

Abstract
Breast cancer is a leading cause of cancer death in women, worldwide. Fortunately, breast cancer is relatively chemosensitive, with recent advances leading to the development of effective therapeutic strategies, significantly increasing disease cure rate. However, disease recurrence and treatment of cases lacking therapeutic molecular targets, such as epidermal growth factor receptor 2 and hormone receptors, referred to as triple-negative breast cancers, still pose major hurdles in the treatment of breast cancer. Thus, novel therapeutic approaches to treat aggressive breast cancers are essential. Blood coagulation factor VII (fVII) is produced in the liver and secreted into the blood stream. Tissue factor (TF), the cellular receptor for fVII, is an integral membrane protein that plays key roles in the extrinsic coagulation cascade. TF is overexpressed in breast cancer tissues. The TF-fVII complex may be formed in the absence of injury, because fVII potentially exists in the tissue fluid within cancer tissues. The active form of this complex (TF-fVIIa) may stimulate the expression of numerous malignant phenotypes in breast cancer cells. Thus, the TF-fVII pathway is a potentially attractive target for breast cancer treatment. To date, a number of studies investigating the mechanisms by which TF-fVII signaling contributes to breast cancer progression, have been conducted. In this review, we summarize the mechanisms controlling TF and fVII synthesis and regulation in breast cancer cells. Our current understanding of the TF-fVII pathway as a mediator of breast cancer progression will be also described. Finally, we will discuss how this knowledge can be applied to the design of future therapeutic strategies.
Keywords: Breast cancer, Blood coagulation, Tissue factor, Coagulation factor VII, Gene regulation, Therapeutic strategy
Core tip: Breast cancer is a worldwide problem. Difficulties with treating the disease and its recurrence persist due, in part, to a lack of therapeutic molecular targets. Blood coagulation factor VII (fVII) is generally produced in the liver. Tissue factor (TF), the cellular receptor for fVII, is an integral membrane protein that plays key roles in the extrinsic coagulation cascade. Formation of the TF-fVII complex causes contributes to the malignant phenotype of breast cancer cells. In this review, we summarize the breast cancer biology associated with the TF-fVII pathway. Further, we will discuss how these mechanisms can be targeted as therapeutics for this aggressive disease.
INTRODUCTION
Breast cancer is a global health problem and remains a common cause of cancer death in women, worldwide[1]. Currently, breast cancer may be treated using multiple chemotherapeutic programs depending on the histologic and molecular classification, such as the presence of hormone (estrogen and progesterone) receptors and/or epidermal growth factor receptor 2 (referred to as ERBB2 or HER2). However, difficulties associated with treating recurrent disease and triple-negative breast cancers that lack expression of apparent therapeutic molecular targets, remain[2,3]. Therefore, a greater understanding of breast cancer biology is essential to translate these findings into novel, therapeutic strategies to combat aggressive breast cancers.
Coagulation factor VII (fVII) is a serine protease component of the extrinsic coagulation cascade, that is primarily synthesized and secreted by hepatocytes[4]. Tissue factor (TF) is a 47-kD cell surface glycoprotein and a cellular receptor of fVII. fVII from blood plasma, associated with TF, gives rise to the activated complex, TF-fVIIa. This triggers a downstream coagulation cascade, eventually resulting in fibrin deposition (Figure 1). Previous studies have identified a correlation between cancer and blood coagulation, known as Trousseau’s syndrome[5]. It is likely that TF-fVII signaling is a major factor underlying this syndrome, although several other molecular mechanisms are possible[5]. Indeed, hypercoagulation is a complication commonly associated with cancer patients and potentially contributes to patient mortality[6]. Venous thromboembolism (VTE) is frequent in ovarian, pancreatic, and liver cancers[7], and breast cancer during chemotherapy[8].
Figure 1.
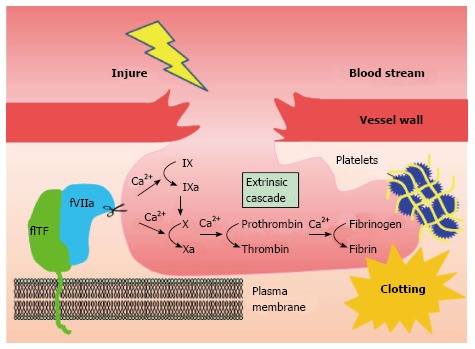
Extrinsic coagulation cascade initiated by the tissue factor-coagulation factor VII pathway. TF-fVIIa complex formation on the plasma membrane triggers an extrinsic coagulation cascade in response to injury. The TF-fVIIa complex located on the cell surface initiates the coagulation cascade by activating factor X via two different (coagulation factor IX-dependent or -independent) routes. This leads to fibrin deposition via the formation of thrombin. Blood coagulation completes by clot formation with other factors, such as platelets and red blood cells. FVII: Coagulation factor VII (fVII); TF: Tissue factor.
Previous studies have revealed that plasma TF levels are elevated in cancer patients, including those with advanced breast cancer[9]. Furthermore, breast cancer cells can release cell membrane-derived particles (generally referred to as microvesicles) with TF under various pathological conditions, leading to the hypercoagulation[10]. A truncated form of TF, derived from alternative mRNA splicing, may be secreted into the blood stream[11]. Therefore, TF-fVIIa complex formation may represent a major cause of thromboembolic events. Numerous studies have also suggested that TF-fVIIa complex formation on the cell surface also contributes to the malignant phenotypes of cancer, including an increase in cell motility, invasiveness, cell survival, and angiogenesis[12,13]. Recently, there is growing experimental evidence to suggest that TF also contributes to tumor initiation[13]. Therefore, therapeutic strategies targeting TF may be advantageous to breast cancer, although the possible impairment of the physiological hemostatic process should be considered.
fVII is thought to penetrate hyperpermeabilized blood vessels around the tumor tissue[14]. fVII may also exist in the lymph[15]. This extravascular fVII may bind to TF, which is present on the surface of cancer cells. Notably, multiple breast cancer cells have been shown to ectopically synthesize fVII[16]. This fVII is functional[16], suggesting that aberrantly synthesized fVII may also contribute to the malignant phenotypes of breast cancer. In this review, we summarize the recent progress in breast cancer biology associated with aberrant coagulation mechanisms. In particular, we focus on the TF-fVII pathway, among the multiple coagulation mechanisms, because breast cancer phenotypes associated with platelets and fibrinolysis have been extensively reviewed[17]. We also describe the mechanisms underlying TF and fVII overexpression and how their functions may be regulated in breast cancer cells. Finally, we discuss potential therapeutic strategies for breast cancer based on our current knowledge of the molecular mechanisms of TF-fVII signaling.
GENERAL BIOLOGY OF TF IN BREAST CANCER CELLS
TF-fVIIa signaling regulates breast cancer phenotypes
TF exists as either full-length or truncated forms, depending on the cell type. Full-length tissue factor (referred to hereafter as TF) is a 47-kDa membrane bound protein, essential for initiation of the extrinsic coagulation cascade (Figure 1). TF is widely, but selectively expressed in normal human tissues[18] including normal breast tissue[19]. In contrast, TF expression is minimally expressed in the liver[18]. Thus, upon injury, the liver may use a coagulation pathway independent of TF-fVII formation[20]. The role of liver TF remains elusive, however, because a recent study revealed that TF expression in mouse hepatocytes significantly contributes to thrombosis during liver injury caused by drug toxicity and hepatocyte transplantation[21].
TF tends to be overexpressed in breast cancer tissues associated with malignant phenotypes[22]. TF was initially classified as a member of the cytokine/growth factor receptor family owing to its amino acid sequence similarity[23,24], suggesting that it may transmit intracellular signals. Indeed, TF-fVIIa is capable of transmitting intracellular signals via multiple pathways, predominantly those involving the activation of protease-activated receptors (PARs) (Figure 2). To date, a number of studies have made significant advances towards our understanding of how TF-fVIIa complex formation contributes to cancer progression[12,13,25]. Until now, many studies concerning the biology of TF-fVIIa-dependent signaling were performed using breast cancer cell lines, possibly owing to their functional dependency on TF signaling[26-29]. Among the breast cancer cell lines, MDA-MB-231 cells are well characterized for their high TF expression, and are frequently used as a TF-dependent breast cancer model. Indeed, previous studies have shown that in vitro and in vivo phenotypes such as motility, invasiveness, and growth of MDA-MB-231 cells are highly TF-dependent[26-30]. Moreover, in these cells, TF has been shown to act as an angiogenic switch, leading to breast tumor development in a spontaneous breast cancer model recapitulating the human disease[31,32].
Figure 2.
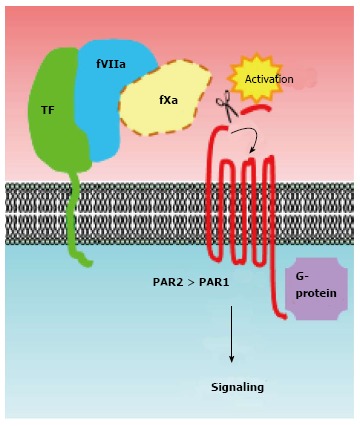
Activation of protease-activated receptors is a major mechanism of tissue factor-coagulation factor VIIa signaling in breast cancer cells. The proteolytic activities of the TF-fVIIa binary complex [potentially (designated as dotted line) ternary complex with fXa] cleave the N-terminal end of PARs. PARs are then activated via intra-molecular binding between the newly created N-terminus and an extracellular loop region of the receptors. Activation of these G-protein-coupled receptors subsequently activates downstream signaling cascades. A number of studies have indicated that PAR2 is crucial for activation of a TF-fVII-driven signaling cascade in breast cancer cells. The role of TF-fVII on PAR1 signaling in breast cancer cells is less evident. FVII: Coagulation factor VII; TF: Tissue factor; PARs: Protease-activated receptors.
