

Abstract
Gene expression levels can be subject to selection. We hypothesized that the age of gene origin is associated with expression constraints, given that it affects the level of gene integration into the functional cellular environment. By studying the genetic variation affecting gene expression levels (cis expression quantitative trait loci [cis-eQTLs]) and protein levels (cis protein QTLs [cis-pQTLs]), we determined that young, primate-specific genes are enriched in cis-eQTLs and cis-pQTLs. Compared to cis-eQTLs of old genes originating before the zebrafish divergence, cis-eQTLs of young genes have a higher effect size, are located closer to the transcription start site, are more significant, and tend to influence genes in multiple tissues and populations. These results suggest that the expression constraint of each gene increases throughout its lifespan. We also detected a positive correlation between expression constraints (approximated by cis-eQTL properties) and coding constraints (approximated by Ka/Ks) and observed that this correlation might be driven by gene age. To uncover factors associated with the increase in gene-age-related expression constraints, we demonstrated that gene connectivity, gene involvement in complex regulatory networks, gene haploinsufficiency, and the strength of posttranscriptional regulation increase with gene age. We also observed an increase in heritability of gene expression levels with age, implying a reduction of the environmental component. In summary, we show that gene age shapes key gene properties during evolution and is therefore an important component of genome function.
Introduction
Variation in gene expression can contribute significantly to phenotypic diversity at the cellular and organismal levels,1,2 thus making it subject to selection.2–6 Negative selection, which fluctuates from relaxed to stringent, is considered to be the primary evolutionary force that acts on maintaining gene expression patterns across mammalian species.7,8 Investigation of intraspecies expression constraints is beneficial because it can uncover an additional class of deleterious regulatory variants that have a low probability of fixation but can segregate in the population.9,10 Selection acting on intraspecies gene expression variation has been investigated with different approaches. Gene expression changes can be estimated directly by the measurement of mRNAs and proteins or indirectly through the analysis of the genetic control of mRNA expression variation or protein abundance variation in cis (cis expression quantitative trait loci [cis-eQTLs] and cis protein QTLs [cis-pQTLs]) and in trans (trans-eQTLs and trans-pQTLs).11–13
Recently, we observed that cis-eQTLs affecting human long intergenic noncoding RNA (lincRNA) genes are more abundant and have a stronger effect than protein-coding genes.14 Because the majority of human lincRNAs are primate-specific genes15 and are therefore significantly younger than the majority of protein-coding genes, we propose that lincRNAs might reflect some common properties of young genes. In order to investigate the changes in expression constraints of protein-coding genes in the context of gene age, we analyzed the variation in expression levels and genetic regulation of expression of both mRNAs and proteins and stratified them by gene age.
We have demonstrated that gene age is associated with a spectrum of traits related to expression variation, distribution, and properties of cis-eQTLs. Our results indicate that (1) young primate-specific protein-coding genes are enriched in cis-eQTLs; (2) cis-eQTLs of young genes have a strong effect size, are located proximally to the transcription start site (TSS), and have highly significant p values; (3) cis-eQTLs of young genes tend to influence genes in multiple tissues and populations; and (4) expression constraints (approximated by cis-eQTLs properties) and coding constraints (approximated by Ka/Ks) of genes correlate with each other and with gene age. We propose that gene expression constraints increase throughout the lifetime of a gene as a result of the gradual integration of the gene into a functional cellular network. We have determined that gene connectivity in coexpression networks, gene involvement in complex regulatory networks, gene haploinsufficiency, and the strength of posttranscriptional regulation increase with the age of a gene and are marked by a depletion of cis-eQTLs. Additionally, we observed that gene-expression heritability increases with gene age, reflecting a diminishing influence of environment on expression level associated with gene age. In summary, our findings suggest that gene expression constraints increase significantly with gene age, reflecting gradual integration of a gene into a functional cellular network and strongly influencing genetic regulation of human gene expression.
Material and Methods
cis-eQTLs from the GenCord Collection
In brief, we collected umbilical cord and cord blood samples of 195 unrelated newborn European individuals in order to derive three cell types: primary fibroblasts, lymphoblastoid cell lines (LCLs), and primary T cells. Genotype and RNA sequencing data were used and analyzed as described previously.11,14 cis-eQTL calls were performed by Spearman rank correlation. The presence or absence of cis-eQTLs for each gene was derived at a 10% false-discovery-rate level from the GenCord study, and 15.9%–26.6% of genes expressed in fibroblasts, LCLs, and T cells were found to have cis-eQTLs (among 13,043, 12,693, and 13,274 genes, expressed in fibroblasts, LCLs, and T cells, respectively, we found 2,433, 3,372, and 2,115 genes, respectively, with cis-eQTLs). The GenCord study has been approved by the ethics committee of the University of Geneva.
Effect Size of cis-eQTLs, Loss-of-Expression cis-eQTLs, and Gain-of-Expression cis-eQTLs
Using ancestral nucleotides from the 1000 Genomes Project, we categorized all cis-eQTL alleles as ancestral (A) or derived (D). We calculated the effect size of each cis-eQTL as the slope of a linear model between the number of derived alleles (AA = 0, AD = 1, DD = 2) and the expression level of the exon used for the cis-eQTL call. Loss-of-expression (LOE) cis-eQTLs have negative slopes, whereas gain-of-expression (GOE) cis-eQTLs have positive slopes.
Gene Age
The age of each gene in the human genome was collected from the human protein-coding gene-age annotation,16 where each gene was coded from 0 (the oldest, originating before the zebrafish divergence) to 12 (the youngest, human-specific genes). A specific branch for each gene was assigned according to a parsimony rule applied to best-to-best matches between human and outgroup genes. This approach is independent of gene-annotation quality of outgroup species and robust to gene translocations.
Permutation Analysis
cis-eQTLs in the GenCord collection were called on the basis of exon expression levels. Because the number of exons can influence the probability that a gene will have cis-eQTLs, we performed a permutation analysis to control for this. In order to simulate an expected distribution of cis-eQTLs in each gene-age category, we coded each exon as 1 if it had a cis-eQTL and as 0 if it did not have a cis-eQTL (across all genes). Thereafter, we randomly shuffled this vector (presence or absence of cis-eQTLs per exon) and reconstructed the presence or absence of cis-eQTLs on the level of each gene as follows: if at least one exon had a cis-eQTL, the gene was coded as 1, and if no exon had a cis-eQTL, the gene was coded as 0. Using this approach, we accounted for gene structure (number of exons per gene). After 100,000 permutations, we obtained an expected distribution of cis-eQTLs under the assumption that the probability of having a cis-eQTL was the same for each exon. This distribution is depicted as dashed lines in Figure 1 for each gene-age category. The observed decrease in the expected fraction of cis-eQTL genes (genes with cis-eQTLs) among young genes (Figure 1) reflects the small number of exons in young genes.
