

Abstract
Catel-Manzke syndrome is characterized by Pierre Robin sequence and a unique form of bilateral hyperphalangy causing a clinodactyly of the index finger. We describe the identification of homozygous and compound heterozygous mutations in TGDS in seven unrelated individuals with typical Catel-Manzke syndrome by exome sequencing. Six different TGDS mutations were detected: c.892A>G (p.Asn298Asp), c.270_271del (p.Lys91Asnfs∗22), c.298G>T (p.Ala100Ser), c.294T>G (p.Phe98Leu), c.269A>G (p.Glu90Gly), and c.700T>C (p.Tyr234His), all predicted to be disease causing. By using haplotype reconstruction we showed that the mutation c.298G>T is probably a founder mutation. Due to the spectrum of the amino acid changes, we suggest that loss of function in TGDS is the underlying mechanism of Catel-Manzke syndrome. TGDS (dTDP-D-glucose 4,6-dehydrogenase) is a conserved protein belonging to the SDR family and probably plays a role in nucleotide sugar metabolism.
Main Text
Catel-Manzke syndrome (MIM 302380) is characterized by Pierre Robin sequence (MIM 261800; HP:0000201)1 and a unique form of bilateral hyperphalangy causing clinodactyly of the index finger (HP:0009467). The condition was first described by Catel2 in 1961 and Manzke3 in 1966. Pierre Robin sequence is defined by micrognathia, obstruction of the airways due to a backward displacement of the tongue base, and, often but not always, cleft palate.4 The bilateral clinodactyly is caused by an accessory bone between the second metacarpal and its corresponding proximal phalanx, resulting in radial deviation of the index finger.5 This is sometimes referred to as Manzke dysostosis. Because not all reported cases have a cleft palate, Manzke suggested replacing the term palatodigital syndrome by the term micrognathia-digital syndrome.5
Previously, 26 individuals from 24 families with typical Catel-Manzke syndrome have been reported2,3,6–23 (reviewed by Manzke et al.5). Ten cases of an atypical form of Catel-Manzke syndrome have been described, including individuals with an extended phenotype20,24–28 and case subjects with unilateral hyperphalangism.15,29,30 Additionally, two cases of Manzke dysostosis without Pierre Robin sequence have been reported.17,31 In one family two affected girls and in a second family two affected boys were observed.12,14 The latter observations and parental consanguinity in another family20 indicate autosomal-recessive inheritance.
We identified homozygous and compound heterozygous mutations in TGDS in seven unrelated individuals with typical Catel-Manzke syndrome by using exome sequencing. Clinical data and mutations are summarized in Table 1.
Table 1.
TGDS Mutations and Clinical Presentation of the Seven Analyzed Individuals with Typical Catel-Manzke Syndrome
| Individual 1 | Individual 2 | Individual 35 | Individual 45 | Individual 52,3,5 | Individual 618 | Individual 720 | |
|---|---|---|---|---|---|---|---|
| Sex | male | female | female | female | male | female | female |
| Consanguinity | none | none | none | none | none | none | none |
| Ethnicity | Cameroon | British/ South American | German | German | German | Dutch | French |
| Age at last exam | 18 months | 3.5 years | 16 months | 19 years | 52 years | 17 years | 28 years |
| Mutation | |||||||
| 1st mutation | c.892A>G (p.Asn298Asp) | c.298G>T (p.Ala100Ser) | c.298G>T (p.Ala100Ser) | c.298G>T (p.Ala100Ser) | c.298G>T (p.Ala100Ser) | c.298G>T (p.Ala100Ser) | c.298G>T (p.Ala100Ser) |
| 2nd mutation | c.270_271del (p.Lys91Asnfs∗22) | c.294T>G (p.Phe98Leu) | – | – | – | c.700T>C (p.Tyr234His) | c.269A>G (p.Glu90Gly) |
| Type | compound heterozygous | compound heterozygous | homozygous | homozygous | homozygous | compound heterozygous | compound heterozygous |
| Clinical Manifestations | |||||||
| Pierre Robin sequence | + | + | + | + | + | + | + |
| Cleft palate | +, V-shaped | +, U-shaped | +, U-shaped | + | +, U-shaped | high arched palate, bifid uvula | +, U-shaped |
| Manzke dysostosis | + | + | + | + | + | + | + |
| Joint hypermobility | nr | nr | nr | nr | nr | + | + |
| Congenital heart defect | – | VSD, spontaneous closure | – | – | – | VSD | – |
| Facial dysmorphism (additional to micrognathia) | low-set ears, prominent anthelices | hypertelorism, upslanted palpebral fissures, thin eyebrows | narrow nostrils, thin arched eyebrows, full cheeks, hypertelorism | mild hypertelorism, narrow nose | nr | thin eyebrows, mild proptosis, long columella, low-set ears, dysplastic helices, small mouth | nr |
| Other skeletal anomalies | short toes, short humeri, short femora | nr | nr | clinodactyly V, genua valga | clinodactyly V, pectus deformity | clinodactyly V, bilateral M. Perthes | brachymetacarpia, scoliosis |
| Other features | adducted thumbs, feet edema, short neck, wide fontanelle | nr | postnatal growth retardation | obstruction of nasolacrimal duct | nr | bronchial hyperreactivity in early childhood | postnatal growth retardation, hearing loss |
Abbreviations are as follows: nr, not reported; VSD, ventricular septal defect.
