

Abstract
Advances in understanding the etiology of Parkinson disease have been driven by the identification of causative mutations in families. Genetic analysis of an Australian family with three males displaying clinical features of early-onset parkinsonism and intellectual disability identified a ∼45 kb deletion resulting in the complete loss of RAB39B. We subsequently identified a missense mutation (c.503C>A [p.Thr168Lys]) in RAB39B in an unrelated Wisconsin kindred affected by a similar clinical phenotype. In silico and in vitro studies demonstrated that the mutation destabilized the protein, consistent with loss of function. In vitro small-hairpin-RNA-mediated knockdown of Rab39b resulted in a reduction in the density of α-synuclein immunoreactive puncta in dendritic processes of cultured neurons. In addition, in multiple cell models, we demonstrated that knockdown of Rab39b was associated with reduced steady-state levels of α-synuclein. Post mortem studies demonstrated that loss of RAB39B resulted in pathologically confirmed Parkinson disease. There was extensive dopaminergic neuron loss in the substantia nigra and widespread classic Lewy body pathology. Additional pathological features included cortical Lewy bodies, brain iron accumulation, tau immunoreactivity, and axonal spheroids. Overall, we have shown that loss-of-function mutations in RAB39B cause intellectual disability and pathologically confirmed early-onset Parkinson disease. The loss of RAB39B results in dysregulation of α-synuclein homeostasis and a spectrum of neuropathological features that implicate RAB39B in the pathogenesis of Parkinson disease and potentially other neurodegenerative disorders.
Main Text
Parkinsonism is a neurological syndrome characterized by tremor, rigidity, balance problems, and a slowing of movement. The most common cause of parkinsonism is Parkinson disease (PD [MIM 168600]), which accounts for up to 70% of this syndrome. PD is a common progressive neurodegenerative disorder with motor symptoms due to the death of dopamine-generating cells, predominantly in the substantia nigra (SN). The pathological hallmark of PD is accumulation of α-synuclein in Lewy bodies and Lewy neurites, although additional pathology (such as neurofibrillary tangles [NFTs]) can be observed.1 Recent genetic studies have driven advances in understanding the molecular pathogenesis of PD, and preclinical discovery projects have investigated compounds that target the identified proteins as a precursor to etiology-based therapeutics. To date, 18 PD-associated loci have been reported, and variants in 13 monogenic or susceptibility genes have been identified.2 Common pathogenic mechanisms associated with these genes include protein turnover, mitochondrial function, and oxidative-stress pathways. However, approximately 90% of individuals with PD do not have a defined genetic etiology. Variants in known genes account for ∼10% of the variation in PD liability, suggesting that variants in additional genes and susceptibility loci remain to be identified.3,4
We identified an Australian kindred with three brothers who presented in childhood with nonprogressive intellectual disability (ID), which included delayed developmental milestones, cognitive impairment, and macrocephaly (Figure 1; Table S1, available online). Subsequently, early-onset parkinsonism (onset prior to 45 years of age) was also apparent, although the clinical progression and presentation varied. The proband developed tremor in late childhood, but the symptoms did not progress to frank parkinsonism. In contrast, his male siblings developed tremor from their late 30s and were diagnosed with L-DOPA-responsive akinetic-rigid PD by their mid-40s. A complete description of the phenotype is presented in Table S1. We collected samples from the Australian family after receiving institutional ethics approval from Royal Childrens Hospital (Melbourne) and written informed consent from participants. Genomic DNA was isolated from whole blood, and primary fibroblast cultures were generated according to standard protocols. SNP array and linkage analysis using a recessive homozygous model did not demonstrate linkage to the autosomes but did identify two ∼10.6 Mb haplotypes shared by the affected brothers at Xp22.2 and Xq27.3–qter (chrX: 3,624,034–14,291,092 and chrX: 145,644,895–tel, respectively; GRCh38/hg38, UCSC Genome Browser; Table S2). Copy-number variation and subsequent PCR analysis identified a ∼45 kb deletion within the Xq haplotype (ClinVar accession number SCV00019029). The deletion segregated with the disease and resulted in the complete deletion of RAB39B (RAB39B, member RAS oncogene family [MIM 300774]) and the last three coding exons of CLIC2 (chloride intracellular channel 2 [MIM 300138]). To assess RAB39B expression, we extracted total RNA from fibroblasts by using the SV Total RNA Isolation System (Promega) and synthesized cDNA with the Transcriptor First Strand cDNA Synthesis Kit (Roche). Consistent with the genomic data, the RAB39B and CLIC2 transcripts were not detected by RT-PCR analysis of fibroblast cells derived from affected individuals (Figure S1).
