 and 10 years of age while urine KS levels peak between ages 0 and 5.(11,13,14) Blood and urine KS levels are higher in Morquio A patients than in age-matched controls under 20 years of age. Urine KS levels remain higher in Morquio A patients than controls after 20 years of age, but blood KS levels tend to be normalized by age 20.(11,13,14) Level of blood and urine KS is correlated with clinical severity at an initial stage, therefore, it is a good prognostic biomarker.
Blood KS directly displays growth, turnover, disruption and/or repair of cartilage where it is mainly synthesized. Measurement of KS in dried blood spot can be convenient for transport of samples and screening purpose. In contrast, urine KS has a broader range of value and may not reflect cartilage condition since urine KS is filtered in kidney and only selected smaller molecules are excreted in urine. Advantage in measuring urine KS is that it is less invasive for the patients.
When Morquio A patients are treated by ERT, it is likely that urine KS level is reduced rapidly since the enzyme is delivered to kidney and digests the KS stored in kidney. Meanwhile, it is unlikely that blood KS level is reduced in a short term unless the enzyme is delivered to the cartilage directly and improves bone lesions. Blood KS could be more important to assess therapeutic effect in bone lesions and association with clinical improvement. Another aspect of KS comes with the unexpected secondary elevation presumably associated with bone lesions in other type of MPS patients. We have explored whether KS can be used as a biomarker to evaluate therapeutic effect of ERT on MPS I patients, leading to the reduction of blood KS after 72 weekly infusions and proved that KS is a good biomarker for other type of MPS as well. (Figure 2)
Figure 2.
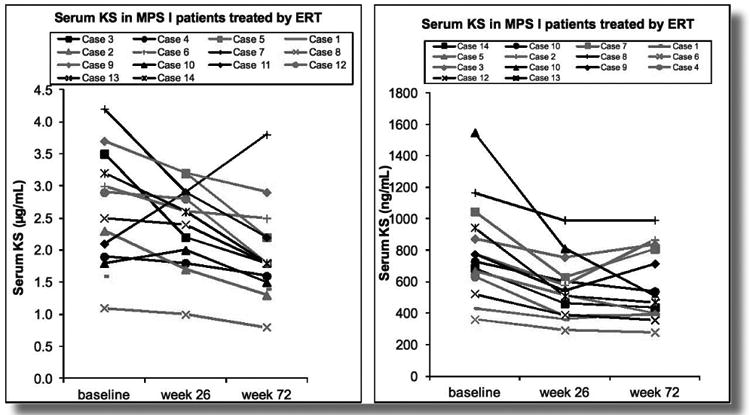
Biochemical profiles in serum of 14 MPS I patients who have undergone ERT (baseline, week 26, week 72). Red line: the patients (cases 1-7) who have undergone ERT from week 1. Black line: the patients who have undergone ERT from week 26 (cases 8-14). Left panel shows the results by MS/MS while right panel shows the results by ELISA. On the average, 25% of KS has been reduced after 72 weekly infusions
The pilot study of a newborn screening for Morquio A by assaying blood KS level with LC-MS/MS is under development supported by NIH fund (1R01HD065767-01). It is important to investigate whether blood KS is already elevated at birth since we can assume when the bone degeneration may start and can prove the concept as a tool for 1st tier newborn screening followed by the enzyme assay.
Overall, determination of KS concentrations provides a potential biomarker in screening the high risk of Morquio A patient, assessing the clinical status at the initial progressive stage, monitoring therapeutic effect and using 1st tier newborn screening followed by the enzyme assay. The scheme of diagnosis of Morquio A is shown in Figure 3.
Figure 3.
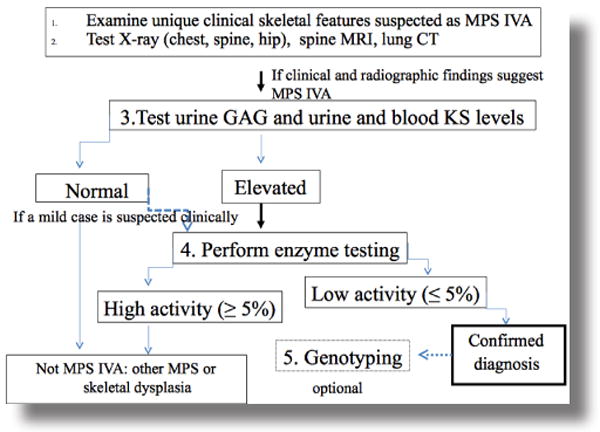
Diagnosis of Morquio A. Observation of careful unique clinical features and systemic skeletal radiographs of the patient will be the first step to suspect Morquio A differentiated from other MPS or skeletal dysplasia. Urine total GAG assay is less reliable, therefore, urine and blood KS test will be preferential. Enzyme assays should not be hesitated once the physician suspects Morquio A in spite of KS assay results. Once newborn screening is available, dried blood spot samples at newborn will be tested and diagnosis of Morquio A could be done prior to appearance of the unique signs and symptoms.
Current and Future Therapies for Morquio A
1) Orthopedic Surgical Procedures
Upper spine: Children with Morquio A have odontoid hypoplasia, ligamentous laxity, incomplete ossification of the anterior and posterior rings of the atlas and extradural GAG deposition, leading to instability of the neck and the spinal cord compression.(5-7, 16, 17) Spinal cord compression is often evident on MRI at the craniocervical junction, as is deposition of storage materials on the tip of the odontoid process.(Figure 4) Untreated upper cervical stenosis and instability can cause irreversible damage to the spinal cord (cervical myelopathy).
Figure 4.
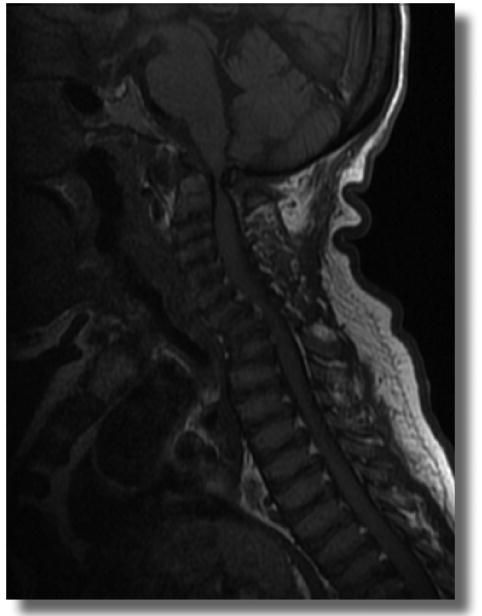
MRI of cervical spine in a patient aged 38 years. The red circle shows the spinal cord compression. Odontoid process does not appear (hypoplasia or anaplasia) and extradural glycosaminoglycan deposition anterior to the spinal cord at C1 level is observed. The patient has severe C1 stenosis. At the age of 16 years, the patient had a stroke which caused paralysis of left leg and became wheelchair bound. During the last few years, the patient had worsening of joint pain, hearing loss, cornea clouding, urinary inconsistency, and abnormal bowel movement suggesting a spinal cord injury.
Paraplegia or sudden death secondary to upper cervical spine instability and cord compression is well-documented serious consequences of Morquio A.(5-7,16,17) Decompression and fusion of the upper cervical spine is required to treat instability and spinal cord compression. Early cervical spine management with prophylactic fusion has been recommended to prevent cervical myelopathy and further complications.(18, 19) Prophylactic fusion was reported to have better neurologic consequences compared with fusions performed after neural compromise. (18, 19) Upper cervical spine fusion provides reliable fusion and a stable neural outcome in patients with Morquio A. The odontoid process usually ossifies after a cervical fusion.