Regulation of TF expression in breast cancer cells
In human cells, the Sp1 transcription factor is a major regulator of the F3 gene encoding TF, under normal conditions. Transcription may be affected by the presence of multiple single nucleotide polymorphisms (SNPs) within the F3 regulatory region and these SNPs have been previously associated with disease characteristics[33-36]. Immunohistochemical analyses demonstrate that TF is highly expressed in breast cancer tissues, in addition to ovarian and pancreatic cancer tissues[9,22]. Although the detailed mechanisms are not clear, transcriptional activation appears to be a major mechanism of TF overexpression.
The mechanisms regulating F3 gene expression are well characterized[37]. Constitutively high F3 gene expression is controlled by multiple transcription factors (Figure 3). It is likely that the aberrant activation of these factors causes higher TF levels in breast cancer cells, given that AP-1 and NFκB are proinflammatory transcription factors which are frequently activated in breast cancer cells[38]. Indeed, previous studies have shown that these transcription factors strongly bind to the F3 gene promoter in MDA-MB-231 cells. However, the promoter is poorly occupied in TF low-expressing MCF-7 cells[39].
Figure 3.
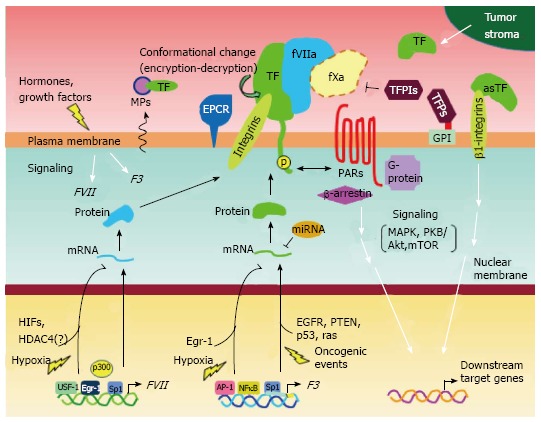
Possible mechanisms of expression and functional regulation of tissue factor and coagulation factor VII in breast cancer cells. Schematic overview summarizing the multiple mechanisms of gene expression, functional tuning, and intracellular signaling in breast cancer cells, as described in this review. It has been suggested that breast cancer cell phenotypes associated with the TF-fVII pathway may be specifically controlled via multiple autocrine and/or paracrine mechanisms, depending on tumor microenvironmental conditions. EGFR: Epidermal growth factor receptor; EPCR: Endothelial protein C receptor; FVII: Coagulation factor VII; TF: Tissue factor.
Breast cancer progression is dependent on sex steroid hormones. It was previously shown that TF expression increases in response to progesterone exposure[40], resulting in breast cancer phenotypes via a TF-dependent pathway[41]. Furthermore, a steady state level of TF mRNA in breast cancer cells may be determined by valance of its positive and negative regulatory mechanisms as it was found that PI3K/Akt and MAPK/ERK signaling pathways inversely regulate TF transcript levels in MDA-MB-231 cells[42].
Inducible gene expression may also account for high TF expression in cancer tissues, although such mechanisms may not necessarily apply to breast cancer cells. For example, F3 gene expression may be regulated by exposure to various environmental stimuli including cytokines[13], growth factors[13], and hypoxia[13], resulting in the activation of AP-1 and NFκB.
It is likely that the tumor microenvironment and the associated blood supply affect the expression level of TF. In addition to serum factors, F3 expression is also influenced by oncogenic events[43-45] (Figure 3). Recent studies have shown that these factors play an important role in the regulation of F3 expression in glioblastoma cells[44,45], and similar mechanisms may also exist in breast cancer cells.
The Egr-1 transcription factor also plays a major role in controlling TF expression[37]. As previously discussed, Sp1 largely controls the basal activation of F3 gene. However, Egr-1 expression may be induced in cancer cells upon various stimuli within the cellular microenvironment. Because Egr-1 and Sp1 share a common DNA binding motif, Egr-1 may subsequently displace Sp1’s occupancy of the F3 gene promoter, thereby enhancing gene expression. A similar mechanism of F3 activation may also apply under conditions of hypoxia (Figure 3). Hypoxia inducible factors (HIFs) are the major transcription factors responsible for adaptation to hypoxic environments within cancer tissues. Interestingly, however, it appears that Egr-1 rather than HIFs, is the major cause of F3 activation under hypoxia, at least in glioblastoma multiforme cells[45].
Recent studies also point to a role for microRNAs in the regulation of TF expression. In one study, miR-19 was shown to bind the 3’-UTR of the TF mRNA, repressing translation in breast cancer cells[46] (Figure 3). This study also demonstrated that miR-19 is highly expressed in a TF low-expressing breast cancer cell line, MCF-7. Thus, regulation by this microRNA can determine TF levels.
Detailed mechanisms of TF-fVIIa signaling
Mechanisms of TF-fVIIa signaling potentially giving rise to malignant phenotypes can be classified as follows. In all cases, however, the TF-fVIIa-PAR axis plays a major role in the regulation of these cancer phenotypes (Figure 2). The first mechanism involves TF-fVIIa binary complex formation. Various studies indicate that the TF-fVIIa complex activates PAR2. This active PAR2 participates in determining the breast cancer phenotype, via activation of the mitogen-activated protein kinase (MAPK) cascade (Figure 3)[27,28,30].
In the second mechanism, TF-fVIIa-dependent signaling may involve coagulation factor X (fX) for signal transmission (Figure 2). The TF-fVIIa complex can produce a TF-fVIIa-fXa complex[26,47,48]. This tertiary complex can cause breast cancer phenotypes via not only activating PAR2-dependent signals, but also by activating PAR1 to trigger thrombin signals[29]. In addition to MAPK, protein kinase B (PKB/Akt) is also involved in a signaling cascade mediated by the TF-fVIIa-fXa ternary complex[48] (Figure 3). This ternary complex may also phosphorylate mTOR to activate components downstream of this signaling cascade in breast cancer cells[29], thereby promoting cell migration. In addition, recent studies using non-breast cancer cells demonstrated that the endothelial protein C receptor (EPCR) supports this ternary complex to transmit signals[49] (Figure 3).
The G protein-independent signaling pathway associated with PAR2 is one candidate mechanism for TF-fVIIa signaling in breast cancer cells[50]. Indeed, enhanced breast cancer cell motility involves the recruitment of a scaffold protein, β-arrestin, to support cellular signaling[51,52] (Figure 3). Which TF-fVIIa signaling mechanism is eventually used by breast cancer cells likely depends on the environmental conditions within the tumor, which may induce the expression of the downstream effectors of angiogenesis[27,53]. Previous studies have shown that connective tissue growth factors such as Cyr61[54], CTGF[54] and the homeodomain DNA binding protein, CUX1[55], are up-regulated in response to TF-fVIIa stimuli, however these experiments were performed using non-breast cancer cell lines.
cDNA microarray analyses in MDA-MB-231 cells also identified various factors down-stream of the TF-fVIIa-PARs axis that may contribute to different breast cancer phenotypes. These studies identified novel target genes regulated by TF-fVIIa signaling, that result in a wound-healing type response, including the chemokine ligand (angiogenic), CXCL1[31,56], anti-apoptotic protein, Birc3[56] and a component of basement membrane, CSF[56].
MODULATORS OF TF FUNCTION IN BREAST CANCER CELLS
Conformational changes modulating TF function
Functional modification is an additional post-transcriptional mechanism regulating TF activity. Multiple mechanisms can control the procoagulant activity of TF by a conformational change referred to as encryption/decryption[57] (Figure 3). Previous studies have shown that disulfide bond isomerization by protein disulfide isomerase (PDI), controls the function of TF as a mediator of coagulation or signaling, in endothelial and keratinocyte cell lines[58]. However, this model appears to be controversial, because recent studies using the same endothelial cell line and MDA-MB-231 cells, indicate that decryption of TF is mediated via its interaction with anionic phospholipids[59]. PDI has also been shown to activate the procoagulant activity of TF via its molecular chaperone activity[60]. In conclusion, it is likely that the procoagulant activity of TF is specific for the decrypted form, while encrypted TF still transmits signals.
Tissue factor pathway inhibitors: negative regulators of TF-fVIIa activity
Tissue factor pathway inhibitors (TFPIs) are known to directly inhibit the enzymatic activity of TF-fVIIa complex[61] (Figure 3).
Tissue factor pathway inhibitors (TFPI-1 and TFPI-2) are endogenous Kunitz-type inhibitors of TF-fVIIa-Xa complex formation, and thereby negatively regulate coagulation[61]. TFPI-1 may exist as two alternatively spliced isoforms, TFPIα and TFPIβ[62]. TFPIα is secreted or attached to the cell membrane via a glycosylphosphatidylinositol anchor, whereas TFPIβ may be expressed as a membrane-bound form[62]. Both forms are primarily synthesized in endothelial cells[63]. Various cell types including macrophages, monocytes, platelets, and fibroblasts also produce TFPI-1[64], thereby contributing to the physiological regulation of bleeding[65]. TFPI-2 is highly expressed in the placenta, but is also synthesized in various normal tissues, including the endothelial cells of various blood vessels[66].
