

Abstract
Epidermal growth factor receptor (EGFR) controls a wide range of cellular processes, and aberrant EGFR signaling as a result of receptor overexpression and/or mutation occurs in many types of cancer. Tumor cells in non-small cell lung cancer (NSCLC) patients that harbor EGFR kinase domain mutations exhibit oncogene addiction to mutant EGFR, which confers high sensitivity to tyrosine kinase inhibitors (TKIs). As patients invariably develop resistance to TKIs, it is important to delineate the cell biological basis of mutant EGFR-induced cellular transformation since components of these pathways can serve as alternate therapeutic targets to preempt or overcome resistance. NSCLC-associated EGFR mutants are constitutively-active and induce ligand-independent transformation in nonmalignant cell lines. Emerging data suggest that a number of factors are critical for the mutant EGFR-dependent tumorigenicity, and bypassing the effects of TKIs on these pathways promotes drug resistance. For example, activation of downstream pathways such as Akt, Erk, STAT3 and Src is critical for mutant EGFR-mediated biological processes. It is now well-established that the potency and spatiotemporal features of cellular signaling by receptor tyrosine kinases such as EGFR, as well as the specific pathways activated, is determined by the nature of endocytic traffic pathways through which the active receptors traverse. Recent evidence indicates that NSCLC-associated mutant EGFRs exhibit altered endocytic trafficking and they exhibit reduced Cbl ubiquitin ligase-mediated lysosomal downregulation. More recent work has shown that mutant EGFRs undergo ligand-independent traffic into the endocytic recycling compartment, a behavior that plays a key role in Src pathway activation and oncogenesis. These studies are beginning to delineate the close nexus between signaling and endocytic traffic of EGFR mutants as a key driver of oncogenic processes. Therefore, in this review, we will discuss the links between mutant EGFR signaling and endocytic properties, and introduce potential mechanisms by which altered endocytic properties of mutant EGFRs may alter signaling and vice versa as well as their implications for NSCLC therapy.
Keywords: Non-small cell lung cancer, Epidermal growth factor receptor, Signaling, Endocytosis, Src, Cbl, Ubiquitination
Core tip: Since their discovery ten years ago, much work has revealed the signaling properties of non-small cell lung cancer-associated mutant epidermal growth factor receptors (EGFRs). While therapeutic options for patients harboring mutants have emerged, these are beset with rapid development of resistance, making it critical that mutant EGFR biology be better understood to design more effective therapies. Emerging data suggests that mutant EGFRs exhibit altered endocytic trafficking, a process critical for the regulation of EGFR signaling. Deregulated endocytic traffic appears to enable mutant EGFRs to activate oncogenic signaling pathways. This review highlights the signaling and endocytic trafficking of mutant EGFR and the intimate link between the two processes.
INTRODUCTION
Epidermal growth factor receptor (EGFR) is a member of the ErbB (avian erythroblastic leukemia virus oncogene homolog) or human EGF receptor (HER) family of receptor tyrosine kinases (RTKs). The ErbB family comprises of EGFR (also known as ErbB-1/HER1), ErbB-2 (neu, HER2), ErbB-3 (HER3) and ErbB-4 (HER4). Studies of EGFR as a model RTK and prototypic oncogene have provided much of our understanding of cellular and molecular mechanisms of RTK function and regulation[1-4]. EGFR is a transmembrane glycoprotein with extracellular domains that bind to ligands such as EGF and transforming growth factor α (TGFα) to promote receptor dimerization and activation of a cytoplasmic tyrosine kinase domain. The resulting phosphorylation of the receptor and downstream signaling proteins mediate the various biological responses downstream[1,5]. In particular, EGFR is known to play crucial roles in cellular proliferation, survival, migration, and differentiation. Indeed, impaired epithelial development in several organs as well as perinatal lethality among EGFR knockout animal models illustrates the essential nature of EGFR in cellular functions[6,7]. Furthermore, oncogenic viruses exploit the EGFR signaling network in many different ways, altering both receptor tyrosine kinase activity and gene expression[8].
The role of aberrant EGFR signaling in oncogenesis has been investigated for many years. A major mechanism for aberrant signaling involves the overexpression of EGFR, found in various epithelial tumors[3]. The cancers where overexpression of EGFR is found include breast cancer, glioblastomas, head-and-neck cancer, non-small cell lung cancer (NSCLC), renal cancer, ovarian cancer, and colon cancer[1,9]. Transgenic studies[10] and in vitro studies, using NIH 3T3 mouse fibroblasts[11], demonstrate that high-level expression of EGFR and EGF ligands can transform cells. Recent studies using genetic deletion of EGFR illustrate the essential role of this receptor in oncogenesis in a pancreatic cancer model[12]. In addition, EGFR activation initiates cytoprotective signaling, enabling tumor cells to become resistant to radiation and chemotherapy[13,14]. Increased expression of EGFR is associated with poorer survival, and EGFR serves as a strong prognostic indicator in many cancer types[15].
In addition to overexpression, recent studies have demonstrated a key oncogenic role of mutant forms of EGFR in driving oncogenesis. EGFR overexpression in glioblastomas is associated with an alternatively-spliced variant, EGFRvIII, lacking the extracellular sequences encoded by exon 2-7 as a result of an 804 base pair in-frame deletion that corresponds to the removal of N-terminal amino acid residues from 6-273[16]. EGFRvIII is expressed in approximately 25% of glioblastomas[17] and in a higher percentage of patients with EGFR amplification[18,19]. This mutant initiates ligand-independent signaling and is transforming in animal models of glioblastoma[20]. Missense point mutations or small in-frame deletions in the kinase domain have been identified in NSCLC and shown to be constitutively active and oncogenic[21-23]. Notably, NSCLC-associated somatic EGFR mutations impart a higher sensitivity to EGFR-directed TKIs such as gefitinib (Iressa) or erlotinib (Tarceva)[3,22]. Because the NSCLC-associated EGFR mutants are constitutively-active and capable of transforming cells, they present fascinating models to study signaling pathways and defects in negative regulatory mechanisms. Therefore, this review will discuss our current understanding of NSCLC-associated mutant EGF receptors and their signaling properties, and the critical links between the endocytic and signaling pathways of mutant EGF receptors.
EGFR MUTATIONS IN NSCLC
Lung cancer is the leading cause of cancer deaths in both men and women in the United States, and NSCLC accounts for about 85% of lung cancers[24]. Studies in gastrointestinal stromal tumors showed that activating mutations of c-KIT gene were associated with clinical responses to small molecule TKI imatinib, and fueled interest to search for similar mutations in EGFR in NSCLC patients that responded favorably to gefitinib and erlotinib[25]. Indeed, in 2004, EGFR mutations associated with gefitinib sensitivity were identified in NSCLC[21,23].
Somatic mutations in the EGFR kinase domain are found in about 10% of NSCLC patients from the United States and about 25% of those from East Asia[26,27]. In-frame deletions in exon 19 (EGFR ∆746-750) and an arginine to leucine mutation at position 858 (EGFR L858R) account for about 90% of these mutations[26]. The mutations confer constitutive activity by disrupting the inactive conformation of the kinase domain of EGFR, and a 20-fold increased TKI binding accounts for their hypersensitivity to TKIs[28]. EGFR mutations in NSCLC have been correlated with gene amplification[29]. Somatic mutations of the ErbB2 kinase domain in NSCLC (in-frame insertions in exon 20) have also been identified in a subset (1.6%) of patients with a similar profile as those that harbor EGFR mutations: never smoker, East Asian ethnicity, and female gender[30]. More recent studies of breast cancer patients identified nine additional somatic mutations among EGFR family members that represent potential TKI therapeutic targets[31].
Multiple factors modify the sensitivity of NSCLC patients with EGFR mutations to EGFR TKIs. For instance, in-frame deletion in exon 19 is more sensitive to erlotinib inhibition than the L858R mutant[32]. Similarly, patients with an in-frame deletion mutant showed better response and longer overall survival with gefitinib or erlotinib treatment than did patients with the L858R mutant[33]. High EGFR gene copy number identified by fluorescence in situ hybridization (FISH) was proposed to be an effective molecular predictor of gefitinib efficacy in advanced NSCLC[34]. However, a meta-analysis has found that EGFR overexpression is not associated with overall survival in NSCLC patients[35]. Increased ErbB2 expression is also associated with increased sensitivity to gefitinib both in the presence[36] and absence of EGFR mutations[37], although phosphorylated-ErbB2 along with total ErbB3 levels have been associated with resistance to gefitinib in head and neck squamous cell carcinoma[38].
Despite their success in a subset of patients, the overall effectiveness of EGFR inhibitor treatment for cancer therapy remains elusive. While erlotinib can be effective for the initial treatment of those with sensitizing EGFR mutations, overall median survival of patients treated with erlotinib vs placebo is only 6.7 mo vs 4.7 mo[24]. Together with the fact that patients with erlotinib reported severe side effects including rash and diarrhea[39], it is vital that alternative and improved regimens be developed to better treat NSCLC.
Further complicating TKI treatment efficacy, patients with drug-sensitive EGFR mutations develop acquired resistance after about 12 mo, and up to 50% of resistant cases can be attributed to a secondary mutation at position 790 (EGFR T790M)[26,40,41]. It is thought that the T790M mutation leads to steric hindrance for erlotinib binding due to the bulky methionine side chain in the ATP-binding pocket[42]. However, another study revealed that the T790M mutation causes drug resistance simply by increasing the affinity for ATP[43].
Amplification of the hepatocyte growth factor receptor tyrosine kinase (MET) has been implicated in drug resistance, presumably by driving ErbB3-dependent activation of phosphoinositide-3 kinase (PI3-K)[44]. MET amplification was reported in about 20% of TKI-resistant patients, sometimes concomitantly with EGFR T790M[44]. In addition, insulin-like growth factor I receptor reportedly interferes with anti-EGFR directed therapies[45], and a block in apoptosis is implicated as one of the mechanisms of TKI resistance[46].
A multigene signature indicative of an epithelial to mesenchymal transition (EMT) was also identified as a determinant of insensitivity to erlotinib[47]. Cells expressing the epithelial cell junction protein E-cadherin show greater sensitivity to EGFR inhibition, whereas cells that have undergone EMT, expressing vimentin or fibronectin, are insensitive[48]. Src-mediated cell signaling has been proposed to be another mechanism for anti-EGFR directed therapy resistance, by bypassing the dependency on EGFR for cell growth and survival[49]. KRAS mutational status also predicts resistance to anti-EGFR directed therapies apparently as cancer cells may no longer need EGFR for survival[50-52].
To circumvent the problem of TKI resistance and to enhance the efficacy of EGFR-directed therapies, alternative strategies are being utilized to enhance the survival rate of cancer patients, albeit with mixed results. Because erlotinib has a lower IC50 than gefitinib against wild-type EGFR, it has been suggested that gefitinib-resistant patients be treated with erlotinib, but such studies have not had much success as erlotinib could not overcome the resistance conferred by the T790M mutation[53]. Phase II clinical trial data also showed that patients with activating EGFR mutations do not respond to monoclonal antibody-based therapy with cetuximab even though they respond to a TKI[54]. However, a different study showed cetuximab to be effective against cells expressing either TKI-sensitive or resistant NSCLC EGFR mutations[55].
Promising results were observed in an animal model using a combination of cetuximab and TKI, which yielded enhanced tumor regression[56]. Cells expressing mutant EGFR also show sensitivity towards an inhibitor of vascular endothelial growth factor receptor-2 (VEGFR-2), ZD6474, indicating that VEGFR-2 may be critically involved in mutant EGFR-mediated cell survival[57]. An irreversible pan-ErbB inhibitor PF00299804 is also a potent inhibitor of gefitinib-resistant EGFR and ErbB2 mutations[58].
It is evident from the current directions of the anti-EGFR directed therapy that it is critical to identify key partners and pathways of EGFR-mediated tumorigenicity and co-target these factors simultaneously. This approach will not only benefit NSCLC, but other cancer types in which EGFR may play a critical role.
MUTANT EGFR SIGNALING
Early studies using NSCLC tumor cell lines indicated that mutant EGFRs are constitutively-active[22,28,59], transform nonmalignant cell lines in a ligand-independent manner[60], and enhance tumor growth in xenograft models[60]. Furthermore, cells harboring mutant EGFRs undergo “oncogene addiction” and require the mutant receptor activity for survival[61]. In transgenic mouse models, reduction in the expression of mutant EGFR or inhibition of its kinase activity caused rapid tumor regression, demonstrating that mutant EGFR is required for tumor maintenance[62].
It is becoming increasingly evident that in addition to oncogene addiction, cells also depend on non-driver oncogenic pathways for survival[63]. It has been found that downstream signaling pathways that are key to mutant EGFR function include Akt, Erk1/2, and STAT3[23,34,64-67]. Sensitivity to growth inhibition by gefitinib is associated with signaling molecules downstream of activated EGFR such as Akt and Erk[68,69], and gefitinib effectively blocked Akt and Erk phosphorylation in gefitinib-sensitive NSCLC cell lines[66].
Transfection studies using the EGFR exon 19 in-frame deletion mutant revealed highly phosphorylated Akt and STAT3 compared to transfection of wild-type EGFR (wtEGFR)[70]. In fact, continued activation of PI3-Kinase signaling by a PIK3CA (PI3-K, catalytic, alpha polypeptide) oncogenic mutant is sufficient to abrogate gefitinib-induced apoptosis in NSCLC-associated EGFR mutant-expressing cells[71]. PI3-K was found to exclusively associate with ErbB3 in gefitinib-sensitive NSCLC cell lines, and, interestingly, the gefitinib-sensitive wtEGFR-expressing NSCLC cell lines showed greater ErbB3 expression than gefitinib-insensitive cell lines[72]. In addition, the expression of NSCLC-associated EGFR mutants correlates with constitutive activation of mammalian target of rapamycin (mTOR) and Erk5 as well as enhanced expression of cyclin D1 and EGR1[73-76].