Some patients who have undergone a successful upper cervical fusion develop symptomatic instability below the fusion mass that requires extension of fusion to lower levels. Thus, instability of distal junction is a major concern at long-term follow-up. It is required that kyphosis of the cervicothoracic and thoracolumbar junctions should be evaluated for spinal stenosis and potential spinal cord compression.
Upper cervical fusion procedure is safe and effective in patients affected by Morquio A to treat or prevent upper cervical instability and neurological involvement although the respiratory status and anesthetic procedure should be performed by a well-trained team with experience in caring for Morquio A patients. Careful long-term observation is advocated for early diagnosis and management of late complications (instability below the fusion level) and a second area of compression.
Lower Extremity: Children with Morquio A have unique waddling gait, a slower walking speed, reduced cadence, and reduced stride length.(20) Over 70% of the patients become wheel-chair bound after teenage.(5) Increased knee flexion and valgus as well as external tibial torsion are noted in stance phase and gait analysis demonstrates increased internal varus moment in the knee. (20)
The hips are either normal or slightly subluxated to start with in patients with Morquio A. (Figure 5) The capital femoral epiphyses are smaller and progressively flatten and may become fragmented over time. Progressive proximal and lateral movement of the femoral heads is present, finally resulting in frank dislocation if no intervention is carried out.(Figure 5) The proximal femurs are in valgus in most patients. The acetabulum is to some extent shallow when compared with normal acetabulum initially and becomes gradually more dysplastic with subluxation of the hip. The acetabular deficiency is mostly antero-superior as noted in a CT scan study.(22) A false acetabulum is often seen when the hips are dislocated. The ossified femoral heads tend to flatten and erode,(Figure 5) and there is widening of the femoral necks in adulthood. A saddle-shaped femoral head may ultimately be observed at skeletal development. The destructive osteoarthritis, which particularly affects the hip joints, is seen as a feature of the skeletal dysplasia in majority of patients, resulting in severe restriction of movement and pain. The macrophage-derived foam cells appear in bone marrow, synovium and ligaments.(Figure 7) Most patients could become wheelchair-bound due to hip dislocation and pain. Ultimately, it leads to requirement of a total hip replacement.
Figure 5.
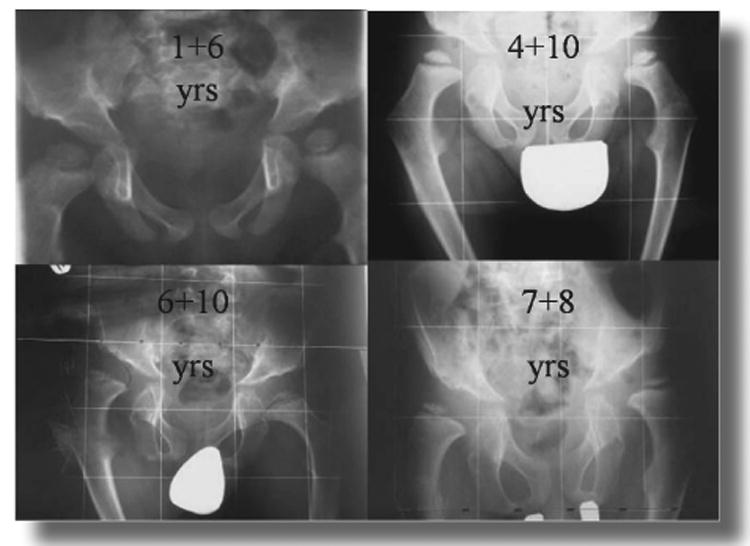
Changes in the hip with age. This figure demonstrates that the hips are mildly subluxated at age one year and 6 months and have mild acetabular dysplasia. The proximal femoral valgus becomes more obvious with time and there is progressive flattening of the capital femoral epiphysis at ages 4 years and 10 months and 6 years and 10 months. There is severe subluxation and acetabular dysplasia at age 7 years and 8 months.
Figure 7.
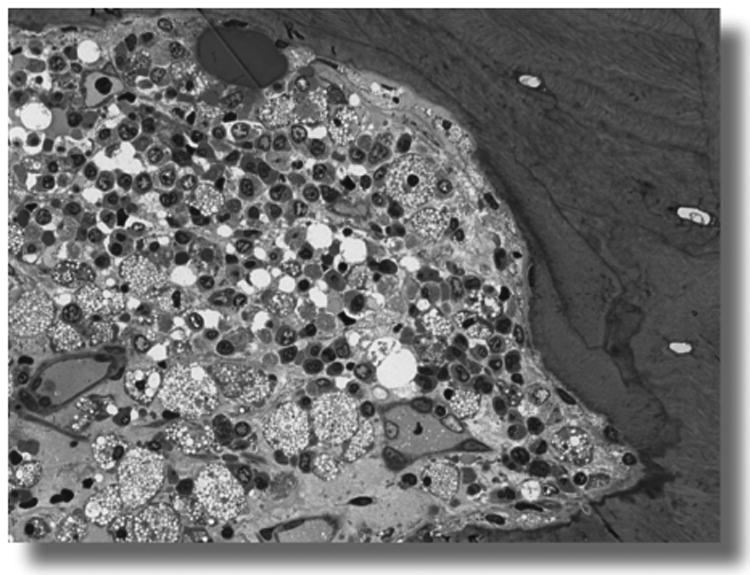
Foam cells in bone marrow of a 20 year-old Morquio A patient. Foam cells are fully vacuolated and observed focally.
When the cartilage of the epiphyses is only partially ossified in patients with MPS IVA, the result is epiphyseal dysplasia. The articular cartilage is abnormal and will degenerate promptly and build up early arthrosis, particularly in the loaded lower extremities. Weight loading and joint instability can delay or inhibit ossification of the epiphyses.
Surgery is often needed in these patients to preserve the hip joint.(Figure 6) Proximal femoral varus and derotation osteotomies and procedures to improve acetabular coverage such as a shelf arthroplasty can stabilize the hip and improve gait mechanics.(20) Recurrent hip deformity is common if the hips are not properly contained. Joint replacement surgery in adults may be needed in cases with severe arthritis and pain.
Figure 6.
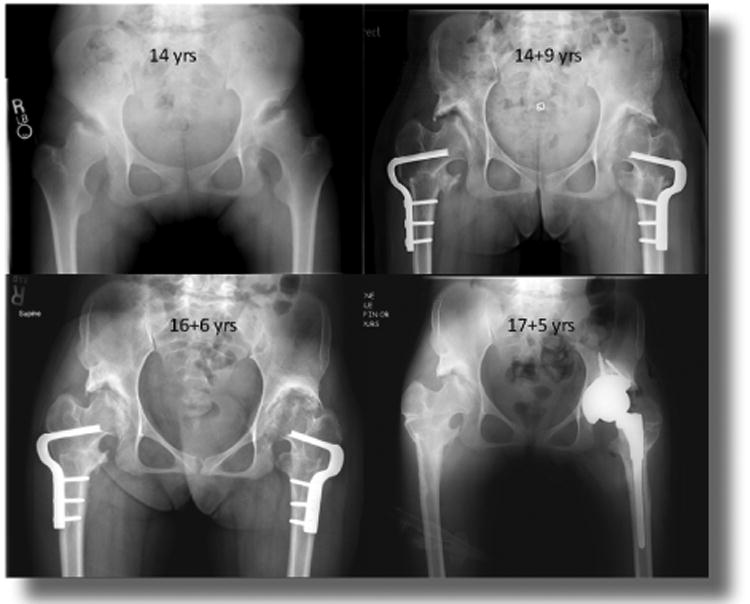
Hip surgery (14 year-old girl with MPS IVA). The patient had severely painful subluxated hips with flattened and irregular femoral heads. She underwent bilateral varus derotation osteotomies of the femur and bilateral shelf acetabuloplasties to cover the femoral heads better. She had transient improvement in her symptoms but two years later had severe pain in the left hip and underwent a total hip replacement. Pathology of tissues is shown in figure 7.