Previous studies have shown that breast cancer tissues may also synthesize TFPI-1[67] and TFPI-2[66], and TFPI-1 expression levels were correlated with disease malignancy. In keeping with this observation, multiple breast cancer cell lines also express TFPIα and TFPIβ[68]. Overexpression of both TFPI-1 isoforms induces apoptosis of breast cancer cells[69]. Downregulation of TFPI-1 increases cell motility and breast cancer cell invasiveness[70], suggesting that TFPI-1 acts as a suppressor of breast cancer phenotypes. In contrast, TFPI-1 may augment certain malignant phenotypes, such as adhesion of bladder cancer cells[71] and invasiveness of breast cancer cells[66], via interaction with the TF-fVIIa complex. Thus, the therapeutic value of TFPIs is unclear, even though these anti-coagulants are aberrantly expressed in breast cancer tissues. Further in vivo studies are therefore required to completely understand the role of TFPIs in breast cancer progression.
POTENTIAL FUNCTIONS OF TF IN BREAST CANCER PROGRESSION
Tumor cell-derived TF
To date, numerous studies have been performed aiming to uncover the role of TF-fVIIa signaling in the malignant phenotypes of breast cancer using multiple cell lines. These cell lines represent useful models, because a number of tumor-associated processes such as motility[28,29], invasiveness[28,29], and survival[48] are TF dependent. MDA-MB-231 is a good model cell line for TF-dependent breast cancer as this cell line synthesizes high levels of TF. Characteristics of this cell line, such as motility and invasiveness, are highly TF dependent in vitro[26-28]. Furthermore, the growth of xenograft tumors derived from MDA-MB-231 cells is dependent on TF activity and subsequent PAR2 activation in vivo[30]. This finding was supported by a PyMT mouse model characterized by spontaneous breast cancer development[31]. These findings are also consistent with earlier studies using colorectal cancer cells, which revealed that TF does not contribute to cell proliferation in vitro[43]. This suggests that host-tumor interactions are essential for the expression of a malignant phenotype in TF-driven breast cancer.
Another potential contributor of TF-driven breast tumor progression is EPCR (Figure 3). As previously discussed, EPCR can regulate TF-fVIIa signaling, and the importance of EPCR in breast cancer progression was recently demonstrated using both human xenograft[72] and spontaneous murine breast tumor development[73,74] models. Accumulating evidence indicates that TF largely contributes to the metastatic potential of breast cancer cells, and a recent study demonstrated that PAR1 signaling in both tumor and host cells is essential for TF-dependent lung metastasis of breast cancer cells[74].
Breast cancer phenotypes regulated by TF-fVIIa complex formation are predominantly dependent on PAR2-dependent signals. However, PAR1 signaling may function to augment breast cancer cell invasiveness and tumorigenesis[29,75-77], irrespective of TF dependency. PAR1 is a receptor of thrombin highly synthesized in metastatic breast cancer cells. Indeed, treatment of breast cancer cells with thrombin increases their PAR1-dependent invasiveness. PAR1 may also act as a receptor for atrix metalloprotease-1, which is released from stromal cells within cancer tissues, and is capable of enhancing MDA-MB-231 cell invasiveness and tumorigenesis[77]. Thus, the function of PAR1 and PAR2 signaling associated with breast cancer progression may vary, according to the cellular microenvironment and relative expression levels.
Exogenous TF
Expression of TF is not limited to tumor parenchyma cells, but is also expressed in the tumor stroma, where it promotes breast cancer metastasis[78]. It was shown that transforming growth factor β released from cancer cells, may stimulate stromal cells to secrete TF, leading to the promotion of breast cancer progression in a paracrine manner[78].
Exogenously synthesized TF in patients may affect a number of breast cancer cell characteristics. It was revealed that a paracrine effect of TF can influence the growth and metastasis of breast cancer cells[79]. Treatment of cells with recombinant TF, mimicking stromal-derived TF (Figure 3), enhances the invasive and proliferative properties of these cells. This occurs via activation of β1-integrins and/or PAR2-dependent signals, followed by inactivation of transcription of the estrogen receptor (ER) gene[79] (Figure 3). The involvement of integrins in TF-driven phenotype expressions of breast cancer cells is similar to the findings that association of TF integrated into plasma membrane of keratinocytes and breast cancer cells bind integrins and may support growth of breast tumor[30]. Furthermore, because ER positivity determines clinically distinct groups of breast cancer patients, ER gene regulation via TF signaling may affect the selection of therapeutic strategies used to treat breast cancer patients.
Previous studies reveal that TF expression within the stromal area of breast cancer tissues is due to production from immune cells[80]. TF levels were closely associated with extravascular fibrin deposition and VEGF expression levels, suggesting that stromal-derived TF contributes to the angiogenic phenotype of breast cancer.
Truncated forms of TF
In addition to full-length TF, alternatively spliced, truncated forms of TF (asTF) also exist in humans[81]. Recently, it was reported that asTF is highly expressed in breast cancer cells and contributes to the malignant phenotype[82]. Similar to TF, asTF binds to integrins and therefore exists on the cell surface (Figure 3). However, the asTF-integrin complex augments cell proliferation, migration and anchorage-independent cell growth in a PAR2-independent manner[82]. These results are in contrast to an earlier report that demonstrated that the growth of breast tumors derived from MDA-MB-231 cells is dependent on PAR2 signaling, downstream of membrane-integrated TF[30]. It is possible that the distinct binding characteristics of these two TF isoforms underlie such differences.
TF EXPRESSION IN BREAST CANCER PATIENTS
TF as a serum component of breast cancer patients
In addition to asTF, breast cancer cells may also shed TF into the bloodstream as a component of microparticles (MPs), derived from plasma membrane in response to multiple stimuli[83,84] (Figure 3). Thus, it is likely that these TF-positive MPs contribute to plasma TF levels associated with clinical parameters[9]. Indeed, TF-positive MPs may be secreted from human breast tumors in a mouse xenograft model, resulting in a coagulation-prone status[10]. However, previous studies indicate that the risk of thrombosis during chemotherapy is independent of TF plasma levels in breast cancer patients, who display high levels of TF[8,85]. Therefore, to date, it is not clear how, and to what extent, plasma TF derived from breast cancer cells, contributes to breast cancer malignancy. Conversely, a recent study demonstrated that circulating tumor cells (CTCs) of breast cancer patients may be detected by labeling cell surface TF[86], suggesting that TF may be used as a diagnostic tool for breast cancer patients.
TF expression and its association with breast cancer patient clinical outcome
Activation of platelets[8] and elevated plasma TF levels[10] may be determinants of the thrombotic events observed in cancer patients. Indeed, VTE post-chemotherapy is a major event in breast cancer patients, prompting an investigation into the relationship between hemostatic markers and thrombosis[8]. These studies revealed that plasma TF levels in patients were significantly elevated compared with those of non-cancerous individuals. However, TF levels did not increase during chemotherapy, indicating that chemotherapy-associated VTE does not correlate with TF, and more likely associated with neutrophil extracellular traps composed of cell free nucleic acids and neutrophils[87].
TF is highly synthesized in breast cancer tissues as revealed by the analysis of clinical samples. In addition, TF is synthesized in the vascular endothelial cells of invasive breast cancer tissues. Thus, TF may be a marker for angiogenic phenotypes in patients[22]. In addition to its expression in breast cancer tissues, TF levels are also increased in the plasma of breast cancer patients[9]. Plasma TF levels were not significantly different between normal and benign tumors. However, TF levels were significantly higher in primary and recurrent cancer patients[9]. Notably, this pattern of TF expression correlated with that observed in cancer tissues. The urine TF levels were also associated with poor prognosis of breast cancer[88]. Taken together, these results suggest that the analysis of plasma and urine TF levels may be used to stratify patients into personalized treatment regimes.
Although the molecular mechanisms of the TF-driven breast cancer phenotypes may be explained by the various cellular events associated with the TF-fVIIa signaling cascade, recent studies indicate that post-translational modifications affecting TF expression also play an important role. Immunohistochemical analyses of breast cancer xenografts and clinical samples reveal that phosphorylation of TF (Figure 3) is correlated with the recurrence and aggressive phenotypes of breast cancer[89]. It was found that the phosphorylation of TF in association with PAR2 expression correlates with breast cancer recurrence[89]. In addition, in vivo studies using TF cytoplasmic domain-deleted mice revealed interplay between the TF cytoplasmic domain and PAR2 signaling, to promote breast cancer by modulating the host angiogenic response[32]. These studies provide a mechanism for the observed clinical association between TF phosphorylation and PAR2 signaling.
ECTOPIC EXPRESSION OF FVII IN BREAST CANCER CELLS
Constitutive expression
Similar to TF, the transcriptional regulation of human FVII has been extensively studied[90-92]. In contrast to TF, biosynthesis of fVII in the mammalian body is limited. The primary site of fVII production is liver. Previous studies demonstrate that the human FVII gene promoter is typically bound by HNF-4 and Sp1 transcription factors, and is therefore highly activated in liver cells. The efficiency of FVII gene expression may be affected by genetic alterations, including SNPs and decanucleotide insertion[93], as in the case of the F3 gene. However, unlike F3, it appears that binding sites for inflammatory transcription factors such as NFκB and AP-1, do not exist within the FVII promoter region. Instead, the activity of FVII may be regulated in response to hormones such as estrogen[94] and insulin[95]. Plasma fVII levels are also known to associate with plasma lipid concentration[96,97]. In addition to the liver, fVII may also be synthesized in monocytes and macrophages[98,99], although the mechanisms of FVII regulation in these non-hepatocytic cells remains unknown.
fVII is primarily synthesized in the liver. However, various cancer cells may ectopically express fVII[16]. Notably, multiple breast cancer cell lines constitutively synthesize high levels of the fVII transcript[16]. Cancer cells with fVII expression exhibit pro-coagulant activity as TF-fVIIa complex is formed on the cell surface, suggesting that aberrantly synthesized fVII may be functionally active and contribute to breast cancer progression. Indeed, fVII expression is frequently observed in breast cancer specimens[100].