Subsequent studies have also identified Src to be critical for cell proliferation, survival and migration of mutant EGFR-expressing cells. In contrast to mutant EGFRs, overexpression of wtEGFR in primary cells is not oncogenic. High levels of exogenous ligands[77] and/or cooperating oncogenic partners are required for the wtEGFR to transform cells. In this regard, Src has been established to cooperate with EGFR and to be an important determinant of EGFR-mediated oncogenesis[78,79]. EGFR and Src overexpression in fibroblast systems led to synergistic increases in EGF-induced DNA synthesis, soft agar colony formation, and tumor formation in nude mice[80]. This cooperativity has also been demonstrated in a model of epithelial cell transformation: loss of polarity in three-dimensional cultures of nonmalignant human mammary epithelial cells as well as their anchorage-independent growth were only seen when both EGFR and Src were co-overexpressed[81]. Consistent with these studies, EGFR and Src are often co-overexpressed in human cancers[82], and enhanced Src activity was observed in NSCLC tissue compared to normal lung tissues[83].
Importantly, mutant EGFR-expressing NSCLC cell lines exhibit increased Src phosphorylation[64], and more Src is associated with mutant EGFR compared to wtEGFR[84,85]. Mutant EGFR-expressing cells were sensitive to Src inhibitors such as Dasatinib, PP1, or SKI-606[64,65], and Src-dependent phosphorylation on EGFR Y845 was required for full phosphorylation of mutant EGFR and downstream signaling molecules such as Akt, Erk and STAT3[85,86]. Interfering with Src-dependent phosphorylation on EGFR Y845 resulted in decreased mutant EGFR-mediated biological processes such as cell transformation and migration[85]. Further studies are needed to identify additional factors and processes critical to mutant EGFR-mediated oncogenic signaling and biological outcomes.
ENDOCYTIC TRAFFIC OF EGFR
In normal cells, EGFR signaling is tightly regulated by endocytic traffic. The ligand-induced EGFR dimerization and activation are associated with rapid internalization from the cell surface into endosomes followed by further traffic into lysosomes, or alternatively into an endocytic recycling compartment from which the receptor returns back to the cell surface (Figure 1). The pathways of endocytic traffic to the lysosome and their functional impact have been substantially elucidated in recent years. Activated EGFR undergoes ubiquitination, which provides a signal for sorting of internalized EGFR into lysosomes for degradation[2,87], a mechanism for signal termination[88,89]. In this context, endocytosed EGFR migrates down a system of heterogeneous compartments that have generally been characterized as “early” or “late” endosomes based on their morphology, kinetics of labeling with endocytic cargo as well as compartment-specific markers[90]. While punctate early endosomes are primarily located towards the cell periphery, the late endosomes are larger and more spherical, and often positioned closer to the nucleus[90]. Furthermore, some late endosomes have a multivesicular appearance by ultrastructure and are referred to as multivesicular bodies (MVBs). The ubiquitinated cargo, such as EGFR, at the outer limiting membrane of MVBs is recognized by a series of protein complexes called the endosomal complex required for transport (ESCRT) 1 to 3, and selectively sorted into invaginating vesicles that bud off to form the internal vesicles of MVB[91]. The MVB subsequently matures and/or fuses into the lysosome where the receptor is degraded by lysosomal hydrolases[91]. Under conditions where EGFR does not undergo ubiquitination, or if ubiquitin chains are cleaved by deubiquitinases[92], the receptor is alternatively sorted into vesicles that traffic along the endocytic recycling pathway back to the cell surface for additional rounds of ligand binding and signaling[93]. Endocytic recycling can take place via the fast/direct recycling from the early endosomes back to the plasma membrane, or via a delayed recycling pathway that involves the perinuclear endocytic recycling compartments (Figure 1)[93,94]. Members of Rab-family of small GTPases play key roles in facilitating alternate sorting itineraries. The delayed recycling pathway utilizes Rab11 as well as Arf6 small GTPase[94]. Notably, members of a family of dynamin-like ATP-binding and EPS15-homology domain-containing (EHD) proteins have been identified as key regulators of the delayed endocytic recycling compartments[95,96].
Figure 1.
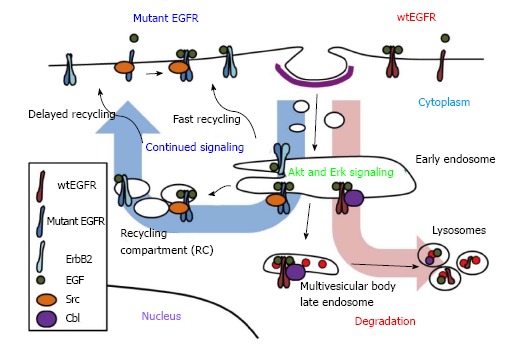
Model of mutant epidermal growth factor receptor endocytic trafficking. Upon ligand binding, activated wtEGFR becomes internalized and localized to endosomes. Internalized EGFR has been linked to Erk and Akt activation. Depending on type of ligand bound, ligand concentration, dimerization partner, mutational statuses and/or availability of other regulators, wtEGFR may recycle to cell surface or be sorted to lysosomes. EGFR bound to EGF is mostly targeted for lysosomes, where it becomes degraded. Sorting of ligand-induced wtEGFR to lysosome is mediated by E3 ubiquitin ligase Cbl which remains attached to receptor throughout endocytosis. Mutant EGFR, however, escapes ligand-induced downregulation through decreased interaction with Cbl, enhanced dimerization with ErbB2, which prefers recycling pathway, and/or constitutive interaction with Src, which antagonizes Cbl. EGFR: Epidermal growth factor receptor.
The endocytic trafficking fates of EGFR are regulated by a complex, and still incompletely understood, molecular machinery. First, the nature of the ligand bound to EGFR can dictate alternate fates of the ligand-receptor complex. In particular, EGF, which forms a more stable complex with EGFR in the low pH environment of endosomes, primarily targets the receptor for ubiquitination and lysosomal degradation; in contrast, other ligands such as TGFα and amphiregulin form less stable complexes with EGFR, induce lower levels of ubiquitination, and promote recycling of the receptor accompanied by more sustained signaling responses[88,97]. The concentration or the availability of ligands is another factor that determines the fate of EGFR degradation as low concentrations of EGF induce clathrin-mediated endocytosis of EGFR for recycling while higher concentrations induce internalization via lipid rafts for degradation[98,99]. The dimerization partner may also affect the regulation of EGFR; for example, overexpression of ErbB2 has been shown to reduce EGFR downregulation by increasing its recycling or decreasing internalization[100,101].
The Cbl (Casitas B-lineage Lymphoma proto-oncogene) family of ubiquitin ligases plays an essential role in promoting ubiquitination and lysosomal degradation of EGFR[102]. Cbl proteins selectively associate with activated EGFR, via phosphorylated tyrosine 1045 (number corresponding to human EGFR) and facilitate the juxtaposition of Cbl-bound ubiquitin conjugating enzymes to facilitate EGFR ubiquitination[102,103]. Once bound, Cbl remains associated with the activated EGFR throughout its endosomal transport to lysosomes[103]. Ubiquitination mediated by the association of Cbl proteins is essential for EGFR sorting into lysosomes (Figure 1). It has also been suggested that Cbl can function as an adaptor protein to recruit Cbl-interacting protein of 85 kDa (CIN85) together with its partner Endophilin A to promote initial internalization of EGFR[104]. However, a more recent study suggested that CIN85 is dispensable for EGFR internalization[105]. Grb2, which interacts with the proline-rich region of Cbl and phosphotyrosine-containing motifs on EGFR, may also mediate EGFR-Cbl complex formation and initiate EGFR internalization by promoting delivery into clathrin-coated pits[104,106,107]. Disruption of Grb2 interactions with EGFR or Grb2 knockdown using a small interfering RNA significantly inhibited the receptor internalization[108,109]. A recent study also demonstrated that the cooperative recruitment of Cbl, in complex with Grb2, to EGFR determines the threshold of ubiquitination of EGFR[110]. Other factors are also known to affect EGFR endocytosis and downregulation. For example, Sprouty2 or activated Cdc42[111], which appear to block Cbl function, as well as suppressor of T-cell receptor signaling (Sts-1/Sts-2), and Cortactin, inhibit efficient EGFR trafficking to the lysosome and block receptor downregulation[107,112,113]. In addition, SNX1, stimulatory G protein subunit (Gαs), and factors important in mediating or regulating ESCRT complex function, such as Hrs (hepatocyte growth factor-regulated tyrosine kinase substrate), signal-transducing adaptor molecule (STAM), tumor susceptibility gene product 101 (TSG101), and other components of ESCRT complexes, are required for the MVB sorting of EGFR and efficient receptor degradation[7,114]. Interfering with Cbl-dependent negative regulatory process prolongs the EGFR activity and enhances the EGFR-mediated cell transformation[87]. Thus, it is clear that endocytic traffic of EGFR plays a critical role in controlling its signaling and regulating its oncogenicity.
ENDOCYTIC TRAFFIC OF MUTANT EGFRS
Mutant EGFRs function as oncogenic drivers in NSCLC and other cancers including glioblastomas. To understand the biological basis of how mutant receptors drive oncogenesis, it is important to gain insights into how the regulatory mechanisms that control EGFR operate in the context of mutant receptors. A key component of EGFR regulation involves the ligand-induced receptor endocytosis which leads to degradation of the receptor and termination of signaling, or to receptor recycling for continued signaling. Because of the radically different outcomes of the alternate endocytic fates, elucidating mechanisms of mutant EGFR endocytic trafficking is fundamentally important to understanding mutant EGFR-driven signaling and oncogenesis, with a potential to improve the EGFR-directed therapies.
Since mutant EGFR exhibits constitutive signaling, it is likely that this is associated with altered endocytic trafficking. Indeed, multiple lines of evidence suggest that mutant EGFRs undergo altered endocytic trafficking compared to the wild-type receptor[115-118]. In this section, we will describe mutant EGFR endocytic trafficking in terms of basal receptor localization, as well as ligand-induced internalization and degradation.
MUTANT EGFR LOCALIZATION AND LIGAND-INDUCED INTERNALIZATION
Mature wtEGFR is primarily localized at the cell surface prior to ligand binding, but becomes internalized upon ligand binding. There are conflicting reports in regards to ligand-induced mutant EGFR internalization compared to that of wtEGFR. It has been reported that EGF-induced internalization of gefitinib-sensitive mutant EGFR expressed on PC9 cell line was faster than that of wtEGFR on gefitinib-insensitive cell lines A549 and QG56[68,119]. Another study, however, reported that mutant EGFR-expressing NSCLC cell lines H1975 and H1650 showed delayed internalization of labeled EGF in comparison to a wtEGFR-expressing cell line H358[116]. Yet another study found that rhodamine-conjugated EGF uptake was comparable between H1299 cell lines permanently transfected with mutant EGFRs or wtEGFR, suggesting that NSCLC EGFR mutation did not affect ligand-induced receptor internalization[120]. Differences in EGF-induced mutant EGFR internalization may be attributed to cell lines used to compare wtEGFR and mutant EGFRs, and underscore the need for more comprehensive and concurrent studies using multiple assays to fully understand if and how the NSCLC-associated mutations of EGFR affect its ligand-induced EGFR internalization.
Compared to the uncertainty of the impact of NSCLC-associated EGFR mutations on ligand-induced internalization, emerging evidence suggests that mutant EGFRs are constitutively internalized. Mutant EGFR ectopically overexpressed in a murine pro-B cell line model was shown to undergo EGF-independent internalization, whereas wtEGFR was primarily localized to the cell surface in the absence of ligand[121]. Another study showed that mutant EGFR in PC9 cell line, but not the wtEGFR, in QG56 cell line was distributed inside the cell[119]. These data suggest that mutant EGFRs may undergo enhanced constitutive internalization compared to wtEGFR. Indeed, unlike wtEGFR, mutants EGFRs showed constitutively intracellular localization and colocalized with endosomal markers[118]. Inhibition of endocytic recycling pathway using monensin resulted in the accumulation of mutant EGFRs in perinuclear vesicles, and mutant EGFRs showed colocalization with various recycling endosomal markers, suggesting that mutant EGFRs undergo altered endocytic trafficking through recycling pathway[118]. Importantly, recycling inhibition delayed the ligand-mediated mutant EGFR degradation and enhanced the mutant EGFR association and colocalization with Src (Figure 1)[118]. These findings support the notion that enhanced endocytic trafficking of mutant EGFRs via the recycling pathway provides a potential compartment where mutant EGFR may engage in preferential interaction with Src and sustained oncogenic signaling.
As for the cellular localization and internalization of EGFRvIII, there remains much confusion. Earlier work has suggested that EGFRvIII expressed in a glioma cell line remains on the plasma membrane even after EGF stimulation, whereas wtEGFR was removed from the cell surface and appeared in perinuclear vesicles corresponding to endosomes and lysosomes[122]. Similarly, EGFRvIII, when transfected into a small cell lung cancer cell line, localizes predominantly at the cell surface[123]. However, confocal microscopic analyses on biopsy samples of human gliomas showed that the subcellular localization of EGFRvIII was identical to that described for wtEGFR; predominant cell membrane expression, with some perinuclear distribution[124]. Future studies will need to delineate the exact subcellular localization and endocytosis of EGFRvIII.
The mechanisms underlying the tendency of NSCLC-associated EGFR mutants to remain constitutively internalized are currently unclear. As mutant EGFRs are constitutively-active, a role for kinase activity in promoting ligand-independent internalization appears plausible. Published studies on the role of kinase activity for internalization of wtEGFR have arrived at opposite conclusions, suggesting either a requirement for or dispensability of the kinase activity for internalization[125-127]. In fact, ligand-induced internalization of EGFR in the presence of TKIs was previously employed by investigators to initiate signaling directly from the endosomes[128]. Rather than the kinase activity per se, it may be the conformational changes associated with activation that expose endocytic motifs in EGFR and permit its internalization[43,60]. Furthermore, activated EGFR recruits adapter proteins such as Epsin and Grb2 that have been shown to promote internalization[129,130]. Given the constitutive activity of mutant EGFRs, these mechanisms may mediate enhanced internalization of mutant compared to wtEGFR. Notably, gefitinib inhibits the ligand-induced internalization of mutant EGFR in gefitinib-sensitive PC9 cell line but does not affect internalization of wtEGFR in gefitinib-insensitive QG56 cell line[119]. Therefore, kinase activity might play a more critical role in the internalization of mutant EGFR compared to wtEGFR. Other studies have shown that EGFR dimerization is critical for wtEGFR internalization[125]. Given that mutant EGFR has been found to be constitutively dimerized[131], dimerization may indeed be critically involved in the constitutive internalization of mutant EGFRs.