Figure 1.
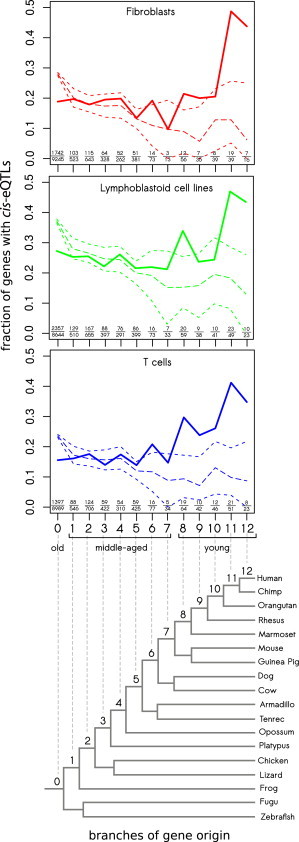
Distribution of cis-eQTL Genes Is Associated with Gene Age
The three upper panels represent changes with gene age in the fraction of cis-eQTL genes in three cell types from the GenCord collection. The bottom panel represents the phylogenetic tree; the branch of origin of each gene is numbered from 0 (oldest) to 12 (youngest; modified from Zhang et al.16). There is a deficit of cis-eQTL genes among old genes (branch 0) and an excess among young genes (branches 8–12). 95% confidence intervals and medians obtained from 100,000 permutations are plotted by short and long dashes, respectively. The number of cis-eQTL genes with the total number of expressed genes is presented for each age category.
cis-pQTLs
Lists of cis-pQTL and non-cis-pQTL genes were obtained from the combined data set of Wu et al.12
Variance in mRNA Expression Levels and Protein Levels
The variance in expression levels of mRNA and protein levels has been estimated for 3,390 genes expressed in LCLs in between three and five human individuals (Table S4 in Khan et al.17). No data for young genes were available in the study.17
Analyses of Matched Gene Pairs
To eliminate the influence of cis-eQTL effect size on the pattern of tissue specificity of cis-eQTL genes, we compared pairs of young and old genes matched by their cis-eQTL effect size. First, for each young gene, we found a middle-aged (or old) gene with a cis-eQTL of the most similar effect size. Second, for each gene of a matched pair, we counted the number of cell types (one to three) from the GenCord collection in which the given gene had a cis-eQTL and calculated the difference (−2 to 2) between the young and middle-aged (or old) gene from each pair. Finally, we obtained nine distributions of the difference (three cell types versus three possible comparisons: middle aged versus old, young versus middle aged, and young versus old).
In the derived-allele-frequency (DAF) analysis, we merged the young and middle-aged genes and then paired them with old genes according to the effect size of their cis-eQTLs.
In the population analysis, we performed a similar comparison of gene pairs matched by effect size. First, we split all cis-eQTL genes into unique (present in only one population) and population shared (present in two or more populations). The genes from the first group were coded as 1, and genes from the second group were coded as 2. Second, for each young cis-eQTL gene, we found an old cis-eQTL gene with a similar effect size. Third, in each matched gene pair, we obtained the difference in the level of population sharing between young and old genes.
In the expression-level analysis, we performed a comparison of gene pairs matched by expression level.
In all analyses, we controlled for the absence of a statistically significant difference (p > 0.05) in cis-eQTL effect sizes (or expression levels) of the matched groups of genes by using the paired Mann-Whitney U test.
Ka/Ks
We used the Ka/Ks values obtained from the human-macaque divergence for each protein-coding gene from Ensembl. Genes with Ka/Ks values greater than 1 were not analyzed.
Binary Logistic Regression Model
The model was run with the “glm” function in R with scaled variables: glm_results = glm(gene_age ∼scale(Ka/Ks) + scale(−log10(p value), family = binomial()). gene_age contains 0s and 1s, which correspond to old and young to middle-aged genes, respectively. The radii of circles were obtained with the “predict” function: probability_of_a_gene_to_be_young_or_middle-aged < - predict(glm_results, type = “response”).
Gene Coexpression Networks and Connectivity
We used the Weighted Gene Co-expression Network Analysis (WGCNA) approach18 implemented in the R package WGCNA version 1.11-3.19 Each node in the network represents a gene, and the weight of the connection between two nodes is a function of the Pearson correlation between the expression levels of the two genes. The connectivity of a gene is defined as the sum of the weights of the node representing this gene.
Genes Involved in Complex Regulatory Networks
Lists of genes involved in complex regulatory networks (discordant genes, described in Pai et al.20) were provided by Athma Pai.
Network Proximity to Haploinsufficient Genes
To derive network proximity to haploinsufficient genes, we first used total connectivity from COXPRESdb21 while considering all gene links with correlation r ≥ 0.3, which gave 3,566,815 links between 15,278 Ensembl genes. Second, using the list of known haploinsufficient genes (297 genes from Dang et al.22), we estimated connectivity to haploinsufficient genes as the number of links with haploinsufficient genes.
Partial Spearman Correlation
Partial Spearman correlations were estimated with R package ppcor. The partial correlation reflects the relationship between two variables while controlling for other variables. Although partial correlation can produce spurious results when applied to noisy data,23 it was our method of choice because gene age in our data set is a categorical variable, and as such, we could not use standard multiple regression analysis. First, we estimated partial Spearman correlation between Ka/Ks and cis-eQTL significance while controlling for gene-age effect. Second, we estimated partial Spearman correlation between gene age and three variables: Ka/Ks, connectivity (total connectivity from COXPRESdb21), and expression level (mean expression level from GenCord cell types with nonzero expression of a given gene) for 9,760 genes. All statistical analyses were performed in R.
Results
To assess the selection acting on the expression levels of different human genes, we took advantage of several expression data sets describing variation in mRNA expression and protein levels, cis-eQTLs, and cis-pQTLs in different tissues and populations. For variation in both mRNA expression and protein levels, we used a data set of 3,390 genes, expressed in LCLs of three to five human individuals.17 For cis-eQTLs, we used the data from our GenCord collection of three cell types (fibroblasts, LCLs, and T cells) from 195 unrelated European individuals11 and cis-eQTLs described in LCLs of 270 individuals from four HapMap populations.24 For cis-pQTLs, we used the data from LCLs of 74 unrelated individuals.12
Variation in mRNA and Protein Expression Levels Decreases with Gene Age
Because gene age can reflect the level of integration of a gene into the functional cellular environment25–28 and can thus be related to gene expression constraints, we stratified all human genes according to their age. Gene age was retrieved from a phylogenetic tree in which the branch of origin of each gene was established16 (Figure 1).