Figure 1.
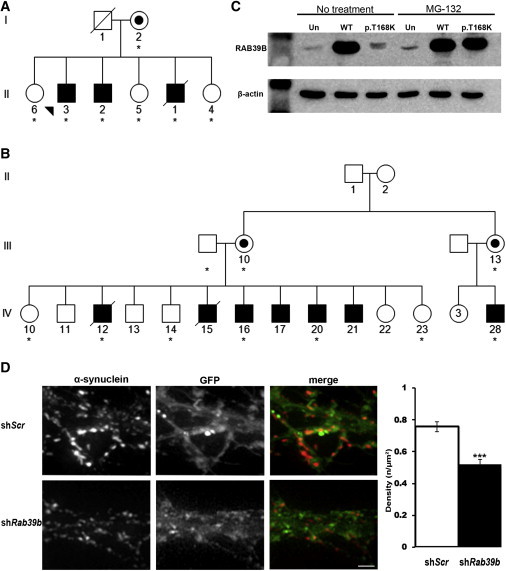
Identification of Mutations in RAB39B
(A and B) Simplified pedigree structure of the Australian (A) and Wisconsin (B) kindreds. Asterisks indicate DNA samples analyzed.
(C) Immunoblot analysis (12162-1-AP, Proteintech; 1:1,000) of RAB39B in BE(2)-M17 neuroblastoma cells grown in the absence or presence of 10 μM MG-132. Abbreviations are as follows: Un, untransfected; WT, wild-type; and p.T168K, RAB39B p.Thr168Lys. β-actin was used for confirming equivalent protein loading (A5441, Sigma-Aldrich; 1:5,000).
(D) Representative images of mouse hippocampal neurons transduced with lentiviral vectors encoding GFP and either scramble (shScr) or shRab39b sequences. Images were captured, and Z-space slices (0.3–0.4 μm) were deconvolved and flattened by maximum projection. ImageJ analysis software (the “Gran filter” plug-in set the size from 1 to infinity) was used to measure α-synuclein density relative to the area of infected dendrites. Quantification showed a significant reduction (0.52 ± 0.03 versus 0.76 ± 0.03, mean ± SEM, p ≤ 0.0005) in the density of α-synuclein immunoreactive puncta in neurons downregulated for Rab39b. The experiment was performed in triplicate (n = 42 neurons scored). The scale bar represents 20 μm.
The phenotype of the Australian kindred is similar to the basal ganglia disorder (Waisman syndrome [MIM 311510]) reported for a Wisconsin kindred5 (Figure 1; Table S1). Members of the defining family included 13 affected males who presented with variable degrees of ID and early-onset parkinsonism. Multipoint linkage analysis of the Wisconsin family previously localized the disease-causing mutation to Xq27.3–qter with a maximum multipoint LOD of 6.75 at the genetic marker F8C (chrX:154,929,351–154,929,630; GRCh38/hg38). The minimal Xq linkage region in the Australian kindred is within the Xq27.3–qter interval and therefore defines the shared critical linkage interval.6 Genomic DNA from individuals of the Wisconsin kindred was kindly provided by Professor Ronald Gregg. Ethics approval was provided by the institutional review board (IRB) at the University of Wisconsin, and informed consent was obtained. Direct sequencing of RAB39B identified a missense mutation (c.503C>A [p.Thr168Lys]; RefSeq accession number NM_171998.2, ClinVar SCV000190018) that segregated with disease (Figure S2) and was not detected in 200 unrelated control individuals or public databases (dbSNP137 and ESP6500). The threonine residue is conserved in evolution (Figure S3), and the mutation is predicted to be damaging by PolyPhen-2 and SIFT. In contrast, no sequence variants were observed by direct sequencing of CLIC2 in the Wisconsin kindred.