Individual 1 is a male infant with healthy, unrelated parents from Cameroon with an unremarkable family history. He was delivered in the 39th week of pregnancy with a birth weight of 2,770 g (−1.5 SD), length of 45.5 cm (−2 SD), and head circumference of 33 cm (−1.6 SD). The diagnosis of Catel-Manzke syndrome was made at the age of 6 days. Intubation and ventilation were required due to a Pierre Robin sequence with severe retrognathia and cleft palate (Figures 1A and 1B). He had bilateral radial deviation and clinodactyly of the second digit and a mild ulnar deviation of the third and fourth finger. Radiographs showed the typical Manzke dysostosis (Figures 1C and 1D). Additionally, he had a wide anterior fontanelle, low set ears with prominent anthelices, a short neck, short humeri and femora, adducted thumbs, edema of the feet and eyelids, and short toes. Radiographs of humeri, femora, and pelvis were normal. When last examined at the age of 18 months, he still had retrognathia and a tracheostomy. However, he had developed well, was able to walk, and had started speaking. His height was 77 cm (−2.7 SD), his head circumference 48.5 cm (+0.2 SD), and his weight 10.2 kg (−0.3 SD).
Figure 1.
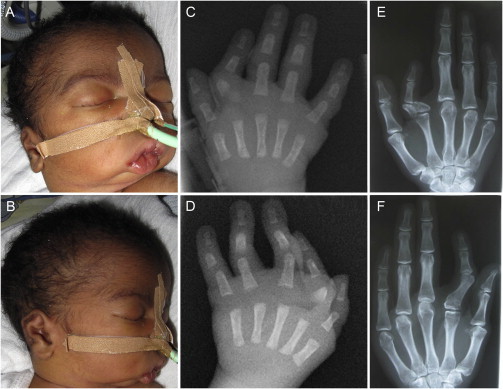
Clinical Features of Individuals 1 and 7
(A and B) Clinical photographs of individual 1 performed at the age of 6 days showing severe micrognathia, a short neck, and low set and dysplastic ears.
(C and D) Hand radiographs of individual 1 at the age of 6 days showing the Manzke dysostosis.
(E and F) Hand radiographs of individual 7 performed at the age of 28 years showing bilateral radial deviation and clinodactyly of the second digit. On the right hand a triangular-shaped bone is inserted between the second metacarpal and its proximal phalanx, and on the left hand the accessory bone and proximal phalanx fused.
Individual 2 is the second child of British and South American nonconsanguineous parents and has a healthy older sister. She was delivered at term after an uncomplicated pregnancy, with birth weight of 3,534 g (+0.3 SD) and head circumference of 35 cm (+0.25 SD). She was noted to have a cleft palate, micrognathia, bilateral hand abnormalities, mild hypertelorism, upslanting palpebral fissures, and thin eyebrows (Figures 2A–2D and 2F). Echocardiogram revealed a ventricular septal defect (VSD), and hand radiographs performed at the age of 3 months showed a triangular-shaped bone inserted between the second metacarpal and its proximal phalanx bilaterally causing radial deviation of the index fingers. On the left hand an additional pin-shaped ossification center was visible (Figures 2E and 2G). She underwent cleft palate repair and jaw distraction procedures. The VSD closed spontaneously, and her development progressed normally. When last seen at the age of 3.5 years, her height was 90 cm (−2.7 SD), head circumference was 49.3 cm (−0.4 SD), and weight was 13.36 kg (+0.2 SD).
Figure 2.
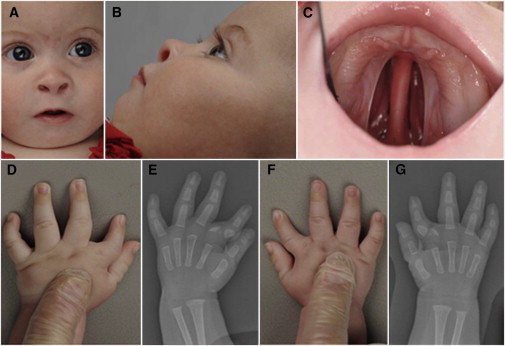
Clinical Features of Individual 2
(A–C) Clinical photographs of individual 2 performed at the age of 5 months showing micrognathia, upslanting palpebral fissures, short nose with anteverted nares, thin eyebrows, and cleft palate.
(D and F) Clinical photographs of both hands of individual 2 showing the clinodactyly of the index fingers, which is more pronounced on the left side.
(E and G) Hand radiographs of individual 2 performed at the age of 3 months showing a triangular-shaped bone inserted between the second metacarpal and its proximal phalanx bilaterally causing radial deviation of the index finger. On the left hand an additional pin-shaped ossification center is visible.
Individuals 3, 4, and 5 correspond to individuals 1, 2, and 3 reported by Manzke and coworkers in 2008 and are of German descent.5 Individual 5 is the individual first described by Catel and Manzke. No DNA samples of the parents of individual 3 were available. Individual 4 was last examined at the age of almost 20 years. She underwent bilateral osteotomy due to genua valga but is otherwise healthy. The mother of individual 4 was included in our study; the father, however, was unavailable for segregation analysis. Individual 5 has two unaffected children from a nonconsanguineous marriage who were included in our study. His unaffected parents were not available for analysis.