We analyzed interindividual variation in both mRNA expression and protein levels by using data from 3,390 genes17 in the context of gene age (see Material and Methods). We found that both mRNA and protein expression variations were more pronounced in younger genes (positive rank correlations between the branch number of origin of gene and mRNA or protein variance: p < 2.1 × 10−7, Spearman’s rho > 0.09; Figure S1A). These results are compatible with the recent finding that the sequence up to 10 kb upstream of the TSS, containing the majority of regulatory elements, is less constrained in young genes than in old genes.28 In order to establish an evolutionary explanation for the variable gene expression levels associated with gene age, we analyzed the genetic component of expression variation by using cis-eQTLs and cis-pQTLs.
Presence and Properties of cis-eQTLs Are Associated with Gene Age
For each gene-age category, we estimated the fraction of cis-eQTL genes and observed the trend that this fraction decreased with gene age: the oldest genes (branch 0) were depleted 2-fold by cis-eQTLs in comparison to the youngest genes (branch 12) (see bold lines in Figure 1). Taking into account that cis-eQTL calls were performed for each exon separately and that young genes are shorter and have fewer exons than old genes,29 we expected the observed trend (Figure 1) to be even more pronounced on the scale of exons. Indeed, comparing fractions of cis-eQTL exons between genes of different ages, we saw a strong positive correlation between the fraction and the branch of origin of the gene (Spearman’s rank correlation rho = 0.90 [fibroblasts], 0.95 [LCLs], and 0.89 [T cells]; all p < 2.2 × 10−16; Figure S1B). Comparing two extreme categories of genes, we observed 6- to 8-fold enrichment of young exons by cis-eQTLs (branch 0 versus branch 12: 4.8% versus 39.3% in fibroblasts, 7.0% versus 42.5% in LCLs, and 4.1% versus 33.0% in T cells; Figure S1B). Because the number of exons can influence the probability that a gene will have a cis-eQTL, we performed a permutation analysis (see Material and Methods). A total of 100,000 permutations revealed that there was a lack of cis-eQTL genes among older genes, originating at the oldest branch (p < 1.0 × 10−5 for branch 0), and an excess of cis-eQTL genes among younger genes, originating at branch 8 and above (p < 0.05 for branches 8 [fibroblasts, LCLs, and T cells], 9 [fibroblasts and T cells], and 10 [T cells]; p ≤ 1.0 × 10−5 for branches 11 [fibroblasts, LCLs, and T cells] and 12 [fibroblasts, LCLs, and T cells]). On the basis of the deviation of observed versus expected fractions of cis-eQTL genes in the three cell types of the GenCord collection (Figure 1), we further categorized genes as old (i.e., those present at the origin of vertebrates [branch 0]), young (i.e., those originating during primate evolution toward human lineage [branches 8–12]), and middle aged (branches 1–7). Notably, despite the semiarbitrary categorization of genes into young, middle aged, and old, the vast majority of our subsequent analyses remained significant when we performed a rank correlation of a property of interest with the branch of origin of genes.
Young genes are expressed at a lower level than old genes.29 In order to eliminate potential bias due to gene expression levels in our observations, we extracted pairs of genes with the same expression level but different age (young, middle aged, and old; see Material and Methods). Thereafter, we analyzed only gene pairs discordant for cis-eQTLs (i.e., one gene had a cis-eQTL, and the other did not) and calculated an asymmetry in the distribution of cis-eQTLs with respect to gene age. We found that the probability that younger versus older genes would have a cis-eQTL was higher than the expected 50% in almost all GenCord cell types and compared categories (Figure S1C). The strongest and most significant effects (p ≤ 0.01, binomial test) were observed in comparisons of young versus old and young versus middle-aged genes in fibroblasts and T cells (Figure S1C). We conclude that irrespective of expression level, young genes have an increased probability of having a cis-eQTL.
Given that variation in protein levels increases among recent-origin genes (Figure S1A), we expected that the fraction of cis-pQTL genes would increase among young genes in a manner similar to that of cis-eQTLs (Figure 1). Despite the small number of recently described cis-pQTLs,12 we observed that young and middle-aged genes together showed more cis-pQTLs than did old genes (one-sided Fisher’s odds ratio = 1.8, p = 0.039; Figure S1D). This association between gene age and cis-eQTLs and cis-pQTLs suggests that young genes can tolerate more expression changes.
If young genes allow extensive variation in expression, we expect that they would be associated with stronger cis-eQTLs. Because strong cis-eQTLs tend to be located close to the TSS and tend to be highly significant,24 we expect that all three characteristics of cis-eQTLs (effect size, location, and p value) will be associated with gene age. By comparing genes of different ages, we observed that young genes had cis-eQTLs with a 1.6- to 2.3-fold higher effect size than old genes (young versus old genes: p = 3.61 × 10−6 [fibroblasts], 0.0002 [LCLs], and 0.0120 [T cells]; young versus middle-aged genes: p = 0.0001 [fibroblasts], 0.0057 [LCLs], and 0.0108 [T cells]; middle-aged versus old genes: p = 0.2032 [fibroblasts], 0.0524 [LCLs], and 0.7698 [T cells]; Mann-Whitney U test; Figure 2A). Additionally, we found that cis-eQTLs of young genes were located closer to the TSS (young versus old genes: p = 0.0492 [fibroblasts], 0.0062 [LCLs], and 0.0193 [T cells]; young versus middle-aged genes: all p > 0.5; middle-aged versus old genes: p = 2.74 × 10−9 [fibroblasts], 2.2 × 10−16 [LCLs], and 3.67 × 10−7 [T cells]; Mann-Whitney U test; Figures 2A and 2B) and were more significant than cis-eQTLs of old and middle-aged genes (young versus old genes: p = 1.83 × 10−8 [fibroblasts], 1.11 × 10−9 [LCLs], and 4.29 × 10−11 [T cells]; young versus middle-aged genes: p = 9.30 × 10−5 [fibroblasts], 9.48 × 10−5 [LCLs], and 1.85 × 10−6 [T cells]; middle-aged versus old genes: p = 8.82 × 10−7 [fibroblasts], 2.48 × 10−8 [LCLs], and 0.0002 [T cells]; Mann-Whitney U test; Figures 2A and 2B).
Figure 2.