The absence of mutations in CLIC2 in the Wisconsin kindred suggests that disruption of RAB39B is the cause of the shared phenotype in both families. It is possible that deletion of CLIC2 in the Australian kindred might act as a disease modifier, although the phenotypic features (seizures and cardiac anomalies) associated with a missense mutation in CLIC2 were not observed.7 X chromosome exome sequencing of affected males from both kindreds confirmed the deleterious RAB39B changes and did not identify any other candidate variants within the linkage regions (Table S3).
In silico modeling of RAB39B was performed with the structure prediction programs MODELER8 and HHpred,9 and protein structures were visualized and superimposed with PyMOL. This analysis suggested that Thr168 is buried within the wild-type protein and interacts with Leu60 in the interswitch region, which undergoes conformational changes upon GTP-GDP exchange10,11 (Figure 2). The mutation introduces a large, positively charged lysine residue that is predicted by multiple algorithms, including ERIS15 and PoPMuSiC,16 to destabilize the protein. We could not directly test this because endogenous RAB39B was not detectable in fibroblast cells; therefore, we generated stable BE(2)-M17 neuroblastoma lines overexpressing wild-type RAB39B and altered (p.Thr168 Lys) RAB39B. The complete RAB39B open reading frame was amplified from human brain cDNA and cloned into the mammalian expression vector pcDNA3.1 (Invitrogen). We used site-directed mutagenesis (QuickChangeII) to generate the p.Thr168Lys altered RAB39B construct and Sanger sequenced all clones to verify that no additional variants were present. RT-PCR analysis confirmed similar expression of the wild-type and altered constructs (data not shown). In contrast, immunoblot analysis revealed high steady-state levels of exogenous wild-type RAB39B but very low levels of exogenous altered RAB39B. Immunoblot and immunofluorescence analysis of the cells after treatment with the proteasome inhibitor MG-132 confirmed that the reduced steady-state level of altered RAB39B was due to rapid turnover of the protein by the ubiquitin proteasome system (Figure 1; Figure S4). These results confirm the in silico modeling suggesting that the altered protein is destabilized and collectively demonstrate that loss of function of RAB39B causes ID and parkinsonism. Previous studies have associated RAB39B mutations with ID17–19 (pedigrees D-23 and MRX72; Table S1). The absence of parkinsonism in these additional families could be due to the individuals’ age at reporting, given that our data suggest that parkinsonism is likely to manifest after the second decade, albeit with some variability in both onset and clinical severity (Table S1). However, the lack of clinical data and inability to re-examine affected individuals mean that it is difficult to determine whether the phenotype associated with loss of RAB39B function represents an age-dependent progression of ID and parkinsonism or a spectrum of heterogeneous phenotypes extending from ID to ID with parkinsonism (see below).
Figure 2.
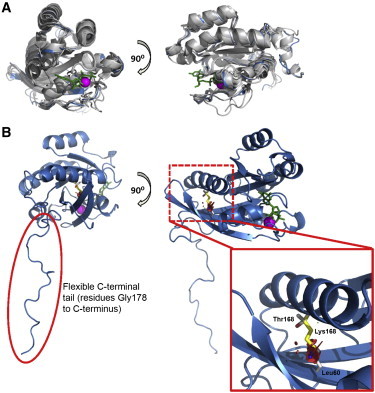
In Silico Modeling of the RAB39B Structure Predicts that p.Thr168Lys Is Destabilizing
(A) We generated a molecular model of the RAB39B structure (blue) by superimposing the structures of several known RAB proteins (gray) available in the Protein Data Bank (YTP1 [PDB 2BCG], RAB1A [PDB 3TKL], RAB11B [PDB 2F9L], RAB18 [PDB 1X3S], RAB23 [PDB 1Z2A], and RAB30 [PDB 2EW1]). GTP (green sticks) and Mg2+ (magenta sphere) are from the structure of RAB5A after superposition.