Some studies also suggest sensitivity to TKI to play a role in ligand-induced EGFR internalization. For example, it has been reported that a H1650 NSCLC cell line rendered gefitinib-resistant showed increased ligand-induced mutant EGFR internalization when compared to the parental gefitinib-sensitive cell line[132]. In contrast, the reverse was true for wtEGFR, as others showed that ligand-induced internalization of wtEGFR in erlotinib-sensitive H292 cells was greater than that in erlotinib-insensitive H1703 cells[133]. Quantification also showed that inhibition of EGF-induced EGFR internalization by erlotinib was greater in sensitive cell line compared to that in the insensitive cells[133]. Further studies are needed to more clearly delineate key determinants of ligand-induced and constitutive mutant EGFR internalization as well as the relationship of these processes with TKI sensitivity vs resistance.
ALTERED LIGAND-INDUCED DEGRADATION OF MUTANT EGFRS
As mentioned in the introduction, lysosomal degradation of EGFR is critically dependent on ubiquitination promoted by Cbl-family ubiquitin ligases. Upon ligand activation and phosphorylation of EGFR, Cbl associates with the phosphorylated (active) receptor and facilitates its ubiquitination[102,134-137]. The Cbl-EGFR association has been shown to persist throughout the endosomal pathway and Cbl-family proteins are essential for the lysosomal sorting step of activated EGFR downregulation[103,134,138]; accordingly, ubiquitin ligase activity-defective Cbl mutants enhance the EGFR recycling[135]. Ubiquitin ligase activity-deficient Cbl itself can become oncogenic due to loss of negative regulatory control on receptor signals[135,139-141]. Depletion of Cbl proteins or expression of mutant forms has clearly shown that lack of Cbl function deregulates EGFR traffic, elevates downstream signaling and promotes epithelial cell migration[134,137,142]. As NSCLC mutant EGFRs appear defective in Cbl-dependent downregulation, it is quite likely that the ensuing recycling and endosomal signaling contribute to the oncogenicity of mutant EGFRs[115-117] (Figure 2).
Figure 2.
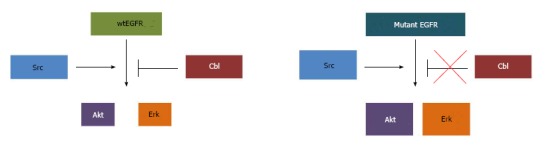
Mutant epidermal growth factor receptor vs wt-epidermal growth factor receptor signaling. While wtEGFR signaling to Akt and Erk is subjected to Cbl-mediated degradation, mutant EGFR cooperates with Src to exaggerate signaling through downstream effectors. EGFR: Epidermal growth factor receptor.
Several studies have examined the association of NSCLC EGFR mutants with Cbl, but have provided conflicting results. Reduced ligand-induced association of mutant EGFR with Cbl, as compared to that of wtEGFR, was reported in NSCLC cell lines H1975 and PC-9 expressing EGFR L858R/T790M or ∆746-750 mutants respectively, as well as in human embryonic kidney and normal lung bronchial epithelial cells made to overexpress EGFR L858R or ∆746-750[116,117,143,144]. However, another study using TGFα as a ligand showed intact and constitutive mutant EGFR-Cbl association in NSCLC cell lines[115].
Similar to conflicting reports on mutant EGFR-Cbl association, the phosphorylation status of the Cbl binding site, EGFR-Y1045, on mutant EGFRs remains unclear[87]. Reverse-phase protein microarray was used to quantify levels of phosphorylation of various EGFR phosphorylation sites on pure tumor cell populations isolated by laser capture microdissection from human lung tumor biopsy specimens[145]. The group found that phosphorylation of EGFR-Y1045 was reduced across patient samples that expressed all classes of mutant EGFRs (inframe deletion mutant, EGFR L858R and H773L/V774M) compared with wtEGFR[145]. Similarly, EGFR L858R and EGFR ∆747-753 mutants expressed in a mouse fibroblast cell line or COS-7 cells showed lower levels of EGFR-Y1045 phosphorylation when compared to wtEGFR, while EGFR ∆746-750 showed hypo-ubiquitination, delayed downregulation, and increased surface retention upon ligand stimulation[146]. Another study, however, showed that when mutant EGFRs were stably expressed in a NSCLC cell line H1299, the mutant EGFRs showed higher basal phosphorylation levels at all tyrosine residues, including Y1045[120]. Intact ligand-induced Y1045 phosphorylation has been observed in other mutant EGFR cell systems, including L858R mutant-expressing and in-frame deletion mutant-expressing non-transformed mouse mammary epithelial cells[23], SF9 insect cells[147], and murine hematopoietic cells[121]. Similarly, endogenous EGFR ∆746-750 or L858R/T790M expressed in NSCLC cell lines H1650 or H1975, respectively showed robust phosphorylation on EGFR Y1045 compared to that in wtEGFR-expressing NSCLC cell line H358[116]. Interestingly, human embryonic kidney 293 cells transfected with EGFR L858R showed intact ligand-induced Y1045 phosphorylation and association with Cbl, whereas EGFR ∆747-753 showed decreased Y1045 phosphorylation and EGFR-Cbl association[55]. The discrepancies among reported results are likely to be due to the difference in EGFR mutation type, different cell type used, and/or the types of cells used as controls to make comparisons. Nonetheless, a consistent picture on Cbl-mutant EGFR association has not emerged, and the role of other Cbl family members remains unclear.
In contrast to a lack of consensus on Y1045 phosphorylation of mutant EGFRs and their association with Cbl, different studies have consistently noted an impairment of ligand-induced ubiquitination and downregulation of mutant EGFRs. It was reported that mutant EGFRs undergo reduced ubiquitination and delayed downregulation upon ligand stimulation in NSCLC cell lines H1650 and H1975, expressing endogenous mutant EGFRs, and in human embryonic kidney cells ectopically overexpressing mutant EGFR[116,143]. Decreased ligand-induced ubiquitination and delayed downregulation were also observed in various NSCLC cell lines expressing endogenous EGFR ∆746-750 or L858R (HCC827 and H3255, respectively), and in normal human bronchial epithelial cells stably expressing EGFR ∆746-750 or L858R[117].
Interestingly, even under conditions that permitted mutant EGFR-Cbl association, mutant EGFR showed decreased ligand-induced ubiquitination and impaired degradation; this correlated with constitutive association of mutant EGFR with the molecular chaperone Hsp90[115]. Constitutive association of mutant EGFR with Hsp90[115,148] may provide a mechanism to impair Cbl-dependent mutant EGFR downregulation. However, Cbl overexpression in HCC827 cell line resulted in enhanced mutant EGFR downregulation, suggesting that mutant EGFRs retain the ability to undergo Cbl-dependent downregulation but the process is less efficient[117]. Among the ErbB family receptors, ErbB2 is known to be stably associated with Hsp90 while EGFR-Hsp90 interaction is transient[149]. It is therefore noteworthy, that heterodimerization with ErbB2 has been identified as a mechanism for the ability of EGFR L858R or EGFR L858R/T790M to avoid ligand-induced downregulation[116] (Figure 1). Previous studies in breast cancer and other models have established that ErbB2 is impaired in downregulation, and its co-overexpression with EGFR inhibits the downregulation of EGFR by increasing the recycling rate of EGFR and/or inhibiting internalization[100,101,150,151]. Indeed, treatment of gefitinib-resistant EGFR L858R/T790M-expressing NSCLC cells with a EGFR/ErbB2 dual TKI, lapatinib, decreased STAT3 activation and this was associated with reduced mutant EGFR-ErbB2 heterodimerization[56]. Combining lapatinib and cetuximab treatment resulted in enhanced cytotoxicity against gefitinib-resistant EGFR L858R/T790M-expressing cells in vitro and in xenograft models in vivo[56]. However, EGFR mutants expressed in Chinese hamster ovary cells were less sensitive to lapatinib, indicated by the levels of autophosphorylation, than wtEGFR[152]. Therefore, the impact of ErbB2 co-expression based on the effects of lapatinib must be considered in the context of the genetic makeup of the cell system used, including the levels of ErbB2 expression. It has also been shown that certain stimuli activate EGFR and promote its internalization but do not induce efficient downregulation. Such stimuli include specific EGFR ligands, such as amphiregulin and TGFα, and exposure to certain chemicals including H2O2 or cigarette smoke[97,153] and the role of such factors in inefficient downregulation of mutant EGFRs in NSCLC needs to be considered.
A number of interacting proteins are thought to affect Cbl-dependent lysosomal trafficking of EGFR. Cbl interacting proteins CD2AP and CIN85 are thought to cooperate with Cbl to promote EGFR endocytosis[113,154], whereas Cool-1, Sprouty and Sts-1/Sts-2 interfere with EGFR downregulation[107,112,113,155]. Therefore, alterations of these components may account for defective Cbl-dependent downregulation of mutant EGFRs even though EGFR Y1045 phosphorylation and association with Cbl remain intact[23,115,116,120].
In addition, there are other factors to be considered for the altered endocytic trafficking of mutant EGFRs. Cdc42-associated tyrosine kinase 1 associates with activated EGFR and is involved in ligand-induced clathrin-coated vesicle-mediated EGFR endocytosis and degradation[156-158]. Rab5 controls endosome fusion and enhances lysosomal degradation of EGFR[159,160], and Rab5 exchange factor GAPex-5 mediates EGFR ubiquitination, lysosomal trafficking and degradation[161]. Alternatively, TBC1D3 enhances EGFR internalization but suppresses Cbl-dependent EGFR ubiquitination and degradation[162]. In addition, STAM1/2, Hrs, Rin1 and ESCRT proteins also regulate EGFR endocytic traffic at various stages[91,163]. Verifying expression levels of proteins critical to Cbl-dependent EGFR downregulation, and/or RNAi-mediated knock-down of proteins implicated in interfering with EGFR downregulation may identify those critical to mutant EGFR endocytic trafficking and provide mechanism of altered endocytic traffic of mutant EGFRs. Given the biological consequences of inefficient mutant EGFR downregulation, elucidation of cell biological and biochemical mechanisms responsible represent a fertile area of future research.
ENDOSOMAL SIGNALING BY EGFR
Aside from the importance of endocytosis as a necessary step in lysosomal downregulation of ligand-activated EGFR, endocytosis has emerged as a requirement for efficient activation of specific downstream signaling pathways. For example, inhibition of the internalization machinery demonstrated that activation of PLCγ1 upon EGF stimulation occurs primarily at the cell surface while activation of Erk and Akt signaling occurs primarily post-internalization[164]. Therefore, oncogenic signaling from mutant EGFRs may result from or be sustained by altered receptor endocytic trafficking. It is now well-established that internalized receptor tyrosine kinases, including EGFR, continue to be active unless degraded, providing a mechanism for persistent signaling as well as activation of distinct pathways through the formation of spatially-distinct signaling complexes[165]. Many studies have shown that Erk activation upon EGF stimulation critically depends on endosomal localization of EGFR; activated EGFR in endosomal compartments participate in the activation of Ras, the upstream activator of Erk signaling[166]. A relationship between accumulation of EGFR in endosomes and enhanced Erk activation is provided by studies in which overexpression of SEF (for similar expression to FGF) enhances EGF-induced EGFR internalization but delays its targeting to lysosome, and results in sustained Erk activation and differentiation in the PC12 rat pheochromocytoma cell model[167]. Knockdown of Cbl in 293 cells[168], or lack of Cbl in Cbl-knockout mouse embryonic fibroblasts[134], delayed exit of ligand-stimulated EGFR out of early endosomes towards lysosomes, and resulted in prolonged Erk activation. Combined knockdown of Cbl and Cbl-b had a similar impact on EGFR traffic and Erk signaling in human mammary epithelial cells[132].
Endosomal localization of not just EGFR but also of Ras signaling cascade proteins is required for Erk activation. Dynamin-regulated endocytosis of activated MEK is required for Erk activation[169]. Analysis of endosomal localization of MEK2-GFP suggests that endosomal localization of MEK2 requires clathrin-dependent endocytosis, the presence of an upstream kinase, RAF, and the catalytic activity of MEK[170]. Erk activation mediated by beta 2-adrenergic receptor transactivation of EGFR is sensitive to clathrin-dependent endocytosis in transfected COS-7 cells[171], although it has been recently reported that clathrin-dependent endocytosis is not required for Erk activation in HeLa cells[170]. MAP kinase signaling may itself affect EGFR endocytic traffic, as activation of p38 MAP kinase induced the internalization of EGFR, suggesting that p38 may provide an important feedback regulatory loop in the regulation of EGFR trafficking and signaling[172]. Recently, it was shown that activated Akt promotes early endosome to lysosome transition and degradation of EGFR by activating PIKfyve (FYVE-containing phosphatidylinositol 3-phosphate 5-kinase)[173]. By utilizing reversible kinase inhibitors to promote ligand-induced internalization into endosomes followed by inhibitor removal, it was established that endosomal EGFR signaling is sufficient to activate major signaling pathways leading to cell survival and proliferation[128,174]. Thus, it is easy to visualize how constitutive internalization into endosomes together with impaired lysosomal downregulation of NSCLC-associated EGFRs can promote endosomal signaling-dependent oncogenic cascades, as further discussed below.
MUTANT EGFR TRAFFIC AND SIGNALING
As discussed above, studies of mutant EGFR signaling pathways have identified Akt, Erk, STATs and Src as critical downstream molecular players. Our studies established that preferential trafficking of mutant EGFRs vs wtEGFR into the endocytic recycling compartment promotes association of mutant EGFRs with their oncogenic partner Src (Figure 1)[118]. The requirement of the major Src phosphorylation site of the mutant EGFRs (Y845) for their ability to transform NIH 3T3 cells supports a role for Src signaling within the endosome in mutant EGFR oncogenic activity[85] (Figure 2). This is consistent with the impact of Src inhibitors on NSCLC in vitro and in animal models[85,175]. Further studies are needed to directly assess if Src-dependent signaling by mutant EGFRs takes place primarily within the endosomal compartments or if endocytic recycling is required to traffic Src to its site of action. In this regard it is notable that inactive Src is primarily localized on lysosomal membranes and an endocytic traffic pathway orchestrated by the ESCRT machinery is required for its traffic to focal adhesions/invadopodia, where Src activity is essential for cell migration and invasion[176,177]. Notably, lysosomal regulatory small GTPase Rab7 and ESCRT1 component TSG101 were shown to be required for un-occupied wtEGFR to enter into the endocytic recycling compartment and to recycle back to the cell surface[178]. It will be of considerable interest in the future to determine if NSCLC-associated mutant EGFRs and c-Src co-traffic through a lysosomal/MVB compartment to enter the endocytic recycling compartment.