Progressive hip subluxation, genu valgum, and ankle valgus often needed surgical procedures. The modalities and results of surgical intervention in the lower extremity in children with Morquio A have recently been described. After shelf acetabuloplasty and varus derotation osteotomy, no recurrent hip subluxation was observed while recurrence after genu valgum correction was common.(21)
2) Enzyme Replacement Therapy (ERT)
ERT is an established and approved strategy of treating MPS including MPS I,(23) MPS II,(14, 25) and MPS VI.(26-29) Clinical trials with ERT in MPS I, II, and VI show limited improvement in joint pain, stiffness, or joint range of motion. Skeletal dysplasia is irreversible by conventional ERT (30-32) since there is little or no evidence that the current ERT directly delivers the enzyme to cartilage and bone lesions in MPS patients. The effectiveness of treatment could be greater if it is introduced early in life.(33)
MPS IVA clinical trial is in progress. It is unlikely that the positive effect of ERT is direct delivery of the enzyme to the cartilage. After six months of preclinical ERT in MPS IVA mice there was little impact on bone pathology.(34) Recent surgical remnant from a 17-year-old Morquio A patient in an extension clinical trial for 3 months did not show any reduction of vacuoles in chondrocytes (preliminary data). The underlying problems associated with progressive skeletal deformity and laxity of joints will not be solved by current ERT with native enzyme. It remains a challenge to achieve significant clinical efficacy for the skeleton, particularly for diseases like MPS IVA. Careful long-term assessment will be required to determine whether the infused enzyme will sufficiently improve skeletal pathology for Morquio A patients. The response to ERT may depend on the starting age of treatment and/or the severity of the clinical condition. In a short term treatment of ERT, most patients have demonstrated decrease of urine KS level but decrease of blood KS level has not been reported yet. Since the origin of urine KS could derive from KS stored or filtered in kidney, reduced urine KS seen in treated Morquio A patients may not reflect improvement in bone pathology at least in the short term. Blood KS derives mainly from the chondrocytes and therefore its reduction may directly reflect bone improvement. It would be of great interest to know how much the blood KS levels will decrease over time.
Degenerative joint changes are not delayed in treated animals since birth, although skeletal pathology is reduced, with more normalized bone dimensions and with more uniform bone density and trabecular pattern.(35) ERT in MPS IVA mouse is effective at reducing the development of pathology in visceral organs but vacuolated chondrocytes in articular and epiphyseal cartilage still remain unsolved.(34)
To improve bone pathology more, two alternative approaches have been tested to combine ERT with a modified enzyme. One is bone targeting strategy investigated for MPS IVA and hypophosphatasia mice and the other one is chemically modified enzyme for MPS VII mouse. Hydroxyapatite (HA) is a major inorganic matrix in bone but is absent in soft tissues. Drugs that attach to HA will be released in the process of the bone resorption and thereby targeting a drug to HA is a potential method for a selective drug delivery to bone. We and others have recently attached this novel bone-targeting peptide to the enzyme (tissue nonspecific alkaline phosphatase), indicating that the tagged enzyme is delivered more specifically to bone than unmodified native enzyme, improving the clinical and pathological consequence of the systemic bone disease, hypophosphatasia.(36, 37) The clinical trial for hypophosphatasia by using bone-targeting system showed substantial achievement of bone pathology with the clinical effect.(38) Human GALNS has also been bioengineered to tag a hexa-glutamate sequence (E6) to its N-terminus (E6-GALNS). This tagged enzyme had markedly prolonged blood clearance, increasing blood levels 20 times higher than that of the untagged enzyme. The bone-targeting enzyme was retained longer in bone. The pathological features in MPS IVA mice treated with the targeting enzyme showed clearance of the storage materials in chondrocytes, especially after 24 weekly injections. These findings suggest that the use of the tagged enzyme enhances delivery and improves pathological effect in MPS IVA mice.(39)
Another strategy is coming from a chemically modified form of ß-glucuronidase (GUS) (PerT-GUS) which escaped clearance by mannose 6-phosphate and mannose receptors. This modified enzyme showed markedly prolonged circulation (over 100 times) compared with native GUS enzyme. To evaluate the effectiveness of long-circulating PerT-GUS in reducing the skeletal pathology, MPS VII mice were treated for 12 weeks with PerT-GUS or native GUS. Reduced storage material and a more organized growth plate were observed in PerT-GUS treated mice compared with native GUS treated mice. Long-circulating PerT-GUS provides a significant impact in rescuing bone lesions.(40)
These modified enzymes have similar property as the long circulating enzyme, in which the higher enzyme activity can be kept in blood and could be accessed to the bone lesions more efficiently and improve the bone pathology.
3) Hematopoietic Stem Cell Therapy (HSCT)
The potential merit of HSCT for MPS is considered to be that the marrow-derived donor macrophages could provide a secreting source of enzyme and gain access to various storage tissues. HSCT has been successfully achieved by correcting the disease course and severity in MPS I, MPS II and MPS VI patients.(41, 42) A major issue of HSCT mainly relies on its adverse effect.(43) The patients need immunosuppression and need steroid following transplantation to protect against graft-versus-host disease. HSCT does not repair advanced skeletal deformities, in spite of improving growth development.(44)
The clinical consequence of HSCT depends upon the following multiple factors; 1) the age and the clinical condition of the recipient at transplantation, 2) prognosis (clinical course) of the recipient, 3) donor type, and 4) preparative regimen. In MPS I, II and VI patients, HSCT results in reversal of visceral organ involvement and improvement of heart function and hearing. However, progress of skeletal features is similar to untreated individuals, and corneal clouding is not diminished. Therefore, HSCT of Morquio A patients will not give a marked direct impact to existing skeletal abnormality. A male Morquio A patient before and after successful allogeneic bone marrow transplantation (BMT) has been recently reported.(6, 7, 8) Five years after BMT the GALNS activity was restored to the level of the enzyme activity of the donor in the lymphocytes of the recipient. The patient showed 1) recovery of ambulation in combination with osteotomies, 2) remission of narrow airway and disappearance of shortage of breath with recovery of pulmonary function, 3) vanishing of snoring, and 5) increase of bone mineral density. Recovery of pulmonary function and bone mineral density last for the first 3 years after BMT. However, restriction of physical activity and hyperlaxity of joints remain unsolved in this patient. The substantial clinical benefits post-BMT in this Morquio A patient indicates that HSCT should be considered as a therapeutic choice for Morquio A patients. We expect that if HSCT is performed for Morquio A patients at an earlier stage, skeletal deformities, restrictive and obstructive airway and growth development should be improved more. Although regimens for HSCT have been advanced substantially for recent years, the procedure may still carry some risk of mortality from infection, graft-versus host disease, and additional complications. For these reasons, HSCT should be chosen in selected cases with careful pre-transplantation counseling and clinical assessment and with systemic longitudinal monitoring of the outcome.