(B) We then used the predicted RAB39B structure (blue) to model the position of the native (Thr168, red) and altered (Lys168, yellow) amino acid. In the native form, Thr168 is predicted to interact with Leu60 within the interswitch region of RAB39B. The internal placement of the large positively charged residue in the p.Thr168Lys protein is predicted to destabilize the protein. The quality of the model was tested with PROCHECK,12 ANOLEA,13 and Verify3D.14
Rab GTPases belong to the Ras superfamily of small GTPases and act as essential regulators of vesicular trafficking. They dynamically localize to distinct intracellular membranes and regulate vesicular transport by recruiting effector proteins.20 The precise localization and function of RAB39B is unknown, but the protein is thought to play a role in synapse formation and maintenance.17,18,21 In support of this, we observed colocalization of endogenous RAB39B with markers of the vesicular-transport pathway, particularly the early endosome in mouse and human neuroblastoma cells (Figure S5). Given the postmortem results (below) and the association between α-synuclein and vesicular-trafficking pathways,22 we tested the effect of downregulation of RAB39B on α-synuclein localization. Mouse hippocampal neurons were prepared and transduced with lentivirus expressing validated Rab39b small hairpin RNA (shRNA) as previously described.17 Fourteen days after transduction, we observed that the density of α-synuclein immunoreactive puncta in the dendritic processes was 30% lower than in the cells transduced with the scramble control shRNA (p ≤ 0.0005; Figure 1; Figure S6). Immunoblot analysis confirmed ∼40% reduction of RAB39B but also demonstrated that α-synuclein levels (detected with the anti-α-synuclein antibody 97/8,23 1:1,000) were reduced by ∼50% (p ≤ 0.05; Figure S6). Similarly, in P19 mouse neuroblastoma cells, immunoblot analysis demonstrated that shRNA-mediated knockdown of Rab39b resulted in ∼50% reduction in α-synuclein steady-state levels (p ≤ 0.005; Figure S6). Although the mechanism remains to be fully defined, these results suggest that downregulation of RAB39B results in dysregulation of α-synuclein homeostasis. We sequenced RAB39B in a cohort of 187 individuals with early-onset PD; they had been previously sequenced and shown not to have mutations in known PD-associated genes, including SNCA (MIM 163890), PARK2 (MIM 602544), DJ1 (MIM 602533), PINK1 (MIM 608309), and LRRK2 (MIM 609007).24 This analysis did not identify any additional variants, suggesting that mutations in RAB39B are not a common cause of early-onset PD.
To determine whether the parkinsonism observed in the affected individuals resulted from PD, we investigated the neuropathology associated with the loss of RAB39B. Individual II:1 died at age 48 years from positional asphyxia. Postmortem neuropathological studies on the brain of II:1 were consistent with PD. The macroscopic findings were unremarkable, and serial coronal sections showed normal cortex, white matter, and ventricles with normal-appearing basal ganglia and thalamus. Cross section of the brain stem showed pallor of the SN and locus coeruleus. SN sections stained with haematoxylin and eosin (H&E) revealed hallmark neuropathological PD features, including loss of pigmented neurons and Lewy bodies in surviving neurons (Figure 3). Immunoreactive staining revealed the presence of α-synuclein-positive Lewy bodies and Lewy neurites in >10% of the surviving neurons. Additional neuropathological features included an abundance of cortical Lewy bodies, which are a pathological feature characteristic of dementia with Lewy bodies (DLB [MIM 127750]; reviewed in27,28). Tau-immunoreactive NFTs were also observed in a small proportion of the surviving pigmented SN neurons (Figure 3). Tau pathology has previously been observed in familial and idiopathic PD, and the tau-encoding gene (microtubule-associated protein tau [MAPT (MIM 157140)]) exists within a PD susceptibility locus. Tau plays a role in iron homeostasis,29–31 and Perl staining revealing a modest accumulation of iron in the SN (Figure 3) was consistent with the slight reduction in T2 signal intensity observed in individual II:1 (Table S1). In addition, analysis of the basal ganglia identified rare axonal spheroids in the white-matter tracts (data not shown), similar to the Wallerian-like degeneration observed in neurodegenerative diseases with impaired axonal transport.32 The additional pathological and clinical features share similarities with other neurodegenerative disorders, the most similar of which was neurodegeneration with brain iron accumulation (NBIA [MIM 234200]). Notably, in rare cases, NBIA can manifest with developmental delay and subsequent early-onset parkinsonism.33 Although MRI of II:3 was normal and II:1 did not show symptoms typical of NBIA,34 genomic DNA from individuals with NBIA was analyzed. Ethics approval was provided by the IRB at Oregon Health & Science University, and informed consent was obtained. Sequence analysis of RAB39B in a cohort of 48 male individuals with NBIA of unknown etiology did not identify any variants.
