

Abstract
The morbidity and mortality attributable to heritable and sporadic carcinomas of the colon are substantial and affect children and adults alike. Despite current colonoscopy screening recommendations colorectal adenocarcinoma (CRC) still accounts for almost 140000 cancer cases yearly. Familial adenomatous polyposis (FAP) is a colon cancer predisposition due to alterations in the adenomatous polyposis coli gene, which is mutated in most CRC. Since the beginning of the genomic era next-generation sequencing analyses of CRC continue to improve our understanding of the genetics of tumorigenesis and promise to expand our ability to identify and treat this disease. Advances in genome sequence analysis have facilitated the molecular diagnosis of individuals with FAP, which enables initiation of appropriate monitoring and timely intervention. Genome sequencing also has potential clinical impact for individuals with sporadic forms of CRC, providing means for molecular diagnosis of CRC tumor type, data guiding selection of tumor targeted therapies, and pharmacogenomic profiles specifying patient specific drug tolerances. There is even a potential role for genomic sequencing in surveillance for recurrence, and early detection, of CRC. We review strategies for diagnostic assessment and management of FAP and sporadic CRC in the current genomic era, with emphasis on the current, and potential for future, impact of genome sequencing on the clinical care of these conditions.
Keywords: Colorectal adenocarcinoma, Familial adenomatous polyposis, Genome sequencing, Personalized medicine, Cancer genomics, Pharmacogenomics, Genomic medicine
Core tip: The era of genomic sequencing is beginning to make significant impact on the diagnosis and management of sporadic and inherited colorectal adenocarcinoma (CRC) such as familial adenomatous polyposis. This review will discuss the current guidelines for diagnosis and management of CRC and how genomic sequencing is enabling earlier definitive diagnosis with associated intensive surveillance and preventative interventions, molecular tumor characterization directing tumor specific therapy, germline patient genome analysis which informs individual drug tolerance and efficacy, and is evolving to develop post-treatment surveillance, with the potential to ultimately decrease the current prevalence and mortality of CRC, sporadic and hereditary.
INTRODUCTION
The annual incidence in the United States of colorectal adenocarcinoma (CRC) is almost 140000[1]. The morbidity and mortality attributable to heritable and sporadic carcinomas of the colon are substantial and affect children and adults alike, with CRC being the third highest cause of cancer mortality among men and women alike. The average individual lifetime risk in the United States of developing sporadic CRC is estimated at 5%, with average onset being over 50 years of age. CRC mortality can be mitigated by early detection and intervention, such as by polypectomy[2-4]. Accordingly, recommendations have been established for surveillance colonoscopy screening, the goal being to detect and remove adenomatous polyps at an early and curable stage.
However, according to the Centers for Disease Control and Prevention, only about 59% of adults aged 50-75 years undergo recommended colonoscopy[5]. Though early detected CRC can be successfully removed, CRC which remains undetected until an advanced stage with metastases remains incurable[6]. The molecular etiology of CRC has been studied extensively, revealing that it develops from an accumulation of genomic mutations. CRC has been associated with mutations in various genes that are altered in other forms of cancer, such as ATM in leukemia and lymphoma, PPP2R1B in lung and breast carcinoma, and MYC in hepatocellular carcinoma[7-11], among others. However, the genes more commonly found mutated in CRC include the APC (approximately 80%), KRAS, SMAD4, and p53, and the cell signaling pathways most commonly impacted by mutations in CRC include the WNT, RAS, TGF-beta, PI3K and P53 pathways[7,12].
While CRC often occurs sporadically, germline mutations in a number of genes can cause syndromes which predispose to the development of CRC. The heritable CRC syndromes are broadly categorized as polyposis associated [including familial adenomatous polyposis (FAP), MUTYH-associated polyposis or MAP, Gardner and Turcot syndromes] and non-polyposis associated (including Lynch, Peutz-Jeghers and PTEN hamartoma tumor syndromes)[13,14]. This review will focus on polyposis associated heritable CRC, and FAP in particular. Inherited mutations in APC cause FAP, which is a colon cancer predisposition syndrome of autosomal dominant inheritance affecting children as early as 9 years of age, with reports of carcinoma in the first decade of life.
Characteristic features of FAP include development of hundreds to thousands of adenomatous polyps beginning in early adolescence, with development of CRC in the absence of treatment. About 7% of patients develop CRC by age 21, about 95% by age 50. In all FAP patients colon cancer is inexorable in the absence of colectomy. Though the classic course of FAP results in CRC, it can also become complicated by non-colonic expressions, in particular gastric and duodenal polyps, and is associated with elevated risk for duodenal, stomach, pancreatic, thyroid, liver, and CNS cancer[15]. While FAP is known to be initiated by a germline mutation of APC, studies have yet to establish whether CRC in FAP requires a similar accumulation of genetic alterations as has been observed in sporadic CRC. Since the beginning of the genomic era[16] at the completion of the Human Genome Project[17], the growth in capacity and availability of genomic sequencing has made it possible to more clearly elucidate the molecular etiology of conditions such as FAP related CRC. Genomic sequencing facilitates the identification of individuals with FAP, enabling and guiding appropriate intervention and is augmenting the ability to characterize CRC tumors on a molecular scale, for selection of targeted therapies that can be personalized per patient tolerance.
FAP DIAGNOSIS
Initial presentation and evaluation
FAP is second most common inherited CRC, with prevalence estimated at 1:10000, and is caused by mutations in APC[15]. Patients will often present with occult blood in the stool, as polyps develop on average by 16 years of age[18]. The average age at identification of CRC in untreated individuals is 39[13]. Clinical diagnosis of classic FAP is established when 100 or more colonic polyps are observed on colonoscopy, or less than 100 colonic polyps are observed in a patient with a family history of FAP[15]. A related syndrome, attenuated FAP, associates with a lower polyp burden (average of 30) and later age at diagnosis of CRC, though it is also caused by APC gene mutations[19]. Identification of a mutation in APC provides molecular confirmation of FAP. The American College of Medical Genetics and Genomics guidelines recommend complete APC gene analysis be considered in any individual with 100 or more colonic polyps, autosomal dominant inheritance and/or extra-colonic manifestations of FAP (e.g., congenital hypertrophy of retinal pigment epithelium, desmoids, gastric fundic gland polyps, among others), when no prior family member has undergone testing[20]. If a familial mutation is identified, targeted APC analysis can be performed[21]. This testing provides clinical confirmation necessary to guide predictive counseling and enable assessment of family members at increased risk, as APC alterations are found in as high as 90% of families with classic FAP. Testing is also important when the patient’s presentation is not completely typical for classic FAP, such has demonstrating a lower than expected polyp burden or later age at onset. In some cases, APC gene analysis will confirm a mutation consistent with attenuated FAP, though APC alterations in the attenuated form are only discovered in 10%-56% of cases[22].
Of particular importance in patients presenting with polyps not clearly identifiable as classic FAP, attenuated FAP, Gardener syndrome, Turcot syndrome, MUTYH associated polyposis (MAP) or one of the nonpolyposis syndromes, are the gene panels made possible by genomic sequencing. Three of these multi-gene sequencing panels are currently offered by Clinical Laboratory Improvement Amendments (CLIA) certified labs in the United States. They are performed with patient blood derived genomic DNA via next-generation sequencing of the coding regions of up to 19 different genes for point mutations associated with various hereditary colon cancer syndromes, are complemented by duplication/deletion analysis of the genes using microarray comparative genomic hybridization or multiplex ligation-dependent probe amplification, and point mutations are confirmed via Sanger sequencing. The OncoGene Dx gene panel offered by GeneDx analyses a total of 18 genes (including APC, ATM, AXIN2, BLM, BMPR1A, CDH1, CHEK2, EPCAM, MLH1, MSH2, MSH6, MUTYH, PMS2, PTEN, SMAD4, STK11, p53, XRCC2) and requires about 4 wk to complete the assay for a new patient (http://www.genedx.com/test-catalog/disorders/colorectal-cancer)[23]. Ambry Genetics’ ColoNext gene panel assays 15 genes (including APC, BMPR1A, CDH1, CHEK2, EPCAM, MLH1, MSH2, MSH6, MUTYH, PMS2, PTEN, SMAD4, STK11, p53) with analysis turnaround time approximately 12-16 wk for new patients (http://www.ambrygen.com/tests/colonext)[24]. The University of Washington Genetics Laboratory offers ColoSeq which sequences the coding regions of 19 genes (APC, AKT1, BMPR1A, CDH1, EPCAM, GALNT12, GREM1, MLH1, MSH2, MSH6, MUTYH, PIK3CA, POLD1, POLE, PMS2, PTEN, SMAD4, STK11, p53), the estimated time to complete assessment is 12 wk for a new patient (http://tests.labmed.washington.edu/COLOSEQ)[25]. Other assays are certain to be introduced. Through these genomic testing panels, patients with an ambiguous presentation can be tested simultaneously for multiple hereditary colon cancer syndromes, decreasing the time to molecular diagnosis and appropriately tailored familial testing and clinical surveillance.