Given the high expression of fVII in breast cancer cells and tissues, the molecular mechanisms of FVII activation were subsequently investigated using breast cancer cells. Binding of HNF-4 to the FVII promoter was shown to be essential for eutopic transcriptional activation. However, HNF-4 is not expressed in breast cancer cells[100], suggesting that other factors are responsible for ectopic activation of FVII. Reporter gene assays revealed that reporter activity is fully activated by the authentic FVII promoter region in breast cancer cells[100]. As expected, the HNF-4 binding site is dispensable for ectopic FVII gene expression in breast cancer cells. Further reporter assays revealed that an Sp1 binding site within the FVII promoter region is crucial for ectopic FVII gene expression. This study further demonstrated that the transcriptional regulators, USF-1 and Egr-1, also regulate ectopic expression of FVII in breast cancer cells[100] (Figure 3).
Histone acetylation of gene promoters also plays a crucial role in the regulation of gene transcription, prompting analysis of such epigenetic modifications at the FVII gene promoter. These studies revealed that the histone acetyltransferases (HATs), p300 and CBP, predominantly occupy the FVII promoter region in breast cancer cells[100]. In contrast, PCAF and SRC-1 HATs were also involved in the regulation of hepatocytic FVII expression[100]. Thus, p300 and CBP may predominantly acetylate histones within the FVII promoter region, followed by accession of transcription factors responsible for transcriptional regulation in breast cancer cells. Conversely, various HATs may be responsible for eutopic FVII regulation.
Inducible expression under hypoxia
FVII transcription is inducible in ovarian cancer cells under hypoxic and hypoxia mimetic (CoCl2 treatment) conditions[16]. To date, the expression of fVII transcripts in response to hypoxia have been tested in several breast cancer cell lines[100,101]. These studies revealed that fVII transcript levels are not enhanced in response to hypoxic stimuli in breast cancer cell lines with high fVII expression[100]. Conversely, fVII mRNA levels were inducible in the breast cancer cell line, MDA-MB-468 under CoCl2 stimuli[101], suggesting a cell-type dependent induction of fVII.
The detailed mechanisms controlling FVII induction under hypoxic conditions were recently defined using ovarian cancer cell lines[102], although it is not clear to what extent these mechanisms are applicable to breast cancer cells. These studies revealed that physical interaction between Sp1 and hypoxia inducible factor-2α (HIF2) may contribute to FVII activation in ovarian cancer cells, although HIF1 also indirectly affects FVII expression (Figure 3). This indicates that the promoter region occupied by HIF2 is devoid of a hypoxia response element (HRE)[16,102], suggesting that HRE-independent mechanisms are responsible for FVII activation under hypoxic conditions. Indeed, chromatin immunoprecipitation analysis with MDA-MB-468 cells revealed that HIF2 predominantly associates with the FVII promoter region[101], as in the case of ovarian cancer cells[16]. Furthermore, this mechanism was synergistically induced following simultaneous exposure of ovarian cancer cells to hypoxic conditions and serum deprivation, via a HDAC4 (a class II histone deacetylase)-dependent pathway[102]. These results suggest that the TF-fVII pathway is controlled by a stress-responsive, transcriptional mechanism, mediated by an HIF2/Sp1/HDAC4 network.
Exogenously supplied fVII vs autonomously produced fVII: Are there any functional differences?
Cell surface-bound TF binds fVII, irrespective of its source (eutopic or ectopic synthesis), raising the question of whether the TF-fVIIa complex functions differently depending on the source of fVII. Previous studies have shown that the plasma concentration of fVII is quite low[4]. Therefore, we can envision that self-production of fVII may facilitate TF-fVII complex formation compared with exogenously expressed fVII. This may be particularly important in hypoxic cancer microenvironments, where the supply of fVII from the bloodstream is likely limited because of poor and aberrant vasculature. In addition, it was recently described that ectopically synthesized fVII can augment the growth of breast cancer cells[101]. This is an unexpected result as it had previously been observed that TF does not contribute to cell proliferation under in vitro cell culture conditions[43]. Indeed, this study demonstrated that proliferation of breast cancer cells with high TF expression was not enhanced by exogenous supply of fVII[101]. To date, however, the mechanisms regulating differential cell growth between cells exposed to exogenously supplied fVII and ectopically synthesized fVII remain unclear. Our knowledge concerning other functional differences in cancer cells associated with differential routes of fVII supply is currently poor; however, this represents an interesting field for future study.
TF-FVIIa SIGNALING AS A THERAPEUTIC TARGET
Potential therapeutic strategies targeting TF-fVIIa signaling
To date, several attempts have been made to inhibit TF-fVIIa activity associated with breast cancer in vitro and in vivo. One simple method used to block TF-fVIIa activity involves treatment with anti-TF antibodies. The use of monoclonal antibodies has been successfully used in breast cancer therapy to target cell surface HER2, and therefore represents a promising strategy[103]. However, the major concern of this strategy is that blocking TF-fVIIa may also impair normal hemostasis, causing bleeding[30]. Previous studies have shown that growth and lung metastasis of orthotopically transplanted MDA-MB-231 cells is profoundly suppressed by successive administration of the humanized anti-TF antibody, CNTO859[104]. In this murine model, the CNTO859 antibody binds to human TF but not rodent TF, and therefore does not preclude normal hemostatic processes. However, the effect of this antibody in humans remains unclear. Similarly, a recent study demonstrated that the tick protein, Ixolaris, binds predominantly to the TF-fVIIa-fX ternary complex, thereby inhibiting downstream signaling involving PAR2 activation and suppressing tumor growth derived from MDA-MB-231mfp cells[105]. However, this protein is unable to suppress the growth of murine breast cancer tumors, because Ixoralis does not bind the murine TF-fVIIa complex[105].
One strategy to overcome the negative effects of TF-targeting antibodies on normal hemostasis is to use an antibody specifically inhibiting TF-fVIIa signaling. To date, we have identified a mouse monoclonal antibody, TF10H10, that fulfils this purpose[30]. Similar to the CNTO859 antibody, growth of xenograft tumors derived from MDA-MB-231 cells was shown to be effectively inhibited following treatment of cancer cells with a TF10H10 antibody prior to inoculation of mice[30].
Inhibition of TF at the transcriptional level represents another strategy to target TF-fVIIa activity. As previously discussed, TF levels are transcriptionally controlled by various transcription factors regulating constitutive and inducible expression. Overexpression and/or functional activation of transcription factors such as Egr-1, NFκB, and AP-1 may be involved in this process. Previous studies have shown that curcumin, a major component of turmeric spice can inhibit binding of these transcription factors to gene promoter regions required for cell survival and invasion activities[106]. Indeed, several studies showed that expression of the F3 gene may be inhibited by this natural pigment in endothelial cells[107,108] and monocytes[109]. Thus, pharmaceutical inhibition by curcumin may suppress aberrant expression of TF in breast cancer cells.
Finally, there have been various attempts to combat breast cancer by targeting TF on the cell surface by increasing target selectivity. In these studies, fVII was conjugated with photosensitizers[110,111], and the effect of this fusion fVII on tumor growth was monitored following injection into mice. Strikingly, tumor volume derived from breast cancer cells was markedly decreased in response to irradiation, compared with negative controls using non-fused fVII. This is likely because of the accumulation of photosensitizing drugs in tumor tissues with high TF expression. A similar approach was tested using fVIIa conjugated to a synthetic curcumin analog instead of photosensitizers[112]. fVIIa successfully delivered curcumin to target breast tumor cells, thereby reducing toxicity and enhancing therapeutic efficacy.
Another interesting approach that may be harnessed to enhance tumor selectivity is the use of prodrugs that can be activated within the tumor microenvironment in a TF-dependent manner[113]. In this study, doxorubicin-based prodrugs conjugated with albumin were used to treat tumors in a murine breast cancer model. These prodrugs penetrated tumor tissues and were predominantly activated by TF-fVIIa activity, because TF is highly expressed on the surface of cancer cells, leading to efficient suppression of breast cancer. Taken together, these studies demonstrate that increasing tumor selectivity of pharmaceutical compounds by targeting TF represents a promising strategy for cancer therapy, although careful control of dosage is necessary to prevent side effects, such as bleeding.
Possible strategies to inhibit ectopic fVII expression
Ectopic expression of fVII contributes to several breast cancer phenotypes in vitro. Thus, in addition to targeting TF expression, inhibition of fVII expression may also represent a potentially valuable therapeutic strategy. Importantly, the success of this strategy would rely on the selective inhibition of ectopic fVII, without inhibiting function of ectopically produced fVII in the liver. Ectopic activation of the FVII promoter in breast cancer cells is associated with binding by p300 and CBP, while the FVII promoter in hepatocytes is occupied by various HATs, suggesting that targeting p300/CBP activities may selectively inhibit FVII expression in breast cancer cells[100]. Curcumin is also capable of blocking the HAT activity of p300/CBP compared with other HATs. Indeed, curcumin markedly reduced fVII transcript levels in breast cancer cells in a dose-dependent manner, while normal expression of FVII in hepatic cells was only weakly impaired[100]. Levels of constitutively expressed TF mRNA were not significantly diminished by curcumin treatment in these cells, consistent with the notion that curcumin specifically inhibits inducible TF expression. Furthermore, anacardic acid, another natural, small compound inhibitor of p300 and PCAF, did not selectively inhibit ectopic FVII expression, highlighting the specificity of curcumin for p300/CBP activity[100]. The effect of curcumin on FVII expression was subsequently confirmed at the protein level. In contrast, HAT activity associated with the FVII promoter in hepatocytes, including p300 and CBP, is heterogeneous. It should be noted however, that selective inhibition of ectopic fVII synthesis was demonstrated using a limited number of cell lines. Therefore hepatocytic fVII synthesis may be considerably impaired by curcumin if p300/CBP is a component of the transcriptional machinery regulating the FVII gene in hepatocytes.