Notably, endocytic traffic-dependent EGFR signaling is also critical for cell migration. The developmentally regulated border cell migration in drosophila requires localized EGFR signaling, and deletion of proteins involved in endocytic traffic of EGFR (Cbl and Rab5 guanine nucleotide exchange factor) disrupted border cell migration[142]. In this system, receptor endocytosis is necessary to help establish ligand gradients and localized RTK signaling, preserving spatial information that is critical to guide migrating cells in a directional manner[142]. Recently, it was shown that TGFα-induced cell migration in mammalian corneal epithelial cells required endocytic recycling of EGFR[179]. Thus, it is plausible that endocytic recycling and endosomal signaling may contribute to cell migration, invasion and metastatic behavior of mutant EGFR-expressing NSCLCs.
NSCLC-associated EGFR mutants exhibit constitutive internalization[118,119,121]. Since hyperactive Erk signaling is a prominent feature of mutant EGFRs in NSCLC[23,66], enhanced endosomal localization of mutant EGFR may provide one of the mechanisms contributing to enhanced Erk activation. Indeed, the upstream activators of Erk signaling pathway have been shown to be associated with endosomal EGFR. Analysis of rat livers following administration of EGF, internalization of EGFR coincided with recruitment of the adaptor protein Shc, and its association with GRB2 and the Ras guanine nucleotide exchange factor, mSOS, and the complex of tyrosine phosphorylated Shc, Grb2 and mSOS was largely present in the endosomal fraction[180]. FRET measurements also indicated that activated EGFR-CFP interacted with YFP-Shc and Grb2-YFP in endosomes[181]. Similarly, inhibition of EGFR internalization through knockdown of Grb2 by RNA interference or use of a dynamin mutant resulted in inhibition of EGF-induced MAPK and Erk activities[106,164].
Akt activation has also been linked to endosome-localized EGFR. Inhibiting internalization through clathrin heavy chain or AP-2 knockdown reduced EGFR-mediated Akt as well as MAPK activation[99]. Initiation of EGFR signaling directly from the endosomes, by allowing EGFR endocytosis in the presence of a washable inhibitor, demonstrated that endosomal signaling of EGFR is sufficient for activating signaling pathways including Erk and Akt, as well as cell survival and proliferation[128,174]. Likewise, ligand-induced trafficking of internalized EGFR from early endosomes to lysosomes was severely delayed in cells lacking presenilin 1 and resulted in prolonged EGFR, Akt and Erk activation[182]. A similar impact of depletion of Cbl proteins has been demonstrated on EGFR-induced Erk and Akt activation[134,137]. Therefore, similar to Erk, Akt activation may also be dependent on EGFR localization in endosomes. Interestingly, some studies indicate that endosomal EGFR does not recruit and activate PI3-K[183,184], suggesting that EGFR-dependent Akt activation may involve additional factors and/or mechanisms. Clathrin-dependent EGFR internalization has also been shown to be essential for STAT3 nuclear translocation and Stat3-dependent gene regulation, and Stat3 co-localizes with labeled EGF in endocytic vesicles[185]. These studies, done on wtEGFR, support the likelihood that constitutively endosome localized mutant EGFRs promote enhanced Map kinase and potentially PI3-kinase and STAT signaling through endosomal signaling. Future investigations targeting this key question are urgently needed.
ROLE OF SRC IN EGFR ENDOCYTOSIS
In addition to its critical involvement in EGFR-mediated oncogenesis[85], Src plays a role in EGFR endocytosis. Active Src has been shown to phosphorylate clathrin heavy chain[186], and overexpression of Src was shown to accelerate clathrin-mediated internalization of EGFR without increasing EGFR degradation in fibroblasts. Interestingly, Src co-overexpression with EGFR in human mammary epithelial cells led to reduction in surface EGFR levels without a decrease in total EGFR levels, together with EGFR hyperactivation, suggesting that overexpressed Src promotes the traffic of surface EGFR into a non-degradative and likely signaling endosomal compartment[187]. Src-mediated tyrosine phosphorylation has been shown to be required for dynamin function in ligand-induced EGFR internalization[188]. Src is also critical for proper ubiquitination and degradation of EGFR; Src activity antagonizes the function of Cbl by mediating its phosphorylation and degradation[189]. In the presence of a Src inhibitor, EGF-induced phosphorylation of Cbl and ubiquitination of EGFR were blocked[190]. Similarly, mouse embryonic fibroblasts with deletion of Src-negative regulator C-terminal Src kinase results in hyperactive Src; suppression of Src family kinases in these cells resulted in delayed EGFR degradation and prolonged EGF-induced activation of Erk1/2[191]. Src-mediated phosphorylation was also shown to antagonize the function ALG-2 interacting protein X (Alix) in receptor tyrosine kinase internalization[192]. A study using Src-GFP fusion protein showed that upon EGF stimulation, Src traffics into endosomal compartments with activated EGFR, and that Src expression and kinase activity prolong the EGFR activation[193]. Src overexpression also induced activation of EGFR and of EGFR-mediated downstream signaling targets Erk and Shc[193]. At a biological level, Src overexpression was found to promote the ability of EGFR overexpression to transform rodent fibroblasts[194], and promote a transformed phenotype in three-dimensional cultures of human mammary epithelial cells[81]. Therefore, constitutive interaction between mutant EGFR and Src[84,85,118] suggests that, in addition to activating EGFR signaling, the hyperactivity of Src in NSCLC may provide a mechanism for delayed ligand-induced association of Cbl with EGFR, and reduced EGFR ubiquitination and/or downregulation[116,117,143,146] (Figure 2). In this scenario, the preferential trafficking of mutant EGFRs to the endocytic recycling compartment[118] may be due, in part, to their constitutive interaction with Src. Future studies are needed to establish if this mechanism can indeed explain the altered trafficking and signaling of mutant EGFRs.
CONCLUSION
Much insight has been gained on naturally occurring NSCLC-associated mutant EGFRs, thanks in large part to studies instigated by their clinical relevance which resulted in targeted therapies with TKIs. However, acquired resistance and other drug interfering mechanisms limit the efficacy of EGFR-directed therapies in NSCLC patients with EGFR mutations. Mutant EGFR biology remains poorly understood, and as yet their biologically-linked and essential traits of signaling and endocytic traffic have not been integrated for mutant EGFRs. The current review summarizes our current understanding of mutant EGFR signaling and traffic and areas where we lack a clear picture, and points to a need for further understanding and integration. It is clear that mutant EGFRs, in addition to attaining constitutive activity, exhibit deregulated endocytic traffic that appears to promote the ability of mutant receptors to signal into oncogenic pathways. Increased understanding of mechanisms that underlie defects in mutant EGFR endocytic traffic could help define novel approaches to refine EGFR-directed therapies by intercepting at key endocytic traffic nodes.
Footnotes
Supported by the NIH grant to Band H, No. CA99163, CA87986, CA105489 and CA116552; a Department of Defense grant to Band H, No. W81WH-11-1-0167; the NIH grant to Band V, No. CA96844 and CA144027; and Department of Defense grant to Band V, No. W81XWH-07-1-0351 and W81XWH-11-1-0171; the Nebraska Department of Health and Human Services LB-506 grant to Band H, No. 2014-01; and the NCI Core Support Grant to the UNMC Buffett Cancer Center; Bielecki TA was a predoctoral trainee under the NCI Institutional Cancer Biology Training Grant, No. CA009476
P- Reviewer: De Petris L, Garfield D, Hida T, Rosell R, Sugawara I S- Editor: Ji FF L- Editor: A E- Editor: Lu YJ
References
- 1.Sebastian S, Settleman J, Reshkin SJ, Azzariti A, Bellizzi A, Paradiso A. The complexity of targeting EGFR signalling in cancer: from expression to turnover. Biochim Biophys Acta. 2006;1766:120–139. doi: 10.1016/j.bbcan.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 2.Sorkin A, Goh LK. Endocytosis and intracellular trafficking of ErbBs. Exp Cell Res. 2009;315:683–696. doi: 10.1016/j.yexcr.2008.07.029. [DOI] [PubMed] [Google Scholar]
- 3.Gschwind A, Fischer OM, Ullrich A. The discovery of receptor tyrosine kinases: targets for cancer therapy. Nat Rev Cancer. 2004;4:361–370. doi: 10.1038/nrc1360. [DOI] [PubMed] [Google Scholar]
- 4.Avraham R, Yarden Y. Feedback regulation of EGFR signalling: decision making by early and delayed loops. Nat Rev Mol Cell Biol. 2011;12:104–117. doi: 10.1038/nrm3048. [DOI] [PubMed] [Google Scholar]
- 5.Wiley HS, Burke PM. Regulation of receptor tyrosine kinase signaling by endocytic trafficking. Traffic. 2001;2:12–18. doi: 10.1034/j.1600-0854.2001.020103.x. [DOI] [PubMed] [Google Scholar]
- 6.Threadgill DW, Dlugosz AA, Hansen LA, Tennenbaum T, Lichti U, Yee D, LaMantia C, Mourton T, Herrup K, Harris RC. Targeted disruption of mouse EGF receptor: effect of genetic background on mutant phenotype. Science. 1995;269:230–234. doi: 10.1126/science.7618084. [DOI] [PubMed] [Google Scholar]
- 7.Miettinen PJ, Berger JE, Meneses J, Phung Y, Pedersen RA, Werb Z, Derynck R. Epithelial immaturity and multiorgan failure in mice lacking epidermal growth factor receptor. Nature. 1995;376:337–341. doi: 10.1038/376337a0. [DOI] [PubMed] [Google Scholar]
- 8.Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol. 2001;2:127–137. doi: 10.1038/35052073. [DOI] [PubMed] [Google Scholar]
- 9.Herbst RS. Review of epidermal growth factor receptor biology. Int J Radiat Oncol Biol Phys. 2004;59:21–26. doi: 10.1016/j.ijrobp.2003.11.041. [DOI] [PubMed] [Google Scholar]
- 10.Wong RW. Transgenic and knock-out mice for deciphering the roles of EGFR ligands. Cell Mol Life Sci. 2003;60:113–118. doi: 10.1007/s000180300007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Di Marco E, Pierce JH, Fleming TP, Kraus MH, Molloy CJ, Aaronson SA, Di Fiore PP. Autocrine interaction between TGF alpha and the EGF-receptor: quantitative requirements for induction of the malignant phenotype. Oncogene. 1989;4:831–838. [PubMed] [Google Scholar]
- 12.Navas C, Hernández-Porras I, Schuhmacher AJ, Sibilia M, Guerra C, Barbacid M. EGF receptor signaling is essential for k-ras oncogene-driven pancreatic ductal adenocarcinoma. Cancer Cell. 2012;22:318–330. doi: 10.1016/j.ccr.2012.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schmidt-Ullrich RK, Contessa JN, Lammering G, Amorino G, Lin PS. ERBB receptor tyrosine kinases and cellular radiation responses. Oncogene. 2003;22:5855–5865. doi: 10.1038/sj.onc.1206698. [DOI] [PubMed] [Google Scholar]
- 14.Sturla LM, Amorino G, Alexander MS, Mikkelsen RB, Valerie K, Schmidt-Ullrichr RK. Requirement of Tyr-992 and Tyr-1173 in phosphorylation of the epidermal growth factor receptor by ionizing radiation and modulation by SHP2. J Biol Chem. 2005;280:14597–14604. doi: 10.1074/jbc.M413287200. [DOI] [PubMed] [Google Scholar]
- 15.Nicholson RI, Gee JM, Harper ME. EGFR and cancer prognosis. Eur J Cancer. 2001;37 Suppl 4:S9–15. doi: 10.1016/s0959-8049(01)00231-3. [DOI] [PubMed] [Google Scholar]
- 16.Fernandes H, Cohen S, Bishayee S. Glycosylation-induced conformational modification positively regulates receptor-receptor association: a study with an aberrant epidermal growth factor receptor (EGFRvIII/DeltaEGFR) expressed in cancer cells. J Biol Chem. 2001;276:5375–5383. doi: 10.1074/jbc.M005599200. [DOI] [PubMed] [Google Scholar]
- 17.Ohka F, Natsume A, Wakabayashi T. Current trends in targeted therapies for glioblastoma multiforme. Neurol Res Int. 2012;2012:878425. doi: 10.1155/2012/878425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ekstrand AJ, James CD, Cavenee WK, Seliger B, Pettersson RF, Collins VP. Genes for epidermal growth factor receptor, transforming growth factor alpha, and epidermal growth factor and their expression in human gliomas in vivo. Cancer Res. 1991;51:2164–2172. [PubMed] [Google Scholar]