Familial genetic testing
For unaffected individuals in families with FAP, testing is not generally offered until about 10-12 years of age, which would be the recommended age to initiate surveillance colonoscopy in an affected individual[21]. Furthermore, efforts are made to identify an affected family member for whom testing has already confirmed an APC mutation, to allow targeted testing in the unaffected individual. If testing is performed in an affected family member and no mutation is found, unaffected individuals must undergo clinical surveillance empirically “as if” they were mutation carriers, because their carrier status cannot be ruled out.
Nondiagnostic and variants of unknown significance challenges
Approximately 90% of APC alterations in FAP introduce a stop codon causing truncation of the resulting protein at the C-terminus, with over 900 different germline APC alterations having been discovered in FAP individuals to date[26-29]. Though clinical genetic testing of the APC gene has a 90% mutation detection rate[30], approximately 10% of classic FAP cases do not have an identifiable mutation in APC, requiring greater reliance on clinical presentation and empiric surveillance screening in all at risk individuals in these families[21,31]. The growing awareness of genetic and genomic testing and its utility in diagnosing FAP has also lead to the observation of non-truncating APC mutations, including missense and silent mutations in the coding sequence and splice-site mutations in less conserved intronic sequence, some of which have been correlated with FAP[29,32,33]. The relevance of these variants of unknown significance (VUS) in some cases can be suggested by in silico prediction algorithms, however in vivo or in vitro functional analyses of these VUS provide more reliable data to predict the functional impact of APC variants. For example, a silent alteration in exon 14 (c.1869G > T) was reported to cause exon skipping due to its impact on splice enhancer sites[34]. Also the p.I1307K missense mutation in APC, though not causing classic FAP, carries a 10%-20% increased lifelong risk of CRC, but the p.E1317Q missense mutation in APC has an, as yet, uncertain role in colon cancer[20]. Though a minority of patients suspected to have FAP are found to carry VUS, with the increasing availability of APC gene testing and broader testing such as whole exome sequencing, its expected the number of patients with APC VUS will grow. Multiple groups have worked to better characterize the biological and clinical significance of these VUS[29,35]. For example, in vitro and in vivo methods of evaluating VUS in MAP are being developed, providing additional information to facilitate characterization of VUS in an effort to clarify the diagnosis of patients suspected to have MAP[36,37]. Further functional evaluation of VUS for FAP, and other hereditary CRC, is a critical step, with the increasing application of genomic sequencing technologies, for improving diagnostic accuracy of APC sequencing for patients with features concerning for FAP.
FAP MANAGEMENT
Surveillance strategies
Individuals with a family history of FAP meet criteria for additional assessment for high-risk syndromes if they present with > 10 adenomas in the same individual, per the most recent NCCN guidelines[38]. Management of FAP includes surveillance colonoscopy every 1-2 years, starting at about 10 years of age. After polyp development is observed, annual colonoscopy is recommended. Colectomy is considered when more than 20 adenomatous polyps develop, when adenomas greater than 1 cm are noted or when concerning histology appears, and is recommended when polyp burden precludes safe colonoscopic surveillance[15]. After colectomy surveillance continues. Annual colonoscopy is recommended if any rectal tissue remains. Upper GI endoscopy is recommended about every 1-3 years, starting at about age 25, as about 20% of patients will eventually require treatment of duodenal adenomas[15,39].
Some correlation between genotype and clinical presentation of classic FAP has been suggested, for example patients with alterations in codon 1309 tend to have an increased number of colon adenomas at an early age, with symptom onset at about 20 years of age. Those with alterations between codons 168 and 1580 had symptom onset about 30 years of age, and those with alterations affecting the 5’ of codon 168 and the 3’ of codon 1580 had symptom onset about 52 years of age[40,41]. Profuse polyposis has been observed (average of 5000 polyps) in patients with mutations in codons 1250-1464[42]. In contrast, attenuated FAP has been associated with the 5’ portion of APC[43], exon 9[43-45] and the distal 3’ portion of the gene[43,46-48], interstitial deletions of chromosome 5q22 including APC[49], partial and whole gene deletions[50] and somatic mosaicism for APC mutations usually associated with classic FAP[51-53]. While these correlations might help predict the course of polyposis in an individual, and suggest more or less aggressive surveillance and intervention strategies, these genotypes are not routinely applied to this purpose at present, though such applications could be clinically significant for management decisions in the future[13].
Treatment options
Certain pharmacological treatments have attempted to abrogate the accumulation of polyps in FAP patients, however surgical resection remains the mainstay of intervention. Chemoprevention strategies, including non-steroidal anti-inflammatory drugs such as celecoxib and sulindac, have been shown to temporarily decrease size or quantity of polyps in FAP patients, however polyps may return while patients are still taking chemopreventive therapy[54]. Currently, no chemopreventative strategy can replace regular surveillance, though this treatment may, in some cases, temporarily delay colectomy[54-56].
Lifelong risk for CRC in FAP individuals approaches 100% by 50 years of age. Because of the significant increase in cancer incidence about the third decade, prophylactic colectomy is often recommended in the second decade[57] . Generally three surgical strategies are available to FAP patients, including total proctocolectomy with ileal pouch anal anastomosis, total abdominal colectomy with ileorectal anastomosis and proctocolectomy with ileostomy[58]. Selection of a surgical strategy takes into consideration rectal polyp burden, personal and familial phenotype, with classic FAP patients receiving proctocolectomy, if possible, due to the increased risk for rectal cancer[58]. After surgical resection, surveillance continues, including endoscopic evaluation of any remaining rectal tissue and endoscopy of the upper gastrointestinal tract, as patients remain at increased risk for tumor formation despite colectomy[57].
Molecular etiology
FAP is caused by germline alterations in APC, a tumor suppressor acting in the WNT signaling cascade[59]. De novo APC mutations cause about 25% of FAP cases[15]. Studies in sporadic CRC show mutations in APC appear to instigate the development of CRC[12]. These studies show that APC acts in the colon to down regulate Wnt signaling by targeting beta catenin for degradation[59]. Wild-type APC protein is an important component of the composite that includes Axin, GSK-3beta and casein kinase 1[59]. This molecular composite regulates the phosphorylation of beta catenin necessary to target it for degradation[59]. When APC is altered, beta catenin increases, translocates to the nucleus, and is thought to coactivate TCF-LEF, which is involved in the transcriptional activation of cell regulatory genes such as c-myc and cyclin D1[59]. The multi-hit hypothesis of CRC tumor development suggests mutation or dysregulation of a number of genes occurs in the evolution of colon cancer, with APC mutations being present earlier in the process and KRAS, SMAD4 and p53 alterations being observed in later stages of cancer development[12,15,60]. As FAP patients carry one germline APC mutation from birth, it is possible that the accumulation of molecular changes leading to malignancy in FAP recapitulates, to some degree, the molecular cascade observed in sporadic CRC (Figure 1), however no comprehensive analysis of the molecular etiology of tumorigenesis in FAP has been reported. Future genomic analyses of FAP may contribute to the understanding of FAP tumorigenesis as well as improving interventional and preventative strategies.
Figure 1.
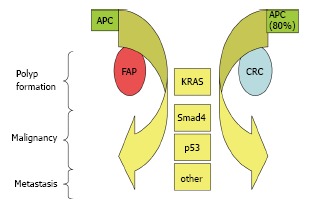
Hypothetical model of familial adenomatous polyposis molecular cascade. APC: Adenomatous polyposis coli; FAP: Familial adenomatous polyposis; CRC: Colorectal cancer; KRAS: Kirsten rat sarcoma viral oncogene homolog; Smad4-SMA: (Small) and MAD (mothers against decapentaplegic) related protein 4.
CRC DIAGNOSIS
Screening techniques-stool based
The morbidity and mortality of CRC can be minimized via prompt detection and appropriate intervention, such as polypectomy. Screening guidelines have been established to monitor individuals based on their estimated risk of developing CRC. An individual is stratified into a risk category based on their age, personal history (adenoma, CRC or inflammatory bowel disease) and family history[38]. For patients of average risk (lifetime risk of approximately 5%), both structural and fecal based screening tests are available. Fecal occult blood tests (FOBT), both guaiac-based and immunochemical, are designed to detect blood in fecal matter as evidence suggestive of possible CRC. These are recommended annually alone, or in combination with flexible sigmoidoscopy every 5 years.
Positive FOBT results should prompt assessment by colonoscopy[38]. Stool DNA tests are an evolving screening option which detect the presence of known CRC related DNA alterations in tumors cells excreted in stool. Single target stool DNA tests appear to have low sensitivity[61], with multi-target stool DNA tests (assessing 21 alterations in genes such as APC, KRAS and p53) detecting up to 52% of CRC with sensitivity ranging from 20%-94% in different trials[62,63]. ColoSure is the only stool DNA test available in the United States[64], nevertheless the FDA has yet to approve stool DNA analysis. While it is not presently acknowledged as a first line assessment tool, additional stool DNA assays are under development[65]. Stool screening tests have the advantage of being non-invasive and not requiring bowel clearance, which can enhance patient adherence to screening recommendations.