4) Substrate Reduction Therapy (SRT)
To penetrate the bone remains an unmet challenge although the involvement of the visceral organs is reversed by the treatment. SRT is one of the potential treatments that could be effective in management of GAGs accumulation. Genistein, a compound from the group of flavonoid compounds, inhibits synthesis and reduces the level of GAG, heparan sulfate (HS), in cultures of fibroblasts of MPS I, II and III patients.(45, 46) Investigational trials on human MPS patients have been conducted and have shown some improvements of neurological symptoms.(47, 48)
Therapeutic effect of genistein on joint function in patients with MPS II was evaluated, suggesting that administration of genistein-rich soy isoflavone extract resulted in improvement of connective tissue elasticity, particularly range of joint motion.(49)
Moreover, other flavonoid compounds, daidzein and kaempferolare, have been proved to be effective to suppress GAG synthesis in vitro.(50)
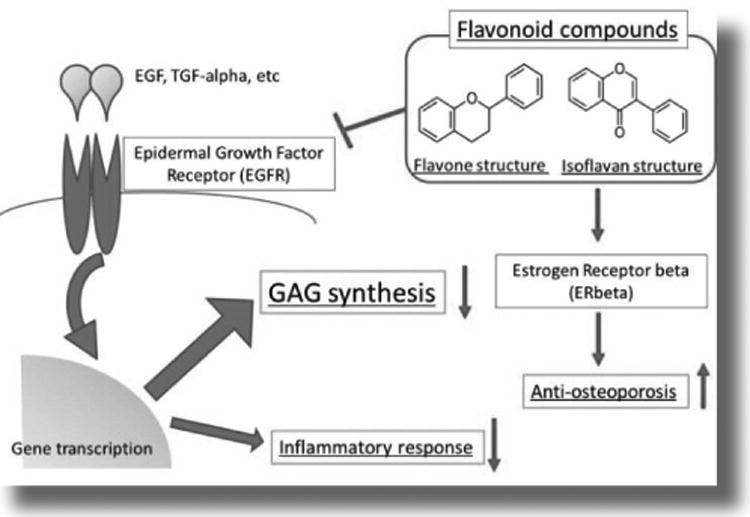
The mechanism of these flavonoid compounds-mediated inhibitions of GAG synthesis is considered to be development of epidermal growth factor (EGF)-dependent pathway.(51-56) Flavonoids are widely distributed in plants functioning as anti-allergic, anti-inflammatory, anti-microbial, anti-cancer, and anti-diarrheal actions. Silibinin, proanthocyanidins, epigallocathechin-3-gallate, theaflavins, resveratorol, quercetin, luteolin, and polymethoxyflavones are classified as the same flavonoid compounds as genistein and are also shown to inhibit of EGFR (epidermal growth factor receptor: Figure 8) in various cancer cells.(57-63) Flavonoid compounds, especially isoflavones, also have estrogen-like effects to protect against the osteoporosis. Until now no report about the effects of flavonoid compounds on KS and C6S accumulation has been published. Therefore, how a group of or a specific flavonoid(s) affect KS and C6S synthesis remain unanswered.
Figure 8. Substrate reduction therapy.
5) Gene Therapy
Gene therapy is another rational approach for treating skeletal diseases, and animal research on other types of MPS is promising. The first study of gene therapy for Morquio A was performed in vitro by using a retrovirus vector.(57) GALNS enzyme activity levels substantially higher than those observed in normal non-transduced cells, leading to the reduction of GAGs accumulation.
Successively, AAV vector has been selected since the AAV vector has several advantages such as a long-term expression, well-characterized serotypes, wide-ranged cell and tissue tropism, low immunogenicity, and experience of preclinical and clinical trials on LSDs with clinical improvements.(58-60)
A targeted drug delivery system can improve gene therapy by altering the natural tropism of viral vectors. Several reports suggest the possibility of AAV vectors.(61-63) Peptide insertion primarily focused on improvement of the transduction rather than in the change of the tropism. Progress has been made in AAV2 vectors with regard to the peptide insertion sites and type of peptides in relation to transduction efficiency and tissue targeting. AAV2 vectors designed for vascular tissue targeting have been constructed by inserting unique peptides, with an increased transduction and specificity for venous endothelial cells and a reduction for HepG2 hepatocytes.(64) This vector indicated significantly higher targeting to the vena cava, demonstrating specificity of the modified vector.(65) Insertion of 28-amino acids ApoE-derived ligand led to a 90-fold increase in the in vitro transduction of pancreatic islet cells, and four-fold increase of expression of human antitrypsin.(62)
However, there has been no report of successful bone-targeting by gene therapy. To address this unmet need, we have developed a method by attaching multiple copies of AAA peptide to the AAV2 vector capsid as follows.
After initial successful demonstration of gene expression in vitro by non-targeting AAV2 vector with CMV promoter,(65) we tested other promoters; the eukaryotic elongation factor 1α (EF1) and the α1-antitrypsin (AAT) promoters. AAT and EF1 promoters allowed similar GALNS enzyme activity levels than those observed with CMV promoter in vitro.(66, 67) We also examined the effects of co-transduction with sulfatase modifying factor 1 (SUMF1) on GALNS activity levels since in vivo studies have shown that the co-expression of other sulfatases with SUMF1 induces a significant elevation of enzyme activity.(68, 69)
GALNS enzyme expression last more than 10 days post-transduction in spite of promoter used. Co-transduction with SUMF1 produced up to a four-fold increase of enzyme activity in Morquio A mouse chondrocytes. In vivo experiments using a Morquio A mouse model showed that after 12 weeks of a single intravenous administration of an AAV vector carrying GALNS cDNA, plasma enzyme activity levels were increased up to 20% when the AAV-GALNS vector was co-administrated with the AAV-SUMF1 vector. GALNS enzyme activity levels in mice infused with the AAV-GALNS and co-administrated AAV-SUMF1 allowed a significant increase in enzyme activity in all the studied tissues. GALNS activities were about 30% of wild-type levels in liver, heart and bone. Since theoretically only 10% of normal levels are required to move from a severe to an attenuated phenotype, these results suggested the potential of AAV gene therapy for the treatment of Morquio A.
Moreover, to target viral capsid to bone, we inserted multiple copies of a short acidic amino acid peptide to the viral capsid. The sequence encoding a stretch of eight Asp acidic amino acids (D8) was inserted immediately after the initial codon of the VP2 protein in the packing plasmid. We evaluated the affinity of AAV2 vector to HA, physical titers and transduction efficiencies, biodistribution and expression level of the gene product at target sites. Preliminary results with the bone-targeting vector compared with unmodified vector showed i) the similar physical properties of the vector or infectious titers; ii) the maximum HA affinity in vitro and higher vector genome copies in bone; iii) targeting to bone, release from HA, transduction in bone cells, and the resultant higher expression of GALNS in bone.
This bone targeting gene therapy will enhance therapeutic efficacy on the bone lesions of Morquio A.
6) Anti-inflammatory Drugs
In Morquio A patients, chronic osteoarthritis is commonly observed in any major joints like hip, knee, wrist and ankle.(Figure 7) To surpress metabolic inflammation by GAG accumulation, two treatments are available: one is to reduce the causative factor, GAG by ERT, gene therapy, SRT, HSCT etc., while the other one is to suppress secondary inflammatory processes by anti-inflammatory (or immunosuppressive) agent. These anti-inflammatory agents have specific mechanisms of action, including inhibiting the action of cytokines, blocking cell-cell interactions, and depleting certain cell types. TNF-α is a dominant proinflammatory cytokine in the pathophysiology of MPS and several biologic agents are approved to treat the autoimmune diseases like rheumatoid arthritis (RA).