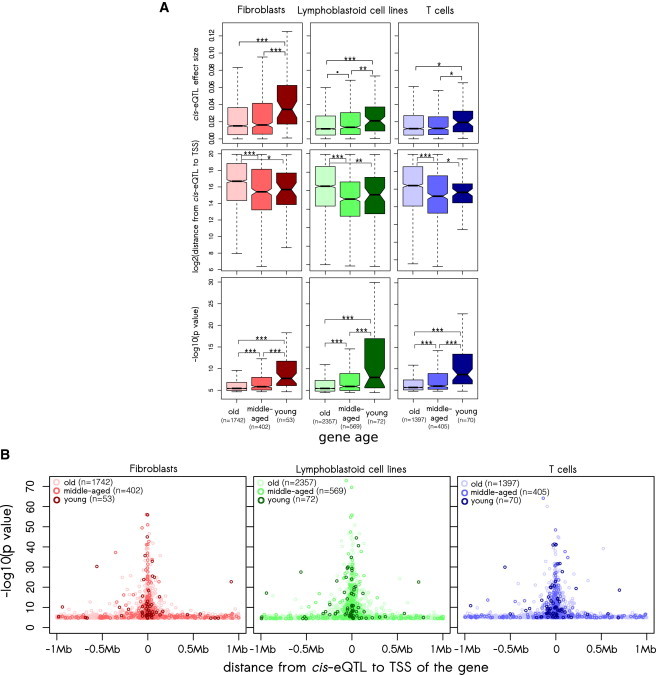
cis-eQTL Properties Are Associated with the Age of the Affected Gene
(A) cis-eQTL effect size (top panel), location (middle panel), and significance (bottom panel) differ among old, middle-aged, and young genes in three cell types from the GenCord collection. Box plots do not show all outliers. The box plot whiskers extend to the most extreme data point, which is not greater than 1.5× the interquartile range from the box. ∗∗∗p < 0.001, ∗∗p < 0.01, ∗p < 0.05, and ⋅p < 0.1.
(B) Distribution of cis-eQTLs of young, middle-aged, and old genes.
The direction of expression changes associated with the derived allele of a cis-eQTL (LOE or GOE) affects the mode of selection pressure.6 To test whether genes of different ages differ in preferential direction of expression changes, we compared the fractions of LOE and GOE cis-eQTLs between old and young genes (see Material and Methods). We observed that young genes were enriched in LOE cis-eQTLs (Figure S2). Considering that young genes evolve under more relaxed expression constraints (Figures 1 and 2) and are thus more prone to accumulation of slightly deleterious mutations, an excess of LOE cis-eQTLs (Figure S2) could indicate that loss of expression is on average more deleterious than gain of expression.
To analyze the tolerance of genes to expression changes in different cell types, we used a subset of ubiquitously expressed genes (genes expressed in at least 90% of samples in all three cell types of the GenCord collection) with cis-eQTLs in at least one cell type. For each gene, we counted the number of cell types with a detectable cis-eQTL, and if there was more than one such cell type, we also calculated the average cis-eQTL effect size, the average cis-eQTL distance to the TSS, and the average cis-eQTL p value. We found that genes with cis-eQTLs detectable in multiple cell types were characterized by cis-eQTLs with a high effect size (the effect size of cis-eQTLs present in three cell types was 1.6× higher than the effect size of cis-eQTLs present in only one cell type), close proximity to the TSS, and a highly significant p value (Figure 3A), which is consistent with a previous observation.30 Additionally, our analysis showed that the genes with cis-eQTLs in multiple tissues were preferentially regulated by LOE cis-eQTLs (Figures S3A and S3B). We noted that genes with cis-eQTLs in multiple tissues and young genes shared similar cis-eQTL properties: these cis-eQTLs were strong, located close to the TSS, highly significant, and predominantly LOE cis-eQTLs (Figure 2A versus Figure 3A; Figure S2 versus Figures S3A and S3B). We hypothesized that genes with cis-eQTLs in multiple tissues could be predominantly young genes, and thus the pattern of cis-eQTL tissue specificity could be shaped by gene age. To test this, we analyzed genes with cis-eQTLs present in one, two, or three cell types in the context of gene age (Figure 3B). We found that the fraction of common cis-eQTL genes (genes with cis-eQTLs in all three cell types) increased from 9% in the oldest genes (branch 0) to 83% in the youngest genes (branch 12) (Figure 3B). Furthermore, there was a significant positive correlation between the branch of gene origin and the fraction of tissue-shared cis-eQTL genes (Spearman’s rho = 0.88, p = 6.33 × 10−5; Figure 3B). Moreover, comparing matched pairs of old and young genes with cis-eQTLs of the same effect size (see Material and Methods), we demonstrated that young genes tended to have cis-eQTLs in more tissues than did old genes (Figure S3C), ruling out the possibility that the increased cis-eQTL effect sizes of young genes explain the higher tissue-shared pattern of cis-eQTLs of young genes. These data suggest that young genes can tolerate expression changes in multiple tissues.
Figure 3.
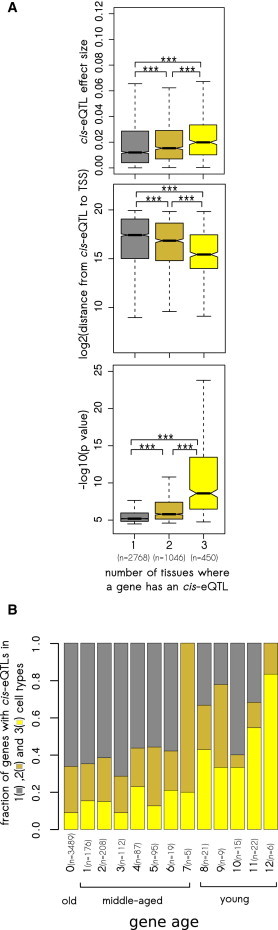
cis-eQTL Tissue Specificity
(A) cis-eQTL effect size (top panel), location (middle panel), and significance (bottom panel) differ among genes with cis-eQTLs in one, two, or three cell types from the GenCord collection. Box plots do not show all outliers. The box plot whiskers extend to the most extreme data point, which is not greater than 1.5× the interquartile range from the box. ∗∗∗p < 0.001.
(B) Tissue specificity of cis-eQTL genes is associated with gene age. A total of 4,264 genes, expressed in all three cell types of the GenCord collection and having a cis-eQTL in at least one cell type, were taken into account.