Screening techniques-colonoscopy
Structural based screening tests include colonoscopy, flexible sigmoidoscopy and computed tomography (CT) colonography. CT colonography, also referred to as virtual colonoscopy has the advantage of being non-invasive and not requiring sedation[38]. Flexible sigmoidoscopy also proceeds without sedation and requires decreased bowel preparation, however is limited to evaluating the distal portion of the colon. In both cases positive findings (lesions greater than 1 cm in the case of flexible sigmoidoscopy) require follow up evaluation by colonoscopy and polypectomy as indicated[38,66]. Colonoscopy remains the most complete screening procedure, permitting visualization of the complete colonic tract and simultaneous polypectomy, and is the gold standard against which other screening methods are measured. Studies have shown colonoscopy to reduce by an estimated 50% the incidence of CRC, and an inverse correlation between colonoscopy use and death from CRC[67-72]. A recent study also found colonoscopy and sigmoidoscopy to be associated with a lower mortality from CRC of the distal colon, and colonoscopy associated with decreased mortality from proximal CRC[69]. Their observations support the ten year assessment interval endorsed by current recommendations for individuals of average risk with a negative colonoscopy, documenting 1164 cases of CRC in individuals without endoscopy compared with 209 cases of CRC in individuals from 3 to 15 years post negative colonoscopy[73].
Screening guidelines
Patients 50 years of age and older, without personal history of polyps, CRC or inflammatory bowel disease and without family history of CRC or advanced adenomatous polyps, are considered at average risk[38]. A positive family history should prompt consideration of a CRC predisposition syndrome. The preferred screening strategy in these individuals is colonoscopy. If no polyps are found, repeat colonoscopy is recommended in 10 years. If polyps are identified, polypectomy is performed and repeat colonscopy recommendations depend on polyp characteristics. Patients with hyperplastic, non-sessile serrated and less than 1 cm polyps should have follow up colonoscopy in 10 years if polyps are left-sided or 5 years if right-sided[38]. Adenomas or sessile serrated polyps are considered low risk if there are no more than 2 tubular polyps less than 1 cm and require repeat colonoscopy in 5 years. Follow up colonoscopy in 3 years is recommended if 3 or more villous polyps are observed, diameter is 1 cm or larger, or they demonstrate high-grade dysplasia[38]. If more than 10 polyps are observed, one of the polyposis syndromes should be considered[38].
CRC MANAGEMENT
Tumor characterization and resection
Current intervention for CRC includes diagnosis, staging, resection, adjuvant chemotherapy, treatment of recurrent disease and ongoing surveillance. Diagnostic determination depends in part on histological assessment of the resected polyp. All adenomatous polyps have some degree of dysplasia, falling on a spectrum from low to high. No specific definition of “high grade” dysplasia has been established, but a number of histological features are assessed in an effort to grade the level of dyplasia, which can include loss of glandular differentiation, cellular and nuclear pleomorphism, nuclear hyperchromatism, loss of nuclear polarity, multi-layered irregular nuclei and loss of mucin, nuclear atypia with prominent nuclei and focal cribriform patterns[74,75]. Polyps with favorable histologic features are graded 1 or 2; those with less favorable histology are assigned grades 3 or 4. Histological grade, features such as angiolymphatic invasion and positive or negative resection margin help guide clinical decisions regarding the need for additional surgical resection. A polyp is considered malignant if cancer is observed infiltrating through the muscularis mucosa into the submucosa, and is designated T1[76]. As nearly one third of CRC in the United States is associated with family clustering, once histological diagnosis of CRC has been confirmed, an individual should be counseled regarding the increased risk of CRC in their first degree relatives[77].
Complete staging of malignant tumors is facilitated by pre-operative colonoscopy and CT scans of chest, abdomen and pelvis, is ultimately accomplished during surgical resection, and is categorized according to the TNM (tumor/node/metastasis) system[76,77]. Staging takes into consideration local invasion of the primary tumor, evidence of lymph node metastasis and evidence of metastasis to other organ sites or the peritoneum, and is considered one of the most important indicators of post treatment outcome[6]. Generally, stage I and II are assigned to lower and higher grade tumors, respectively, without nodal metastasis, stage III tumors have lymph node metastasis and stage IV tumors have metastasis to other organ sites or the peritoneum, with stages II, III and IV having additional subclassifications[76,78]. Though 20% of CRC is metastatic at presentation, 80% is localized to the colon wall or lymph nodes and surgical resection can be curative for localized CRC, accordingly treatment for CRC is primarily surgical. For resectable colon cancer, colectomy with regional lymph removal is the preferred surgical strategy, with the extent of colectomy based in part on tumor location as well as family history of polyposis[77].
Adjuvant therapy and surveillance
For stage I CRC, post resection adjuvant chemotherapy is not recommended, however various studies have shown a role for adjuvant therapy in more advanced CRC. Current recommendations are for patients with high-risk stage II and stage III CRC to receive 6 mo of adjuvant chemotherapy after primary surgical resection, and for patients with low-risk stage II CRC to be considered for adjuvant chemotherapy, enrolled in a clinical trial or observed without adjuvant therapy[77,79]. The specific chemotherapeutic regimens have been reviewed recently in detail[77,80]. These recommendations are derived from various studies which have shown survival benefit in patients with resected early stage CRC who received adjuvant chemotherapy, with most of the benefit being seen with stage III CRC, but not with node negative stage II disease, suggesting the benefit is increased in patients at higher risk related to nodal status[81-83]. Chemotherapy also has a role in patients presenting with more advanced CRC, including stage IV metastatic CRC. In individuals diagnosed with unresectable or medically inoperable CRC, chemotherapy is recommended in an effort to convert the lesion to a resectable state, and can be used to convert unresectable metastases, such as those in the liver, to resectable lesions, with the particular chemotherapeutic agents having been recently reviewed[77,84,85].
Posttreatment surveillance includes application of a variety of tools in an effort to discover any recurrence that is potentially curatively resectable. These tools include serial history and physical examination by a physician, CEA testing, colonoscopy and in some cases CT scans of the chest, abdomen and pelvis[77]. The advantages of more intensive surveillance regimens for patients with stage II and III CRC have been shown[86-88] and current recommendations for patients with successfully treated stage I-III CRC include history and physical examination every 3-6 mo for 2 years, CEA testing every 3-6 mo for 2 years and colonoscopy 3-6 mo post-resection (if not performed preoperatively)[77]. Surveillance colonoscopy is repeated based on findings (3 years if normal and 1 year if concerning adenomatous polyp removed), with CEA, history and physical exam spaced to every 6 mo to complete the first 5 years of posttreatment surveillance[77].
Patients with a history of CRC have a particularly high risk of another cancer within 2 years after resection, and recommended surveillance frequencies for the first 5 years post treatment vary with stage of CRC and patient characteristics such as age of onset and history of hereditary CRC[80]. Chest, abdominal and pelvic CT scans are recommended yearly for 3-5 years for stage II-III CRC patients at high risk for recurrence and every 3-6 mo for 2 years spaced to every 6-12 mo for a total of 5 years for individuals with stage IV CRC[77]. If disease recurrence is observed, the steps of diagnosis, staging, treatment via resection and/or adjuvant chemotherapy are revisited as with a primary presentation, with the potential complications of a more advanced presentation and chemotherapy resistant lesions.
CRC genomics
In the more than ten years since the completion of the Human Genome Project[17], advances in the capacity, speed and accuracy of genomic DNA sequencing have rapidly increased our knowledge of the molecular basis of multiple diseases, and ushered in the genomic era[16]. These powerful tools of genomic analysis have been recently trained on CRC, the results of which are just beginning to exert what is expected to be a substantial effect on the diagnosis and treatment of this disease.
Genome scale sequencing (including whole exome and whole genome sequencing) of over 200 CRC samples was recently completed by The Cancer Genome Atlas Network (TCGA), with a primary goal of characterizing somatic mutations in these lesions. The 24 genes which they found to be significantly mutated included somatic alterations in genes known to act in CRC, namely APC, TP53, SMAD4, PIK3CA and KRAS (Figure 1), as well as ARID1A, SOX9 and FAM123B[7]. They observed significantly different somatic mutations rates among the tumors assessed, classifying them into two categories: hypermutated and non-hypermutated. As a potential etiology for the elevated mutation rate, they tested and found 77% of hypermutated tumors to have elevated levels of micro-satellite instability (MSI), which is caused by DNA mismatch repair (MMR) deficiency[77], and can occur due to deleterious mutations to the genes MLH1, MLH3 MSH2, MSH3, MSH5 and PMS2[7]. In fact, in the majority of these same hypermutated lesions with high MSI were found evidence of epigenetic silencing of MLH1 and frameshift/nonsense/missense mutations in MLH1, MLH3 MSH2, MSH3, MSH5 and PMS2[7]. They proposed that the higher survival rate of patients with high MSI-related cancers, with these tumors being hypermutated, the mutation rate may be a prognostic indicator[7].