The effect of anti-TNF-α (infliximab) therapy was assessed in MPS VI rats. Early treatment in the presymptomatic period inhibited the elevation of TNF-α, RANKL and other inflammatory factors in the blood, articular chondrocytes and synovial fibroblasts.(70) The number of apoptotic articular chondrocytes was reduced and was not different from healthy control rats. However, there was no impact on bone growth or mobility since stored GAGs still remained in chondrocytes of the growth plate. The efficacy of ERT alone and combined treatment using ERT and anti-TNF-α drug (specific monoclonal antibody against TNF- α: CNTO1081) was also tested.(71) Both treatments markedly reduced serum levels of TNF-α and RANKL, although only combined treatment reduced TNF-α in the articular cartilage. Analysis of cultured articular chondrocytes showed that combination therapy restored collagen IIA1 expression and reduced expression of apoptotic markers. Only combined therapy suppressed hyperplasia of synovial cells into underlying bone and clinical effects on other organs that are not accessible to the enzyme (e.g., cartilage). (71)
A anti-inflammatory treatment should be evaluated in MPS IVA alone or in combined therapy with ERT, HSCT, or gene therapy. (72)
Conclusion
Families and their patients should be provided personalised management including genetic counseling, supportive therapies, physiotherapies, orthopedic interventions, by a well-trained and experienced team including experienced anesthesiologists. what does this mean? Is something missing?. Communication with other MPS IVA families, patients, support groups, and the Morquio Foundation (www.morquio.com) as well as participation of the annual educational Morquio Symposium should provide the opportunity to access knowledge of the disease and increase the exchange of experiences of those who live with Morquio disease. (Figure 8)
Acknowledgments
This work was supported by grants from the Austrian MPS Society, National MPS Society and International Morquio Organization (Carol Ann Foundation). W.S.S. was also supported by National Institutes of Health grant GM34182. S.T. and R.M. were supported by National Institutes of Health grant P20GM103464. S.T. and A.M were supported by National Institutes of Health grant 1R01HD065767-01. CJAD and LAB are supported by The Administrative Department of Science, Technology and Innovation COLCIENCIAS (Colombia) grant 120356933205 (ID PPTA 5174). The content of the article has not been influenced by the sponsors. Editorial assistance to the manuscript was provided by Michelle Stofa at Nemours/Alfred I. duPont Hospital for Children.
References
- 1.Neufeld EF, Muenzer J. The mucopolysaccharidoses. In: Scriver CR, Beaudet AL, Sly WS, Valle D, Childs B, Kinzler KW, Vogelstein B, editors. The Metabolic and Molecular Bases of inherited Disease. 8th. McGraw-Hill; New York: 2001. pp. 3421–3452. [Google Scholar]
- 2.Tomatsu S, Orii KO, Vogler C, Nakayama J, Levy B, Grubb JH, Gutierrez MA, Shim S, Yamaguchi S, Nishioka T, Montaño AM, Noguchi A, Orii T, Kondo N, Sly WS. Mouse model for Galns-/- produced by targeted disruption of the gene defective in Morquio A disease. Hum Mol Genet. 2003;12:3349–3358. doi: 10.1093/hmg/ddg366. [DOI] [PubMed] [Google Scholar]
- 3.Tomatsu S, Gutiérrez MA, Nishioka T, Yamada M, Yamada M, Tosaka Y, Grubb JH, Montano AM, Vieira MB, Trandafirescu GG, Pena OM, Yamaguchi S, Orii KO, Orii T, Noguchi A, Laybauer L. Development of MPS IVA mouse (Galns tm(hC79mC76)slu ) tolerant hGALNS. Hum Mol Genet. 2005;14:3321–3336. doi: 10.1093/hmg/ddi364. [DOI] [PubMed] [Google Scholar]
- 4.Tomatsu S, Vogler C, Montaño AM, Gutierrez M, Oikawa H, Dung VC, Orii T, Noguchi A, Sly WS. Murine model (Galns(tm(C76S)slu)) of MPS IVA with missense mutation at the active site cysteine conserved among sulfatase proteins. Mol Genet Metab. 2007;91:251–258. doi: 10.1016/j.ymgme.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 5.Montaño AM, Tomatsu S, Gottesman GS, Smith M, Orii T. International Morquio A Registry: clinical manifestation and natural course of Morquio A disease. J Inherit Metab Dis. 2007;30:165–174. doi: 10.1007/s10545-007-0529-7. [DOI] [PubMed] [Google Scholar]
- 6.Tomatsu S, Montaño AM, Oikawa H, Smith M, Barrera L, Chinen Y, Thacker MM, Mackenzie WG, Suzuki Y, Orii T. Mucopolysaccharidosis type IVA (Morquio A disease): clinical review and current treatment. Cur Pharm Biotech. 2011;12:931–945. doi: 10.2174/138920111795542615. [DOI] [PubMed] [Google Scholar]
- 7.Tomatsu S, Mackenzie WG, Theroux MC, Mason RW, Thacker MM, Shaffer TH, Montaño AM, Rowan D, Sly W, Alméciga-Díaz CJ, Barrera LA, Chinen Y, Yasuda E, Ruhnke K, Suzuki Y, Orii T. Current and emerging treatments and surgical interventions for Morquio A Syndrome: A review. Research and Reports in Endocrine Disorders. 2012;2:65–77. doi: 10.2147/RRED.S37278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chinen Y, Higa T, Tomatsu S, Suzuki Y, Orii T, Hyakuna N. Long-term eherapeutic efficacy of allogenic bone marrow transplantation in a patient with mucopolysaccharidosis IVA. MGM reports. 2014;1:31–41. doi: 10.1016/j.ymgmr.2013.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tomatsu S, Okamura K, Taketani T, Orii KO, Nishioka T, Gutierrez MA, Velez-Castrillon S, Fachel AA, Grubb JH, Cooper A, Thornley M, Wraith E, Barrera LA, Giugliani R, Schwartz IV, Frenking GS, Beck M, Kircher SG, Paschke E, Yamaguchi S, Ullrich K, Isogai K, Suzuki Y, Orii T, Kondo N, Creer M, Noguchi A. Development and testing of new screening method for keratan sulfate in mucopolysaccharidosis IVA. Pediatr Res. 2004;55:592–597. doi: 10.1203/01.PDR.0000113767.60140.E9. [DOI] [PubMed] [Google Scholar]
- 10.Oguma T, Tomatsu S, Okazaki O. Analytical method for determination of disaccharides derived from keratan sulfates in human serum and plasma by high-performance liquid chromatography/turbo-ionspray ionization tandem mass spectrometry. Biomed Chromatogr. 2007;21:356–362. doi: 10.1002/bmc.760. [DOI] [PubMed] [Google Scholar]
- 11.Tomatsu S, Montaño AM, Oguma T, Dung VC, Oikawa H, de Carvalho TG, Gutiérrez ML, Yamaguchi S, Suzuki Y, Fukushi M, Kida K, Kubota M, Barrera L, Orii T. Validation of keratan sulfate level in Mucopolysaccharidosis IVA by liquid tandem mass spectrometry method. J Inherit Metab Dis. 2010;33(Suppl 3):S35–42. doi: 10.1007/s10545-009-9013-x. [DOI] [PubMed] [Google Scholar]