SUMMARY AND PERSPECTIVES
In this review, we describe various therapeutic strategies based on our current understanding of breast cancer biology associated with the TF-fVII pathway. The TF-fVIIa pathway in breast cancer cells may be regulated via multiple molecular mechanisms (summarized in Figure 3), enabling us to envisage a number of possible therapeutic strategies. Indeed, accumulating evidence suggests that TF is a promising target in breast cancer. Many breast cancer cell lines constitutively and perhaps inducibly express fVII. Given that TF-fVIIa signaling is a major mechanism underlying breast cancer-associated malignant phenotypes, strategies targeting ectopic fVII expression may also be considered in future therapeutic designs. How ectopic fVII expression affects breast cancer progression in vivo, however, remains an important question. Previous studies have shown that curcumin selectively inhibits ectopic fVII synthesis by removing p300/CBP from the FVII promoter region. Furthermore, animal studies have shown that curcumin can cure cardiovascular diseases[114,115] and cancer[116] by targeting p300 activity. Therefore, anti-p300 strategies using curcumin may be clinically applicable, without posing significant toxicity. Based on this, it may be of interest to investigate anti-p300/CBP strategies in fVII-expressing breast cancer models. It should be noted however, that anti-p300/CBP strategies may be compromised because HATs can also be targeted to the hepatocytic FVII promoter. The identification of novel molecular targets, specifically associated with ectopic fVII synthesis in breast cancer cells is therefore critical.
From a clinical point of view, the identification of relationships between fVII expression and various clinical parameters, such as chemoresistance, relapse, and overall survival, is essential to predict which patients may benefit from anti-TF-fVIIa treatment. Finally, many issues concerning the biology of ectopic fVII synthesis in breast cancer cells remain unresolved. Why do breast cancer cells tend to synthesize more fVII compared with other cancer cells? How does ectopically expressed fVII associate with TF to express the TF-fVIIa complex on the cell surface? It will be intriguing to investigate whether ectopically expressed fVII is equivalent to ectopically expressed fVII. A more detailed understanding of how ectopic fVII expression is regulated, and how it can contribute to breast cancer biology, is essential for translating our current knowledge to anti-breast cancer strategies.
Footnotes
P- Reviewer: McCarty OJT S- Editor: Wen LL L- Editor: A E- Editor: Lu YJ
References
- 1.Alvarez RH. Present and future evolution of advanced breast cancer therapy. Breast Cancer Res. 2010;12 Suppl 2:S1. doi: 10.1186/bcr2572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Foulkes WD, Smith IE, Reis-Filho JS. Triple-negative breast cancer. N Engl J Med. 2010;363:1938–1948. doi: 10.1056/NEJMra1001389. [DOI] [PubMed] [Google Scholar]
- 3.Crown J, O’Shaughnessy J, Gullo G. Emerging targeted therapies in triple-negative breast cancer. Ann Oncol. 2012;23 Suppl 6:vi56–vi65. doi: 10.1093/annonc/mds196. [DOI] [PubMed] [Google Scholar]
- 4.Furie B, Furie BC. The molecular basis of blood coagulation. Cell. 1988;53:505–518. doi: 10.1016/0092-8674(88)90567-3. [DOI] [PubMed] [Google Scholar]
- 5.Varki A. Trousseau’s syndrome: multiple definitions and multiple mechanisms. Blood. 2007;110:1723–1729. doi: 10.1182/blood-2006-10-053736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.ten Cate H, Falanga A. Overview of the postulated mechanisms linking cancer and thrombosis. Pathophysiol Haemost Thromb. 2008;36:122–130. doi: 10.1159/000175150. [DOI] [PubMed] [Google Scholar]
- 7.Iodice S, Gandini S, Löhr M, Lowenfels AB, Maisonneuve P. Venous thromboembolic events and organ-specific occult cancers: a review and meta-analysis. J Thromb Haemost. 2008;6:781–788. doi: 10.1111/j.1538-7836.2008.02928.x. [DOI] [PubMed] [Google Scholar]
- 8.Kirwan CC, McDowell G, McCollum CN, Kumar S, Byrne GJ. Early changes in the haemostatic and procoagulant systems after chemotherapy for breast cancer. Br J Cancer. 2008;99:1000–1006. doi: 10.1038/sj.bjc.6604620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ueno T, Toi M, Koike M, Nakamura S, Tominaga T. Tissue factor expression in breast cancer tissues: its correlation with prognosis and plasma concentration. Br J Cancer. 2000;83:164–170. doi: 10.1054/bjoc.2000.1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Davila M, Amirkhosravi A, Coll E, Desai H, Robles L, Colon J, Baker CH, Francis JL. Tissue factor-bearing microparticles derived from tumor cells: impact on coagulation activation. J Thromb Haemost. 2008;6:1517–1524. doi: 10.1111/j.1538-7836.2008.02987.x. [DOI] [PubMed] [Google Scholar]
- 11.van den Berg YW, van den Hengel LG, Myers HR, Ayachi O, Jordanova E, Ruf W, Spek CA, Reitsma PH, Bogdanov VY, Versteeg HH. Alternatively spliced tissue factor induces angiogenesis through integrin ligation. Proc Natl Acad Sci USA. 2009;106:19497–19502. doi: 10.1073/pnas.0905325106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ruf W, Disse J, Carneiro-Lobo TC, Yokota N, Schaffner F. Tissue factor and cell signalling in cancer progression and thrombosis. J Thromb Haemost. 2011;9 Suppl 1:306–315. doi: 10.1111/j.1538-7836.2011.04318.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Milsom C, Rak J. Tissue factor and cancer. Pathophysiol Haemost Thromb. 2008;36:160–176. doi: 10.1159/000175154. [DOI] [PubMed] [Google Scholar]
- 14.McDonald DM, Baluk P. Significance of blood vessel leakiness in cancer. Cancer Res. 2002;62:5381–5385. [PubMed] [Google Scholar]
- 15.Miller GJ, Howarth DJ, Attfield JC, Cooke CJ, Nanjee MN, Olszewski WL, Morrissey JH, Miller NE. Haemostatic factors in human peripheral afferent lymph. Thromb Haemost. 2000;83:427–432. [PubMed] [Google Scholar]
- 16.Koizume S, Jin MS, Miyagi E, Hirahara F, Nakamura Y, Piao JH, Asai A, Yoshida A, Tsuchiya E, Ruf W, et al. Activation of cancer cell migration and invasion by ectopic synthesis of coagulation factor VII. Cancer Res. 2006;66:9453–9460. doi: 10.1158/0008-5472.CAN-06-1803. [DOI] [PubMed] [Google Scholar]
- 17.Lal I, Dittus K, Holmes CE. Platelets, coagulation and fibrinolysis in breast cancer progression. Breast Cancer Res. 2013;15:207. doi: 10.1186/bcr3425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Drake TA, Morrissey JH, Edgington TS. Selective cellular expression of tissue factor in human tissues. Implications for disorders of hemostasis and thrombosis. Am J Pathol. 1989;134:1087–1097. [PMC free article] [PubMed] [Google Scholar]
- 19.Chen Z, Sager R. Differential expression of human tissue factor in normal mammary epithelial cells and in carcinomas. Mol Med. 1995;1:153–160. [PMC free article] [PubMed] [Google Scholar]
- 20.Mackman N. Tissue-specific hemostasis in mice. Arterioscler Thromb Vasc Biol. 2005;25:2273–2281. doi: 10.1161/01.ATV.0000183884.06371.52. [DOI] [PubMed] [Google Scholar]
- 21.Sullivan BP, Kopec AK, Joshi N, Cline H, Brown JA, Bishop SC, Kassel KM, Rockwell C, Mackman N, Luyendyk JP. Hepatocyte tissue factor activates the coagulation cascade in mice. Blood. 2013;121:1868–1874. doi: 10.1182/blood-2012-09-455436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Contrino J, Hair G, Kreutzer DL, Rickles FR. In situ detection of tissue factor in vascular endothelial cells: correlation with the malignant phenotype of human breast disease. Nat Med. 1996;2:209–215. doi: 10.1038/nm0296-209. [DOI] [PubMed] [Google Scholar]
- 23.Bazan JF. Structural design and molecular evolution of a cytokine receptor superfamily. Proc Natl Acad Sci. 1990;87:6934–6938. doi: 10.1073/pnas.87.18.6934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Martin DM, Boys CW, Ruf W. Tissue factor: molecular recognition and cofactor function. FASEB J. 1995;9:852–859. doi: 10.1096/fasebj.9.10.7615155. [DOI] [PubMed] [Google Scholar]
- 25.van den Berg YW, Osanto S, Reitsma PH, Versteeg HH. The relationship between tissue factor and cancer progression: insights from bench and bedside. Blood. 2012;119:924–932. doi: 10.1182/blood-2011-06-317685. [DOI] [PubMed] [Google Scholar]
- 26.Jiang X, Bailly MA, Panetti TS, Cappello M, Konigsberg WH, Bromberg ME. Formation of tissue factor-factor VIIa-factor Xa complex promotes cellular signaling and migration of human breast cancer cells. J Thromb Haemost. 2004;2:93–101. doi: 10.1111/j.1538-7836.2004.00545.x. [DOI] [PubMed] [Google Scholar]