Increasing understanding of the role of MMR in tumorigenesis has already impacted the clinical approach to CRC. Current recommendations include assessment of new CRC for evidence of MMR deficiency for patients younger than 50 years old, though many centers assess for MMR deficiency, and sometime MSI, on all patients with CRC. This is done for two reasons: (1) it can be used as a screening tool to identify individuals at risk to have Lynch syndrome, causing hereditary colon and endometrial cancer[14], for whom genomic sequence analysis for mutations in MLH1, MSH2, MSH6 or PMS2 would be diagnostic; (2) deficiency in tumor MMR (as measured by protein immunohistochemistry) or high MSI tumor status is suggested to indicate decreased likelihood to metastasize[80] and be a prognostic indicator of more favorable outcome[89,90].
The application of genome sequencing technology has also led to an evolving array of clinical tools to augment the diagnosis and treatment of CRC (Table 1). For example, multigene assays have been developed to provide prognostic and predictive information about individual CRC tumors, as well having the potential to guide tumor specific therapeutic choices. Several such multigene panels are currently available (Oncotype DX Colon, ColoPrint, ColDX), simultaneously assessing 18 genes or more, using the data to predict an individual tumor’s risk of recurrence[80]. While early trials have found such multigene panels can help predict recurrence risk for stage I-III CRC, they do not appear to predict the benefit of adjuvant therapy[80], and further studies are necessary before such genomic assessment tools will have clinical relevance for choice of adjuvant therapy.
Table 1.
Evolving genomic tools for management of colon cancer
| Detection | Diagnosis | Management | |
| Clinically available | Fecal occult blood test | Single gene sequencing (APC) Multigene panel next generation sequencing | Targeted gene analysis for therapeutic contraindications (KRAS, UGT1A1) |
| Research basis | Fecal genomic DNA analysis | Tumor genome sequencing for prognosis | |
| Future application | Cell free genomic DNA sequencing | Development of gene pathway directed therapeutics (e.g., small molecule inhibitors) |
APC: Adenomatous polyposis coli; KRAS: Kirsten rat sarcoma viral oncogene homolog; UGT1A1: Uridine diphosphate-glucuronosyl transferase 1A1.
With detailed genomic information now more easily obtainable for individual tumors, the potential for delivering treatments more specifically targeted to a tumor’s molecular signature is becoming a reality. For example, MSI observed in CRC tumors by the TCGA study, has been found to be a potential predictor of benefit from adjuvant therapy, with fluoropyrimidine specifically[89]. MSI in CRC has been shown to be a predictor of more favorable outcome, and studies suggest high MSI, or deficient MMR, to be a marker predicting decreased benefit, and potential deleterious effect, of adjuvant treatment with fluoropyrimidine alone in individuals with stage II CRC[89,90]. It is currently recommended that MMR tumor analysis be considered for individuals with stage II disease and planned fluoropyrimidine adjuvant treatment alone[77].
In another example, TCGA genomic analysis of CRC further confirmed the presence of a significant number of KRAS mutations (in 43% of non-hypermutable CRC), consistent with its role in the molecular etiology of CRC (Figure 1)[7]. Additional studies have shown up to 40% of CRC tumors have alterations in codons 12 and 13 of exon 2 in the coding region of KRAS[91,92]. These particular KRAS alterations have been shown to be predictive of a lack of response to specific chemotherapies, anti-EGFR drugs cetuximab and panitumumab in particular[93-97], and the FDA has stated that these drugs should not be used for management of CRC with these specific KRAS alterations [Cetuximab (package insert). Branchburg, NJ: ImClone Systems Incorporated; 2009; Vectibix (package insert). Thousand Oaks, CA: Amgen Inc.; 2009].
Furthermore, downstream of activated KRAS, the BRAF gene protein product is activated. BRAF mutations were observed in the TCGA CRC genomic analysis (46% of hypermutated tumors)[7] and up to 9% of CRC contain the BRAF gene V600E mutation[98]. Retrospective studies have suggested that mutated BRAF, in the presence of wild-type KRAS, also confers a lower response rate to cetuximab[99]. For this reason, patients with stage IV CRC whose tumor has tested wild-type for KRAS, should have the option of BRAF tumor genotyping, in an effort to avoid potentially ineffective therapy choices[77]. With these potential opportunities to positively impact the selection of chemotherapeutics on a tumor specific basis, genotyping of CRC tumor tissue in all individuals with metastatic CRC diagnosed as stage IV is now strongly recommended (Table 1)[77].
Pharmacogenomic data represents another advance in CRC therapy with the ability to personalize the selection of agents for CRC treatment to each patient’s tolerance. Germline genomic sequencing of individual CRC patients is enabling the identification of individuals who have increased susceptibility to the side effects of particular drugs as well as increased or decreased metabolism of specific pharmacological agents. Each of these characteristics can positively, or negatively, impact the efficacy of a selected therapeutic agent in treating the patient’s CRC. Data is growing to provide clinical guidance in the selection of chemotherapeutics for CRC based in part on a patient’s germline genomic sequence, to avoid excessively toxic drugs and promote optimal dosing. For example, patients with a particular variant of the uridine diphosphate-glucuronosyl transferase 1A1 (UGT1A1) gene are at higher risk of developing neutropenia and diarrhea when treated with irinotecan for CRC[100-102]. Accordingly, the FDA currently recommends genotyping of patients under consideration of irinotecan treatment for CRC prior to initiation (Table 1)[103].
Genomic analysis of CRC by TCGA continued the expansion of our understanding of the molecular etiology of CRC by identifying somatic mutations in novel genes not previously associated with CRC, such as SOX9[7]. Their analysis also found a majority of recurrent CRC mutations could be grouped into several major cellular networks, specifically the WNT, MAPK, PI3K, TGF-beta and p53 pathways. For example, though alterations in APC, a component of the WNT pathway commonly altered in CRC, was mutated in 81% of non-hypermutated and 53% of hypermutated CRC, the overall WNT signaling pathway was altered in 93% of non-hypermutated and 97% of hypermutated CRC[7]. This observation suggests that almost all CRC are driven in part by a very similar molecular mechanism to that which initiates CRC in patients with FAP (Figure 1). This also suggests that pathway or network-level convergence would be an important methodology whereby the functional impact of non-synonymous point mutations in CRC might be predicted[104]. The TCGA data also suggest various potential targets for therapeutic intervention including proteins in the WNT, RTK-RAS and PI3K pathways which could be targets for inhibition[7]. Some of these targets are already under investigation and have shown initial potential (Table 1), such as inhibitors of WNT signaling and beta-catenin inhibitors[105-107].
The diagnosis of CRC, both at initial presentation and during surveillance for recurrance, has yet to be significantly impacted by genomic sequencing applications. However, the developing ability to more accurately sequence cell free genomic DNA isolated from the serum, as has been done for prenatal diagnostics[108], and the detection of significant levels of tumor DNA in the blood of cancer patients[109], suggest there is significant potential for genomic advances in the diagnosis of CRC (Table 1). Many exciting discoveries remain on the horizon for the diagnosis and managment of CRC as the genomic era continues to unfold.
CONCLUSION
We have discussed the current diagnosis and management of sporadic and FAP related CRC, and the initial impacts genomic sequencing is having on these diseases. The diagnosis of FAP continues to rely on the performance of a thorough patient history and physical examination, which includes a detailed familial medical history. Clinical diagnosis via colonoscopy provides the gold standard for identifying the physical manifestations of FAP related CRC. Genomic sequencing has begun to manifest its impact on the diagnosis of FAP in the availability of APC gene sequencing and next generation sequencing based multi-gene panels, through which patients without a significant family history or with an ambiguous presentation, can be tested simultaneously for several hereditary cancer syndromes, minimizing the time to confirmed diagnosis and initiation of recommended surveillance protocols, testing of at risk family members. Molecular testing of at risk but asymptomatic family members has the added benefit of confirming individuals as being unaffected by an APC gene mutation, and allowing them to safely avoid unnecessary colonoscopic surveillance beyond that recommended for individuals of average risk[38]. With the increasing availability of these genomic sequencing assays to assess the molecular status of APC, a small but growing number of VUS are accumulating, which complicates the diagnostic capacity of these tests and leaves patients having to undergo empiric intensive FAP CRC screening protocols in the absence of a definitive “positive” or “negative” result. Further functional characterization of VUS remains an important area of research to improve the accuracy and applicability of diagnostic genomic sequencing.