- 12.Tomatsu S, Montaño AM, DunG VC, Ohashi A, Oikawa H, Oguma T, Orii T, Barrera L, Sly WS. Enhancement of drug delivery: enzyme replacement therapy for murine Morquio A syndrome. Mol Ther. 2010;18:1094–10102. doi: 10.1038/mt.2010.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hintze JP, Tomatsu S, Fujii T, Montaño AM, Yamaguchi S, Suzuki Y, Fukushi M, Ishimaru T, Orii T. Comparison of liquid chromatography-tandem mass spectrometry and sandwich ELISA for determination of keratan sulfate in plasma and urine. Biomark Insights. 2011;6:69–78. doi: 10.4137/BMI.S7451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Martell LA, Cunico RL, Ohh J, Fulkerson W, Furneaux R, Foehr ED. Validation of an LC-MS/MS assay for detecting relevant disaccharides from keratan sulfate as a biomarker for Morquio A syndrome. Bioanalysis. 2011;3:1855–1866. doi: 10.4155/bio.11.172. [DOI] [PubMed] [Google Scholar]
- 15.Hendriksz CJ, Al-Jawad M, Berger KI, Hawley SM, Lawrence R, Mc Ardle C, Summers CG, Wright E, Braunlin E. Clinical overview and treatment options for non-skeletal manifestations of mucopolysaccharidosis type IVA. J Inherit Metab Dis. 2013;36:309–322. doi: 10.1007/s10545-012-9459-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stevens JM, Kendall BE, Crockard HA, Ransford A. The odontoid process in Morquio-Brailsford's disease. The effects of occipitocervical fusion. J Bone Joint Surg Br. 1991;73:851–858. doi: 10.1302/0301-620X.73B5.1910048. [DOI] [PubMed] [Google Scholar]
- 17.Hughes DG, Chadderton RD, Cowie RA, Wraith JE, Jenkins JP. MRI of the brain and craniocervical junction in Morquio's disease. Neuroradiology. 1997;39:381–385. doi: 10.1007/s002340050429. [DOI] [PubMed] [Google Scholar]
- 18.Lipson SJ. Dysplasia of the odontoid process in Morquio's syndrome causing quadriparesis. J Bone Joint Surg Am. 1997;59:340–344. [PubMed] [Google Scholar]
- 19.Ransford AO, Crockard HA, Stevens JM, Modaghegh S. Occipito-atlanto-axial fusion in Morquio-Brailsford syndrome. A ten-year experience J Bone Joint Surg Br. 1996;78:307–313. [PubMed] [Google Scholar]
- 20.Dhawale AA, Church C, Henley J, Holmes L, Jr, Thacker MM, Mackenzie WG, Miller F. Gait pattern and lower extremity alignment in children with Morquio syndrome. Pediatr Orthop B. 2013;22:59–62. doi: 10.1097/BPB.0b013e32835a0e6d. [DOI] [PubMed] [Google Scholar]
- 21.Dhawale AA, Thacker MM, Belthur MV, Rogers K, Bober MB, Mackenzie WG. The Lower Extremity in Morquio Syndrome. J Pediatr Orthop. 2012;32:534–540. doi: 10.1097/BPO.0b013e318259fe57. [DOI] [PubMed] [Google Scholar]
- 22.Borowski A, Thacker MM, Mackenzie WG, Littleton AG, Grissom L. The use of computed tomography to assess acetabular morphology in Morquio-Brailsford syndrome. J Pediatr Orthop. 2007;27:893–897. doi: 10.1097/bpo.0b013e31815a6007. [DOI] [PubMed] [Google Scholar]
- 23.Kakkis ED, Muenzer J, Tiller GE, Waber L, Belmont J, Passage M, Izykowski B, Phillips J, Doroshow R, Walot I, Hoft R, Neufeld EF. Enzyme-replacement therapy in mucopolysaccharidosis I. N Engl J Med. 2001;344:182–188. doi: 10.1056/NEJM200101183440304. [DOI] [PubMed] [Google Scholar]
- 24.Muenzer J, Wraith JE, Beck M, Giugliani R, Harmatz P, Eng CM, Vellodi A, Martin R, Ramaswami U, Gucsavas-Calikoglu M, Vijayaraghavan S, Wendt S, Puga AC, Ulbrich B, Shinawi M, Cleary M, Piper D, Conway AM, Kimura A. A phase II/III clinical study of enzyme replacement therapy with idursulfase in mucopolysaccharidosis II (Hunter syndrome) Genet Med. 2006;8:465–473. doi: 10.1097/01.gim.0000232477.37660.fb. [DOI] [PubMed] [Google Scholar]
- 25.Muenzer J, Lamsa JC, Garcia A, Dacosta J, Garcia J, Treco DA. Enzyme replacement therapy in mucopolysaccharidosis type II (Hunter syndrome): a preliminary report. Acta Paediatr Suppl. 2002;91:98–99. doi: 10.1111/j.1651-2227.2002.tb03115.x. [DOI] [PubMed] [Google Scholar]
- 26.Harmatz P, Whitley CB, Waber L, Pais R, Steiner R, Plecko B, Kaplan P, Simon J, Butensky E, Hopwood JJ. Enzyme replacement therapy in mucopolysaccharidosis VI (Maroteaux-Lamy syndrome) J Pediatr. 2004;144:574–580. doi: 10.1016/j.jpeds.2004.03.018. [DOI] [PubMed] [Google Scholar]
- 27.Harmatz P, Ketteridge D, Giugliani R, Guffon N, Teles EL, Miranda MC, Yu ZF, Swiedler SJ, Hopwood JJ. MPS VI Study Group. Direct comparison of measures of endurance, mobility, and joint function during enzyme-replacement therapy of mucopolysaccharidosis VI (Maroteaux-Lamy syndrome): results after 48 weeks in a phase 2 open-label clinical study of recombinant human N-acetylgalactosamine 4-sulfatase. Pediatrics. 2005;115:e681–689. doi: 10.1542/peds.2004-1023. [DOI] [PubMed] [Google Scholar]
- 28.Harmatz P, Giugliani R, Schwartz I, Guffon N, Teles EL, Miranda MC, Wraith JE, Beck M, Arash L, Scarpa M, Yu ZF, Wittes J, Berger KI, Newman MS, Lowe AM, Kakkis E, Swiedler SJ. MPS VI Phase 3 Study Group. Enzyme replacement therapy for mucopolysaccharidosis VI: a phase 3, randomized, double-blind, placebo-controlled, multinational study of recombinant human N-acetylgalactosamine 4-sulfatase (recombinant human arylsulfatase B or rhASB) and follow-on, open-label extension study. J Pediatr. 2006;148:533–539. doi: 10.1016/j.jpeds.2005.12.014. [DOI] [PubMed] [Google Scholar]
- 29.Harmatz P, Giugliani R, Schwartz IV, Guffon N, Teles EL, Miranda MC, Wraith JE, Beck M, Arash L, Scarpa M, Ketteridge D, Hopwood JJ, Plecko B, Steiner R, Whitley CB, Kaplan P, Yu ZF, Swiedler SJ, Decker C. MPS VI Study Group. Long-term follow-up of endurance and safety outcomes during enzyme replacement therapy for mucopolysaccharidosis VI: Final results of three clinical studies of recombinant human N-acetylgalactosamine 4-sulfatase. Mol Genet Metab. 2008;94:469–475. doi: 10.1016/j.ymgme.2008.04.001. [DOI] [PubMed] [Google Scholar]
- 30.Giugliani R, Federhen A, Rojas MV, Vieira T, Artigalas O, Pinto LL, Azevedo AC, Acosta A, Bonfim C, Lourenco CM, Kim CA, Horovitz D, Bonfim D, Norato D, Marinho D, Palhares D, Santos ES, Ribeiro E, Valadares E, Guarany F, de Lucca GR, Pimentel H, de Souza IN, Correa J, Sr, Fraga JC, Goes JE, Cabral JM, Simionato J, Llerena J, Jr, Jardim L, Giuliani L, da Silva LC, Santos ML, Moreira MA, Kerstenetzky M, Ribeiro M, Ruas N, Barrios P, Aranda P, Honjo R, Boy R, Costa R, Souza C, Alcantara FF, Avilla SG, Fagondes S, Martins AM. Mucopolysaccharidosis I, II, and VI: Brief review and guidelines for treatment. Genetics and molecular biology. 2010;33:589–604. doi: 10.1590/S1415-47572010005000093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Harmatz P. Enzyme replacement therapy with galsulfase for mucopolysaccharidosis VI: clinical facts and figures. Turkish Journal of Pediatrics. 2010;52:443–449. [PubMed] [Google Scholar]