Individual 6 is the second child of healthy, unrelated Dutch parents and is now a 17-year-old adolescent who was already reported by Kant and coworkers in 1998.18 Growth and development were normal. She now has thin eyebrows, mild proptosis, low-set ears with dysplastic helices, a long columella, and a small mouth with high palate and slightly bifid uvula. She developed Perthes disease affecting the right hip at the age of 9 years and the left hip at the age of about 13 years. Her right leg is longer because of incomplete recovery of the left hip. She has subluxation of both knees, hypermobile elbow joints, and broad feet. Her older brother is unaffected.
Individual 7 is a woman who was reported as individual 3 by Nizon and coworkers in 2012 (Figures 1E and 1F).20 She has healthy, unrelated Northern French parents, who were not available for segregation analysis.
Individuals of family 1 (proband and parents), family 2 (proband and parents), family 4 (proband and mother), family 5 (proband and children), and the affected individual of family 7 were subjected to exome sequencing after approval of the ethical boards of the Charité, the Christian-Albrecht-University, and the University of Paris Decartes. The probands and/or parents gave their written consent for genetic testing and publication of images. High-throughput sequencing and data processing were performed in NGS-core facilities of the respective universities or collaborating service providers.
Eight exomes from individuals 2, 4, 5, and their relatives were captured with SeqCap EZ Human Exome Library version 2.0 kit (Roche NimbleGen). The finished libraries were sequenced on an Illumina HiSeq 2000 sequencing instrument (Illumina) with a paired-end 2 × 100 bp protocol. On average, this resulted in 6.7 Gb of mapped unique sequences with a mean coverage of 80×, i.e., 20× coverage for 91% of target sequences and 80× coverage for 85% of target sequences. The CCG VARBANK pipeline v.2.1 was used for data analysis and filtering with bwa-aln plus hg19 for mapping of reads in combination with the GATK toolkit for base quality score recalibration (BQSR), local realignment, and variant quality score recalibration (VQSR). GATK and samtools mpileup were used to call SNVs and short indels. Software developed in-house was used to annotate variants on a functional level (see Web Resources). Four DNA samples, including individual 1, his parents, and individual 7, were subjected to exome analysis with SureSelect All Exon Kit V2 (Agilent) for targeted enrichment followed by sequencing with Illumina’s HiSeq system (paired-end 2 × 100 bp). Sequence reads were mapped to the haploid human reference genome (hg19) using Novoalign (Novocraft Technologies). Single-nucleotide variants (SNVs) and short insertions and deletions (indels) were called with GATK toolkit according to the best practice guidelines and resulted in a high-quality exome variant set.32–34 The variant annotation on a functional level was performed with Jannovar and GeneTalk was used for filtering and further data analysis.35,36
Because a separate analysis of the families did not yield a single candidate gene, we combined all affected individuals into a case group and selected control subjects that matched the ethnicity and data quality of the cases for filtering. We assumed that the prevalence of the disorder is below 1 in 1,000,000 and has a high penetrance. We considered a recessive as well as a dominant mode of inheritance. Because none of the families had affected individuals in more than one generation, we filtered for potential de novo mutations when analyzing the dominant model of inheritance and could not identify a candidate gene. For the analysis of a recessive mode of inheritance, we first filtered the sequence variants in the samples based on genotype frequency data from large population genetics studies.37,38 We removed sequence variants that occurred in a homozygous state in more than one individual of the healthy controls. Alleles that had a heterozygous state in the affected individuals were removed if their allele frequency was above 0.01 in healthy controls.
The analysis of the autosomal-recessive model of inheritance yielded three candidate genes: MUC4 (MIM 158372), MUC6 (MIM 158374), and TGDS. Because mucin genes are known to be highly variable, we focused on TGDS as the most likely candidate gene. The following variants in TGDS (RefSeq accession number NM_014305.2) were identified: c.892A>G (p.Asn298Asp), c.270_271del (p.Lys91Asnfs∗22), c.298G>T (p.Ala100Ser), c.294T>G (p.Phe98Leu), and c.269A>G (p.Glu90Gly). We analyzed two additional individuals with Catel-Manzke syndrome by Sanger sequencing of all coding exons of TGDS and identified the homozygous variant c.298G>T in individual 3 and compound heterozygosity for the variants c.700T>C (p.Tyr234His); c.298G>T in individual 6. In total, six different variants (five missense and one nonsense) in TGDS were detected (Table 1). All candidate mutations were validated by ABI Sanger sequencing (sequencing primers are available upon request). Sanger sequencing demonstrated a biparental mode of inheritance of the compound heterozygous mutations in the parents except individual 7 whose parents were not available. We analyzed seven individuals with Catel-Manzke syndrome and all of them carried homozygous or compound heterozygous mutations in TGDS.
The function prediction algorithm MutationTaster39 classified all variants as disease causing. SIFT40 predicted only the amino acid change p.Phe98Leu and p.Tyr234His to be “tolerated,” and PolyPhen-241 predicted only the amino acid change p.Phe98Leu to be “benign.” All other alterations were also predicted to be “probably damaging” or “damaging.” The differences in prediction outputs are most likely due to different protein sequence alignments.42 The positions Ala100 and Tyr234 are completely conserved throughout evolution including E. coli (Figure 3B). The position Glu90 is conserved in zebrafish, the position Phe98 in C. elegans, and the position Asn298 in frog (not shown). Altogether we classify all substitutions to be disease causing. The amino acid change p.Lys91Asnfs∗22 is predicted to cause nonsense-mediated decay due to the truncation of the protein with the loss of 229 aa residues (out of a total of 350) at the C terminus.