Sporadic CRC has also begun to benefit from the tools of genome sequencing analysis. Recent whole exome and whole genome sequencing of hundreds of CRCs have confirmed the presence of the canonical genetic mutations contributing to its pathogenesis, while also identifying novel genes with potential roles in CRC tumor development[7]. The genomic era has begun to contribute new tools to facilitate both diagnosis and guide management of sporadic CRC. Fecal derived DNA sequencing assays are being tested as a precursor to colonoscopic assessment, providing a vast amount more information than its antecedent the fecal occult blood test, though these remain unready at this time for clinical application. Direct genomic analysis of patient CRC tumors is being actively pursued in the research setting, with growing evidence that such information may provide prognostic and predictive information about clinical course and response to intervention, with the current standard of care already requiring certain CRC genes be assessed prior to treatment with specific chemotherapeutic agents[77]. Germline genomic sequencing of CRC patients has the potential to allow personalization of treatment to patient tolerances, and some pharmacogenomic studies have already lead to screening of patients prior to initiation of specific regimens in order to avoid deleterious side effects. These advances, brought about in the short time since the advent of the genomic era[16], have already significantly impacted the clinical management of sporadic and FAP related CRC, with the promise of further discoveries on the horizon with the potential to ultimately decrease the current prevalence and mortality of CRC, both sporadic and hereditary.
Footnotes
Supported by The Child Health Research Institute and the Stanford CTSA, No. UL1 TR000093
P- Reviewer: Nishiyama M, Sugimura H S- Editor: Ji FF L- Editor: A E- Editor: Lu YJ
References
- 1.Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64:9–29. doi: 10.3322/caac.21208. [DOI] [PubMed] [Google Scholar]
- 2.Levin B, Lieberman DA, McFarland B, Smith RA, Brooks D, Andrews KS, Dash C, Giardiello FM, Glick S, Levin TR, et al. Screening and surveillance for the early detection of colorectal cancer and adenomatous polyps, 2008: a joint guideline from the American Cancer Society, the US Multi-Society Task Force on Colorectal Cancer, and the American College of Radiology. CA Cancer J Clin. 2008;58:130–160. doi: 10.3322/CA.2007.0018. [DOI] [PubMed] [Google Scholar]
- 3.Rex DK, Johnson DA, Anderson JC, Schoenfeld PS, Burke CA, Inadomi JM. American College of Gastroenterology guidelines for colorectal cancer screening 2009 [corrected] Am J Gastroenterol. 2009;104:739–750. doi: 10.1038/ajg.2009.104. [DOI] [PubMed] [Google Scholar]
- 4.US Preventive Services Task Force. Screening for colorectal cancer: U.S. Preventive Services Task Force recommendation statement. Ann Intern Med. 2008;149:627–637. doi: 10.7326/0003-4819-149-9-200811040-00243. [DOI] [PubMed] [Google Scholar]
- 5.Centers for Disease Control and Prevention (CDC) Cancer screening-United States, 2010. MMWR Morb Mortal Wkly Rep. 2012;61:41–45. [PubMed] [Google Scholar]
- 6.O‘Connell JB, Maggard MA, Ko CY. Colon cancer survival rates with the new American Joint Committee on Cancer sixth edition staging. J Natl Cancer Inst. 2004;96:1420–1425. doi: 10.1093/jnci/djh275. [DOI] [PubMed] [Google Scholar]
- 7.Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–337. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang SS, Esplin ED, Li JL, Huang L, Gazdar A, Minna J, Evans GA. Alterations of the PPP2R1B gene in human lung and colon cancer. Science. 1998;282:284–287. doi: 10.1126/science.282.5387.284. [DOI] [PubMed] [Google Scholar]
- 9.Esplin ED, Ramos P, Martinez B, Tomlinson GE, Mumby MC, Evans GA. The glycine 90 to aspartate alteration in the Abeta subunit of PP2A (PPP2R1B) associates with breast cancer and causes a deficit in protein function. Genes Chromosomes Cancer. 2006;45:182–190. doi: 10.1002/gcc.20284. [DOI] [PubMed] [Google Scholar]
- 10.Sablina AA, Chen W, Arroyo JD, Corral L, Hector M, Bulmer SE, DeCaprio JA, Hahn WC. The tumor suppressor PP2A Abeta regulates the RalA GTPase. Cell. 2007;129:969–982. doi: 10.1016/j.cell.2007.03.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zimonjic DB, Popescu NC. Role of DLC1 tumor suppressor gene and MYC oncogene in pathogenesis of human hepatocellular carcinoma: potential prospects for combined targeted therapeutics (review) Int J Oncol. 2012;41:393–406. doi: 10.3892/ijo.2012.1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Kinzler KW. Cancer genome landscapes. Science. 2013;339:1546–1558. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jasperson KW, Burt RW. APC-Associated Polyposis Conditions. In: Pagon RA, Adam MP, Ardinger HH, Bird TD, Dolan CR, et al., editors. Seattle (WA): University of Washington, Seattle, 1993-; 2014. [Google Scholar]
- 14.Kohlmann W, Gruber SB. Lynch Syndrome. In: Pagon RA, Adam MP, Ardinger HH, Bird TD, Dolan CR, et al., editors. Seattle (WA): University of Washington, Seattle, 1993-; 2014. [Google Scholar]
- 15.Jasperson KW, Tuohy TM, Neklason DW, Burt RW. Hereditary and familial colon cancer. Gastroenterology. 2010;138:2044–2058. doi: 10.1053/j.gastro.2010.01.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guttmacher AE, Collins FS. Welcome to the genomic era. N Engl J Med. 2003;349:996–998. doi: 10.1056/NEJMe038132. [DOI] [PubMed] [Google Scholar]
- 17.Collins FS, Green ED, Guttmacher AE, Guyer MS. A vision for the future of genomics research. Nature. 2003;422:835–847. doi: 10.1038/nature01626. [DOI] [PubMed] [Google Scholar]
- 18.Petersen GM, Slack J, Nakamura Y. Screening guidelines and premorbid diagnosis of familial adenomatous polyposis using linkage. Gastroenterology. 1991;100:1658–1664. doi: 10.1016/0016-5085(91)90666-9. [DOI] [PubMed] [Google Scholar]
- 19.Nielsen M, Hes FJ, Nagengast FM, Weiss MM, Mathus-Vliegen EM, Morreau H, Breuning MH, Wijnen JT, Tops CM, Vasen HF. Germline mutations in APC and MUTYH are responsible for the majority of families with attenuated familial adenomatous polyposis. Clin Genet. 2007;71:427–433. doi: 10.1111/j.1399-0004.2007.00766.x. [DOI] [PubMed] [Google Scholar]
- 20.Hegde M, Ferber M, Mao R, Samowitz W, Ganguly A. ACMG technical standards and guidelines for genetic testing for inherited colorectal cancer (Lynch syndrome, familial adenomatous polyposis, and MYH-associated polyposis) Genet Med. 2014;16:101–116. doi: 10.1038/gim.2013.166. [DOI] [PubMed] [Google Scholar]
- 21.Lynch PM. When and how to perform genetic testing for inherited colorectal cancer syndromes. J Natl Compr Canc Netw. 2013;11:1577–1583. doi: 10.6004/jnccn.2013.0181. [DOI] [PubMed] [Google Scholar]
- 22.Grover S, Kastrinos F, Steyerberg EW, Cook EF, Dewanwala A, Burbidge LA, Wenstrup RJ, Syngal S. Prevalence and phenotypes of APC and MUTYH mutations in patients with multiple colorectal adenomas. JAMA. 2012;308:485–492. doi: 10.1001/jama.2012.8780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grover S GeneDx. OncoGene Dx. Available from: http: //www.genedx.com/test-catalog/disorders/colorectal-cancer.
- 24.Ambry Genetics. ColoNext. Available from: http: //www.ambrygen.com/tests/colonext.
- 25.University of Washington Genetics Laboratory. ColoSeq. Available from: http: //tests.labmed.washington.edu/COLOSEQ.