Figure 3.
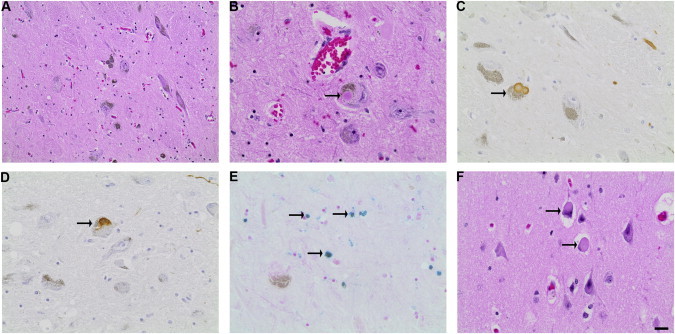
Neuropathology Associated with Loss of RAB39B
CNS postmortem tissue from II:1 was collected at autopsy, and microscopic examination was performed on H&E-stained sections. To detect PD-associated proteins, we applied immunohistochemistry to 5 μm formalin-fixed paraffin-embedded sections as previously described.25 Primary antibodies utilized were anti-α-synuclein (97/8; 1:1,000)23 and anti-tau (A0024, Dako; 1:500). CNS nonhaem iron (Fe2+ and Fe3+) was detected with a previously described Perl’s methodology.26 The scale bar represents 50 μm (A) or 20 μm (B–F).
(A–E) SN sections show (A) neuronal loss and pigment incontinence (H&E), (B) intraneuronal Lewy bodies (H&E), (C) α-synuclein-reactive Lewy bodies and neurites, (D) tau-immunoreactive intraneuronal NFTs, and (E) extracellular iron deposition (Perl’s stain).
(F) Neocortical Lewy bodies were identified by H&E staining.
In conclusion, genetic studies have demonstrated that loss of RAB39B causes pathologically defined PD, and functional studies have provided additional evidence for pathogenicity. Our results link the loss of a single gene involved in neuronal organization and synaptic function to the early manifestation of both ID and neurodegeneration and suggest that the loss of RAB39B dysregulates α-synuclein. For the two families we ascertained and clinically characterized, there appears to be a canonical age-dependent progression, namely ID first and then a slowly progressive basal ganglia disorder that advances after puberty. This was observed or reported in all 16 affected males in the Australian and Wisconsin families. However, given the lack of clinical data for the other two families previously described to be affected by RAB39B mutations,17 it is unclear whether this age-dependent phenotype predominates or whether heterogeneous phenotypes extending from ID to ID with parkinsonism are associated with loss of RAB39B function. This issue will be resolved by future studies of additional individuals with mutations in RAB39B.
The proposed role of RAB39B in vesicular trafficking identifies a potential disease mechanism that is distinct from pathways associated with genes in which mutations are currently known to cause familial early-onset PD. Previous in vitro studies have demonstrated that α-synuclein-mediated deficits in vesicular trafficking can be ameliorated by the overexpression of several RAB proteins35,36 but have not shown that loss of a specific RAB can cause PD. Current studies are further investigating how loss of RAB39B might cause the observed in vitro deficits in localization and reduced steady-state levels of α-synuclein but in vivo accumulation of significant α-synuclein pathology at end-stage disease. It is possible that in simple cell models with efficient protein-metabolism pathways, the “mislocalized” α-synuclein is rapidly turned over and thus leads to reduced steady-state levels. However, protein-turnover pathways are compromised in individuals with PD;37 therefore, mislocalized α-synuclein might not be turned over efficiently, and as the disease progresses, the protein could accumulate and be incorporated into the protein aggregates that define PD.