In order to dissect the selective constraints acting on cis-eQTL variants, we estimated their time of origin in the human population. Assuming that the primary mode of selection on cis-eQTLs is negative selection, we hypothesized that the oldest cis-eQTLs would be the most neutral. We approximated the time of origin of cis-eQTL variants by the DAFs (the higher the DAF, the older the allele)31 and by their level of sharing among different populations (the more sharing between different populations, the older the allele).32 First, we compared the DAF of GenCord cis-eQTLs (ancestral allele information was obtained from the European population of the 1000 Genomes Consortium) between genes of different ages. Because detection of a cis-eQTL depends on its effect size and minor allele frequency (MAF), we considered pairs of genes of different ages but matched by their cis-eQTL effect size (see Material and Methods). We did not observe significant trends when comparing young genes with middle-aged and old genes, probably as a result of the small sample size (the numbers of young genes with a reconstructed ancestral state of cis-eQTLs were 32 [fibroblasts], 47 [LCLs], and 40 [T cells] in the GenCord collection). However, when we combined young and middle-aged genes, the expected trend was revealed: the increased DAF of cis-eQTLs of young and middle-aged genes versus cis-eQTLs of old genes in two of three cell types of the GenCord collection (p = 0.010 [fibroblasts], 0.037 [LCLs], and 0.640 [T cells]; number of analyzed pairs of genes = 289 [fibroblasts], 374 [LCLs], and 289 [T cells]; one-sided paired Mann-Whitney U test). Second, we analyzed cis-eQTLs from four different populations24 and categorized ubiquitously expressed genes (genes expressed in all four populations) according to the number of populations (one to four) in which a gene had a cis-eQTL. We observed an excess of population-shared cis-eQTL genes (genes with cis-eQTLs in two or more populations) among young genes (Spearman’s rho = 0.61, p = 0.035; Figure S3D). Analyzing pairs of young and old genes matched by effect size (see Material and Methods), we observed that cis-eQTLs of young genes tended to be more population shared than those of old genes (expected mean = 0, observed mean = 0.19, p = 0.035, Wilcoxon test; Figure S3E). These results are compatible with our hypothesis that cis-eQTLs of young genes are older than cis-eQTLs of old genes, indicating that selection against variation in the expression level of young genes is less effective.
Coding and Expression Constraints are Congruent and Associated with Gene Age
In the analyses above, we determined that cis-eQTL properties are associated with gene age, which suggests an increase in expression constraints with gene age. These results are complementary to a model of increased coding constraints,26 which postulates relaxed coding constraints of young genes and thus explains the inverse correlation between the rate of coding sequence evolution and the age of genes. Using our data set, we confirmed the positive correlation between the branch of gene origin and Ka/Ks (Spearman’s rho = 0.34, p < 2.2 × 10−16). To investigate whether coding and regulatory constraints act in concordance with each other or independently, we examined the correlation between expression constraints, approximated by cis-eQTLs, and coding constraints, approximated by Ka/Ks (see Material and Methods). We observed that genes with cis-eQTLs (or cis-pQTLs) had 7%–15% (or 43%) higher Ka/Ks values than genes without cis-eQTLs (or cis-pQTLs) (Figure 4A; Figure S4A), suggesting that expression and coding constraints evolve in parallel. We also observed a gradual increase in Ka/Ks across genes with cis-eQTLs in zero, one, two, or three cell types of the GenCord collection (Figure S4B). Together with Figure 3A, this confirms the parallel dynamics of Ka/Ks and cis-eQTLs. We then split a subset of genes with cis-eQTLs into weak and strong according to the median effect size (0.0152 [fibroblasts], 0.0121 [LCLs], and 0.0118 [T cells]), distal or proximal according to the median distance of cis-eQTLs to the TSS (85,271 bp [fibroblasts], 75,213 bp [LCLs], and 76,706 bp [T cells]), and lowly or highly significant according to the median Spearman’s p value (−log10(p value) = 5.5031 [fibroblasts], 5.4810 [LCLs], and 5.6882 [T cells]). We found that genes with strong, proximal, and highly significant cis-eQTLs had 13%–43% increased Ka/Ks values (Figure 4B). Combined, these results demonstrate a concordance between expression and coding constraints of genes. Because gene age is determined on a scale of hundreds of millions of years, Ka/Ks is determined on a scale of dozens of millions of years, and cis-eQTL variants are determined on a scale of under one million years, we hypothesized that gene age, through gene-age-dependent factors, influences Ka/Ks and expression constraints simultaneously and thus shapes the correlation between them.
Figure 4.
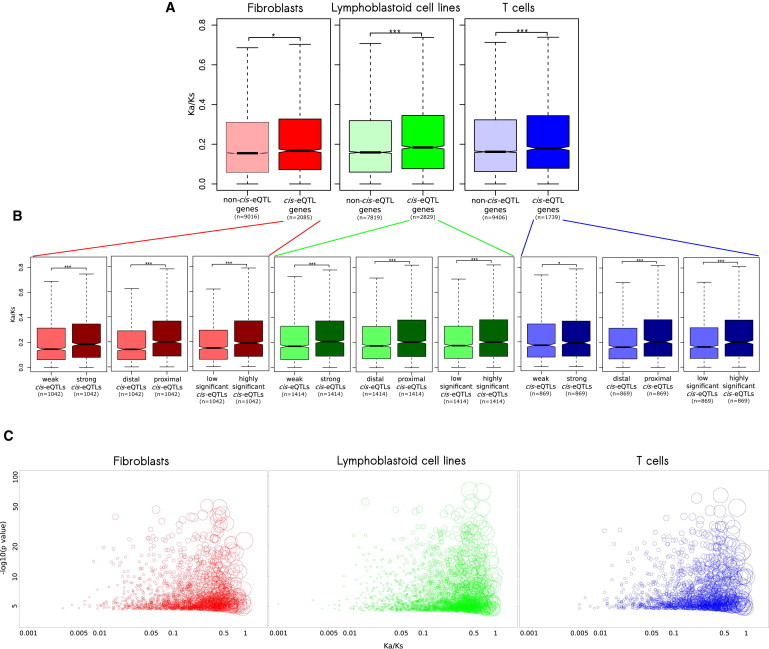
Coding and Expression Constraints Are Correlated
(A) cis-eQTL genes have higher Ka/Ks values than non-cis-eQTL genes in all three cell types from the GenCord collection.
(B) Properties of cis-eQTLs are associated with Ka/Ks of the affected gene in all three cell types from the GenCord collection: strong cis-eQTLs (with an effect size higher than the median), proximal cis-eQTLs (whose distance to the TSS is shorter than the median distance), and highly significant cis-eQTLs (with a −log10(p value) higher than the median) affect genes with high Ka/Ks values.
(C) Gene age is significantly associated with both coding (Ka/Ks) and expression (−log10(p value)) constraints in all three cell types from the GenCord collection. Circle size represents the probability that a gene is young or middle aged. Genes with Ka/Ks values of 0 are not shown.
Box plots do not show all outliers. The box plot whiskers extend to the most extreme data point, which is not greater than 1.5× the interquartile range from the box. ∗∗∗p < 0.001, ∗∗p < 0.01, ∗p < 0.05, and ⋅p < 0.1.