- 26.Pineda M, González S, Lázaro C, Blanco I, Capellá G. Detection of genetic alterations in hereditary colorectal cancer screening. Mutat Res. 2010;693:19–31. doi: 10.1016/j.mrfmmm.2009.11.002. [DOI] [PubMed] [Google Scholar]
- 27.Castellsagué E, González S, Nadal M, Campos O, Guinó E, Urioste M, Blanco I, Frebourg T, Capellá G. Detection of APC gene deletions using quantitative multiplex PCR of short fluorescent fragments. Clin Chem. 2008;54:1132–1140. doi: 10.1373/clinchem.2007.101006. [DOI] [PubMed] [Google Scholar]
- 28.Nieuwenhuis MH, Mathus-Vliegen LM, Slors FJ, Griffioen G, Nagengast FM, Schouten WR, Kleibeuker JH, Vasen HF. Genotype-phenotype correlations as a guide in the management of familial adenomatous polyposis. Clin Gastroenterol Hepatol. 2007;5:374–378. doi: 10.1016/j.cgh.2006.12.014. [DOI] [PubMed] [Google Scholar]
- 29.Kaufmann A, Vogt S, Uhlhaas S, Stienen D, Kurth I, Hameister H, Mangold E, Kötting J, Kaminsky E, Propping P, et al. Analysis of rare APC variants at the mRNA level: six pathogenic mutations and literature review. J Mol Diagn. 2009;11:131–139. doi: 10.2353/jmoldx.2009.080129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Giardiello FM, Brensinger JD, Petersen GM. AGA technical review on hereditary colorectal cancer and genetic testing. Gastroenterology. 2001;121:198–213. doi: 10.1053/gast.2001.25581. [DOI] [PubMed] [Google Scholar]
- 31.Markey K, Axel L, Ahnen D. Basic concepts for genetic testing in common hereditary colorectal cancer syndromes. Curr Gastroenterol Rep. 2002;4:404–413. doi: 10.1007/s11894-002-0011-5. [DOI] [PubMed] [Google Scholar]
- 32.Azzopardi D, Dallosso AR, Eliason K, Hendrickson BC, Jones N, Rawstorne E, Colley J, Moskvina V, Frye C, Sampson JR, et al. Multiple rare nonsynonymous variants in the adenomatous polyposis coli gene predispose to colorectal adenomas. Cancer Res. 2008;68:358–363. doi: 10.1158/0008-5472.CAN-07-5733. [DOI] [PubMed] [Google Scholar]
- 33.Heinimann K, Thompson A, Locher A, Furlanetto T, Bader E, Wolf A, Meier R, Walter K, Bauerfeind P, Marra G, et al. Nontruncating APC germ-line mutations and mismatch repair deficiency play a minor role in APC mutation-negative polyposis. Cancer Res. 2001;61:7616–7622. [PubMed] [Google Scholar]
- 34.Montera M, Piaggio F, Marchese C, Gismondi V, Stella A, Resta N, Varesco L, Guanti G, Mareni C. A silent mutation in exon 14 of the APC gene is associated with exon skipping in a FAP family. J Med Genet. 2001;38:863–867. doi: 10.1136/jmg.38.12.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Aretz S, Uhlhaas S, Sun Y, Pagenstecher C, Mangold E, Caspari R, Möslein G, Schulmann K, Propping P, Friedl W. Familial adenomatous polyposis: aberrant splicing due to missense or silent mutations in the APC gene. Hum Mutat. 2004;24:370–380. doi: 10.1002/humu.20087. [DOI] [PubMed] [Google Scholar]
- 36.Goto M, Shinmura K, Nakabeppu Y, Tao H, Yamada H, Tsuneyoshi T, Sugimura H. Adenine DNA glycosylase activity of 14 human MutY homolog (MUTYH) variant proteins found in patients with colorectal polyposis and cancer. Hum Mutat. 2010;31:E1861–E1874. doi: 10.1002/humu.21363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shinmura K, Goto M, Tao H, Matsuura S, Matsuda T, Sugimura H. Impaired suppressive activities of human MUTYH variant proteins against oxidative mutagenesis. World J Gastroenterol. 2012;18:6935–6942. doi: 10.3748/wjg.v18.i47.6935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Burt RW, Cannon JA, David DS, Early DS, Ford JM, Giardiello FM, Halverson AL, Hamilton SR, Hampel H, Ismail MK, et al. Colorectal cancer screening. J Natl Compr Canc Netw. 2013;11:1538–1575. doi: 10.6004/jnccn.2013.0180. [DOI] [PubMed] [Google Scholar]
- 39.Lynch HT, de la Chapelle A. Hereditary colorectal cancer. N Engl J Med. 2003;348:919–932. doi: 10.1056/NEJMra012242. [DOI] [PubMed] [Google Scholar]
- 40.Friedl W, Caspari R, Sengteller M, Uhlhaas S, Lamberti C, Jungck M, Kadmon M, Wolf M, Fahnenstich J, Gebert J, et al. Can APC mutation analysis contribute to therapeutic decisions in familial adenomatous polyposis? Experience from 680 FAP families. Gut. 2001;48:515–521. doi: 10.1136/gut.48.4.515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bertario L, Russo A, Sala P, Varesco L, Giarola M, Mondini P, Pierotti M, Spinelli P, Radice P. Multiple approach to the exploration of genotype-phenotype correlations in familial adenomatous polyposis. J Clin Oncol. 2003;21:1698–1707. doi: 10.1200/JCO.2003.09.118. [DOI] [PubMed] [Google Scholar]
- 42.Nagase H, Miyoshi Y, Horii A, Aoki T, Ogawa M, Utsunomiya J, Baba S, Sasazuki T, Nakamura Y. Correlation between the location of germ-line mutations in the APC gene and the number of colorectal polyps in familial adenomatous polyposis patients. Cancer Res. 1992;52:4055–4057. [PubMed] [Google Scholar]
- 43.Sieber OM, Segditsas S, Knudsen AL, Zhang J, Luz J, Rowan AJ, Spain SL, Thirlwell C, Howarth KM, Jaeger EE, et al. Disease severity and genetic pathways in attenuated familial adenomatous polyposis vary greatly but depend on the site of the germline mutation. Gut. 2006;55:1440–1448. doi: 10.1136/gut.2005.087106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Soravia C, Berk T, Madlensky L, Mitri A, Cheng H, Gallinger S, Cohen Z, Bapat B. Genotype-phenotype correlations in attenuated adenomatous polyposis coli. Am J Hum Genet. 1998;62:1290–1301. doi: 10.1086/301883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.van der Luijt RB, Vasen HF, Tops CM, Breukel C, Fodde R, Meera Khan P. APC mutation in the alternatively spliced region of exon 9 associated with late onset familial adenomatous polyposis. Hum Genet. 1995;96:705–710. doi: 10.1007/BF00210303. [DOI] [PubMed] [Google Scholar]
- 46.Friedl W, Meuschel S, Caspari R, Lamberti C, Krieger S, Sengteller M, Propping P. Attenuated familial adenomatous polyposis due to a mutation in the 3’ part of the APC gene. A clue for understanding the function of the APC protein. Hum Genet. 1996;97:579–584. doi: 10.1007/BF02281864. [DOI] [PubMed] [Google Scholar]
- 47.van der Luijt RB, Meera Khan P, Vasen HF, Breukel C, Tops CM, Scott RJ, Fodde R. Germline mutations in the 3’ part of APC exon 15 do not result in truncated proteins and are associated with attenuated adenomatous polyposis coli. Hum Genet. 1996;98:727–734. doi: 10.1007/s004390050293. [DOI] [PubMed] [Google Scholar]
- 48.Walon C, Kartheuser A, Michils G, Smaers M, Lannoy N, Ngounou P, Mertens G, Verellen-Dumoulin C. Novel germline mutations in the APC gene and their phenotypic spectrum in familial adenomatous polyposis kindreds. Hum Genet. 1997;100:601–605. doi: 10.1007/s004390050560. [DOI] [PubMed] [Google Scholar]
- 49.Pilarski RT, Brothman AR, Benn P, Shulman Rosengren S. Attenuated familial adenomatous polyposis in a man with an interstitial deletion of chromosome arm 5q. Am J Med Genet. 1999;86:321–324. doi: 10.1002/(sici)1096-8628(19991008)86:4<321::aid-ajmg4>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 50.Nielsen M, Bik E, Hes FJ, Breuning MH, Vasen HF, Bakker E, Tops CM, Weiss MM. Genotype-phenotype correlations in 19 Dutch cases with APC gene deletions and a literature review. Eur J Hum Genet. 2007;15:1034–1042. doi: 10.1038/sj.ejhg.5201871. [DOI] [PubMed] [Google Scholar]
- 51.Friedl W, Aretz S. Familial adenomatous polyposis: experience from a study of 1164 unrelated german polyposis patients. Hered Cancer Clin Pract. 2005;3:95–114. doi: 10.1186/1897-4287-3-3-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Aretz S, Stienen D, Friedrichs N, Stemmler S, Uhlhaas S, Rahner N, Propping P, Friedl W. Somatic APC mosaicism: a frequent cause of familial adenomatous polyposis (FAP) Hum Mutat. 2007;28:985–992. doi: 10.1002/humu.20549. [DOI] [PubMed] [Google Scholar]