Figure 3.
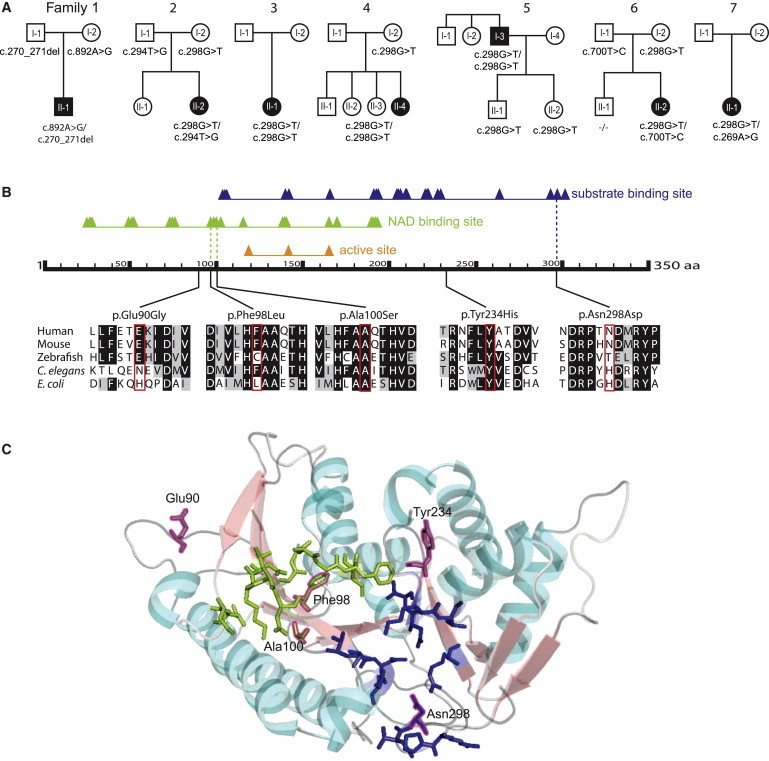
Segregation Analysis of the TGDS Variants and Predicted Effects on the Protein
(A) Pedigree structures and TGDS genotypes for the seven families.
(B) Schematic of human TGDS showing the predicted domains, the position of the amino acid changes, and the conservation. RefSeq accession numbers are as follows: human, NM_014305.2; mouse, NP_083854; zebrafish, AAH_66615; E. coli, WP_001710460; C. elegans, NP_508390. The predicted domains are based on the Conserved Domain Database. The triangles show conserved positions. The alignment was created with BoxShade and ClustalW. The colored background of the aa in the different species indicates the degree of conservation (black, conserved; gray, similarity in properties; white, not conserved, no similarity in properties).
(C) Predicted position of the amino acid changes in the structure of TGDS in Pyrococcus horikoshii. The structure of TGDS is shown as a model created with PyMol. The substrate binding domain is shown in blue, and the NAD binding domain is in light green. The positions aligning with the amino acid changes in TGDS in our cohort are framed in magenta. Ala100 and Phe98 are localized in the NAD-binding domain, and Asn298 is in the substrate binding domain. Glu90 and Tyr234 are localized in two of the loops.
Except for c.298G>T and c.270_271del, all identified variants were not previously reported in the databases. The missense mutation c.298G>T is a known variant, rs140430952, and was observed in a heterozygous state in 9 out of 4,300 healthy individuals of European descent that were analyzed in the National Heart, Lung, and Blood Institute (NHLBI) Exome Sequencing Project (ESP) and in 40 out of 61,377 individuals that were analyzed by the Exome Aggregation Consortium (ExAC). The variant c.270_271del was observed in a heterozygous state in 2 out of 2,132 individuals of African descent in the ESP data and in 4 out of 61,447 individuals in the ExAC data. However, both sequence variants, c.298T and c.270_271, did not occur in a homozygous state in healthy control subjects, supporting our hypothesis that these alleles are pathogenic.
If we assume a carrier rate of 0.002 for c.298T in the general European population, we would expect a homozygous individual in 1 out of 1,000,000, which is in good agreement with the estimated prevalence for Catel-Manzke syndrome. The relatively high frequency of c.298T in the European population might be due to a founder effect. We extracted sequence variants around c.298 from the available exome data for all affected individuals and estimated the haplotypes with the statistical software package PHASE v.2.1.43 Based on ten informative markers, we could reconstruct a 50 kb haplotype that is shared by all individuals with the pathogenic allele c.298T, supporting our hypothesis of a founder mutation (Tables S1–S3 available online).
TGDS (dTDP-D-glucose 4,6-dehydratase/growth-inhibiting protein 21) is an evolutionarily conserved 350 aa protein belonging to the short-chain dehydrogenase/reductase (SDR) family. It shares a sequence identity of 35% with its E. coli ortholog,44 which catalyzes the synthesis of dTDP-4-oxo-6-deoxy-D-glucose from dTDP-glucose as part of the rhamnose pathway. Because rhamnose, a common component of the cell wall and the capsule of many pathogenic bacteria, does not exist in humans,45 the specific function of TGDS and dTDP-4-oxo-6-deoxy-D-glucose in humans is unknown.