- 27.Hjortoe GM, Petersen LC, Albrektsen T, Sorensen BB, Norby PL, Mandal SK, Pendurthi UR, Rao LV. Tissue factor-factor VIIa-specific up-regulation of IL-8 expression in MDA-MB-231 cells is mediated by PAR-2 and results in increased cell migration. Blood. 2004;103:3029–3037. doi: 10.1182/blood-2003-10-3417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Morris DR, Ding Y, Ricks TK, Gullapalli A, Wolfe BL, Trejo J. Protease-activated receptor-2 is essential for factor VIIa and Xa-induced signaling, migration, and invasion of breast cancer cells. Cancer Res. 2006;66:307–314. doi: 10.1158/0008-5472.CAN-05-1735. [DOI] [PubMed] [Google Scholar]
- 29.Jiang X, Zhu S, Panetti TS, Bromberg ME. Formation of tissue factor-factor VIIa-factor Xa complex induces activation of the mTOR pathway which regulates migration of human breast cancer cells. Thromb Haemost. 2008;100:127–133. doi: 10.1160/TH07-12-0722. [DOI] [PubMed] [Google Scholar]
- 30.Versteeg HH, Schaffner F, Kerver M, Petersen HH, Ahmed J, Felding-Habermann B, Takada Y, Mueller BM, Ruf W. Inhibition of tissue factor signaling suppresses tumor growth. Blood. 2008;111:190–199. doi: 10.1182/blood-2007-07-101048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Versteeg HH, Schaffner F, Kerver M, Ellies LG, Andrade-Gordon P, Mueller BM, Ruf W. Protease-activated receptor (PAR) 2, but not PAR1, signaling promotes the development of mammary adenocarcinoma in polyoma middle T mice. Cancer Res. 2008;68:7219–7227. doi: 10.1158/0008-5472.CAN-08-0419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schaffner F, Versteeg HH, Schillert A, Yokota N, Petersen LC, Mueller BM, Ruf W. Cooperation of tissue factor cytoplasmic domain and PAR2 signaling in breast cancer development. Blood. 2010;116:6106–6113. doi: 10.1182/blood-2010-06-289314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Arnaud E, Barbalat V, Nicaud V, Cambien F, Evans A, Morrison C, Arveiler D, Luc G, Ruidavets JB, Emmerich J, et al. Polymorphisms in the 5’ regulatory region of the tissue factor gene and the risk of myocardial infarction and venous thromboembolism: the ECTIM and PATHROS studies. Etude Cas-Témoins de l’Infarctus du Myocarde. Paris Thrombosis case-control Study. Arterioscler Thromb Vasc Biol. 2000;20:892–898. doi: 10.1161/01.atv.20.3.892. [DOI] [PubMed] [Google Scholar]
- 34.Terry CM, Kling SJ, Cheang KI, Hoidal JR, Rodgers GM. Polymorphisms in the 5’-UTR of the tissue factor gene are associated with altered expression in human endothelial cells. J Thromb Haemost. 2004;2:1351–1358. doi: 10.1111/j.1538-7836.2004.00770.x. [DOI] [PubMed] [Google Scholar]
- 35.DE Gaetano M, Quacquaruccio G, Pezzini A, Latella MC, DI Castelnuovo A, Del Zotto E, Padovani A, Lichy C, Grond-Ginsbach C, Gattone M, et al. Tissue factor gene polymorphisms and haplotypes and the risk of ischemic vascular events: four studies and a meta-analysis. J Thromb Haemost. 2009;7:1465–1471. doi: 10.1111/j.1538-7836.2009.03541.x. [DOI] [PubMed] [Google Scholar]
- 36.Isada A, Konno S, Hizawa N, Tamari M, Hirota T, Harada M, Maeda Y, Hattori T, Takahashi A, Nishimura M. A functional polymorphism (-603A --& gt; G) in the tissue factor gene promoter is associated with adult-onset asthma. J Hum Genet. 2010;55:167–174. doi: 10.1038/jhg.2010.4. [DOI] [PubMed] [Google Scholar]
- 37.Mackman N. Regulation of the tissue factor gene. FASEB J. 1995;9:883–889. doi: 10.1096/fasebj.9.10.7615158. [DOI] [PubMed] [Google Scholar]
- 38.Benz CC, Yau C. Ageing, oxidative stress and cancer: paradigms in parallax. Nat Rev Cancer. 2008;8:875–879. doi: 10.1038/nrc2522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhou JN, Ljungdahl S, Shoshan MC, Swedenborg J, Linder S. Activation of tissue-factor gene expression in breast carcinoma cells by stimulation of the RAF-ERK signaling pathway. Mol Carcinog. 1998;21:234–243. [PubMed] [Google Scholar]
- 40.Kato S, Pinto M, Carvajal A, Espinoza N, Monso C, Sadarangani A, Villalon M, Brosens JJ, White JO, Richer JK, et al. Progesterone increases tissue factor gene expression, procoagulant activity, and invasion in the breast cancer cell line ZR-75-1. J Clin Endocrinol Metab. 2005;90:1181–1188. doi: 10.1210/jc.2004-0857. [DOI] [PubMed] [Google Scholar]
- 41.Henriquez S, Calderon C, Quezada M, Oliva B, Bravo ML, Aranda E, Kato S, Cuello MA, Gutiérrez J, Quest AF, et al. Progesterone utilizes distinct membrane pools of tissue factor to increase coagulation and invasion and these effects are inhibited by TFPI. J Cell Physiol. 2011;226:3278–3285. doi: 10.1002/jcp.22689. [DOI] [PubMed] [Google Scholar]
- 42.Hu C, Huang L, Gest C, Xi X, Janin A, Soria C, Li H, Lu H. Opposite regulation by PI3K/Akt and MAPK/ERK pathways of tissue factor expression, cell-associated procoagulant activity and invasiveness in MDA-MB-231 cells. J Hematol Oncol. 2012;5:16. doi: 10.1186/1756-8722-5-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yu JL, May L, Lhotak V, Shahrzad S, Shirasawa S, Weitz JI, Coomber BL, Mackman N, Rak JW. Oncogenic events regulate tissue factor expression in colorectal cancer cells: implications for tumor progression and angiogenesis. Blood. 2005;105:1734–1741. doi: 10.1182/blood-2004-05-2042. [DOI] [PubMed] [Google Scholar]
- 44.Rong Y, Post DE, Pieper RO, Durden DL, Van Meir EG, Brat DJ. PTEN and hypoxia regulate tissue factor expression and plasma coagulation by glioblastoma. Cancer Res. 2005;65:1406–1413. doi: 10.1158/0008-5472.CAN-04-3376. [DOI] [PubMed] [Google Scholar]
- 45.Rong Y, Hu F, Huang R, Mackman N, Horowitz JM, Jensen RL, Durden DL, Van Meir EG, Brat DJ. Early growth response gene-1 regulates hypoxia-induced expression of tissue factor in glioblastoma multiforme through hypoxia-inducible factor-1-independent mechanisms. Cancer Res. 2006;66:7067–7074. doi: 10.1158/0008-5472.CAN-06-0346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang X, Yu H, Lou JR, Zheng J, Zhu H, Popescu NI, Lupu F, Lind SE, Ding WQ. MicroRNA-19 (miR-19) regulates tissue factor expression in breast cancer cells. J Biol Chem. 2011;286:1429–1435. doi: 10.1074/jbc.M110.146530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Riewald M, Ruf W. Mechanistic coupling of protease signaling and initiation of coagulation by tissue factor. Proc Natl Acad Sci USA. 2001;98:7742–7747. doi: 10.1073/pnas.141126698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jiang X, Guo YL, Bromberg ME. Formation of tissue factor-factor VIIa-factor Xa complex prevents apoptosis in human breast cancer cells. Thromb Haemost. 2006;96:196–201. [PubMed] [Google Scholar]
- 49.Disse J, Petersen HH, Larsen KS, Persson E, Esmon N, Esmon CT, Teyton L, Petersen LC, Ruf W. The endothelial protein C receptor supports tissue factor ternary coagulation initiation complex signaling through protease-activated receptors. J Biol Chem. 2011;286:5756–5767. doi: 10.1074/jbc.M110.201228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kumar P, Lau CS, Mathur M, Wang P, DeFea KA. Differential effects of beta-arrestins on the internalization, desensitization and ERK1/2 activation downstream of protease activated receptor-2. Am J Physiol Cell Physiol. 2007;293:C346–C357. doi: 10.1152/ajpcell.00010.2007. [DOI] [PubMed] [Google Scholar]
- 51.Ge L, Shenoy SK, Lefkowitz RJ, DeFea K. Constitutive protease-activated receptor-2-mediated migration of MDA MB-231 breast cancer cells requires both beta-arrestin-1 and -2. J Biol Chem. 2004;279:55419–55424. doi: 10.1074/jbc.M410312200. [DOI] [PubMed] [Google Scholar]
- 52.Zoudilova M, Kumar P, Ge L, Wang P, Bokoch GM, DeFea KA. Beta-arrestin-dependent regulation of the cofilin pathway downstream of protease-activated receptor-2. J Biol Chem. 2007;282:20634–20646. doi: 10.1074/jbc.M701391200. [DOI] [PubMed] [Google Scholar]
- 53.Liu Y, Mueller BM. Protease-activated receptor-2 regulates vascular endotherial growth factor expression in MDA-MB-231 cells via MAPK pathways. Biochem Biophys Res Commun. 2006;344:1263–1270. doi: 10.1016/j.bbrc.2006.04.005. [DOI] [PubMed] [Google Scholar]
- 54.Pendurthi UR, Allen KE, Ezban M, Rao LV. Factor VIIa and thrombin induce the expression of Cyr61 and connective tissue growth factor, extracellular matrix signaling proteins that could act as possible downstream mediators in factor VIIa x tissue factor-induced signal transduction. J Biol Chem. 2000;275:14632–14641. doi: 10.1074/jbc.275.19.14632. [DOI] [PubMed] [Google Scholar]