In order to test the relationship among gene age, expression, and coding constraints explicitly, we performed binary logistic regression, where gene age was coded as 1 if it was young or middle aged and coded as 0 if it was old. We merged categories of young and middle-aged genes given that we considered Ka/Ks values obtained from a comparison of human and macaque; therefore, Ka/Ks values were only available for a very small number of young genes. In our model, gene age was a function of two variables: Ka/Ks and cis-eQTL significance (see Material and Methods). For each gene, we estimated the probability of being young or middle aged as
| (Equation 1) |
We observed that both Ka/Ks and −log10(cis-eQTL p value) were significantly associated with gene age (see Table 1).
Table 1.
Coefficients of the Binary Multiple Logistic Regression
| α | β | γ | |
|---|---|---|---|
| Fibroblasts | −1.667∗∗∗ | 0.604∗∗∗ | 0.198∗∗∗ |
| LCLs | −1.695∗∗∗ | 0.710∗∗∗ | 0.224∗∗∗ |
| T cells | −1.441∗∗∗ | 0.618∗∗∗ | 0.137∗ |
∗∗∗p < 0.001, ∗p < 0.05.
It is noteworthy that both β and γ coefficients are positive, indicating that young and middle-aged genes are characterized by both increased Ka/Ks and highly significant cis-eQTLs. The absolute magnitude of the coefficients revealed that Ka/Ks is about three to four times more important as a predictor of gene age but that the influence of cis-eQTL p values is also significant. To visualize the results of the binary logistic regression, we represented each gene as a circle in which the center corresponds to the gene-specific Ka/Ks and cis-eQTL p value and in which the radius represents the predicted probability that a gene is young or middle aged (Figure 4C). Our data showed that the radius of each circle increased with Ka/Ks and cis-eQTL significance (Figure 4C). The associations between expression and coding constraints (Figures 4A and 4B), and both of them with gene age (Figure 4C), confirm that overall functional constraints of genes can be driven by gene age. If gene age is an important determinant of the correlation between expression and coding constraints, we expect that this correlation will become significantly weaker after we control for gene-age effect. Indeed, the magnitude of Spearman’s rho between Ka/Ks and cis-eQTL significance (−log10(p value)) decreased to 20%–37% after we accounted for the branch of origin of genes: pairwise Spearman’s rho values without the effect of gene age were 0.10 (fibroblasts), 0.08 (LCLs), and 0.10 (T cells) (all p < 7.0 × 10−5), whereas Spearman’s rho values with control for gene-age effect were 0.08 (fibroblasts), 0.05 (LCLs), 0.08 (T cells) (all p < 0.01).
Gene-Age-Dependent Evolutionary Constraints
In all previous analyses, we used gene age as an evolutionary metric that reflects the level of integration of genes into the cellular environment. In the following section, we uncover and describe particular gene-age-dependent properties that affect gene expression constraints more directly.
It has been shown that young genes have fewer physical interactions in yeast species33,34 and are regulated by fewer genes in humans.28 To estimate the level of integration of each gene product into cellular metabolism, we reconstructed a gene coexpression network for each cell type of the GenCord collection, estimated the connectivity of each gene as the number of genes it is coexpressed with (see Material and Methods), and then correlated this metric with gene age. Our analysis showed that the connectivity increased with gene age. We observed a negative rank correlation between the branch of gene origin and connectivity (Spearman’s rho < −0.10 and p < 2.2 × 10−16 for all GenCord cell types) and significant differences in connectivity among old, middle-aged, and young genes (p < 0.006 for all GenCord cell types, Mann-Whitney U test; Figure 5A). Correspondingly, non-cis-eQTL genes had higher connectivity than cis-eQTL genes (p < 3.9 × 10−7 for all GenCord cell types, Mann-Whitney U test; Figure S5A).
Figure 5.
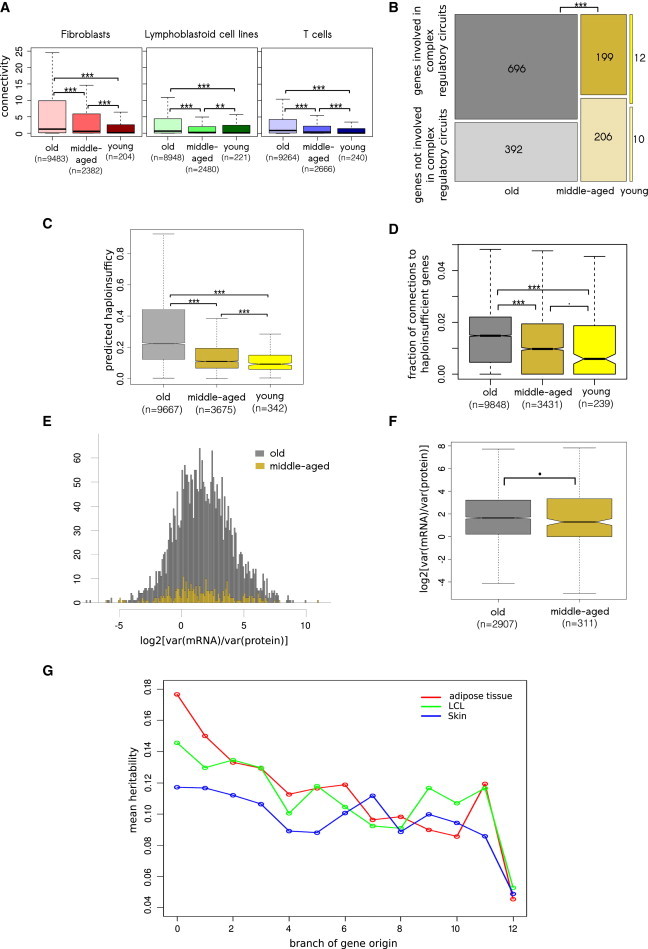
Gene Properties Are Associated with Gene Age
(A) Estimated for each cell type from the GenCord collection, connectivity of genes in coexpression networks increases with gene age.
(B) Old genes are enriched in genes involved in complex regulatory circuits.
(C) Predicted haploinsufficiency probabilities increases with gene age.
(D) The fraction of connections with haploinsufficient genes increases with gene age.
(E) Distribution of the ratio of variances of mRNA to protein levels for old and middle-aged genes.
(F) The ratio of variances of mRNA to protein levels tends to be higher for old genes than for middle-aged genes.
(G) Gene expression heritability, derived for three tissues of the MuTHER data set, increases with gene age. The mean heritability is plotted for each gene-age category.
Box plots do not show all outliers. The box plot whiskers extend to the most extreme data point, which is not greater than 1.5× the interquartile range from the box. ∗∗∗p < 0.001, ∗∗p < 0.01, ∗p < 0.05, and ⋅p < 0.1.