SDRs are structured as a one-domain Rossman fold that typically consists of a central beta sheet flanked by three alpha helices on each side. The beta sheet and the alpha helices show high conservation, although there are differences at the loops that are involved in substrate binding.46 According to the NCBI Conserved Domain Database, human TGDS is predicted to contain an active site, a NADP binding site, a substrate binding site, and a homodimer interface (Figure 3B). The amino acid changes p.Ala100Ser and p.Phe98Leu are located in the NAD binding domain, the amino acid change p.Asn298Asp is in the substrate binding site (Figures 3B and 3C), and we presume that the substitutions impair the functions of the respective domains. The amino acid changes p.Glu90Gly and Tyr234His, which are predicted to be located in two of the loops, possibly change the substrate binding potential of TGDS (Figure 3C). This could be explained by the loss of the negatively charged, hydrophilic properties of glutamine by substitution with the hydrophobic amino acid glycine and the loss of the aromatic, hydrophobic properties of tyrosine by substitution with the positively charged, hydrophilic amino acid histidine. Because of its proximity, Tyr243His could also affect the catalytic site.
More than 70 SDR genes exist in humans and have roles in steroid hormone, prostaglandin, and retinoid metabolisms, as well as extracellular matrix formation.47 The closest paralog of TGDS in the human genome is UXS1 (UGD; UDP-glucoronate decarboxylase 1), which shares an identity of 27% at the protein level (UXS1 isoform 1, RefSeq NP_ 001240804 [MIM 609749]). Mice homozygous for a null allele of UXS1 die prenatally.48 UXS1 encodes a 425 aa enzyme localized in the perinuclear Golgi compartment and catalyzes the synthesis of UDP-xylose of UDP-glucoronate, required in glycosaminoglycan (GAG) synthesis on proteoglycans. UXS1 does not have any dehydratase activity, emphasizing that UXS1 and TGDS are probably involved in different enzymatic processes.49
The features of Catel-Manzke syndrome demonstrate clinical overlap with Desbuquois dysplasia 1 (DBQD1 [MIM 251450]), caused by mutations in CANT150 (MIM 613165), Temtamy brachydactyly syndrome (TPBS [MIM 605282]), caused by mutations in CHSY151 (MIM 601883), and chondrodysplasia with joint dislocations, GPAPP type (GPAPP deficiency [MIM 614078]), caused by mutations in IMPAD152 (MIM 614010). Extra ossification centers distal to the second metacarpal50 and micrognathia have been described for several individuals with Desbuquois dysplasia 1. Hyperphalangism of digits 1–3 is a constant feature of Temtamy brachydactyly syndrome and some individuals have a cleft palate.53 IMPAD1 mutations were also identified in two individuals classified as Catel-Manzke-like with micrognathia and cleft palate, extra ossification centers, and additional features similar to GPAPP deficiency.20
Both CANT1 and CHSY1 are involved in proteoglycan synthesis.54,55 IMPAD1 is involved in proteoglycan sulfation.52 Based on the clinical overlap to these disorders and the homology to UXS1, it is plausible that TGDS could also be involved in either proteoglycan synthesis or sulfation. Additional features like congenital heart defects, joint hyperlaxity, pectus deformity, and scoliosis indicate that alterations in TGDS have a general effect on connective tissue, although milder than alterations in IMPAD1, CANT1, and CHSY1. Due to the spectrum of the amino acid substitutions, we suggest that loss of function in TGDS is the underlying mechanism of Catel-Manzke syndrome. Because none of the individuals carries two nonsense mutations, the missense mutations are probably hypofunctional alleles that do not cause complete loss of function.
In summary, we identified homozygous and compound heterozygous mutations in TGDS as disease causing in a cohort of individuals with Catel-Manzke syndrome. By using haplotype reconstruction, we showed that the mutation c.298G>T is probably a founder mutation. Because of the clinical overlap of Catel-Manzke syndrome with a spectrum of disorders caused by defects in proteoglycan synthesis or sulfation, we suppose that TGDS could also be involved in nucleotide sugar metabolism.
Acknowledgments
We would like to thank the families for their collaboration and contribution to this project, David Genevieve for his support, as well as Randi Kroll, Fabienne Pritsch, Gundula Leschik, Catrin Janetzki, Christian Becker, and Elisabeth Kirst for their technical assistance. We are indebted to Ammi Grahn and Emile Van Schaftingen for performing biochemical assays. U.K. received funding from the EU (EURO-CDG). Exome sequencing analysis for individual 7 has been supported by a GIS maladies rares funding A11144KS. The major part of exome data was analyzed on a high-performance computer cluster of the Regional Computing Centre of Cologne (RRZK), grant-aided by the Deutsche Forschungsgemeinschaft (DFG). P.N. is a founder, CEO, and shareholder of ATLAS Biolabs GmbH, which is a service provider for genomic analyses. P.M.K. is a founder, CEO, and shareholder of GeneTalk and received a grant from the DFG (KR3895/1-1). S.M. was supported by grants from the Deutsche Forschungsgemeinschaft and the Max Planck Foundation.
Contributor Information
Nadja Ehmke, Email: nadja.ehmke@charite.de.