- 19.Aldape KD, Ballman K, Furth A, Buckner JC, Giannini C, Burger PC, Scheithauer BW, Jenkins RB, James CD. Immunohistochemical detection of EGFRvIII in high malignancy grade astrocytomas and evaluation of prognostic significance. J Neuropathol Exp Neurol. 2004;63:700–707. doi: 10.1093/jnen/63.7.700. [DOI] [PubMed] [Google Scholar]
- 20.Dunn GP, Rinne ML, Wykosky J, Genovese G, Quayle SN, Dunn IF, Agarwalla PK, Chheda MG, Campos B, Wang A, et al. Emerging insights into the molecular and cellular basis of glioblastoma. Genes Dev. 2012;26:756–784. doi: 10.1101/gad.187922.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Paez JG, Jänne PA, Lee JC, Tracy S, Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–1500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 22.Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat SM, Supko JG, Haluska FG, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 23.Sordella R, Bell DW, Haber DA, Settleman J. Gefitinib-sensitizing EGFR mutations in lung cancer activate anti-apoptotic pathways. Science. 2004;305:1163–1167. doi: 10.1126/science.1101637. [DOI] [PubMed] [Google Scholar]
- 24.Johnson JR, Cohen M, Sridhara R, Chen YF, Williams GM, Duan J, Gobburu J, Booth B, Benson K, Leighton J, et al. Approval summary for erlotinib for treatment of patients with locally advanced or metastatic non-small cell lung cancer after failure of at least one prior chemotherapy regimen. Clin Cancer Res. 2005;11:6414–6421. doi: 10.1158/1078-0432.CCR-05-0790. [DOI] [PubMed] [Google Scholar]
- 25.Johnson BE, Jänne PA. Epidermal growth factor receptor mutations in patients with non-small cell lung cancer. Cancer Res. 2005;65:7525–7529. doi: 10.1158/0008-5472.CAN-05-1257. [DOI] [PubMed] [Google Scholar]
- 26.Balak MN, Gong Y, Riely GJ, Somwar R, Li AR, Zakowski MF, Chiang A, Yang G, Ouerfelli O, Kris MG, et al. Novel D761Y and common secondary T790M mutations in epidermal growth factor receptor-mutant lung adenocarcinomas with acquired resistance to kinase inhibitors. Clin Cancer Res. 2006;12:6494–6501. doi: 10.1158/1078-0432.CCR-06-1570. [DOI] [PubMed] [Google Scholar]
- 27.Dearden S, Stevens J, Wu YL, Blowers D. Mutation incidence and coincidence in non small-cell lung cancer: meta-analyses by ethnicity and histology (mutMap) Ann Oncol. 2013;24:2371–2376. doi: 10.1093/annonc/mdt205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yun CH, Boggon TJ, Li Y, Woo MS, Greulich H, Meyerson M, Eck MJ. Structures of lung cancer-derived EGFR mutants and inhibitor complexes: mechanism of activation and insights into differential inhibitor sensitivity. Cancer Cell. 2007;11:217–227. doi: 10.1016/j.ccr.2006.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Okabe T, Okamoto I, Tamura K, Terashima M, Yoshida T, Satoh T, Takada M, Fukuoka M, Nakagawa K. Differential constitutive activation of the epidermal growth factor receptor in non-small cell lung cancer cells bearing EGFR gene mutation and amplification. Cancer Res. 2007;67:2046–2053. doi: 10.1158/0008-5472.CAN-06-3339. [DOI] [PubMed] [Google Scholar]
- 30.Shigematsu H, Takahashi T, Nomura M, Majmudar K, Suzuki M, Lee H, Wistuba II, Fong KM, Toyooka S, Shimizu N, et al. Somatic mutations of the HER2 kinase domain in lung adenocarcinomas. Cancer Res. 2005;65:1642–1646. doi: 10.1158/0008-5472.CAN-04-4235. [DOI] [PubMed] [Google Scholar]
- 31.Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Carey KD, Garton AJ, Romero MS, Kahler J, Thomson S, Ross S, Park F, Haley JD, Gibson N, Sliwkowski MX. Kinetic analysis of epidermal growth factor receptor somatic mutant proteins shows increased sensitivity to the epidermal growth factor receptor tyrosine kinase inhibitor, erlotinib. Cancer Res. 2006;66:8163–8171. doi: 10.1158/0008-5472.CAN-06-0453. [DOI] [PubMed] [Google Scholar]
- 33.Jackman DM, Yeap BY, Sequist LV, Lindeman N, Holmes AJ, Joshi VA, Bell DW, Huberman MS, Halmos B, Rabin MS, et al. Exon 19 deletion mutations of epidermal growth factor receptor are associated with prolonged survival in non-small cell lung cancer patients treated with gefitinib or erlotinib. Clin Cancer Res. 2006;12:3908–3914. doi: 10.1158/1078-0432.CCR-06-0462. [DOI] [PubMed] [Google Scholar]
- 34.Cappuzzo F, Hirsch FR, Rossi E, Bartolini S, Ceresoli GL, Bemis L, Haney J, Witta S, Danenberg K, Domenichini I, et al. Epidermal growth factor receptor gene and protein and gefitinib sensitivity in non-small-cell lung cancer. J Natl Cancer Inst. 2005;97:643–655. doi: 10.1093/jnci/dji112. [DOI] [PubMed] [Google Scholar]
- 35.Nakamura H, Kawasaki N, Taguchi M, Kabasawa K. Survival impact of epidermal growth factor receptor overexpression in patients with non-small cell lung cancer: a meta-analysis. Thorax. 2006;61:140–145. doi: 10.1136/thx.2005.042275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lynch TJ, Adjei AA, Bunn PA, Eisen TG, Engelman J, Goss GD, Haber DA, Heymach JV, Jänne PA, Johnson BE, et al. Summary statement: novel agents in the treatment of lung cancer: advances in epidermal growth factor receptor-targeted agents. Clin Cancer Res. 2006;12:4365s–4371s. doi: 10.1158/1078-0432.CCR-06-1005. [DOI] [PubMed] [Google Scholar]
- 37.Hirata A, Hosoi F, Miyagawa M, Ueda S, Naito S, Fujii T, Kuwano M, Ono M. HER2 overexpression increases sensitivity to gefitinib, an epidermal growth factor receptor tyrosine kinase inhibitor, through inhibition of HER2/HER3 heterodimer formation in lung cancer cells. Cancer Res. 2005;65:4253–4260. doi: 10.1158/0008-5472.CAN-04-2748. [DOI] [PubMed] [Google Scholar]
- 38.Erjala K, Sundvall M, Junttila TT, Zhang N, Savisalo M, Mali P, Kulmala J, Pulkkinen J, Grenman R, Elenius K. Signaling via ErbB2 and ErbB3 associates with resistance and epidermal growth factor receptor (EGFR) amplification with sensitivity to EGFR inhibitor gefitinib in head and neck squamous cell carcinoma cells. Clin Cancer Res. 2006;12:4103–4111. doi: 10.1158/1078-0432.CCR-05-2404. [DOI] [PubMed] [Google Scholar]
- 39.Cusatis G, Gregorc V, Li J, Spreafico A, Ingersoll RG, Verweij J, Ludovini V, Villa E, Hidalgo M, Sparreboom A, et al. Pharmacogenetics of ABCG2 and adverse reactions to gefitinib. J Natl Cancer Inst. 2006;98:1739–1742. doi: 10.1093/jnci/djj469. [DOI] [PubMed] [Google Scholar]
- 40.Riely GJ, Pao W, Pham D, Li AR, Rizvi N, Venkatraman ES, Zakowski MF, Kris MG, Ladanyi M, Miller VA. Clinical course of patients with non-small cell lung cancer and epidermal growth factor receptor exon 19 and exon 21 mutations treated with gefitinib or erlotinib. Clin Cancer Res. 2006;12:839–844. doi: 10.1158/1078-0432.CCR-05-1846. [DOI] [PubMed] [Google Scholar]
- 41.Pao W, Miller VA, Politi KA, Riely GJ, Somwar R, Zakowski MF, Kris MG, Varmus H. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005;2:e73. doi: 10.1371/journal.pmed.0020073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kobayashi S, Boggon TJ, Dayaram T, Jänne PA, Kocher O, Meyerson M, Johnson BE, Eck MJ, Tenen DG, Halmos B. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005;352:786–792. doi: 10.1056/NEJMoa044238. [DOI] [PubMed] [Google Scholar]
- 43.Yun CH, Mengwasser KE, Toms AV, Woo MS, Greulich H, Wong KK, Meyerson M, Eck MJ. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc Natl Acad Sci USA. 2008;105:2070–2075. doi: 10.1073/pnas.0709662105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen J, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316:1039–1043. doi: 10.1126/science.1141478. [DOI] [PubMed] [Google Scholar]
- 45.Tao Y, Pinzi V, Bourhis J, Deutsch E. Mechanisms of disease: signaling of the insulin-like growth factor 1 receptor pathway--therapeutic perspectives in cancer. Nat Clin Pract Oncol. 2007;4:591–602. doi: 10.1038/ncponc0934. [DOI] [PubMed] [Google Scholar]
- 46.Deng J, Shimamura T, Perera S, Carlson NE, Cai D, Shapiro GI, Wong KK, Letai A. Proapoptotic BH3-only BCL-2 family protein BIM connects death signaling from epidermal growth factor receptor inhibition to the mitochondrion. Cancer Res. 2007;67:11867–11875. doi: 10.1158/0008-5472.CAN-07-1961. [DOI] [PubMed] [Google Scholar]
- 47.Yauch RL, Januario T, Eberhard DA, Cavet G, Zhu W, Fu L, Pham TQ, Soriano R, Stinson J, Seshagiri S, et al. Epithelial versus mesenchymal phenotype determines in vitro sensitivity and predicts clinical activity of erlotinib in lung cancer patients. Clin Cancer Res. 2005;11:8686–8698. doi: 10.1158/1078-0432.CCR-05-1492. [DOI] [PubMed] [Google Scholar]
- 48.Thomson S, Buck E, Petti F, Griffin G, Brown E, Ramnarine N, Iwata KK, Gibson N, Haley JD. Epithelial to mesenchymal transition is a determinant of sensitivity of non-small-cell lung carcinoma cell lines and xenografts to epidermal growth factor receptor inhibition. Cancer Res. 2005;65:9455–9462. doi: 10.1158/0008-5472.CAN-05-1058. [DOI] [PubMed] [Google Scholar]
- 49.Lu Y, Li X, Liang K, Luwor R, Siddik ZH, Mills GB, Mendelsohn J, Fan Z. Epidermal growth factor receptor (EGFR) ubiquitination as a mechanism of acquired resistance escaping treatment by the anti-EGFR monoclonal antibody cetuximab. Cancer Res. 2007;67:8240–8247. doi: 10.1158/0008-5472.CAN-07-0589. [DOI] [PubMed] [Google Scholar]
- 50.Lièvre A, Bachet JB, Le Corre D, Boige V, Landi B, Emile JF, Côté JF, Tomasic G, Penna C, Ducreux M, et al. KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res. 2006;66:3992–3995. doi: 10.1158/0008-5472.CAN-06-0191. [DOI] [PubMed] [Google Scholar]
- 51.Sheridan C. EGFR inhibitors embrace KRAS. Nat Biotechnol. 2008;26:839–840. doi: 10.1038/nbt0808-839. [DOI] [PubMed] [Google Scholar]
- 52.Pao W, Wang TY, Riely GJ, Miller VA, Pan Q, Ladanyi M, Zakowski MF, Heelan RT, Kris MG, Varmus HE. KRAS mutations and primary resistance of lung adenocarcinomas to gefitinib or erlotinib. PLoS Med. 2005;2:e17. doi: 10.1371/journal.pmed.0020017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Costa DB, Nguyen KS, Cho BC, Sequist LV, Jackman DM, Riely GJ, Yeap BY, Halmos B, Kim JH, Jänne PA, et al. Effects of erlotinib in EGFR mutated non-small cell lung cancers with resistance to gefitinib. Clin Cancer Res. 2008;14:7060–7067. doi: 10.1158/1078-0432.CCR-08-1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mukohara T, Engelman JA, Hanna NH, Yeap BY, Kobayashi S, Lindeman N, Halmos B, Pearlberg J, Tsuchihashi Z, Cantley LC, et al. Differential effects of gefitinib and cetuximab on non-small-cell lung cancers bearing epidermal growth factor receptor mutations. J Natl Cancer Inst. 2005;97:1185–1194. doi: 10.1093/jnci/dji238. [DOI] [PubMed] [Google Scholar]
- 55.Doody JF, Wang Y, Patel SN, Joynes C, Lee SP, Gerlak J, Rolser RL, Li Y, Steiner P, Bassi R, et al. Inhibitory activity of cetuximab on epidermal growth factor receptor mutations in non small cell lung cancers. Mol Cancer Ther. 2007;6:2642–2651. doi: 10.1158/1535-7163.MCT-06-0506. [DOI] [PubMed] [Google Scholar]
- 56.Kim HP, Han SW, Kim SH, Im SA, Oh DY, Bang YJ, Kim TY. Combined lapatinib and cetuximab enhance cytotoxicity against gefitinib-resistant lung cancer cells. Mol Cancer Ther. 2008;7:607–615. doi: 10.1158/1535-7163.MCT-07-2068. [DOI] [PubMed] [Google Scholar]
- 57.Arao T, Fukumoto H, Takeda M, Tamura T, Saijo N, Nishio K. Small in-frame deletion in the epidermal growth factor receptor as a target for ZD6474. Cancer Res. 2004;64:9101–9104. doi: 10.1158/0008-5472.CAN-04-2360. [DOI] [PubMed] [Google Scholar]
- 58.Engelman JA, Zejnullahu K, Gale CM, Lifshits E, Gonzales AJ, Shimamura T, Zhao F, Vincent PW, Naumov GN, Bradner JE, et al. PF00299804, an irreversible pan-ERBB inhibitor, is effective in lung cancer models with EGFR and ERBB2 mutations that are resistant to gefitinib. Cancer Res. 2007;67:11924–11932. doi: 10.1158/0008-5472.CAN-07-1885. [DOI] [PubMed] [Google Scholar]
- 59.Zhang X, Gureasko J, Shen K, Cole PA, Kuriyan J. An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell. 2006;125:1137–1149. doi: 10.1016/j.cell.2006.05.013. [DOI] [PubMed] [Google Scholar]
- 60.Greulich H, Chen TH, Feng W, Jänne PA, Alvarez JV, Zappaterra M, Bulmer SE, Frank DA, Hahn WC, Sellers WR, et al. Oncogenic transformation by inhibitor-sensitive and -resistant EGFR mutants. PLoS Med. 2005;2:e313. doi: 10.1371/journal.pmed.0020313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sharma SV, Bell DW, Settleman J, Haber DA. Epidermal growth factor receptor mutations in lung cancer. Nat Rev Cancer. 2007;7:169–181. doi: 10.1038/nrc2088. [DOI] [PubMed] [Google Scholar]