Gene expression constraints might depend not only on the number of coexpressed genes but also on the complexity of gene interactions. By comparing a list of genes potentially involved in complex regulatory circuits20 with the remaining genes, we observed that genes involved in complex regulatory circuits were enriched among old genes (Fisher’s odds ratio = 1.82, p = 2.746 × 10−7; Figure 5B). Using data from the GenCord LCL (the same cell type that Pai and colleagues20 used to derive the list of genes involved into complex regulatory circuits), we demonstrated that genes involved in complex regulation had a deficit of cis-eQTLs (Fisher’s odds ratio = 0.75, p = 0.0311; Figure S5B) and a lower cis-eQTL effect size (median effect sizes were 0.008 and 0.013, p = 0.0102, Mann-Whitney U test; Figure S5C).
We next hypothesized that the increase in gene connectivity and involvement in complex regulatory pathways could be correlated with haploinsufficiency. Haploinsufficiency is defined as the occurrence of abnormal phenotypes when only one functional gene copy is present. Thus, haploinsufficiency is directly related to gene expression constraints. Using the predicted haploinsufficiency probabilities of human genes,35 we found that the probability of being haploinsufficient was indeed higher for old genes: there was a negative rank correlation between the branch of gene origin and the predicted haploinsufficiency (Spearman’s rho = −0.37, p < 2.2 × 10−16; pairwise differences in haploinsufficiency among old, middle-aged, and young genes were significant: all p < 2.4 × 10−6, Mann-Whitney U test, Figure 5C). Additionally, we found that haploinsufficiency probabilities were higher for non-cis-eQTL genes than for cis-eQTL genes for all cell types of the GenCord collection (p < 1.3 × 10−5, Mann-Whitney U test; Figure S5D).
Estimated haploinsufficiency depends on four variables: network proximity of a given gene to annotated haploinsufficient genes, expression level during embryogenesis, Ka/Ks, and connectivity.35 For two of the four traits (Ka/Ks and connectivity), we have already shown associations with gene age (Figures 4C and 5A); thus, the correlation between haploinsufficiency and gene age (Figure 5C) cannot be considered a completely new and independent finding. Therefore, we also analyzed the network proximity to haploinsufficient genes, which is the most important predictor for haploinsufficiency probabilities.35 Using a large coexpression database (COXPRESdb21) and a list of human haploinsufficient genes,22 for each gene we derived the total number of coexpressed genes, the number of coexpressed haploinsufficient genes, and the fraction of haploinsufficient genes among all coexpressed genes (see Material and Methods). Using a negative rank correlation between the branch of gene origin and total connectivity, we found that the total connectivity was higher for old genes (p < 2.2 × 10−16 and Spearman’s rho = −0.15) and observed significant differences in connectivity between old, middle-aged, and young genes (all p < 8.6 × 10−5, Mann-Whitney U test; Figure S5E). The connectivity with haploinsufficient genes also increased with gene age. We observed a negative rank correlation between the branch of gene origin and connectivity with haploinsufficient genes (p < 2.2 × 10−16, Spearman’s rho = −0.18) and significant differences in connectivity to haploinsufficient genes between old, middle-aged, and young genes (all p < 0.0004, Mann-Whitney U test; Figure S5E). Importantly, the fraction of connections to haploinsufficient genes also increased with gene age, which was demonstrated by (1) a negative rank correlation between the branch of gene origin and the fraction (p < 2.2 × 10−16, Spearman’s rho = −0.16), (2) significant differences in the fractions between old and young genes (p < 2.2 × 10−16, Mann-Whitney U test; Figure 5D) and between old and middle-aged genes (p < 2.2 × 10−16, Mann-Whitney U test; Figure 5D), and (3) a marginally significant difference between middle-aged and young genes (p = 0.054, Mann-Whitney U test; Figure 5D). This reveals that, in parallel with the increase in the total number of connections, the fraction of connections with haploinsufficient genes also increases with gene age and thus multiplies expression constraints of old genes.
Using available data comprising both the mRNA expression levels and protein levels for more than 3,000 genes,17 we investigated the additional contribution of posttranscriptional constraints, which can be derived as the ratio of coefficients of variation on mRNA and protein levels. First, we noticed that the variation in the expression level of proteins was lower than the variation in the mRNA expression level—the log2 ratio of the coefficients of variation of mRNA to protein levels was significantly higher than expected (observed median = 1.63, expected mu = 0, p < 2.2 × 10−16, Wilcoxon test; Figure 5E). This is consistent with protein expression buffering due to the stronger evolutionary constraints acting on protein than on mRNA levels.17 Because there are no young genes in the data set of Khan et al., we compared old and middle-aged genes (marked by gray and biege, respectively, in Figure 5E). We noted that the median ratio for middle-aged genes was marginally significantly smaller than the median ratio for old genes (1.29 versus 1.64, p = 0.07, Mann-Whitney U test; Figures 5E and 5F). We hypothesized that the strength of buffering can increase with gene age as a result of the acquisition of additional posttranscriptional constraints. To test this hypothesis, we split the distribution of log2 ratios (Figure 5E) into 643 bins (five genes in each bin) ordered from left to right. We estimated the probability of finding at least one middle-age gene in each bin as a function of the numerical order of the bin (1–643). Using logistic regression (the “glm” function in R), we found that the probability significantly (p = 1.94 × 10−7) decreased from 50% (an average of one middle-aged gene per ten genes) on the left side of the distribution to 35% (an average of one middle-aged gene per 15 genes) on the right side of the distribution. Thus, using a relatively small subset of genes with information on both mRNA expression levels and protein levels, we revealed a significant trend suggesting that posttranscriptional modifications become more abundant and stronger with gene age.
So far, we have demonstrated that the regulation of the expression level of old genes is more constrained, suggesting that the expression level of old genes is less affected by segregating regulatory variants. To compare the relative impact of genetic and environmental factors on variation of the expression level of different genes, we analyzed the narrow-sense heritability of gene expression, which was estimated in three tissues of 856 female twins from the MuTHER project:36 h2 = Va / (Va + Ve), where Va is the additive genetic component and Ve is the environmental component of variation in gene expression. For all three tissues of the MuTHER data set, we observed a strong increase in gene expression heritability with gene age (negative rank correlation between the branch of gene origin and heritability: all p < 2.0 × 10−16, all Spearman’s rho < −0.09, Figure 5G). This result is compatible with recently demonstrated increased heritability of housekeeping (i.e., older) versus nonhousekeeping (i.e., younger) genes.37,38 The high heritability of old genes (Figure 5G) together with the low total variation in expression level of old genes (Va + Ve; Figure S1A) can be explained by the decreased effect of environment on the expression level of old genes (Ve; Figure 6). It is important to emphasize that heritability is based on all genetic variants segregating in a population (common cis-eQTLs with MAF > 5%, rare cis-eQTLs with MAF < 5%, and trans-eQTLs), among which trans-eQTLs are the main determinants of the heritability.36 In this study, we only analyzed common cis-eQTLs and demonstrated that the genetic component of variation based on these cis-eQTLs is increased in young genes. The impact of rare cis-eQTLs and trans-eQTLs on the genetic component of variation of old and young genes is still unknown and could be the subject of future investigations (gray dashed line in Figure 6).