Stefan Mundlos, Email: stefan.mundlos@charite.de.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes, http://browser.1000genomes.org
BoxShade Server, http://embnet.vital-it.ch/software/BOX_form.html
ClustalW Server, http://embnet.vital-it.ch/software/ClustalW.html
Ensembl Genome Browser, http://www.ensembl.org/index.html
ExAC Browser, http://exac.broadinstitute.org/
GeneTalk, http://www.gene-talk.de/
Human Phenotype Ontology (HPO), http://www.human-phenotype-ontology.org/
MGI Batch Query, http://www.informatics.jax.org/batch
MutationTaster, http://www.mutationtaster.org/
NCBI Conserved Domains, http://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
PolyPhen-2, http://www.genetics.bwh.harvard.edu/pph2/
Protein BLAST, http://www.ncbi.nlm.nih.gov/BLAST/Blast.cgi?PAGE=Proteins
PyMOL, http://www.pymol.org
SIFT Human Protein, http://sift.jcvi.org/www/SIFT_enst_submit.html
UCSC Genome Browser, http://genome.ucsc.edu
varbank, https://varbank.ccg.uni-koeln.de
References
- 1.Robinson P.N., Köhler S., Bauer S., Seelow D., Horn D., Mundlos S. The Human Phenotype Ontology: a tool for annotating and analyzing human hereditary disease. Am. J. Hum. Genet. 2008;83:610–615. doi: 10.1016/j.ajhg.2008.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Catel W. G. Thieme; Stuttgart: 1961. Differentialdiagnose von Krankheitssymptomen bei Kindern und Jugendlichen. [Google Scholar]
- 3.Manzke H. [Symmetrical hyperphalangy of the second finger by a supplementary metacarpus bone] Fortschr. Geb. Rontgenstr. Nuklearmed. 1966;105:425–427. [PubMed] [Google Scholar]
- 4.Sheffield L.J., Reiss J.A., Strohm K., Gilding M. A genetic follow-up study of 64 patients with the Pierre Robin complex. Am. J. Med. Genet. 1987;28:25–36. doi: 10.1002/ajmg.1320280105. [DOI] [PubMed] [Google Scholar]
- 5.Manzke H., Lehmann K., Klopocki E., Caliebe A. Catel-Manzke syndrome: two new patients and a critical review of the literature. Eur. J. Med. Genet. 2008;51:452–465. doi: 10.1016/j.ejmg.2008.03.005. [DOI] [PubMed] [Google Scholar]
- 6.Farnsworth P.B., Pacik P.T. Glossoptotic hypoxia and micrognathia—the Pierre Robin syndrome reviewed. Early recognition and prompt surgical treatment is important for survival. Clin. Pediatr. (Phila.) 1971;10:600–606. doi: 10.1177/000992287101001029. [DOI] [PubMed] [Google Scholar]
- 7.Gorlin R.J., Cervenka J., Pruzansky S. Facial clefting and its syndromes. Birth Defects Orig. Artic. Ser. 1971;7:3–49. [PubMed] [Google Scholar]
- 8.Holthusen W. [The Pierre Robin syndrome: unusual associated developmental defects] Ann. Radiol. (Paris) 1972;15:253–262. [PubMed] [Google Scholar]
- 9.Silengo M.C., Franceschini P., Cerutti A., Fabris C. Pierre Robin syndrome with hyperphalangism-clinodactylysm of the index finger: a possible new palato-digital syndrome. Pediatr. Radiol. 1977;6:178–180. doi: 10.1007/BF00972113. [DOI] [PubMed] [Google Scholar]
- 10.Gewitz M., Dinwiddie R., Yuille T., Hill F., Carter C.O. Cleft palate and accessory metacarpal of index finger syndrome: possible familial occurrence. J. Med. Genet. 1978;15:162–164. doi: 10.1136/jmg.15.2.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Klug M.S., Ketchum L.D., Lipsey J.H. Symmetric hyperphalangism of the index finger in the palatodigital syndrome: a case report. J. Hand Surg. Am. 1983;8:599–603. doi: 10.1016/s0363-5023(83)80135-x. [DOI] [PubMed] [Google Scholar]
- 12.Brude E. Pierre Robin sequence and hyperphalangy—a genetic entity (Catel-Manzke syndrome) Eur. J. Pediatr. 1984;142:222–223. doi: 10.1007/BF00442455. [DOI] [PubMed] [Google Scholar]
- 13.Thompson E.M., Winter R.M., Williams M.J. A male infant with the Catel-Manzke syndrome and dislocatable knees. J. Med. Genet. 1986;23:271–274. doi: 10.1136/jmg.23.3.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dignan P.S., Martin L.W., Zenni E.J., Jr. Pierre Robin anomaly with an accessory metacarpal of the index fingers. The Catel-Manzke syndrome. Clin. Genet. 1986;29:168–173. doi: 10.1111/j.1399-0004.1986.tb01244.x. [DOI] [PubMed] [Google Scholar]
- 15.Skinner S.A., Sr., Flannery D.B. Catel-Manzke Syndrome. Proc. Greenwood Genetic Center. 1989;8:60–63. [Google Scholar]
- 16.Bernd L., Martini A.K., Schiltenwolf M., Graf J. [Hyperphalangia in Pierre-Robin syndrome] Z. Orthop. Ihre Grenzgeb. 1990;128:463–465. doi: 10.1055/s-2008-1039597. [DOI] [PubMed] [Google Scholar]
- 17.Buck-Gramcko D. Chuchvill Livingstone; London: 1998. Congenital Malformations of the Hand and Forearm. [DOI] [PubMed] [Google Scholar]