- 53.Hes FJ, Nielsen M, Bik EC, Konvalinka D, Wijnen JT, Bakker E, Vasen HF, Breuning MH, Tops CM. Somatic APC mosaicism: an underestimated cause of polyposis coli. Gut. 2008;57:71–76. doi: 10.1136/gut.2006.117796. [DOI] [PubMed] [Google Scholar]
- 54.Giardiello FM, Hamilton SR, Krush AJ, Piantadosi S, Hylind LM, Celano P, Booker SV, Robinson CR, Offerhaus GJ. Treatment of colonic and rectal adenomas with sulindac in familial adenomatous polyposis. N Engl J Med. 1993;328:1313–1316. doi: 10.1056/NEJM199305063281805. [DOI] [PubMed] [Google Scholar]
- 55.Lynch PM. Chemoprevention with special reference to inherited colorectal cancer. Fam Cancer. 2008;7:59–64. doi: 10.1007/s10689-007-9158-4. [DOI] [PubMed] [Google Scholar]
- 56.Chan AT. Aspirin and familial adenomatous polyposis: coming full circle. Cancer Prev Res (Phila) 2011;4:623–627. doi: 10.1158/1940-6207.CAPR-11-0157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Burt RW, Barthel JS, Dunn KB, David DS, Drelichman E, Ford JM, Giardiello FM, Gruber SB, Halverson AL, Hamilton SR, et al. NCCN clinical practice guidelines in oncology. Colorectal cancer screening. J Natl Compr Canc Netw. 2010;8:8–61. doi: 10.6004/jnccn.2010.0003. [DOI] [PubMed] [Google Scholar]
- 58.Guillem JG, Wood WC, Moley JF, Berchuck A, Karlan BY, Mutch DG, Gagel RF, Weitzel J, Morrow M, Weber BL, et al. ASCO/SSO review of current role of risk-reducing surgery in common hereditary cancer syndromes. J Clin Oncol. 2006;24:4642–4660. doi: 10.1200/JCO.2005.04.5260. [DOI] [PubMed] [Google Scholar]
- 59.Fearon ER. Molecular genetics of colorectal cancer. Annu Rev Pathol. 2011;6:479–507. doi: 10.1146/annurev-pathol-011110-130235. [DOI] [PubMed] [Google Scholar]
- 60.Phelps RA, Chidester S, Dehghanizadeh S, Phelps J, Sandoval IT, Rai K, Broadbent T, Sarkar S, Burt RW, Jones DA. A two-step model for colon adenoma initiation and progression caused by APC loss. Cell. 2009;137:623–634. doi: 10.1016/j.cell.2009.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Osborn NK, Ahlquist DA. Stool screening for colorectal cancer: molecular approaches. Gastroenterology. 2005;128:192–206. doi: 10.1053/j.gastro.2004.10.041. [DOI] [PubMed] [Google Scholar]
- 62.Imperiale TF, Ransohoff DF, Itzkowitz SH, Turnbull BA, Ross ME. Fecal DNA versus fecal occult blood for colorectal-cancer screening in an average-risk population. N Engl J Med. 2004;351:2704–2714. doi: 10.1056/NEJMoa033403. [DOI] [PubMed] [Google Scholar]
- 63.Ahlquist DA, Sargent DJ, Loprinzi CL, Levin TR, Rex DK, Ahnen DJ, Knigge K, Lance MP, Burgart LJ, Hamilton SR, et al. Stool DNA and occult blood testing for screen detection of colorectal neoplasia. Ann Intern Med. 2008;149:441–450, W81. doi: 10.7326/0003-4819-149-7-200810070-00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ned RM, Melillo S, Marrone M. Fecal DNA testing for Colorectal Cancer Screening: the ColoSure™ test. PLoS Curr. 2011;3:RRN1220. doi: 10.1371/currents.RRN1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ahlquist DA, Zou H, Domanico M, Mahoney DW, Yab TC, Taylor WR, Butz ML, Thibodeau SN, Rabeneck L, Paszat LF, et al. Next-generation stool DNA test accurately detects colorectal cancer and large adenomas. Gastroenterology. 2012;142:248–256; quiz e25-e26. doi: 10.1053/j.gastro.2011.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Whitlock EP, Lin JS, Liles E, Beil TL, Fu R. Screening for colorectal cancer: a targeted, updated systematic review for the U.S. Preventive Services Task Force. Ann Intern Med. 2008;149:638–658. doi: 10.7326/0003-4819-149-9-200811040-00245. [DOI] [PubMed] [Google Scholar]
- 67.Rabeneck L, Paszat LF, Saskin R, Stukel TA. Association between colonoscopy rates and colorectal cancer mortality. Am J Gastroenterol. 2010;105:1627–1632. doi: 10.1038/ajg.2010.83. [DOI] [PubMed] [Google Scholar]
- 68.Citarda F, Tomaselli G, Capocaccia R, Barcherini S, Crespi M. Efficacy in standard clinical practice of colonoscopic polypectomy in reducing colorectal cancer incidence. Gut. 2001;48:812–815. doi: 10.1136/gut.48.6.812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jacob BJ, Moineddin R, Sutradhar R, Baxter NN, Urbach DR. Effect of colonoscopy on colorectal cancer incidence and mortality: an instrumental variable analysis. Gastrointest Endosc. 2012;76:355–364.e1. doi: 10.1016/j.gie.2012.03.247. [DOI] [PubMed] [Google Scholar]
- 70.Winawer SJ, Zauber AG, Ho MN, O’Brien MJ, Gottlieb LS, Sternberg SS, Waye JD, Schapiro M, Bond JH, Panish JF. Prevention of colorectal cancer by colonoscopic polypectomy. The National Polyp Study Workgroup. N Engl J Med. 1993;329:1977–1981. doi: 10.1056/NEJM199312303292701. [DOI] [PubMed] [Google Scholar]
- 71.Manser CN, Bachmann LM, Brunner J, Hunold F, Bauerfeind P, Marbet UA. Colonoscopy screening markedly reduces the occurrence of colon carcinomas and carcinoma-related death: a closed cohort study. Gastrointest Endosc. 2012;76:110–117. doi: 10.1016/j.gie.2012.02.040. [DOI] [PubMed] [Google Scholar]
- 72.Müller AD, Sonnenberg A. Prevention of colorectal cancer by flexible endoscopy and polypectomy. A case-control study of 32,702 veterans. Ann Intern Med. 1995;123:904–910. doi: 10.7326/0003-4819-123-12-199512150-00002. [DOI] [PubMed] [Google Scholar]
- 73.Nishihara R, Wu K, Lochhead P, Morikawa T, Liao X, Qian ZR, Inamura K, Kim SA, Kuchiba A, Yamauchi M, et al. Long-term colorectal-cancer incidence and mortality after lower endoscopy. N Engl J Med. 2013;369:1095–1105. doi: 10.1056/NEJMoa1301969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bujanda L, Cosme A, Gil I, Arenas-Mirave JI. Malignant colorectal polyps. World J Gastroenterol. 2010;16:3103–3111. doi: 10.3748/wjg.v16.i25.3103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Appelman HD. Con: High-grade dysplasia and villous features should not be part of the routine diagnosis of colorectal adenomas. Am J Gastroenterol. 2008;103:1329–1331. doi: 10.1111/j.1572-0241.2008.02005_3.x. [DOI] [PubMed] [Google Scholar]
- 76.Edge SB, American Joint Committee on Cancer, American Cancer Society. AJCC cancer staging handbook: from the AJCC cancer staging manual. 7th ed. New York: Springer; 2010. [Google Scholar]
- 77.Benson AB, Arnoletti JP, Bekaii-Saab T, Chan E, Chen YJ, Choti MA, Cooper HS, Dilawari RA, Engstrom PF, Enzinger PC, et al. Colon cancer. J Natl Compr Canc Netw. 2011;9:1238–1290. doi: 10.6004/jnccn.2011.0104. [DOI] [PubMed] [Google Scholar]
- 78.Compton CC, Greene FL. The staging of colorectal cancer: 2004 and beyond. CA Cancer J Clin. 2004;54:295–308. doi: 10.3322/canjclin.54.6.295. [DOI] [PubMed] [Google Scholar]
- 79.Des Guetz G, Uzzan B, Morere JF, Perret G, Nicolas P. Duration of adjuvant chemotherapy for patients with non-metastatic colorectal cancer. Cochrane Database Syst Rev. 2010;1:CD007046. doi: 10.1002/14651858.CD007046.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Benson AB, Bekaii-Saab T, Chan E, Chen YJ, Choti MA, Cooper HS, Engstrom PF, Enzinger PC, Fakih MG, Fenton MJ, et al. Localized colon cancer, version 3.2013: featured updates to the NCCN Guidelines. J Natl Compr Canc Netw. 2013;11:519–528. doi: 10.6004/jnccn.2013.0069. [DOI] [PubMed] [Google Scholar]
- 81.Efficacy of adjuvant fluorouracil and folinic acid in colon cancer. International Multicentre Pooled Analysis of Colon Cancer Trials (IMPACT) investigators. Lancet. 1995;345:939–944. [PubMed] [Google Scholar]
- 82.Efficacy of adjuvant fluorouracil and folinic acid in B2 colon cancer. International Multicentre Pooled Analysis of B2 Colon Cancer Trials (IMPACT B2) Investigators. J Clin Oncol. 1999;17:1356–1363. [PubMed] [Google Scholar]