- 32.Tylki-Szymanska A, Marucha J, Jurecka A, Syczewska M, Czartoryska B. Efficacy of recombinant human alpha-L-iduronidase (laronidase) on restricted range of motion of upper extremities in mucopolysaccharidosis type I patients. J Inherit Metab Dis. 2010;33:151–157. doi: 10.1007/s10545-010-9059-9. [DOI] [PubMed] [Google Scholar]
- 33.Tylki-Szymanska A, Jurecka A, Zuber Z, Rozdzynska A, Marucha J, Czartoryska B. Enzyme replacement therapy for mucopolysaccharidosis II from 3 months of age: 3-year follow-up. ActaPaediatr. 2012;101:e42–47. doi: 10.1111/j.1651-2227.2011.02385.x. [DOI] [PubMed] [Google Scholar]
- 34.Tomatsu S, Montaño AM, Ohashi A, Oikawa H, Oguma T, Dung VC, Nishioka T, Orii T, Sly WS. Enzyme replacement therapy in a murine model of Morquio A syndrome. Hum Mol Genet. 2008;17:815–824. doi: 10.1093/hmg/ddm353. [DOI] [PubMed] [Google Scholar]
- 35.Simonaro CM, D'Angelo M, Haskins ME, Schuchman EH. Joint and bone disease in mucopolysaccharidoses VI and VII: identification of new therapeutic targets and biomarkers using animal models. Pediatric Research. 2005;57:701–707. doi: 10.1203/01.PDR.0000156510.96253.5A. [DOI] [PubMed] [Google Scholar]
- 36.Nishioka T, Tomatsu S, Gutierrez MA, Miyamoto K, Trandafirescu GG, Lopez PL, Grubb JH, Kanai R, Kobayashi H, Yamaguchi S, Gottesman GS, Cahill R, Noguchi A, Sly WS. Enhancement of drug delivery to bone: characterization of human tissue-nonspecific alkaline phosphatase tagged with an acidic oligopeptide. Mol Genet Metab. 2006;88:244–255. doi: 10.1016/j.ymgme.2006.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Millán JL, Narisawa S, Lemire I, Loisel TP, Boileau G, Leonard P, Gramatikova S, Terkeltaub R, Camacho NP, McKee MD, Crine P, Whyte MP. Enzyme replacement therapy for murine hypophosphatasia. J Bone Miner Res. 2008;23:777–787. doi: 10.1359/JBMR.071213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Whyte MP, Greenberg CR, Salman NJ, Bober MB, McAlister WH, Wenkert D, Van Sickle BJ, Simmons JH, Edgar TS, Bauer ML, Hamdan MA, Bishop N, Lutz RE, McGinn M, Craig S, Moore JN, Taylor JW, Cleveland RH, Cranley WR, Lim R, Thacher TD, Mayhew JE, Downs M, Millán JL, Skrinar AM, Crine P, Landy H. Enzyme-replacement therapy in life-threatening hypophosphatasia. N Engl J Med. 2011;8(366):904–913. doi: 10.1056/NEJMoa1106173. [DOI] [PubMed] [Google Scholar]
- 39.Tomatsu S, Montaño AM, DunG VC, Ohashi A, Oikawa H, Oguma T, Orii T, Barrera L, Sly WS. Enhancement of drug delivery: enzyme replacement therapy for murine Morquio A syndrome. Mol Ther. 2012;18:1094–1102. doi: 10.1038/mt.2010.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rowan DJ, Tomatsu S, Grubb JH, Haupt B, Montaño AM, Oikawa H, Sosa AC, Chen A, Sly WS. Long circulating enzyme replacement therapy rescues bone pathology in mucopolysaccharidosis VII murine model. Mol Genet Metab. 2012;107:161–72. doi: 10.1016/j.ymgme.2012.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tolar J, Grewal SS, Bjoraker KJ, Whitley CB, Shapiro EG, Charnas L, Orchard PJ. Combination of enzyme replacement and hematopoietic stem cell transplantation as therapy for Hurler syndrome. Bone Marrow Transplant. 2008;41:531–535. doi: 10.1038/sj.bmt.1705934. [DOI] [PubMed] [Google Scholar]
- 42.Tanaka A, Okuyama T, Suzuki Y, Sakai N, Takakura H, Sawada T, Tanaka T, Otomo T, Ohashi T, Ishige-Wada M, Yabe H, Ohura T, Suzuki N, Kato K, Adachi S, Kobayashi R, Mugishima H, Kato S. Long-term efficacy of hematopoietic stem cell transplantation on brain involvement in patients with mucopolysaccharidosis type II: A nationwide survey in Japan. Mol Genet Metab. 2012;107:513–520. doi: 10.1016/j.ymgme.2012.09.004. [DOI] [PubMed] [Google Scholar]
- 43.Turbeville S, Nicely H, Rizzo JD, Pedersen TL, Orchard PJ, Horwitz ME, Horwitz EM, Veys P, Bonfim C, Al-Seraihy A. Clinical outcomes following hematopoietic stem cell transplantation for the treatment of mucopolysaccharidosis VI. Mol Genet Metab. 2011;102:111–115. doi: 10.1016/j.ymgme.2010.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Khanna G, Van Heest AE, Agel J, Bjoraker K, Grewal S, Abel S, Krivit W, Peters C, Orchard PJ. Analysis of factors affecting development of carpal tunnel syndrome in patients with Hurler syndrome after hematopoietic cell transplantation. Bone Marrow Transplant. 2007;39:331–334. doi: 10.1038/sj.bmt.1705586. [DOI] [PubMed] [Google Scholar]
- 45.Piotrowska E, Jakóbkiewicz-Banecka J, Barańska S, Tylki-Szymańska A, Czartoryska B, Wegrzyn A, Wegrzyn G. Genistein-mediated inhibition of glycosaminoglycan synthesis as a basis for gene expression-targeted isoflavone therapy for mucopolysaccharidoses. Eur J Hum Genet. 2006;14:846–852. doi: 10.1038/sj.ejhg.5201623. [DOI] [PubMed] [Google Scholar]
- 46.Jakóbkiewicz-Banecka J, Piotrowska E, Narajczyk M, Barańska S, Wegrzyn G. Genistein-mediated inhibition of glycosaminoglycan synthesis, which corrects storage in cells of patients suffering from mucopolysaccharidoses, acts by influencing an epidermal growth factor-dependent pathway. J Biomed Sci. 2009;16:26. doi: 10.1186/1423-0127-16-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Delgadillo V, O'Callaghan Mdel M, Artuch R, Montero R, Pineda M. Genistein supplementation in patients affected by Sanfilippo disease. J Inherit Metab Dis. 2011;34:1039–1044. doi: 10.1007/s10545-011-9342-4. [DOI] [PubMed] [Google Scholar]
- 48.Marucha J, Tylki-Szymańska A, Jakóbkiewicz-Banecka J, Piotrowska E, Kloska A, Czartoryska B, Węgrzyn G. Improvement in the range of joint motion in seven patients with mucopolysaccharidosis type II during experimental gene expression-targeted isoflavone therapy (GET IT) Am J Med Genet A. 2011;155A:2257–2262. doi: 10.1002/ajmg.a.34146. [DOI] [PubMed] [Google Scholar]
- 49.Kloska A, Jakóbkiewicz-Banecka J, Narajczyk M, Banecka-Majkutewicz Z, Węgrzyn G. Effects of flavonoids on glycosaminoglycan synthesis: implications for substrate reduction therapy in Sanfilippo disease and other mucopolysaccharidoses. Metab Brain Dis. 2011;26:1–8. doi: 10.1007/s11011-011-9233-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liang L, Li L, Zeng J, Gao Y, Chen YL, Wang ZQ, Wang XY, Chang LS, He D. Inhibitory effect of silibinin on EGFR signal-induced renal cell carcinoma progression via suppression of the EGFR/MMP-9 signaling pathway. Oncol Rep. 2012;28:999–1005. doi: 10.3892/or.2012.1874. [DOI] [PubMed] [Google Scholar]