The broader pathology of iron accumulation, NFTs, and axonal spheroids is similar to that reported for a range of neurodegenerative conditions. However, the abundance of both brainstem and cortical Lewy bodies suggests that RAB39B and/or associated pathways might directly contribute to the pathogenic mechanisms underlying dementia disorders such as DLB. Further studies, including the development of animal models, will be important for understanding the underlying pathogenic mechanism(s) of RAB39B dysfunction and identifying potential targeted therapeutic interventions.
Acknowledgments
We thank the families for their participation in this study and the generous support of the Lefroy and Handbury families. This work was funded in part by Australian National Health and Medical Research Council (NHMRC) program grant 490037 to D.J.A. and M.B., NHMRC project grant APP1041860 to P.J.L., Parkinson’s Disease Foundation grant PDF-IRG-1220 to P.J.L. and G.R.W., and the project GENCODYS (grant 241995 to V.M.K.), which was funded by the European Union Framework Programme 7. P.J.L. was supported by an NHMRC Career Development Fellowship (APP1032364), and M.B. was supported by an Australian Research Council Future Fellowship (FT100100764). This work was made possible through Victorian State Government Operational Infrastructure Support and the NHMRC Independent Medical Research Institutes Infrastructure Support Scheme.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
ClinVar, http://www.ncbi.nlm.nih.gov/clinvar/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
PyMOL, http://www.pymol.org/
Accession Numbers
The ClinVar accession numbers for the RAB39B variants reported in this paper are SCV000190018 and SCV000190929.
References
- 1.Galpern W.R., Lang A.E. Interface between tauopathies and synucleinopathies: a tale of two proteins. Ann. Neurol. 2006;59:449–458. doi: 10.1002/ana.20819. [DOI] [PubMed] [Google Scholar]
- 2.Klein C., Westenberger A. Genetics of Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012;2:a008888. doi: 10.1101/cshperspect.a008888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Do C.B., Tung J.Y., Dorfman E., Kiefer A.K., Drabant E.M., Francke U., Mountain J.L., Goldman S.M., Tanner C.M., Langston J.W. Web-based genome-wide association study identifies two novel loci and a substantial genetic component for Parkinson’s disease. PLoS Genet. 2011;7:e1002141. doi: 10.1371/journal.pgen.1002141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Keller M.F., Saad M., Bras J., Bettella F., Nicolaou N., Simón-Sánchez J., Mittag F., Büchel F., Sharma M., Gibbs J.R., International Parkinson’s Disease Genomics Consortium (IPDGC) Wellcome Trust Case Control Consortium 2 (WTCCC2) Using genome-wide complex trait analysis to quantify ‘missing heritability’ in Parkinson’s disease. Hum. Mol. Genet. 2012;21:4996–5009. doi: 10.1093/hmg/dds335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Laxova R., Brown E.S., Hogan K., Hecox K., Opitz J.M. An X-linked recessive basal ganglia disorder with mental retardation. Am. J. Med. Genet. 1985;21:681–689. doi: 10.1002/ajmg.1320210409. [DOI] [PubMed] [Google Scholar]
- 6.Gregg R.G., Metzenberg A.B., Hogan K., Sekhon G., Laxova R. Waisman syndrome, a human X-linked recessive basal ganglia disorder with mental retardation: localization to Xq27.3-qter. Genomics. 1991;9:701–706. doi: 10.1016/0888-7543(91)90363-j. [DOI] [PubMed] [Google Scholar]
- 7.Takano K., Liu D., Tarpey P., Gallant E., Lam A., Witham S., Alexov E., Chaubey A., Stevenson R.E., Schwartz C.E. An X-linked channelopathy with cardiomegaly due to a CLIC2 mutation enhancing ryanodine receptor channel activity. Hum. Mol. Genet. 2012;21:4497–4507. doi: 10.1093/hmg/dds292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eswar N., Webb B., Marti-Renom M.A., Madhusudhan M.S., Eramian D., Shen M.Y., Pieper U., Sali A. Comparative protein structure modeling using Modeller. Curr. Protoc. Bioinformatics. 2006;Chapter 5 doi: 10.1002/0471250953.bi0506s15. Unit 5.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hildebrand A., Remmert M., Biegert A., Söding J. Fast and accurate automatic structure prediction with HHpred. Proteins. 2009;77(Suppl 9):128–132. doi: 10.1002/prot.22499. [DOI] [PubMed] [Google Scholar]