- 18.Kant S.G., Oudshoorn A., Gi C.V., Zonderland H.M., Van Haeringen A. The Catel-Manzke syndrome in a female infant. Genet. Couns. 1998;9:187–190. [PubMed] [Google Scholar]
- 19.Puri R.D., Phadke S.R. Catel-Manzke syndrome without cleft palate: a case report. Clin. Dysmorphol. 2003;12:279–281. doi: 10.1097/00019605-200310000-00015. [DOI] [PubMed] [Google Scholar]
- 20.Nizon M., Alanay Y., Tuysuz B., Kiper P.O., Geneviève D., Sillence D., Huber C., Munnich A., Cormier-Daire V. IMPAD1 mutations in two Catel-Manzke like patients. Am. J. Med. Genet. A. 2012;158A:2183–2187. doi: 10.1002/ajmg.a.35504. [DOI] [PubMed] [Google Scholar]
- 21.Kiraz A., Tubas F., Ekinci Y., Dögen M.E., Varli M. A patient with hyperphalangism: the milder phenotype of Catel-Manzke syndrome. Clin. Dysmorphol. 2013;22:169–171. doi: 10.1097/MCD.0000000000000010. [DOI] [PubMed] [Google Scholar]
- 22.Gorlin R.J., Cohen M.M., Hennekam R.C.M. Oxford University Press; Oxford: 2001. Syndromes of the Head and Neck. [Google Scholar]
- 23.Kapoor S., Ghosh V., Dhua A., Aggarwal S.K. Cystic hygroma and hirsutism in a child with Catel-Manzke syndrome. Clin. Dysmorphol. 2011;20:117–120. doi: 10.1097/MCD.0b013e3283458946. [DOI] [PubMed] [Google Scholar]
- 24.Stevenson R.E., Taylor H.A., Jr., Burton O.M., Hearn H.B., 3rd A digitopalatal syndrome with associated anomalies of the heart, face, and skeleton. J. Med. Genet. 1980;17:238–242. doi: 10.1136/jmg.17.3.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sundaram V., Taysi K., Hartmann A.F., Jr., Shackelford G.D., Keating J.P. Hyperphalangy and clinodactyly of the index finger with Pierre Robin anomaly: Catel-Manzke syndrome. A case report and review of the literature. Clin. Genet. 1982;21:407–410. doi: 10.1111/j.1399-0004.1982.tb01395.x. [DOI] [PubMed] [Google Scholar]
- 26.Wilson G.N., King T.E., Brookshire G.S. Index finger hyperphalangy and multiple anomalies: Catel-Manzke syndrome? Am. J. Med. Genet. 1993;46:176–179. doi: 10.1002/ajmg.1320460215. [DOI] [PubMed] [Google Scholar]
- 27.Clarkson J.H., Homfray T., Heron C.W., Moss A.L. Catel-Manzke syndrome: a case report of a female with severely malformed hands and feet. An extension of the phenotype or a new syndrome? Clin. Dysmorphol. 2004;13:237–240. [PubMed] [Google Scholar]
- 28.Kiper P.O., Utine G.E., Boduroğlu K., Alanay Y. Catel-Manzke syndrome: a clinical report suggesting autosomal recessive inheritance. Am. J. Med. Genet. A. 2011;155A:2288–2292. doi: 10.1002/ajmg.a.34163. [DOI] [PubMed] [Google Scholar]
- 29.Oh S.H., Park K.E., Park Y.B., Yang S.K., Jr. A case of Catel Manzke syndrome. J. Korean Pediatr. Soc. 1999;42:1154–1158. [Google Scholar]
- 30.Lipson A., Beuhler B., Bartley J., Walsh D., Yu J., O’Halloran M., Webster W. Maternal hyperphenylalaninemia fetal effects. J. Pediatr. 1984;104:216–220. doi: 10.1016/s0022-3476(84)80995-6. [DOI] [PubMed] [Google Scholar]
- 31.Dudin A., Abdelshafi M., Rambaud-Cousson A. Choledochal cyst associated with rare hand malformation. Am. J. Med. Genet. 1995;56:161–163. doi: 10.1002/ajmg.1320560209. [DOI] [PubMed] [Google Scholar]
- 32.Li H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics. 2011;27:2987–2993. doi: 10.1093/bioinformatics/btr509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Heinrich V., Kamphans T., Stange J., Parkhomchuk D., Hecht J., Dickhaus T., Robinson P.N., Krawitz P.M. Estimating exome genotyping accuracy by comparing to data from large scale sequencing projects. Genome Med. 2013;5:69. doi: 10.1186/gm473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.DePristo M.A., Banks E., Poplin R., Garimella K.V., Maguire J.R., Hartl C., Philippakis A.A., del Angel G., Rivas M.A., Hanna M. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jäger M., Wang K., Bauer S., Smedley D., Krawitz P., Robinson P.N. Jannovar: a java library for exome annotation. Hum. Mutat. 2014;35:548–555. doi: 10.1002/humu.22531. [DOI] [PubMed] [Google Scholar]
- 36.Kamphans T., Krawitz P.M. GeneTalk: an expert exchange platform for assessing rare sequence variants in personal genomes. Bioinformatics. 2012;28:2515–2516. doi: 10.1093/bioinformatics/bts462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Abecasis G.R., Auton A., Brooks L.D., DePristo M.A., Durbin R.M., Handsaker R.E., Kang H.M., Marth G.T., McVean G.A., 1000 Genomes Project Consortium An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tennessen J.A., Bigham A.W., O’Connor T.D., Fu W., Kenny E.E., Gravel S., McGee S., Do R., Liu X., Jun G., Broad GO. Seattle GO. NHLBI Exome Sequencing Project Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science. 2012;337:64–69. doi: 10.1126/science.1219240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schwarz J.M., Rödelsperger C., Schuelke M., Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods. 2010;7:575–576. doi: 10.1038/nmeth0810-575. [DOI] [PubMed] [Google Scholar]