- 62.Politi K, Zakowski MF, Fan PD, Schonfeld EA, Pao W, Varmus HE. Lung adenocarcinomas induced in mice by mutant EGF receptors found in human lung cancers respond to a tyrosine kinase inhibitor or to down-regulation of the receptors. Genes Dev. 2006;20:1496–1510. doi: 10.1101/gad.1417406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Luo J, Solimini NL, Elledge SJ. Principles of cancer therapy: oncogene and non-oncogene addiction. Cell. 2009;136:823–837. doi: 10.1016/j.cell.2009.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhang J, Kalyankrishna S, Wislez M, Thilaganathan N, Saigal B, Wei W, Ma L, Wistuba II, Johnson FM, Kurie JM. SRC-family kinases are activated in non-small cell lung cancer and promote the survival of epidermal growth factor receptor-dependent cell lines. Am J Pathol. 2007;170:366–376. doi: 10.2353/ajpath.2007.060706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Song L, Morris M, Bagui T, Lee FY, Jove R, Haura EB. Dasatinib (BMS-354825) selectively induces apoptosis in lung cancer cells dependent on epidermal growth factor receptor signaling for survival. Cancer Res. 2006;66:5542–5548. doi: 10.1158/0008-5472.CAN-05-4620. [DOI] [PubMed] [Google Scholar]
- 66.Janmaat ML, Rodriguez JA, Gallegos-Ruiz M, Kruyt FA, Giaccone G. Enhanced cytotoxicity induced by gefitinib and specific inhibitors of the Ras or phosphatidyl inositol-3 kinase pathways in non-small cell lung cancer cells. Int J Cancer. 2006;118:209–214. doi: 10.1002/ijc.21290. [DOI] [PubMed] [Google Scholar]
- 67.Haura EB, Zheng Z, Song L, Cantor A, Bepler G. Activated epidermal growth factor receptor-Stat-3 signaling promotes tumor survival in vivo in non-small cell lung cancer. Clin Cancer Res. 2005;11:8288–8294. doi: 10.1158/1078-0432.CCR-05-0827. [DOI] [PubMed] [Google Scholar]
- 68.Ono M, Hirata A, Kometani T, Miyagawa M, Ueda S, Kinoshita H, Fujii T, Kuwano M. Sensitivity to gefitinib (Iressa, ZD1839) in non-small cell lung cancer cell lines correlates with dependence on the epidermal growth factor (EGF) receptor/extracellular signal-regulated kinase 1/2 and EGF receptor/Akt pathway for proliferation. Mol Cancer Ther. 2004;3:465–472. [PubMed] [Google Scholar]
- 69.Han SW, Hwang PG, Chung DH, Kim DW, Im SA, Kim YT, Kim TY, Heo DS, Bang YJ, Kim NK. Epidermal growth factor receptor (EGFR) downstream molecules as response predictive markers for gefitinib (Iressa, ZD1839) in chemotherapy-resistant non-small cell lung cancer. Int J Cancer. 2005;113:109–115. doi: 10.1002/ijc.20550. [DOI] [PubMed] [Google Scholar]
- 70.Akca H, Tani M, Hishida T, Matsumoto S, Yokota J. Activation of the AKT and STAT3 pathways and prolonged survival by a mutant EGFR in human lung cancer cells. Lung Cancer. 2006;54:25–33. doi: 10.1016/j.lungcan.2006.06.007. [DOI] [PubMed] [Google Scholar]
- 71.Engelman JA, Mukohara T, Zejnullahu K, Lifshits E, Borrás AM, Gale CM, Naumov GN, Yeap BY, Jarrell E, Sun J, et al. Allelic dilution obscures detection of a biologically significant resistance mutation in EGFR-amplified lung cancer. J Clin Invest. 2006;116:2695–2706. doi: 10.1172/JCI28656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Engelman JA, Jänne PA, Mermel C, Pearlberg J, Mukohara T, Fleet C, Cichowski K, Johnson BE, Cantley LC. ErbB-3 mediates phosphoinositide 3-kinase activity in gefitinib-sensitive non-small cell lung cancer cell lines. Proc Natl Acad Sci USA. 2005;102:3788–3793. doi: 10.1073/pnas.0409773102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jiang J, Greulich H, Jänne PA, Sellers WR, Meyerson M, Griffin JD. Epidermal growth factor-independent transformation of Ba/F3 cells with cancer-derived epidermal growth factor receptor mutants induces gefitinib-sensitive cell cycle progression. Cancer Res. 2005;65:8968–8974. doi: 10.1158/0008-5472.CAN-05-1829. [DOI] [PubMed] [Google Scholar]
- 74.Kobayashi S, Shimamura T, Monti S, Steidl U, Hetherington CJ, Lowell AM, Golub T, Meyerson M, Tenen DG, Shapiro GI, et al. Transcriptional profiling identifies cyclin D1 as a critical downstream effector of mutant epidermal growth factor receptor signaling. Cancer Res. 2006;66:11389–11398. doi: 10.1158/0008-5472.CAN-06-2318. [DOI] [PubMed] [Google Scholar]
- 75.Conde E, Angulo B, Tang M, Morente M, Torres-Lanzas J, Lopez-Encuentra A, Lopez-Rios F, Sanchez-Cespedes M. Molecular context of the EGFR mutations: evidence for the activation of mTOR/S6K signaling. Clin Cancer Res. 2006;12:710–717. doi: 10.1158/1078-0432.CCR-05-1362. [DOI] [PubMed] [Google Scholar]
- 76.Maegawa M, Arao T, Yokote H, Matsumoto K, Kudo K, Tanaka K, Kaneda H, Fujita Y, Ito F, Nishio K. EGFR mutation up-regulates EGR1 expression through the ERK pathway. Anticancer Res. 2009;29:1111–1117. [PubMed] [Google Scholar]
- 77.Di Fiore PP, Pierce JH, Fleming TP, Hazan R, Ullrich A, King CR, Schlessinger J, Aaronson SA. Overexpression of the human EGF receptor confers an EGF-dependent transformed phenotype to NIH 3T3 cells. Cell. 1987;51:1063–1070. doi: 10.1016/0092-8674(87)90592-7. [DOI] [PubMed] [Google Scholar]
- 78.Bromann PA, Korkaya H, Courtneidge SA. The interplay between Src family kinases and receptor tyrosine kinases. Oncogene. 2004;23:7957–7968. doi: 10.1038/sj.onc.1208079. [DOI] [PubMed] [Google Scholar]
- 79.Bjorge JD, Jakymiw A, Fujita DJ. Selected glimpses into the activation and function of Src kinase. Oncogene. 2000;19:5620–5635. doi: 10.1038/sj.onc.1203923. [DOI] [PubMed] [Google Scholar]
- 80.Maa MC, Leu TH, McCarley DJ, Schatzman RC, Parsons SJ. Potentiation of epidermal growth factor receptor-mediated oncogenesis by c-Src: implications for the etiology of multiple human cancers. Proc Natl Acad Sci USA. 1995;92:6981–6985. doi: 10.1073/pnas.92.15.6981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Dimri M, Naramura M, Duan L, Chen J, Ortega-Cava C, Chen G, Goswami R, Fernandes N, Gao Q, Dimri GP, et al. Modeling breast cancer-associated c-Src and EGFR overexpression in human MECs: c-Src and EGFR cooperatively promote aberrant three-dimensional acinar structure and invasive behavior. Cancer Res. 2007;67:4164–4172. doi: 10.1158/0008-5472.CAN-06-2580. [DOI] [PubMed] [Google Scholar]
- 82.Silva CM. Role of STATs as downstream signal transducers in Src family kinase-mediated tumorigenesis. Oncogene. 2004;23:8017–8023. doi: 10.1038/sj.onc.1208159. [DOI] [PubMed] [Google Scholar]
- 83.Masaki T, Igarashi K, Tokuda M, Yukimasa S, Han F, Jin YJ, Li JQ, Yoneyama H, Uchida N, Fujita J, et al. pp60c-src activation in lung adenocarcinoma. Eur J Cancer. 2003;39:1447–1455. doi: 10.1016/s0959-8049(03)00276-4. [DOI] [PubMed] [Google Scholar]
- 84.Yang S, Park K, Turkson J, Arteaga CL. Ligand-independent phosphorylation of Y869 (Y845) links mutant EGFR signaling to stat-mediated gene expression. Exp Cell Res. 2008;314:413–419. doi: 10.1016/j.yexcr.2007.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Chung BM, Dimri M, George M, Reddi AL, Chen G, Band V, Band H. The role of cooperativity with Src in oncogenic transformation mediated by non-small cell lung cancer-associated EGF receptor mutants. Oncogene. 2009;28:1821–1832. doi: 10.1038/onc.2009.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Fu YN, Yeh CL, Cheng HH, Yang CH, Tsai SF, Huang SF, Chen YR. EGFR mutants found in non-small cell lung cancer show different levels of sensitivity to suppression of Src: implications in targeting therapy. Oncogene. 2008;27:957–965. doi: 10.1038/sj.onc.1210684. [DOI] [PubMed] [Google Scholar]
- 87.Marmor MD, Yarden Y. Role of protein ubiquitylation in regulating endocytosis of receptor tyrosine kinases. Oncogene. 2004;23:2057–2070. doi: 10.1038/sj.onc.1207390. [DOI] [PubMed] [Google Scholar]
- 88.Conner SD, Schmid SL. Regulated portals of entry into the cell. Nature. 2003;422:37–44. doi: 10.1038/nature01451. [DOI] [PubMed] [Google Scholar]
- 89.Wiley HS. Trafficking of the ErbB receptors and its influence on signaling. Exp Cell Res. 2003;284:78–88. doi: 10.1016/s0014-4827(03)00002-8. [DOI] [PubMed] [Google Scholar]
- 90.Katzmann DJ, Odorizzi G, Emr SD. Receptor downregulation and multivesicular-body sorting. Nat Rev Mol Cell Biol. 2002;3:893–905. doi: 10.1038/nrm973. [DOI] [PubMed] [Google Scholar]
- 91.Raiborg C, Stenmark H. The ESCRT machinery in endosomal sorting of ubiquitylated membrane proteins. Nature. 2009;458:445–452. doi: 10.1038/nature07961. [DOI] [PubMed] [Google Scholar]
- 92.Mizuno E, Iura T, Mukai A, Yoshimori T, Kitamura N, Komada M. Regulation of epidermal growth factor receptor down-regulation by UBPY-mediated deubiquitination at endosomes. Mol Biol Cell. 2005;16:5163–5174. doi: 10.1091/mbc.E05-06-0560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Maxfield FR, McGraw TE. Endocytic recycling. Nat Rev Mol Cell Biol. 2004;5:121–132. doi: 10.1038/nrm1315. [DOI] [PubMed] [Google Scholar]
- 94.Grant BD, Donaldson JG. Pathways and mechanisms of endocytic recycling. Nat Rev Mol Cell Biol. 2009;10:597–608. doi: 10.1038/nrm2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.George M, Ying G, Rainey MA, Solomon A, Parikh PT, Gao Q, Band V, Band H. Shared as well as distinct roles of EHD proteins revealed by biochemical and functional comparisons in mammalian cells and C. elegans. BMC Cell Biol. 2007;8:3. doi: 10.1186/1471-2121-8-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Naslavsky N, Caplan S. EHD proteins: key conductors of endocytic transport. Trends Cell Biol. 2011;21:122–131. doi: 10.1016/j.tcb.2010.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Roepstorff K, Grandal MV, Henriksen L, Knudsen SL, Lerdrup M, Grøvdal L, Willumsen BM, van Deurs B. Differential effects of EGFR ligands on endocytic sorting of the receptor. Traffic. 2009;10:1115–1127. doi: 10.1111/j.1600-0854.2009.00943.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sigismund S, Woelk T, Puri C, Maspero E, Tacchetti C, Transidico P, Di Fiore PP, Polo S. Clathrin-independent endocytosis of ubiquitinated cargos. Proc Natl Acad Sci USA. 2005;102:2760–2765. doi: 10.1073/pnas.0409817102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Sigismund S, Argenzio E, Tosoni D, Cavallaro E, Polo S, Di Fiore PP. Clathrin-mediated internalization is essential for sustained EGFR signaling but dispensable for degradation. Dev Cell. 2008;15:209–219. doi: 10.1016/j.devcel.2008.06.012. [DOI] [PubMed] [Google Scholar]
- 100.Offterdinger M, Bastiaens PI. Prolonged EGFR signaling by ERBB2-mediated sequestration at the plasma membrane. Traffic. 2008;9:147–155. doi: 10.1111/j.1600-0854.2007.00665.x. [DOI] [PubMed] [Google Scholar]
- 101.Hendriks BS, Wiley HS, Lauffenburger D. HER2-mediated effects on EGFR endosomal sorting: analysis of biophysical mechanisms. Biophys J. 2003;85:2732–2745. doi: 10.1016/s0006-3495(03)74696-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Mohapatra B, Ahmad G, Nadeau S, Zutshi N, An W, Scheffe S, Dong L, Feng D, Goetz B, Arya P, et al. Protein tyrosine kinase regulation by ubiquitination: critical roles of Cbl-family ubiquitin ligases. Biochim Biophys Acta. 2013;1833:122–139. doi: 10.1016/j.bbamcr.2012.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.de Melker AA, van der Horst G, Calafat J, Jansen H, Borst J. c-Cbl ubiquitinates the EGF receptor at the plasma membrane and remains receptor associated throughout the endocytic route. J Cell Sci. 2001;114:2167–2178. doi: 10.1242/jcs.114.11.2167. [DOI] [PubMed] [Google Scholar]
- 104.Haglund K, Sigismund S, Polo S, Szymkiewicz I, Di Fiore PP, Dikic I. Multiple monoubiquitination of RTKs is sufficient for their endocytosis and degradation. Nat Cell Biol. 2003;5:461–466. doi: 10.1038/ncb983. [DOI] [PubMed] [Google Scholar]
- 105.Rønning SB, Pedersen NM, Madshus IH, Stang E. CIN85 regulates ubiquitination and degradative endosomal sorting of the EGF receptor. Exp Cell Res. 2011;317:1804–1816. doi: 10.1016/j.yexcr.2011.05.016. [DOI] [PubMed] [Google Scholar]
- 106.Huang F, Sorkin A. Growth factor receptor binding protein 2-mediated recruitment of the RING domain of Cbl to the epidermal growth factor receptor is essential and sufficient to support receptor endocytosis. Mol Biol Cell. 2005;16:1268–1281. doi: 10.1091/mbc.E04-09-0832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Stang E, Blystad FD, Kazazic M, Bertelsen V, Brodahl T, Raiborg C, Stenmark H, Madshus IH. Cbl-dependent ubiquitination is required for progression of EGF receptors into clathrin-coated pits. Mol Biol Cell. 2004;15:3591–3604. doi: 10.1091/mbc.E04-01-0041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Huang F, Khvorova A, Marshall W, Sorkin A. Analysis of clathrin-mediated endocytosis of epidermal growth factor receptor by RNA interference. J Biol Chem. 2004;279:16657–16661. doi: 10.1074/jbc.C400046200. [DOI] [PubMed] [Google Scholar]