- 10.Khan A.R., Ménétrey J. Structural biology of Arf and Rab GTPases’ effector recruitment and specificity. Structure. 2013;21:1284–1297. doi: 10.1016/j.str.2013.06.016. [DOI] [PubMed] [Google Scholar]
- 11.Park H.H. Structural basis of membrane trafficking by rab family small g protein. Int. J. Mol. Sci. 2013;14:8912–8923. doi: 10.3390/ijms14058912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Laskowski R.A., Moss D.S., Thornton J.M. Main-chain bond lengths and bond angles in protein structures. J. Mol. Biol. 1993;231:1049–1067. doi: 10.1006/jmbi.1993.1351. [DOI] [PubMed] [Google Scholar]
- 13.Melo F., Devos D., Depiereux E., Feytmans E. ANOLEA: a www server to assess protein structures. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1997;5:187–190. [PubMed] [Google Scholar]
- 14.Eisenberg D., Lüthy R., Bowie J.U. VERIFY3D: assessment of protein models with three-dimensional profiles. Methods Enzymol. 1997;277:396–404. doi: 10.1016/s0076-6879(97)77022-8. [DOI] [PubMed] [Google Scholar]
- 15.Yin S., Ding F., Dokholyan N.V. Eris: an automated estimator of protein stability. Nat. Methods. 2007;4:466–467. doi: 10.1038/nmeth0607-466. [DOI] [PubMed] [Google Scholar]
- 16.Dehouck Y., Kwasigroch J.M., Gilis D., Rooman M. PoPMuSiC 2.1: a web server for the estimation of protein stability changes upon mutation and sequence optimality. BMC Bioinformatics. 2011;12:151. doi: 10.1186/1471-2105-12-151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Giannandrea M., Bianchi V., Mignogna M.L., Sirri A., Carrabino S., D’Elia E., Vecellio M., Russo S., Cogliati F., Larizza L. Mutations in the small GTPase gene RAB39B are responsible for X-linked mental retardation associated with autism, epilepsy, and macrocephaly. Am. J. Hum. Genet. 2010;86:185–195. doi: 10.1016/j.ajhg.2010.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vanmarsenille L., Giannandrea M., Fieremans N., Verbeeck J., Belet S., Raynaud M., Vogels A., Männik K., Õunap K., Jacqueline V. Increased dosage of RAB39B affects neuronal development and could explain the cognitive impairment in male patients with distal Xq28 copy number gains. Hum. Mutat. 2014;35:377–383. doi: 10.1002/humu.22497. [DOI] [PubMed] [Google Scholar]
- 19.Russo S., Cogliati F., Cavalleri F., Cassitto M.G., Giglioli R., Toniolo D., Casari G., Larizza L. Mapping to distal Xq28 of nonspecific X-linked mental retardation MRX72: linkage analysis and clinical findings in a three-generation Sardinian family. Am. J. Med. Genet. 2000;94:376–382. doi: 10.1002/1096-8628(20001023)94:5<376::aid-ajmg6>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 20.Hutagalung A.H., Novick P.J. Role of Rab GTPases in membrane traffic and cell physiology. Physiol. Rev. 2011;91:119–149. doi: 10.1152/physrev.00059.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cheng H., Ma Y., Ni X., Jiang M., Guo L., Ying K., Xie Y., Mao Y. Isolation and characterization of a human novel RAB (RAB39B) gene. Cytogenet. Genome Res. 2002;97:72–75. doi: 10.1159/000064047. [DOI] [PubMed] [Google Scholar]
- 22.Eisbach S.E., Outeiro T.F. Alpha-synuclein and intracellular trafficking: impact on the spreading of Parkinson’s disease pathology. J. Mol. Med. 2013;91:693–703. doi: 10.1007/s00109-013-1038-9. [DOI] [PubMed] [Google Scholar]