- 83.Gill S, Loprinzi CL, Sargent DJ, Thomé SD, Alberts SR, Haller DG, Benedetti J, Francini G, Shepherd LE, Francois Seitz J, et al. Pooled analysis of fluorouracil-based adjuvant therapy for stage II and III colon cancer: who benefits and by how much? J Clin Oncol. 2004;22:1797–1806. doi: 10.1200/JCO.2004.09.059. [DOI] [PubMed] [Google Scholar]
- 84.Berger AC, Sigurdson ER, LeVoyer T, Hanlon A, Mayer RJ, Macdonald JS, Catalano PJ, Haller DG. Colon cancer survival is associated with decreasing ratio of metastatic to examined lymph nodes. J Clin Oncol. 2005;23:8706–8712. doi: 10.1200/JCO.2005.02.8852. [DOI] [PubMed] [Google Scholar]
- 85.Benson AB, Bekaii-Saab T, Chan E, Chen YJ, Choti MA, Cooper HS, Engstrom PF, Enzinger PC, Fakih MG, Fenton MJ, et al. Metastatic colon cancer, version 3.2013: featured updates to the NCCN Guidelines. J Natl Compr Canc Netw. 2013;11:141–152; quiz 152. doi: 10.6004/jnccn.2013.0022. [DOI] [PubMed] [Google Scholar]
- 86.Desch CE, Benson AB, Somerfield MR, Flynn PJ, Krause C, Loprinzi CL, Minsky BD, Pfister DG, Virgo KS, Petrelli NJ. Colorectal cancer surveillance: 2005 update of an American Society of Clinical Oncology practice guideline. J Clin Oncol. 2005;23:8512–8519. doi: 10.1200/JCO.2005.04.0063. [DOI] [PubMed] [Google Scholar]
- 87.Figueredo A, Rumble RB, Maroun J, Earle CC, Cummings B, McLeod R, Zuraw L, Zwaal C. Follow-up of patients with curatively resected colorectal cancer: a practice guideline. BMC Cancer. 2003;3:26. doi: 10.1186/1471-2407-3-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Renehan AG, Egger M, Saunders MP, O’Dwyer ST. Impact on survival of intensive follow up after curative resection for colorectal cancer: systematic review and meta-analysis of randomised trials. BMJ. 2002;324:813. doi: 10.1136/bmj.324.7341.813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ribic CM, Sargent DJ, Moore MJ, Thibodeau SN, French AJ, Goldberg RM, Hamilton SR, Laurent-Puig P, Gryfe R, Shepherd LE, et al. Tumor microsatellite-instability status as a predictor of benefit from fluorouracil-based adjuvant chemotherapy for colon cancer. N Engl J Med. 2003;349:247–257. doi: 10.1056/NEJMoa022289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sargent DJ, Marsoni S, Monges G, Thibodeau SN, Labianca R, Hamilton SR, French AJ, Kabat B, Foster NR, Torri V, et al. Defective mismatch repair as a predictive marker for lack of efficacy of fluorouracil-based adjuvant therapy in colon cancer. J Clin Oncol. 2010;28:3219–3226. doi: 10.1200/JCO.2009.27.1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Roth AD, Tejpar S, Delorenzi M, Yan P, Fiocca R, Klingbiel D, Dietrich D, Biesmans B, Bodoky G, Barone C, et al. Prognostic role of KRAS and BRAF in stage II and III resected colon cancer: results of the translational study on the PETACC-3, EORTC 40993, SAKK 60-00 trial. J Clin Oncol. 2010;28:466–474. doi: 10.1200/JCO.2009.23.3452. [DOI] [PubMed] [Google Scholar]
- 92.Amado RG, Wolf M, Peeters M, Van Cutsem E, Siena S, Freeman DJ, Juan T, Sikorski R, Suggs S, Radinsky R, et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol. 2008;26:1626–1634. doi: 10.1200/JCO.2007.14.7116. [DOI] [PubMed] [Google Scholar]
- 93.Van Cutsem E, Köhne CH, Hitre E, Zaluski J, Chang Chien CR, Makhson A, D’Haens G, Pintér T, Lim R, Bodoky G, et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N Engl J Med. 2009;360:1408–1417. doi: 10.1056/NEJMoa0805019. [DOI] [PubMed] [Google Scholar]
- 94.Bokemeyer C, Bondarenko I, Makhson A, Hartmann JT, Aparicio J, de Braud F, Donea S, Ludwig H, Schuch G, Stroh C, et al. Fluorouracil, leucovorin, and oxaliplatin with and without cetuximab in the first-line treatment of metastatic colorectal cancer. J Clin Oncol. 2009;27:663–671. doi: 10.1200/JCO.2008.20.8397. [DOI] [PubMed] [Google Scholar]
- 95.Baselga J, Rosen N. Determinants of RASistance to anti-epidermal growth factor receptor agents. J Clin Oncol. 2008;26:1582–1584. doi: 10.1200/JCO.2007.15.3700. [DOI] [PubMed] [Google Scholar]
- 96.Karapetis CS, Khambata-Ford S, Jonker DJ, O’Callaghan CJ, Tu D, Tebbutt NC, Simes RJ, Chalchal H, Shapiro JD, Robitaille S, et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med. 2008;359:1757–1765. doi: 10.1056/NEJMoa0804385. [DOI] [PubMed] [Google Scholar]
- 97.Dahabreh IJ, Terasawa T, Castaldi PJ, Trikalinos TA. Systematic review: Anti-epidermal growth factor receptor treatment effect modification by KRAS mutations in advanced colorectal cancer. Ann Intern Med. 2011;154:37–49. doi: 10.7326/0003-4819-154-1-201101040-00006. [DOI] [PubMed] [Google Scholar]
- 98.Tol J, Nagtegaal ID, Punt CJ. BRAF mutation in metastatic colorectal cancer. N Engl J Med. 2009;361:98–99. doi: 10.1056/NEJMc0904160. [DOI] [PubMed] [Google Scholar]
- 99.De Roock W, Claes B, Bernasconi D, De Schutter J, Biesmans B, Fountzilas G, Kalogeras KT, Kotoula V, Papamichael D, Laurent-Puig P, et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. Lancet Oncol. 2010;11:753–762. doi: 10.1016/S1470-2045(10)70130-3. [DOI] [PubMed] [Google Scholar]
- 100.Innocenti F, Undevia SD, Iyer L, Chen PX, Das S, Kocherginsky M, Karrison T, Janisch L, Ramírez J, Rudin CM, et al. Genetic variants in the UDP-glucuronosyltransferase 1A1 gene predict the risk of severe neutropenia of irinotecan. J Clin Oncol. 2004;22:1382–1388. doi: 10.1200/JCO.2004.07.173. [DOI] [PubMed] [Google Scholar]
- 101.Iyer L, Das S, Janisch L, Wen M, Ramírez J, Karrison T, Fleming GF, Vokes EE, Schilsky RL, Ratain MJ. UGT1A1*28 polymorphism as a determinant of irinotecan disposition and toxicity. Pharmacogenomics J. 2002;2:43–47. doi: 10.1038/sj.tpj.6500072. [DOI] [PubMed] [Google Scholar]
- 102.Hoskins JM, Goldberg RM, Qu P, Ibrahim JG, McLeod HL. UGT1A1*28 genotype and irinotecan-induced neutropenia: dose matters. J Natl Cancer Inst. 2007;99:1290–1295. doi: 10.1093/jnci/djm115. [DOI] [PubMed] [Google Scholar]
- 103.Wheeler HE, Maitland ML, Dolan ME, Cox NJ, Ratain MJ. Cancer pharmacogenomics: strategies and challenges. Nat Rev Genet. 2013;14:23–34. doi: 10.1038/nrg3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kim TM, Lee SH, Chung YJ. Clinical applications of next-generation sequencing in colorectal cancers. World J Gastroenterol. 2013;19:6784–6793. doi: 10.3748/wjg.v19.i40.6784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Chen B, Dodge ME, Tang W, Lu J, Ma Z, Fan CW, Wei S, Hao W, Kilgore J, Williams NS, et al. Small molecule-mediated disruption of Wnt-dependent signaling in tissue regeneration and cancer. Nat Chem Biol. 2009;5:100–107. doi: 10.1038/nchembio.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ewan K, Pajak B, Stubbs M, Todd H, Barbeau O, Quevedo C, Botfield H, Young R, Ruddle R, Samuel L, et al. A useful approach to identify novel small-molecule inhibitors of Wnt-dependent transcription. Cancer Res. 2010;70:5963–5973. doi: 10.1158/0008-5472.CAN-10-1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sack U, Walther W, Scudiero D, Selby M, Aumann J, Lemos C, Fichtner I, Schlag PM, Shoemaker RH, Stein U. S100A4-induced cell motility and metastasis is restricted by the Wnt/β-catenin pathway inhibitor calcimycin in colon cancer cells. Mol Biol Cell. 2011;22:3344–3354. doi: 10.1091/mbc.E10-09-0739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kitzman JO, Snyder MW, Ventura M, Lewis AP, Qiu R, Simmons LE, Gammill HS, Rubens CE, Santillan DA, Murray JC, et al. Noninvasive whole-genome sequencing of a human fetus. Sci Transl Med. 2012;4:137ra76. doi: 10.1126/scitranslmed.3004323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Benesova L, Belsanova B, Suchanek S, Kopeckova M, Minarikova P, Lipska L, Levy M, Visokai V, Zavoral M, Minarik M. Mutation-based detection and monitoring of cell-free tumor DNA in peripheral blood of cancer patients. Anal Biochem. 2013;433:227–234. doi: 10.1016/j.ab.2012.06.018. [DOI] [PubMed] [Google Scholar]