- 40.Kumar P., Henikoff S., Ng P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009;4:1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 41.Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S., Sunyaev S.R. A method and server for predicting damaging missense mutations. Nat. Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Karchin R. Next generation tools for the annotation of human SNPs. Brief. Bioinform. 2009;10:35–52. doi: 10.1093/bib/bbn047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stephens M., Smith N.J., Donnelly P. A new statistical method for haplotype reconstruction from population data. Am. J. Hum. Genet. 2001;68:978–989. doi: 10.1086/319501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 45.Giraud M.F., Naismith J.H. The rhamnose pathway. Curr. Opin. Struct. Biol. 2000;10:687–696. doi: 10.1016/s0959-440x(00)00145-7. [DOI] [PubMed] [Google Scholar]
- 46.Persson B., Kallberg Y. Classification and nomenclature of the superfamily of short-chain dehydrogenases/reductases (SDRs) Chem. Biol. Interact. 2013;202:111–115. doi: 10.1016/j.cbi.2012.11.009. [DOI] [PubMed] [Google Scholar]
- 47.Persson B., Kallberg Y., Bray J.E., Bruford E., Dellaporta S.L., Favia A.D., Duarte R.G., Jörnvall H., Kavanagh K.L., Kedishvili N. The SDR (short-chain dehydrogenase/reductase and related enzymes) nomenclature initiative. Chem. Biol. Interact. 2009;178:94–98. doi: 10.1016/j.cbi.2008.10.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Blake J.A., Bult C.J., Eppig J.T., Kadin J.A., Richardson J.E., Mouse Genome Database Group The Mouse Genome Database: integration of and access to knowledge about the laboratory mouse. Nucleic Acids Res. 2014;42:D810–D817. doi: 10.1093/nar/gkt1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Moriarity J.L., Hurt K.J., Resnick A.C., Storm P.B., Laroy W., Schnaar R.L., Snyder S.H. UDP-glucuronate decarboxylase, a key enzyme in proteoglycan synthesis: cloning, characterization, and localization. J. Biol. Chem. 2002;277:16968–16975. doi: 10.1074/jbc.M109316200. [DOI] [PubMed] [Google Scholar]
- 50.Huber C., Oulès B., Bertoli M., Chami M., Fradin M., Alanay Y., Al-Gazali L.I., Ausems M.G., Bitoun P., Cavalcanti D.P. Identification of CANT1 mutations in Desbuquois dysplasia. Am. J. Hum. Genet. 2009;85:706–710. doi: 10.1016/j.ajhg.2009.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li Y., Laue K., Temtamy S., Aglan M., Kotan L.D., Yigit G., Canan H., Pawlik B., Nürnberg G., Wakeling E.L. Temtamy preaxial brachydactyly syndrome is caused by loss-of-function mutations in chondroitin synthase 1, a potential target of BMP signaling. Am. J. Hum. Genet. 2010;87:757–767. doi: 10.1016/j.ajhg.2010.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vissers L.E., Lausch E., Unger S., Campos-Xavier A.B., Gilissen C., Rossi A., Del Rosario M., Venselaar H., Knoll U., Nampoothiri S. Chondrodysplasia and abnormal joint development associated with mutations in IMPAD1, encoding the Golgi-resident nucleotide phosphatase, gPAPP. Am. J. Hum. Genet. 2011;88:608–615. doi: 10.1016/j.ajhg.2011.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Temtamy S.A., Meguid N.A., Ismail S.I., Ramzy M.I. A new multiple congenital anomaly, mental retardation syndrome with preaxial brachydactyly, hyperphalangism, deafness and orodental anomalies. Clin. Dysmorphol. 1998;7:249–255. doi: 10.1097/00019605-199810000-00003. [DOI] [PubMed] [Google Scholar]
- 54.Nizon M., Huber C., De Leonardis F., Merrina R., Forlino A., Fradin M., Tuysuz B., Abu-Libdeh B.Y., Alanay Y., Albrecht B. Further delineation of CANT1 phenotypic spectrum and demonstration of its role in proteoglycan synthesis. Hum. Mutat. 2012;33:1261–1266. doi: 10.1002/humu.22104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kitagawa H., Uyama T., Sugahara K. Molecular cloning and expression of a human chondroitin synthase. J. Biol. Chem. 2001;276:38721–38726. doi: 10.1074/jbc.M106871200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.