- 23.Culvenor J.G., McLean C.A., Cutt S., Campbell B.C., Maher F., Jäkälä P., Hartmann T., Beyreuther K., Masters C.L., Li Q.X. Non-Abeta component of Alzheimer’s disease amyloid (NAC) revisited. NAC and alpha-synuclein are not associated with Abeta amyloid. Am. J. Pathol. 1999;155:1173–1181. doi: 10.1016/s0002-9440(10)65220-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mellick G.D., Siebert G.A., Funayama M., Buchanan D.D., Li Y., Imamichi Y., Yoshino H., Silburn P.A., Hattori N. Screening PARK genes for mutations in early-onset Parkinson’s disease patients from Queensland, Australia. Parkinsonism Relat. Disord. 2009;15:105–109. doi: 10.1016/j.parkreldis.2007.11.016. [DOI] [PubMed] [Google Scholar]
- 25.Fodero-Tavoletti M.T., Smith D.P., McLean C.A., Adlard P.A., Barnham K.J., Foster L.E., Leone L., Perez K., Cortés M., Culvenor J.G. In vitro characterization of Pittsburgh compound-B binding to Lewy bodies. J. Neurosci. 2007;27:10365–10371. doi: 10.1523/JNEUROSCI.0630-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Meguro R., Asano Y., Odagiri S., Li C., Shoumura K. Cellular and subcellular localizations of nonheme ferric and ferrous iron in the rat brain: a light and electron microscopic study by the perfusion-Perls and -Turnbull methods. Arch. Histol. Cytol. 2008;71:205–222. doi: 10.1679/aohc.71.205. [DOI] [PubMed] [Google Scholar]
- 27.Huang Y., Halliday G. Can we clinically diagnose dementia with Lewy bodies yet? Transl. Neurodegener. 2013;2:4. doi: 10.1186/2047-9158-2-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mrak R.E., Griffin W.S. Dementia with Lewy bodies: Definition, diagnosis, and pathogenic relationship to Alzheimer’s disease. Neuropsychiatr. Dis. Treat. 2007;3:619–625. [PMC free article] [PubMed] [Google Scholar]
- 29.Simón-Sánchez J., Schulte C., Bras J.M., Sharma M., Gibbs J.R., Berg D., Paisan-Ruiz C., Lichtner P., Scholz S.W., Hernandez D.G. Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat. Genet. 2009;41:1308–1312. doi: 10.1038/ng.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lei P., Ayton S., Finkelstein D.I., Spoerri L., Ciccotosto G.D., Wright D.K., Wong B.X., Adlard P.A., Cherny R.A., Lam L.Q. Tau deficiency induces parkinsonism with dementia by impairing APP-mediated iron export. Nat. Med. 2012;18:291–295. doi: 10.1038/nm.2613. [DOI] [PubMed] [Google Scholar]
- 31.Wray S., Lewis P.A. A tangled web - tau and sporadic Parkinson’s disease. Front. Psychiatry. 2010;1:150. doi: 10.3389/fpsyt.2010.00150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Coleman M.P., Freeman M.R. Wallerian degeneration, wld(s), and nmnat. Annu. Rev. Neurosci. 2010;33:245–267. doi: 10.1146/annurev-neuro-060909-153248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gregory A., Polster B.J., Hayflick S.J. Clinical and genetic delineation of neurodegeneration with brain iron accumulation. J. Med. Genet. 2009;46:73–80. doi: 10.1136/jmg.2008.061929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schipper H.M. Neurodegeneration with brain iron accumulation - clinical syndromes and neuroimaging. Biochim. Biophys. Acta. 2012;1822:350–360. doi: 10.1016/j.bbadis.2011.06.016. [DOI] [PubMed] [Google Scholar]
- 35.Cooper A.A., Gitler A.D., Cashikar A., Haynes C.M., Hill K.J., Bhullar B., Liu K., Xu K., Strathearn K.E., Liu F. Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson’s models. Science. 2006;313:324–328. doi: 10.1126/science.1129462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gitler A.D., Bevis B.J., Shorter J., Strathearn K.E., Hamamichi S., Su L.J., Caldwell K.A., Caldwell G.A., Rochet J.C., McCaffery J.M. The Parkinson’s disease protein alpha-synuclein disrupts cellular Rab homeostasis. Proc. Natl. Acad. Sci. USA. 2008;105:145–150. doi: 10.1073/pnas.0710685105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cook C., Stetler C., Petrucelli L. Disruption of protein quality control in Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012;2:a009423. doi: 10.1101/cshperspect.a009423. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.