- 109.Wang Z, Moran MF. Requirement for the adapter protein GRB2 in EGF receptor endocytosis. Science. 1996;272:1935–1939. doi: 10.1126/science.272.5270.1935. [DOI] [PubMed] [Google Scholar]
- 110.Sigismund S, Algisi V, Nappo G, Conte A, Pascolutti R, Cuomo A, Bonaldi T, Argenzio E, Verhoef LG, Maspero E, et al. Threshold-controlled ubiquitination of the EGFR directs receptor fate. EMBO J. 2013;32:2140–2157. doi: 10.1038/emboj.2013.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Schmidt MH, Husnjak K, Szymkiewicz I, Haglund K, Dikic I. Cbl escapes Cdc42-mediated inhibition by downregulation of the adaptor molecule betaPix. Oncogene. 2006;25:3071–3078. doi: 10.1038/sj.onc.1209329. [DOI] [PubMed] [Google Scholar]
- 112.Kowanetz K, Crosetto N, Haglund K, Schmidt MH, Heldin CH, Dikic I. Suppressors of T-cell receptor signaling Sts-1 and Sts-2 bind to Cbl and inhibit endocytosis of receptor tyrosine kinases. J Biol Chem. 2004;279:32786–32795. doi: 10.1074/jbc.M403759200. [DOI] [PubMed] [Google Scholar]
- 113.Lynch DK, Winata SC, Lyons RJ, Hughes WE, Lehrbach GM, Wasinger V, Corthals G, Cordwell S, Daly RJ. A Cortactin-CD2-associated protein (CD2AP) complex provides a novel link between epidermal growth factor receptor endocytosis and the actin cytoskeleton. J Biol Chem. 2003;278:21805–21813. doi: 10.1074/jbc.M211407200. [DOI] [PubMed] [Google Scholar]
- 114.Stern KA, Visser Smit GD, Place TL, Winistorfer S, Piper RC, Lill NL. Epidermal growth factor receptor fate is controlled by Hrs tyrosine phosphorylation sites that regulate Hrs degradation. Mol Cell Biol. 2007;27:888–898. doi: 10.1128/MCB.02356-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Yang S, Qu S, Perez-Tores M, Sawai A, Rosen N, Solit DB, Arteaga CL. Association with HSP90 inhibits Cbl-mediated down-regulation of mutant epidermal growth factor receptors. Cancer Res. 2006;66:6990–6997. doi: 10.1158/0008-5472.CAN-06-1042. [DOI] [PubMed] [Google Scholar]
- 116.Shtiegman K, Kochupurakkal BS, Zwang Y, Pines G, Starr A, Vexler A, Citri A, Katz M, Lavi S, Ben-Basat Y, et al. Defective ubiquitinylation of EGFR mutants of lung cancer confers prolonged signaling. Oncogene. 2007;26:6968–6978. doi: 10.1038/sj.onc.1210503. [DOI] [PubMed] [Google Scholar]
- 117.Padrón D, Sato M, Shay JW, Gazdar AF, Minna JD, Roth MG. Epidermal growth factor receptors with tyrosine kinase domain mutations exhibit reduced Cbl association, poor ubiquitylation, and down-regulation but are efficiently internalized. Cancer Res. 2007;67:7695–7702. doi: 10.1158/0008-5472.CAN-07-0484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Chung BM, Raja SM, Clubb RJ, Tu C, George M, Band V, Band H. Aberrant trafficking of NSCLC-associated EGFR mutants through the endocytic recycling pathway promotes interaction with Src. BMC Cell Biol. 2009;10:84. doi: 10.1186/1471-2121-10-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Nishimura Y, Bereczky B, Ono M. The EGFR inhibitor gefitinib suppresses ligand-stimulated endocytosis of EGFR via the early/late endocytic pathway in non-small cell lung cancer cell lines. Histochem Cell Biol. 2007;127:541–553. doi: 10.1007/s00418-007-0281-y. [DOI] [PubMed] [Google Scholar]
- 120.Chen YR, Fu YN, Lin CH, Yang ST, Hu SF, Chen YT, Tsai SF, Huang SF. Distinctive activation patterns in constitutively active and gefitinib-sensitive EGFR mutants. Oncogene. 2006;25:1205–1215. doi: 10.1038/sj.onc.1209159. [DOI] [PubMed] [Google Scholar]
- 121.Choi SH, Mendrola JM, Lemmon MA. EGF-independent activation of cell-surface EGF receptors harboring mutations found in gefitinib-sensitive lung cancer. Oncogene. 2007;26:1567–1576. doi: 10.1038/sj.onc.1209957. [DOI] [PubMed] [Google Scholar]
- 122.Huang HS, Nagane M, Klingbeil CK, Lin H, Nishikawa R, Ji XD, Huang CM, Gill GN, Wiley HS, Cavenee WK. The enhanced tumorigenic activity of a mutant epidermal growth factor receptor common in human cancers is mediated by threshold levels of constitutive tyrosine phosphorylation and unattenuated signaling. J Biol Chem. 1997;272:2927–2935. doi: 10.1074/jbc.272.5.2927. [DOI] [PubMed] [Google Scholar]
- 123.Damstrup L, Wandahl Pedersen M, Bastholm L, Elling F, Skovgaard Poulsen H. Epidermal growth factor receptor mutation type III transfected into a small cell lung cancer cell line is predominantly localized at the cell surface and enhances the malignant phenotype. Int J Cancer. 2002;97:7–14. doi: 10.1002/ijc.1572. [DOI] [PubMed] [Google Scholar]
- 124.Wikstrand CJ, McLendon RE, Friedman AH, Bigner DD. Cell surface localization and density of the tumor-associated variant of the epidermal growth factor receptor, EGFRvIII. Cancer Res. 1997;57:4130–4140. [PubMed] [Google Scholar]
- 125.Wang Q, Villeneuve G, Wang Z. Control of epidermal growth factor receptor endocytosis by receptor dimerization, rather than receptor kinase activation. EMBO Rep. 2005;6:942–948. doi: 10.1038/sj.embor.7400491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Schmidt MH, Furnari FB, Cavenee WK, Bögler O. Epidermal growth factor receptor signaling intensity determines intracellular protein interactions, ubiquitination, and internalization. Proc Natl Acad Sci USA. 2003;100:6505–6510. doi: 10.1073/pnas.1031790100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Sorkina T, Huang F, Beguinot L, Sorkin A. Effect of tyrosine kinase inhibitors on clathrin-coated pit recruitment and internalization of epidermal growth factor receptor. J Biol Chem. 2002;277:27433–27441. doi: 10.1074/jbc.M201595200. [DOI] [PubMed] [Google Scholar]
- 128.Wang Y, Pennock S, Chen X, Wang Z. Endosomal signaling of epidermal growth factor receptor stimulates signal transduction pathways leading to cell survival. Mol Cell Biol. 2002;22:7279–7290. doi: 10.1128/MCB.22.20.7279-7290.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Kazazic M, Bertelsen V, Pedersen KW, Vuong TT, Grandal MV, Rødland MS, Traub LM, Stang E, Madshus IH. Epsin 1 is involved in recruitment of ubiquitinated EGF receptors into clathrin-coated pits. Traffic. 2009;10:235–245. doi: 10.1111/j.1600-0854.2008.00858.x. [DOI] [PubMed] [Google Scholar]
- 130.Martinu L, Santiago-Walker A, Qi H, Chou MM. Endocytosis of epidermal growth factor receptor regulated by Grb2-mediated recruitment of the Rab5 GTPase-activating protein RN-tre. J Biol Chem. 2002;277:50996–51002. doi: 10.1074/jbc.M204869200. [DOI] [PubMed] [Google Scholar]
- 131.Sakai K, Arao T, Shimoyama T, Murofushi K, Sekijima M, Kaji N, Tamura T, Saijo N, Nishio K. Dimerization and the signal transduction pathway of a small in-frame deletion in the epidermal growth factor receptor. FASEB J. 2006;20:311–313. doi: 10.1096/fj.05-4034fje. [DOI] [PubMed] [Google Scholar]
- 132.Kwak EL, Sordella R, Bell DW, Godin-Heymann N, Okimoto RA, Brannigan BW, Harris PL, Driscoll DR, Fidias P, Lynch TJ, et al. Irreversible inhibitors of the EGF receptor may circumvent acquired resistance to gefitinib. Proc Natl Acad Sci USA. 2005;102:7665–7670. doi: 10.1073/pnas.0502860102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Huang DH, Su L, Peng XH, Zhang H, Khuri FR, Shin DM, Chen ZG. Quantum dot-based quantification revealed differences in subcellular localization of EGFR and E-cadherin between EGFR-TKI sensitive and insensitive cancer cells. Nanotechnology. 2009;20:225102. doi: 10.1088/0957-4484/20/22/225102. [DOI] [PubMed] [Google Scholar]
- 134.Duan L, Miura Y, Dimri M, Majumder B, Dodge IL, Reddi AL, Ghosh A, Fernandes N, Zhou P, Mullane-Robinson K, et al. Cbl-mediated ubiquitinylation is required for lysosomal sorting of epidermal growth factor receptor but is dispensable for endocytosis. J Biol Chem. 2003;278:28950–28960. doi: 10.1074/jbc.M304474200. [DOI] [PubMed] [Google Scholar]
- 135.Levkowitz G, Waterman H, Zamir E, Kam Z, Oved S, Langdon WY, Beguinot L, Geiger B, Yarden Y. c-Cbl/Sli-1 regulates endocytic sorting and ubiquitination of the epidermal growth factor receptor. Genes Dev. 1998;12:3663–3674. doi: 10.1101/gad.12.23.3663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Lill NL, Douillard P, Awwad RA, Ota S, Lupher ML, Miyake S, Meissner-Lula N, Hsu VW, Band H. The evolutionarily conserved N-terminal region of Cbl is sufficient to enhance down-regulation of the epidermal growth factor receptor. J Biol Chem. 2000;275:367–377. doi: 10.1074/jbc.275.1.367. [DOI] [PubMed] [Google Scholar]
- 137.Duan L, Raja SM, Chen G, Virmani S, Williams SH, Clubb RJ, Mukhopadhyay C, Rainey MA, Ying G, Dimri M, et al. Negative regulation of EGFR-Vav2 signaling axis by Cbl ubiquitin ligase controls EGF receptor-mediated epithelial cell adherens junction dynamics and cell migration. J Biol Chem. 2011;286:620–633. doi: 10.1074/jbc.M110.188086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Umebayashi K, Stenmark H, Yoshimori T. Ubc4/5 and c-Cbl continue to ubiquitinate EGF receptor after internalization to facilitate polyubiquitination and degradation. Mol Biol Cell. 2008;19:3454–3462. doi: 10.1091/mbc.E07-10-0988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Andoniou CE, Thien CB, Langdon WY. Tumour induction by activated abl involves tyrosine phosphorylation of the product of the cbl oncogene. EMBO J. 1994;13:4515–4523. doi: 10.1002/j.1460-2075.1994.tb06773.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Bonita DP, Miyake S, Lupher ML, Langdon WY, Band H. Phosphotyrosine binding domain-dependent upregulation of the platelet-derived growth factor receptor alpha signaling cascade by transforming mutants of Cbl: implications for Cbl’s function and oncogenicity. Mol Cell Biol. 1997;17:4597–4610. doi: 10.1128/mcb.17.8.4597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Dikic I, Schmidt MH. Malfunctions within the Cbl interactome uncouple receptor tyrosine kinases from destructive transport. Eur J Cell Biol. 2007;86:505–512. doi: 10.1016/j.ejcb.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 142.Jékely G, Sung HH, Luque CM, Rørth P. Regulators of endocytosis maintain localized receptor tyrosine kinase signaling in guided migration. Dev Cell. 2005;9:197–207. doi: 10.1016/j.devcel.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 143.Hosaka T, Inoue F, Ando K, Ishida H, Kusumoto S, Sugiyama T, Shirai T, Okuda K, Hirose T, Ohnishi T, et al. Mutant epidermal growth factor receptor undergoes less protein degradation due to diminished binding to c-Cbl. Anticancer Res. 2007;27:2253–2263. [PubMed] [Google Scholar]
- 144.Walsh AM, Lazzara MJ. Regulation of EGFR trafficking and cell signaling by Sprouty2 and MIG6 in lung cancer cells. J Cell Sci. 2013;126:4339–4348. doi: 10.1242/jcs.123208. [DOI] [PubMed] [Google Scholar]
- 145.VanMeter AJ, Rodriguez AS, Bowman ED, Jen J, Harris CC, Deng J, Calvert VS, Silvestri A, Fredolini C, Chandhoke V, et al. Laser capture microdissection and protein microarray analysis of human non-small cell lung cancer: differential epidermal growth factor receptor (EGPR) phosphorylation events associated with mutated EGFR compared with wild type. Mol Cell Proteomics. 2008;7:1902–1924. doi: 10.1074/mcp.M800204-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Furukawa M, Nagatomo I, Kumagai T, Yamadori T, Takahashi R, Yoshimura M, Yoneda T, Takeda Y, Goya S, Matsuoka H, et al. Gefitinib-sensitive EGFR lacking residues 746-750 exhibits hypophosphorylation at tyrosine residue 1045, hypoubiquitination, and impaired endocytosis. DNA Cell Biol. 2007;26:178–185. doi: 10.1089/dna.2006.0573. [DOI] [PubMed] [Google Scholar]
- 147.Mulloy R, Ferrand A, Kim Y, Sordella R, Bell DW, Haber DA, Anderson KS, Settleman J. Epidermal growth factor receptor mutants from human lung cancers exhibit enhanced catalytic activity and increased sensitivity to gefitinib. Cancer Res. 2007;67:2325–2330. doi: 10.1158/0008-5472.CAN-06-4293. [DOI] [PubMed] [Google Scholar]