- 51.Sun Q, Prasad R, Rosenthal E, Katiyar SK. Grape seed proanthocyanidins inhibit the invasive potential of head and neck cutaneous squamous cell carcinoma cells by targeting EGFR expressionand epithelial-to-mesenchymal transition. BMC Complement Altern Med. 2011;11:134. doi: 10.1186/1472-6882-11-134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lim YC, Cha YY. 2011 Epigallocatechin-3-gallate induces growth inhibition and apoptosis of human anaplastic thyroid carcinoma cells through suppression of EGFR/ERK pathway and cyclin B1/CDK1 complex. J Surg Oncol. 2011;104:776–780. doi: 10.1002/jso.21999. [DOI] [PubMed] [Google Scholar]
- 53.Phromnoi K, Prasad S, Gupta SC, Kannappan R, Reuter S, Limtrakul P, Aggarwal BB. Dihydroxypentamethoxyflavone down-regulates constitutive and inducible signal transducers and activators of transcription-3 through the induction of tyrosine phosphatase SHP-1. Mol Pharmacol. 2011;80:889–899. doi: 10.1124/mol.111.073676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mizuno H, Cho YY, Zhu F, Ma WY, Bode AM, Yang CS, Ho CT, Dong Z. Theaflavin-3, 3'-digallate induces epidermal growth factor receptor downregulation. Mol Carcinog. 2006;45:204–212. doi: 10.1002/mc.20174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lee LT, Huang YT, Hwang JJ, Lee PP, Ke FC, Nair MP, Kanadaswam C, Lee MT. Blockade of the epidermal growth factor receptor tyrosine kinase activity by quercetin and luteolin leads to growth inhibition and apoptosis of pancreatic tumor cells. Anticancer Res. 2002;22:1615–1627. [PubMed] [Google Scholar]
- 56.Godichaud S, Si-Tayeb K, Augé N, Desmoulière A, Balabaud C, Payrastre B, Nègre-Salvayre A, Rosenbaum J. The grape-derived polyphenol resveratrol differentially affects epidermal and platelet-derived growth factor signaling in human liver myofibroblasts. Int J Biochem Cell Biol. 2006;38:629–637. doi: 10.1016/j.biocel.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 57.Toietta G, Severini GM, Traversari C, Tomatsu S, Sukegawa K, Fukuda S, Kondo N, Tortora P, Bordignon C. Various cells retrovirally transduced with N-acetylgalactosoamine-6-sulfate sulfatase correct Morquio skin fibroblasts in vitro. Hum Gene Ther. 2001;12:2007–2016. doi: 10.1089/104303401753204571. [DOI] [PubMed] [Google Scholar]
- 58.Hacein-Bey-Abina S, Garrigue A, Wang GP, Soulier J, Lim A, Morillon E, Clappier E, Caccavelli L, Delabesse E, Beldjord K, Asnafi V, MacIntyre E, Dal Cortivo L, Radford I, Brousse N, Sigaux F, Moshous D, Hauer J, Borkhardt A, Belohradsky BH, Wintergerst U, Velez MC, Leiva L, Sorensen R, Wulffraat N, Blanche S, Bushman FD, Fischer A, Cavazzana-Calvo M. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J Clin Invest. 2008;118:3132–3142. doi: 10.1172/JCI35700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Alexander IE, Cunningham SC, Logan GJ, Christodoulou J. Potential of AAV vectors in the treatment of metabolic disease. Gene Ther. 2008;15:831–839. doi: 10.1038/gt.2008.64. [DOI] [PubMed] [Google Scholar]
- 60.Carter BJ. Adeno-associated virus vectors in clinical trials. Hum Gene Ther. 2005;16:541–550. doi: 10.1089/hum.2005.16.541. [DOI] [PubMed] [Google Scholar]
- 61.Büning H, Ried MU, Perabo L, Gerner FM, Huttner NA, Enssle J, Hallek M. Receptor targeting of adeno-associated virus vectors. Gene Ther. 2003;10:1142–1151. doi: 10.1038/sj.gt.3301976. [DOI] [PubMed] [Google Scholar]
- 62.Loiler SA, Conlon TJ, Song S, Tang Q, Warrington KH, Agarwal A, Kapturczak M, Li C, Ricordi C, Atkinson MA, Muzyczka N, Flotte TR. Targeting recombinant adeno-associated virus vectors to enhance gene transfer to pancreatic islets and liver. Gene Ther. 2003;10:1551–1558. doi: 10.1038/sj.gt.3302046. [DOI] [PubMed] [Google Scholar]
- 63.Choi VW, McCarty DM, Samulski RJ. AAV hybrid serotypes: improved vectors for gene delivery. Curr Gene Ther. 2005;5:299–310. doi: 10.2174/1566523054064968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.White K, Büning H, Kritz A, Janicki H, McVey J, Perabo L, Murphy G, Odenthal M, Work LM, Hallek M, Nicklin SA, Baker AH. Engineering adeno-associated virus 2 vectors for targeted gene delivery to atherosclerotic lesions. Gene Ther. 2008;15:443–451. doi: 10.1038/sj.gt.3303077. [DOI] [PubMed] [Google Scholar]
- 65.Gutiérrez MA, García-Vallejo F, Tomatsu S, Ceron F, Almeciga-Diaz CJ, Dominguez MC, Barrera LA. Construction of an adenoassociated, viral derived, expression vector to correct the genetic defect in Morquio A disease. Biomedica. 2008;28:448–459. [PubMed] [Google Scholar]
- 66.Alméciga-Díaz CJ, Rueda-Paramo MA, Espejo AJ, Echeverri OY, Montano A, Tomatsu S, Barrera LA. Effect of elongation factor 1 alpha promoter and SUMF1 over in vitro expression of N-acetylgalactosamine-6-sulfate sulfatase. Mol Biol Rep. 2009;36:1863–1870. doi: 10.1007/s11033-008-9392-3. [DOI] [PubMed] [Google Scholar]
- 67.Almeciga-Diaz CJ, Montano AM, Tomatsu S, Barrera LA. Adeno-associated virus gene transfer in Morquio A disease - effect of promoters and sulfatase-modifying factor 1. FEBS J. 2010;277:3608–3619. doi: 10.1111/j.1742-4658.2010.07769.x. [DOI] [PubMed] [Google Scholar]
- 68.Fraldi A, Biffi A, Lombardi A, Visigalli I, Pepe S, Settembre C, Nusco E, Auricchio A, Naldini L, Ballabio A, Cosma MP. SUMF1 enhances sulfatase activities in vivo in five sulfatase deficiencies. Biochem J. 2007;403:305–312. doi: 10.1042/BJ20061783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fraldi A, Biffi A, Lombardi A, Visigalli I, Pepe S, Settembre C, Nusco E, Auricchio A, Naldini L, Ballabio A, Cosma MP. SUMF1 enhances sulfatase activities in vivo in five sulfatase deficiencies. Biochem J. 2007;403:305–312. doi: 10.1042/BJ20061783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Simonaro CM, Ge Y, Eliyahu E, He X, Jepsen KJ, Schuchman EH. Involvement of the Toll-like receptor 4 pathway and use of TNF-alpha antagonists for treatment of the mucopolysaccharidoses. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:222–227. doi: 10.1073/pnas.0912937107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Eliyahu E, Wolfson T, Ge Y, Jepsen KJ, Schuchman EH, Simonaro CM. Anti-TNF-alpha therapy enhances the effects of enzyme replacement therapy in rats with mucopolysaccharidosis type VI. PloS one. 2011;6:e22447. doi: 10.1371/journal.pone.0022447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Schuchman EH, Ge Y, Lai A, Borisov Y, Faillace M, Eliyahu E, He X, Iatridis J, Vlassara H, Striker G, Simonaro CM. Pentosan polysulfate: a novel therapy for the mucopolysaccharidoses. PLoS One. 2013;8:e54459. doi: 10.1371/journal.pone.0054459. [DOI] [PMC free article] [PubMed] [Google Scholar]