- 55.Wilson BJ, Harada R, LeDuy L, Hollenberg MD, Nepveu A. CUX1 transcription factor is a downstream effector of the proteinase-activated receptor 2 (PAR2) J Biol Chem. 2009;284:36–45. doi: 10.1074/jbc.M803808200. [DOI] [PubMed] [Google Scholar]
- 56.Albrektsen T, Sørensen BB, Hjortø GM, Fleckner J, Rao LV, Petersen LC. Transcriptional program induced by factor VIIa-tissue factor, PAR1 and PAR2 in MDA-MB-231 cells. J Thromb Haemost. 2007;5:1588–1597. doi: 10.1111/j.1538-7836.2007.02603.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bach RR. Tissue factor encryption. Arterioscler Thromb Vasc Biol. 2006;26:456–461. doi: 10.1161/01.ATV.0000202656.53964.04. [DOI] [PubMed] [Google Scholar]
- 58.Ahamed J, Versteeg HH, Kerver M, Chen VM, Mueller BM, Hogg PJ, Ruf W. Disulfide isomerization switches tissue factor from coagulation to cell signaling. Proc Natl Acad Sci USA. 2006;103:13932–13937. doi: 10.1073/pnas.0606411103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pendurthi UR, Ghosh S, Mandal SK, Rao LV. Tissue factor activation: is disulfide bond switching a regulatory mechanism? Blood. 2007;110:3900–3908. doi: 10.1182/blood-2007-07-101469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Versteeg HH, Ruf W. Tissue factor coagulant function is enhanced by protein-disulfide isomerase independent of oxidoreductase activity. J Biol Chem. 2007;282:25416–25424. doi: 10.1074/jbc.M702410200. [DOI] [PubMed] [Google Scholar]
- 61.Bajaj MS, Birktoft JJ, Steer SA, Bajaj SP. Structure and biology of tissue factor pathway inhibitor. Thromb Haemost. 2001;86:959–972. [PubMed] [Google Scholar]
- 62.Broze GJ, Girard TJ. Tissue factor pathway inhibitor: structure-function. Front Biosci (Landmark Ed) 2012;17:262–280. doi: 10.2741/3926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bajaj MS, Kuppuswamy MN, Manepalli AN, Bajaj SP. Transcriptional expression of tissue factor pathway inhibitor, thrombomodulin and von Willebrand factor in normal human tissues. Thromb Haemost. 1999;82:1047–1052. [PubMed] [Google Scholar]
- 64.Werling RW, Zacharski LR, Kisiel W, Bajaj SP, Memoli VA, Rousseau SM. Distribution of tissue factor pathway inhibitor in normal and malignant human tissues. Thromb Haemost. 1993;69:366–369. [PubMed] [Google Scholar]
- 65.Maroney SA, Cooley BC, Ferrel JP, Bonesho CE, Nielsen LV, Johansen PB, Hermit MB, Petersen LC, Mast AE. Absence of hematopoietic tissue factor pathway inhibitor mitigates bleeding in mice with hemophilia. Proc Natl Acad Sci USA. 2012;109:3927–3931. doi: 10.1073/pnas.1119858109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wojtukiewicz MZ, Sierko E, Zimnoch L, Kozlowski L, Kisiel W. Immunohistochemical localization of tissue factor pathway inhibitor-2 in human tumor tissue. Thromb Haemost. 2003;90:140–146. [PubMed] [Google Scholar]
- 67.Sierko E, Wojtukiewicz MZ, Zimnoch L, Kisiel W. Expression of tissue factor pathway inhibitor (TFPI) in human breast and colon cancer tissue. Thromb Haemost. 2010;103:198–204. doi: 10.1160/TH09-06-0416. [DOI] [PubMed] [Google Scholar]
- 68.Stavik B, Tinholt M, Sletten M, Skretting G, Sandset PM, Iversen N. TFPIα and TFPIβ are expressed at the surface of breast cancer cells and inhibit TF-FVIIa activity. J Hematol Oncol. 2013;6:5. doi: 10.1186/1756-8722-6-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Stavik B, Skretting G, Sletten M, Sandset PM, Iversen N. Overexpression of both TFPIα and TFPIβ induces apoptosis and expression of genes involved in the death receptor pathway in breast cancer cells. Mol Carcinog. 2010;49:951–963. doi: 10.1002/mc.20679. [DOI] [PubMed] [Google Scholar]
- 70.Stavik B, Skretting G, Aasheim HC, Tinholt M, Zernichow L, Sletten M, Sandset PM, Iversen N. Downregulation of TFPI in breast cancer cells induces tyrosine phosphorylation signaling and increases metastatic growth by stimulating cell motility. BMC Cancer. 2011;11:357. doi: 10.1186/1471-2407-11-357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fischer EG, Riewald M, Huang HY, Miyagi Y, Kubota Y, Mueller BM, Ruf W. Tumor cell adhesion and migration supported by interaction of a receptor-protease complex with its inhibitor. J Clin Invest. 1999;104:1213–1221. doi: 10.1172/JCI7750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Keshava S, Kothari H, Rao LV, Pendurthi UR. Influence of endothelial cell protein C receptor on breast cancer development. J Thromb Haemost. 2013;11:2062–2065. doi: 10.1111/jth.12402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Schaffner F, Yokota N, Carneiro-Lobo T, Kitano M, Schaffer M, Anderson GM, Mueller BM, Esmon CT, Ruf W. Endothelial protein C receptor function in murine and human breast cancer development. PLoS One. 2013;8:e61071. doi: 10.1371/journal.pone.0061071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yokota N, Zarpellon A, Chakrabarty S, Bogdanov VY, Gruber A, Castellino FJ, Mackman N, Ellies LG, Weiler H, Ruggeri ZM, et al. Contributions of thrombin targets to tissue factor-dependent metastasis in hyperthrombotic mice. J Thromb Haemost. 2014;12:71–81. doi: 10.1111/jth.12442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kamath L, Meydani A, Foss F, Kuliopulos A. Signaling from protease-activated receptor-1 inhibits migration and invasion of breast cancer cells. Cancer Res. 2001;61:5933–5940. [PubMed] [Google Scholar]
- 76.Booden MA, Eckert LB, Der CJ, Trejo J. Persistent signaling by dysregulated thrombin receptor trafficking promotes breast carcinoma cell invasion. Mol Cell Biol. 2004;24:1990–1999. doi: 10.1128/MCB.24.5.1990-1999.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Boire A, Covic L, Agarwal A, Jacques S, Sherifi S, Kuliopulos A. PAR1 is a matrix metalloprotease-1 receptor that promotes invasion and tumorigenesis of breast cancer cells. Cell. 2005;120:303–313. doi: 10.1016/j.cell.2004.12.018. [DOI] [PubMed] [Google Scholar]
- 78.Vrana JA, Stang MT, Grande JP, Getz MJ. Expression of tissue factor in tumor stroma correlates with progression to invasive human breast cancer: paracrine regulation by carcinoma cell-derived members of the transforming growth factor beta family. Cancer Res. 1996;56:5063–5070. [PubMed] [Google Scholar]
- 79.Collier ME, Li C, Ettelaie C. Influence of exogenous tissue factor on estrogen receptor alpha expression in breast cancer cells: involvement of beta1-integrin, PAR2, and mitogen-activated protein kinase activation. Mol Cancer Res. 2008;6:1807–1818. doi: 10.1158/1541-7786.MCR-08-0109. [DOI] [PubMed] [Google Scholar]
- 80.Shoji M, Hancock WW, Abe K, Micko C, Casper KA, Baine RM, Wilcox JN, Danave I, Dillehay DL, Matthews E, et al. Activation of coagulation and angiogenesis in cancer: immunohistochemical localization in situ of clotting proteins and vascular endothelial growth factor in human cancer. Am J Pathol. 1998;152:399–411. [PMC free article] [PubMed] [Google Scholar]
- 81.Bogdanov VY, Balasubramanian V, Hathcock J, Vele O, Lieb M, Nemerson Y. Alternatively spliced human tissue factor: a circulating, soluble, thrombogenic protein. Nat Med. 2003;9:458–462. doi: 10.1038/nm841. [DOI] [PubMed] [Google Scholar]
- 82.Kocatürk B, Van den Berg YW, Tieken C, Mieog JS, de Kruijf EM, Engels CC, van der Ent MA, Kuppen PJ, Van de Velde CJ, Ruf W, et al. Alternatively spliced tissue factor promotes breast cancer growth in a β1 integrin-dependent manner. Proc Natl Acad Sci USA. 2013;110:11517–11522. doi: 10.1073/pnas.1307100110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Amin C, Mackman N, Key NS. Microparticles and cancer. Pathophysiol Haemost Thromb. 2008;36:177–183. doi: 10.1159/000175155. [DOI] [PubMed] [Google Scholar]
- 84.Geddings JE, Mackman N. Tumor-derived tissue factor-positive microparticles and venous thrombosis in cancer patients. Blood. 2013;122:1873–1880. doi: 10.1182/blood-2013-04-460139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mukherjee SD, Swystun LL, Mackman N, Wang JG, Pond G, Levine MN, Liaw PC. Impact of chemotherapy on thrombin generation and on the protein C pathway in breast cancer patients. Pathophysiol Haemost Thromb. 2010;37:88–97. doi: 10.1159/000324166. [DOI] [PubMed] [Google Scholar]
- 86.Tormoen GW, Cianchetti FA, Bock PE, McCarty OJ. Development of coagulation factor probes for the identification of procoagulant circulating tumor cells. Front Oncol. 2012;2:110. doi: 10.3389/fonc.2012.00110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Van Den Berg YW, Reitsma PH. Not exclusively tissue factor: neutrophil extracellular traps provide another link between chemotherapy and thrombosis. J Thromb Haemost. 2011;9:2311–2312. doi: 10.1111/j.1538-7836.2011.04502.x. [DOI] [PubMed] [Google Scholar]