- 148.Shimamura T, Lowell AM, Engelman JA, Shapiro GI. Epidermal growth factor receptors harboring kinase domain mutations associate with the heat shock protein 90 chaperone and are destabilized following exposure to geldanamycins. Cancer Res. 2005;65:6401–6408. doi: 10.1158/0008-5472.CAN-05-0933. [DOI] [PubMed] [Google Scholar]
- 149.Xu W, Yuan X, Xiang Z, Mimnaugh E, Marcu M, Neckers L. Surface charge and hydrophobicity determine ErbB2 binding to the Hsp90 chaperone complex. Nat Struct Mol Biol. 2005;12:120–126. doi: 10.1038/nsmb885. [DOI] [PubMed] [Google Scholar]
- 150.Shankaran H, Zhang Y, Opresko L, Resat H. Quantifying the effects of co-expressing EGFR and HER2 on HER activation and trafficking. Biochem Biophys Res Commun. 2008;371:220–224. doi: 10.1016/j.bbrc.2008.04.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Worthylake R, Opresko LK, Wiley HS. ErbB-2 amplification inhibits down-regulation and induces constitutive activation of both ErbB-2 and epidermal growth factor receptors. J Biol Chem. 1999;274:8865–8874. doi: 10.1074/jbc.274.13.8865. [DOI] [PubMed] [Google Scholar]
- 152.Gilmer TM, Cable L, Alligood K, Rusnak D, Spehar G, Gallagher KT, Woldu E, Carter HL, Truesdale AT, Shewchuk L, et al. Impact of common epidermal growth factor receptor and HER2 variants on receptor activity and inhibition by lapatinib. Cancer Res. 2008;68:571–579. doi: 10.1158/0008-5472.CAN-07-2404. [DOI] [PubMed] [Google Scholar]
- 153.Khan EM, Lanir R, Danielson AR, Goldkorn T. Epidermal growth factor receptor exposed to cigarette smoke is aberrantly activated and undergoes perinuclear trafficking. FASEB J. 2008;22:910–917. doi: 10.1096/fj.06-7729com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Soubeyran P, Kowanetz K, Szymkiewicz I, Langdon WY, Dikic I. Cbl-CIN85-endophilin complex mediates ligand-induced downregulation of EGF receptors. Nature. 2002;416:183–187. doi: 10.1038/416183a. [DOI] [PubMed] [Google Scholar]
- 155.Feng Q, Baird D, Peng X, Wang J, Ly T, Guan JL, Cerione RA. Cool-1 functions as an essential regulatory node for EGF receptor- and Src-mediated cell growth. Nat Cell Biol. 2006;8:945–956. doi: 10.1038/ncb1453. [DOI] [PubMed] [Google Scholar]
- 156.Shen F, Lin Q, Gu Y, Childress C, Yang W. Activated Cdc42-associated kinase 1 is a component of EGF receptor signaling complex and regulates EGF receptor degradation. Mol Biol Cell. 2007;18:732–742. doi: 10.1091/mbc.E06-02-0142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Yang W, Lo CG, Dispenza T, Cerione RA. The Cdc42 target ACK2 directly interacts with clathrin and influences clathrin assembly. J Biol Chem. 2001;276:17468–17473. doi: 10.1074/jbc.M010893200. [DOI] [PubMed] [Google Scholar]
- 158.Hopper NA, Lee J, Sternberg PW. ARK-1 inhibits EGFR signaling in C. elegans. Mol Cell. 2000;6:65–75. [PubMed] [Google Scholar]
- 159.Chen X, Wang Z. Regulation of epidermal growth factor receptor endocytosis by wortmannin through activation of Rab5 rather than inhibition of phosphatidylinositol 3-kinase. EMBO Rep. 2001;2:842–849. doi: 10.1093/embo-reports/kve179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Stenmark H, Vitale G, Ullrich O, Zerial M. Rabaptin-5 is a direct effector of the small GTPase Rab5 in endocytic membrane fusion. Cell. 1995;83:423–432. doi: 10.1016/0092-8674(95)90120-5. [DOI] [PubMed] [Google Scholar]
- 161.Su X, Kong C, Stahl PD. GAPex-5 mediates ubiquitination, trafficking, and degradation of epidermal growth factor receptor. J Biol Chem. 2007;282:21278–21284. doi: 10.1074/jbc.M703725200. [DOI] [PubMed] [Google Scholar]
- 162.Wainszelbaum MJ, Charron AJ, Kong C, Kirkpatrick DS, Srikanth P, Barbieri MA, Gygi SP, Stahl PD. The hominoid-specific oncogene TBC1D3 activates Ras and modulates epidermal growth factor receptor signaling and trafficking. J Biol Chem. 2008;283:13233–13242. doi: 10.1074/jbc.M800234200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Kong C, Su X, Chen PI, Stahl PD. Rin1 interacts with signal-transducing adaptor molecule (STAM) and mediates epidermal growth factor receptor trafficking and degradation. J Biol Chem. 2007;282:15294–15301. doi: 10.1074/jbc.M611538200. [DOI] [PubMed] [Google Scholar]
- 164.Vieira AV, Lamaze C, Schmid SL. Control of EGF receptor signaling by clathrin-mediated endocytosis. Science. 1996;274:2086–2089. doi: 10.1126/science.274.5295.2086. [DOI] [PubMed] [Google Scholar]
- 165.Burke P, Schooler K, Wiley HS. Regulation of epidermal growth factor receptor signaling by endocytosis and intracellular trafficking. Mol Biol Cell. 2001;12:1897–1910. doi: 10.1091/mbc.12.6.1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Haugh JM, Huang AC, Wiley HS, Wells A, Lauffenburger DA. Internalized epidermal growth factor receptors participate in the activation of p21(ras) in fibroblasts. J Biol Chem. 1999;274:34350–34360. doi: 10.1074/jbc.274.48.34350. [DOI] [PubMed] [Google Scholar]
- 167.Ren Y, Cheng L, Rong Z, Li Z, Li Y, Zhang X, Xiong S, Hu J, Fu XY, Chang Z. hSef potentiates EGF-mediated MAPK signaling through affecting EGFR trafficking and degradation. Cell Signal. 2008;20:518–533. doi: 10.1016/j.cellsig.2007.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Baldys A, Göoz M, Morinelli TA, Lee MH, Raymond JR, Luttrell LM, Raymond JR. Essential role of c-Cbl in amphiregulin-induced recycling and signaling of the endogenous epidermal growth factor receptor. Biochemistry. 2009;48:1462–1473. doi: 10.1021/bi801771g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Kranenburg O, Verlaan I, Moolenaar WH. Dynamin is required for the activation of mitogen-activated protein (MAP) kinase by MAP kinase kinase. J Biol Chem. 1999;274:35301–35304. doi: 10.1074/jbc.274.50.35301. [DOI] [PubMed] [Google Scholar]
- 170.Galperin E, Sorkin A. Endosomal targeting of MEK2 requires RAF, MEK kinase activity and clathrin-dependent endocytosis. Traffic. 2008;9:1776–1790. doi: 10.1111/j.1600-0854.2008.00788.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Maudsley S, Pierce KL, Zamah AM, Miller WE, Ahn S, Daaka Y, Lefkowitz RJ, Luttrell LM. The beta(2)-adrenergic receptor mediates extracellular signal-regulated kinase activation via assembly of a multi-receptor complex with the epidermal growth factor receptor. J Biol Chem. 2000;275:9572–9580. doi: 10.1074/jbc.275.13.9572. [DOI] [PubMed] [Google Scholar]
- 172.Vergarajauregui S, San Miguel A, Puertollano R. Activation of p38 mitogen-activated protein kinase promotes epidermal growth factor receptor internalization. Traffic. 2006;7:686–698. doi: 10.1111/j.1600-0854.2006.00420.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Er EE, Mendoza MC, Mackey AM, Rameh LE, Blenis J. AKT facilitates EGFR trafficking and degradation by phosphorylating and activating PIKfyve. Sci Signal. 2013;6:ra45. doi: 10.1126/scisignal.2004015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Pennock S, Wang Z. Stimulation of cell proliferation by endosomal epidermal growth factor receptor as revealed through two distinct phases of signaling. Mol Cell Biol. 2003;23:5803–5815. doi: 10.1128/MCB.23.16.5803-5815.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Rothschild SI, Gautschi O, Haura EB, Johnson FM. Src inhibitors in lung cancer: current status and future directions. Clin Lung Cancer. 2010;11:238–242. doi: 10.3816/CLC.2010.n.030. [DOI] [PubMed] [Google Scholar]
- 176.Tu C, Ortega-Cava CF, Winograd P, Stanton MJ, Reddi AL, Dodge I, Arya R, Dimri M, Clubb RJ, Naramura M, et al. Endosomal-sorting complexes required for transport (ESCRT) pathway-dependent endosomal traffic regulates the localization of active Src at focal adhesions. Proc Natl Acad Sci USA. 2010;107:16107–16112. doi: 10.1073/pnas.1009471107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Sandilands E, Frame MC. Endosomal trafficking of Src tyrosine kinase. Trends Cell Biol. 2008;18:322–329. doi: 10.1016/j.tcb.2008.05.004. [DOI] [PubMed] [Google Scholar]
- 178.Rush JS, Ceresa BP. RAB7 and TSG101 are required for the constitutive recycling of unliganded EGFRs via distinct mechanisms. Mol Cell Endocrinol. 2013;381:188–197. doi: 10.1016/j.mce.2013.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.McClintock JL, Ceresa BP. Transforming growth factor-{alpha} enhances corneal epithelial cell migration by promoting EGFR recycling. Invest Ophthalmol Vis Sci. 2010;51:3455–3461. doi: 10.1167/iovs.09-4386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Di Guglielmo GM, Baass PC, Ou WJ, Posner BI, Bergeron JJ. Compartmentalization of SHC, GRB2 and mSOS, and hyperphosphorylation of Raf-1 by EGF but not insulin in liver parenchyma. EMBO J. 1994;13:4269–4277. doi: 10.1002/j.1460-2075.1994.tb06747.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Sorkin A. Internalization of the epidermal growth factor receptor: role in signalling. Biochem Soc Trans. 2001;29:480–484. doi: 10.1042/bst0290480. [DOI] [PubMed] [Google Scholar]
- 182.Repetto E, Yoon IS, Zheng H, Kang DE. Presenilin 1 regulates epidermal growth factor receptor turnover and signaling in the endosomal-lysosomal pathway. J Biol Chem. 2007;282:31504–31516. doi: 10.1074/jbc.M704273200. [DOI] [PubMed] [Google Scholar]
- 183.Haugh JM, Meyer T. Active EGF receptors have limited access to PtdIns(4,5)P(2) in endosomes: implications for phospholipase C and PI 3-kinase signaling. J Cell Sci. 2002;115:303–310. doi: 10.1242/jcs.115.2.303. [DOI] [PubMed] [Google Scholar]
- 184.Várnai P, Rother KI, Balla T. Phosphatidylinositol 3-kinase-dependent membrane association of the Bruton’s tyrosine kinase pleckstrin homology domain visualized in single living cells. J Biol Chem. 1999;274:10983–10989. doi: 10.1074/jbc.274.16.10983. [DOI] [PubMed] [Google Scholar]
- 185.Bild AH, Turkson J, Jove R. Cytoplasmic transport of Stat3 by receptor-mediated endocytosis. EMBO J. 2002;21:3255–3263. doi: 10.1093/emboj/cdf351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Wilde A, Beattie EC, Lem L, Riethof DA, Liu SH, Mobley WC, Soriano P, Brodsky FM. EGF receptor signaling stimulates SRC kinase phosphorylation of clathrin, influencing clathrin redistribution and EGF uptake. Cell. 1999;96:677–687. doi: 10.1016/s0092-8674(00)80578-4. [DOI] [PubMed] [Google Scholar]
- 187.Helikar T, Kochi N, Kowal B, Dimri M, Naramura M, Raja SM, Band V, Band H, Rogers JA. A comprehensive, multi-scale dynamical model of ErbB receptor signal transduction in human mammary epithelial cells. PLoS One. 2013;8:e61757. doi: 10.1371/journal.pone.0061757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Ahn S, Kim J, Lucaveche CL, Reedy MC, Luttrell LM, Lefkowitz RJ, Daaka Y. Src-dependent tyrosine phosphorylation regulates dynamin self-assembly and ligand-induced endocytosis of the epidermal growth factor receptor. J Biol Chem. 2002;277:26642–26651. doi: 10.1074/jbc.M201499200. [DOI] [PubMed] [Google Scholar]
- 189.Yokouchi M, Kondo T, Sanjay A, Houghton A, Yoshimura A, Komiya S, Zhang H, Baron R. Src-catalyzed phosphorylation of c-Cbl leads to the interdependent ubiquitination of both proteins. J Biol Chem. 2001;276:35185–35193. doi: 10.1074/jbc.M102219200. [DOI] [PubMed] [Google Scholar]
- 190.Kassenbrock CK, Hunter S, Garl P, Johnson GL, Anderson SM. Inhibition of Src family kinases blocks epidermal growth factor (EGF)-induced activation of Akt, phosphorylation of c-Cbl, and ubiquitination of the EGF receptor. J Biol Chem. 2002;277:24967–24975. doi: 10.1074/jbc.M201026200. [DOI] [PubMed] [Google Scholar]
- 191.Kasai A, Shima T, Okada M. Role of Src family tyrosine kinases in the down-regulation of epidermal growth factor signaling in PC12 cells. Genes Cells. 2005;10:1175–1187. doi: 10.1111/j.1365-2443.2005.00909.x. [DOI] [PubMed] [Google Scholar]
- 192.Schmidt MH, Dikic I, Bögler O. Src phosphorylation of Alix/AIP1 modulates its interaction with binding partners and antagonizes its activities. J Biol Chem. 2005;280:3414–3425. doi: 10.1074/jbc.M409839200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Donepudi M, Resh MD. c-Src trafficking and co-localization with the EGF receptor promotes EGF ligand-independent EGF receptor activation and signaling. Cell Signal. 2008;20:1359–1367. doi: 10.1016/j.cellsig.2008.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Biscardi JS, Ishizawar RC, Silva CM, Parsons SJ. Tyrosine kinase signalling in breast cancer: epidermal growth factor receptor and c-Src interactions in breast cancer. Breast Cancer Res. 2000;2:203–210. doi: 10.1186/bcr55. [DOI] [PMC free article] [PubMed] [Google Scholar]