- 88.Lwaleed BA, Chisholm M, Francis JL. Urinary tissue factor levels in patients with breast and colorectal cancer. J Pathol. 1999;187:291–294. doi: 10.1002/(SICI)1096-9896(199902)187:3<291::AID-PATH213>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 89.Rydén L, Grabau D, Schaffner F, Jönsson PE, Ruf W, Belting M. Evidence for tissue factor phosphorylation and its correlation with protease-activated receptor expression and the prognosis of primary breast cancer. Int J Cancer. 2010;126:2330–2340. doi: 10.1002/ijc.24921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Greenberg D, Miao CH, Ho WT, Chung DW, Davie EW. Liver-specific expression of the human factor VII gene. Proc Natl Acad Sci USA. 1995;92:12347–12351. doi: 10.1073/pnas.92.26.12347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Erdmann D, Heim J. Orphan nuclear receptor HNF-4 binds to the human coagulation factor VII promoter. J Biol Chem. 1995;270:22988–22996. doi: 10.1074/jbc.270.39.22988. [DOI] [PubMed] [Google Scholar]
- 92.Pollak ES, Hung HL, Godin W, Overton GC, High KA. Functional characterization of the human factor VII 5’-flanking region. J Biol Chem. 1996;271:1738–1747. doi: 10.1074/jbc.271.3.1738. [DOI] [PubMed] [Google Scholar]
- 93.Kudaravalli R, Tidd T, Pinotti M, Ratti A, Santacroce R, Margaglione M, Dallapiccola B, Bernardi F, Fortina P, Devoto M, et al. Polymorphic changes in the 5’ flanking region of factor VII have a combined effect on promoter strength. Thromb Haemost. 2002;88:763–767. [PubMed] [Google Scholar]
- 94.Di Bitondo R, Hall AJ, Peake IR, Iacoviello L, Winship PR. Oestrogenic repression of human coagulation factor VII expression mediated through an oestrogen response element sequence motif in the promoter region. Hum Mol Genet. 2002;11:723–731. doi: 10.1093/hmg/11.7.723. [DOI] [PubMed] [Google Scholar]
- 95.Jackson AA, Cronin KR, Zachariah R, Carew JA. CCAAT/enhancer-binding protein-beta participates in insulin-responsive expression of the factor VII gene. J Biol Chem. 2007;282:31156–31165. doi: 10.1074/jbc.M704694200. [DOI] [PubMed] [Google Scholar]
- 96.Mennen LI, Schouten EG, Grobbee DE, Kluft C. Coagulation factor VII, dietary fat and blood lipids: a review. Thromb Haemost. 1996;76:492–499. [PubMed] [Google Scholar]
- 97.Miller GJ, Martin JC, Mitropoulos KA, Esnouf MP, Cooper JA, Morrissey JH, Howarth DJ, Tuddenham EG. Activation of factor VII during alimentary lipemia occurs in healthy adults and patients with congenital factor XII or factor XI deficiency, but not in patients with factor IX deficiency. Blood. 1996;87:4187–4196. [PubMed] [Google Scholar]
- 98.Chapman HA, Allen CL, Stone OL, Fair DS. Human alveolar macrophages synthesize factor VII in vitro. Possible role in interstitial lung disease. J Clin Invest. 1985;75:2030–2037. doi: 10.1172/JCI111922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wilcox JN, Noguchi S, Casanova J. Extrahepatic synthesis of factor VII in human atherosclerotic vessels. Arterioscler Thromb Vasc Biol. 2003;23:136–141. doi: 10.1161/01.atv.0000043418.84185.3c. [DOI] [PubMed] [Google Scholar]
- 100.Koizume S, Yokota N, Miyagi E, Hirahara F, Nakamura Y, Sakuma Y, Yoshida A, Kameda Y, Tsuchiya E, Ruf W, et al. Hepatocyte nuclear factor-4-independent synthesis of coagulation factor VII in breast cancer cells and its inhibition by targeting selective histone acetyltransferases. Mol Cancer Res. 2009;7:1928–1936. doi: 10.1158/1541-7786.MCR-09-0372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Koizume S, Miyagi Y. Ectopic synthesis of coagulation factor VII in breast cancer cells: Mechanisms, functional correlates, and potential for a new therapeutic target. Breast Cancer: Current and Alternative Therapeutic Modalities. In: Gunduz E, Gunduz M, editors. Croatia: INTECH; 2011. pp. 197–212. [Google Scholar]
- 102.Koizume S, Ito S, Miyagi E, Hirahara F, Nakamura Y, Sakuma Y, Osaka H, Takano Y, Ruf W, Miyagi Y. HIF2α-Sp1 interaction mediates a deacetylation-dependent FVII-gene activation under hypoxic conditions in ovarian cancer cells. Nucleic Acids Res. 2012;40:5389–5401. doi: 10.1093/nar/gks201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Strebhardt K, Ullrich A. Paul Ehrlich’s magic bullet concept: 100 years of progress. Nat Rev Cancer. 2008;8:473–480. doi: 10.1038/nrc2394. [DOI] [PubMed] [Google Scholar]
- 104.Ngo CV, Picha K, McCabe F, Millar H, Tawadros R, Tam SH, Nakada MT, Anderson GM. CNTO 859, a humanized anti-tissue factor monoclonal antibody, is a potent inhibitor of breast cancer metastasis and tumor growth in xenograft models. Int J Cancer. 2007;120:1261–1267. doi: 10.1002/ijc.22426. [DOI] [PubMed] [Google Scholar]
- 105.Carneiro-Lobo TC, Schaffner F, Disse J, Ostergaard H, Francischetti IM, Monteiro RQ, Ruf W. The tick-derived inhibitor Ixolaris prevents tissue factor signaling on tumor cells. J Thromb Haemost. 2012;10:1849–1858. doi: 10.1111/j.1538-7836.2012.04864.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Goel A, Aggarwal BB. Curcumin, the golden spice from Indian saffron, is a chemosensitizer and radiosensitizer for tumors and chemoprotector and radioprotector for normal organs. Nutr Cancer. 2010;62:919–930. doi: 10.1080/01635581.2010.509835. [DOI] [PubMed] [Google Scholar]
- 107.Bierhaus A, Zhang Y, Quehenberger P, Luther T, Haase M, Müller M, Mackman N, Ziegler R, Nawroth PP. The dietary pigment curcumin reduces endothelial tissue factor gene expression by inhibiting binding of AP-1 to the DNA and activation of NF-kappa B. Thromb Haemost. 1997;77:772–782. [PubMed] [Google Scholar]
- 108.Pendurthi UR, Williams JT, Rao LV. Inhibition of tissue factor gene activation in cultured endothelial cells by curcumin. Suppression of activation of transcription factors Egr-1, AP-1, and NF-kappa B. Arterioscler Thromb Vasc Biol. 1997;17:3406–3413. doi: 10.1161/01.atv.17.12.3406. [DOI] [PubMed] [Google Scholar]
- 109.Xia L, Zhou H, Hu L, Xie H, Wang T, Xu Y, Liu J, Zhang X, Yan J. Both NF-κB and c-Jun/AP-1 involved in anti-β2GPI/β2GPI-induced tissue factor expression in monocytes. Thromb Haemost. 2013;109:643–651. doi: 10.1160/TH12-09-0655. [DOI] [PubMed] [Google Scholar]
- 110.Hu Z, Rao B, Chen S, Duanmu J. Targeting tissue factor on tumour cells and angiogenic vascular endothelial cells by factor VII-targeted verteporfin photodynamic therapy for breast cancer in vitro and in vivo in mice. BMC Cancer. 2010;10:235. doi: 10.1186/1471-2407-10-235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Duanmu J, Cheng J, Xu J, Booth CJ, Hu Z. Effective treatment of chemoresistant breast cancer in vitro and in vivo by a factor VII-targeted photodynamic therapy. Br J Cancer. 2011;104:1401–1409. doi: 10.1038/bjc.2011.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Shoji M, Sun A, Kisiel W, Lu YJ, Shim H, McCarey BE, Nichols C, Parker ET, Pohl J, Mosley CA, et al. Targeting tissue factor-expressing tumor angiogenesis and tumors with EF24 conjugated to factor VIIa. J Drug Target. 2008;16:185–197. doi: 10.1080/10611860801890093. [DOI] [PubMed] [Google Scholar]
- 113.Liu Y, Jiang P, Capkova K, Xue D, Ye L, Sinha SC, Mackman N, Janda KD, Liu C. Tissue factor-activated coagulation cascade in the tumor microenvironment is critical for tumor progression and an effective target for therapy. Cancer Res. 2011;71:6492–6502. doi: 10.1158/0008-5472.CAN-11-1145. [DOI] [PubMed] [Google Scholar]
- 114.Morimoto T, Sunagawa Y, Kawamura T, Takaya T, Wada H, Nagasawa A, Komeda M, Fujita M, Shimatsu A, Kita T, et al. The dietary compound curcumin inhibits p300 histone acetyltransferase activity and prevents heart failure in rats. J Clin Invest. 2008;118:868–878. doi: 10.1172/JCI33160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Li HL, Liu C, de Couto G, Ouzounian M, Sun M, Wang AB, Huang Y, He CW, Shi Y, Chen X, et al. Curcumin prevents and reverses murine cardiac hypertrophy. J Clin Invest. 2008;118:879–893. doi: 10.1172/JCI32865. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 116.Shah S, Prasad S, Knudsen KE. Targeting pioneering factor and hormone receptor cooperative pathways to suppress tumor progression. Cancer Res. 2012;72:1248–1259. doi: 10.1158/0008-5472.CAN-11-0943. [DOI] [PMC free article] [PubMed] [Google Scholar]