Figure 6.
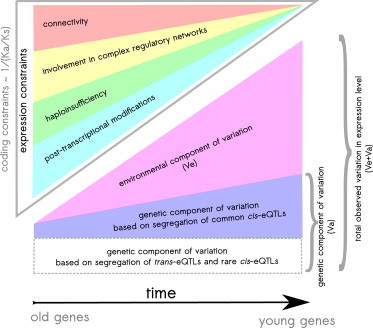
The Model of Increased Expression Constraints
Expression constraints of genes increase with gene age and are driven by increased connectivity, involvement in complex regulatory networks, haploinsufficiency, and posttranscriptional modifications. Both environmental and genetic components of variation in expression level diminish with gene age, reflecting an increase in gene expression constraints. The genetic component of expression variation based on common cis-eQTLs diminishes with gene age. The dynamic of rare cis-eQTLs and trans-eQTLs with gene age is still unknown and is represented as a rectangular gray dashed section. The scheme also emphasizes the increased heritability of old genes: [Va / (Va + Ve) for old genes] > [Va / (Va + Ve) for young genes].
Discussion
In this study, we investigated changes in human gene expression constraints in the context of age of gene origin. We have shown that (1) young genes have an excess of cis-eQTLs and cis-pQTLs (Figure 1; Figure S1D), (2) cis-eQTLs of young genes are stronger, located closer to the TSS, and more significant than cis-eQTLs of old genes (Figure 2), and (3) compared to cis-eQTLs of old genes, cis-eQTLs of young genes affect different tissues (Figure 3), are present in more populations (Figure S3), and involve older derived alleles. We conclude that all of these properties of cis-eQTLs mark low expression constraints of young genes and that the loss of cis-eQTLs with gene age reflects an increase in expression constraints throughout the lifespan of each gene.
It has been shown previously that the time of gene origin is associated with many functional traits of eukaryotic genes.39,40 In summary, all previously published studies demonstrate that gene age is associated with the strength of purifying selection acting on gene coding constraints, gene expression constraints, and the correlation of these constraints. The coding constraints of young genes are more relaxed, leading to fast26,29,41 and variable25,42 rates of sequence evolution of young genes. The expression constraints of genes were investigated with gene duplicates, which are the most common mechanism of origin of new genes. It has been concluded that the dosage-balanced hypothesis43 determines the main selection forces influencing fixation of gene duplications.44,45 For example, dosage-sensitive genes, which include those encoding transcription factors, are retained after whole-genome duplications but tend to be lost after small-scale duplications.46–48 In mammals, interspecies expression divergence also tends to be lower in highly expressed and tissue-specific genes.49 The correlation between coding and expression constraints and the main causative factors driving this correlation is a long-standing puzzle.50,51 In yeast, both expression and coding divergences increase after gene duplication,52 supporting the observation that expression similarity and the fraction of shared eQTLs decrease with the age of duplicated genes.53 The strong and stable correlation between interspecies expression divergence and interspecies sequence divergence has also been described in mammals,51,54 and it has been suggested that the correlation could at least partially represent a default evolutionary state.51
Our results complement and extend previous findings toward the level of genetic polymorphisms. We demonstrate that expression constraints increase with gene age and contribute to the congruent evolution between coding and expression constraints. Because gene age is associated with both coding and expression constraints (Figure 4), we propose that the correlation between coding and expression constraints can be maintained by age-dependent purifying selection, which affects both types of constraints simultaneously. Thus, gene age can be an evolutionary proxy for the level of functional constraints of a gene. Taking into account our own results and the results of other studies elucidating associations between gene age and expression level,29 between gene age and coding constraints,26 between connectivity and coding constraints,55,56 and between gene age and connectivity,28 we propose an integrative point of view that gene age shapes all of these correlations. To infer the direction of causality, we can use the time scales of different analyzed traits, among which gene age is the oldest variable and is thus most likely the causative one. To test whether the correlation between gene age and two key variables (Ka/Ks and connectivity) persists after the consideration of gene expression level,57 we performed partial Spearman correlations (see Material and Methods) and observed that gene age continues to affect these variables (Figure S6).
Given that gene expression constraints, estimated from human polymorphisms (with the average age of variants under one million years), reflect the time of origin of genes (with timescales of dozens or hundreds of million years), we can conclude that the acquisition of new functional constraints is a very long process that has most likely not reached its limit, even for the oldest human genes.28 To pinpoint the driving factors, we demonstrated that gene connectivity, gene involvement in complex regulatory networks, network proximity to haploinsufficient genes, and the strength of posttranscriptional regulation increase with gene age (Figure 5). This provides a mechanistic explanation for the model of increased expression constraints (Figure 6). The increased heritability of the expression level of old genes (Figure 5G), which implies a reduced effect of environmental factors on the expression level of old genes, further supports the model of increased expression constraints.
Interestingly, the expression of young genes is less constrained not only among different individuals of the same developmental stage, as demonstrated in our study, but also among different developmental stages. Recently, Domazet-Lošo and Tautz40 demonstrated that changes in the transcriptome during ontogeny are mainly driven by young genes and that the expression of old genes is more or less stable across all ontogenetic stages. The oldest transcriptome is expressed during the most critical and conserved stage of ontogenesis, whereas reproductively active animals show the youngest transcriptome. Recent findings showing a positive correlation between the earliest expression stage during development and expression divergence51 can also be explained by gene age, given that young genes tend to be expressed later and have less constrained, and therefore more divergent, expression levels.
Because the distribution and properties of cis-eQTLs can be explained by purifying selection, which is stronger for old genes, cis-eQTLs could be considered a class of slightly deleterious regulatory variants. The excess of such variants in young genes demonstrated here and the excess of complex human diseases associated with genes of recent origin58 could explain the significant overlap between cis-eQTL genes and genes associated with complex human diseases.59–61 The high susceptibility of young genes to slightly deleterious regulatory and coding variants might provide evidence that young genes have an increased mutational load and therefore cumulatively affect human fitness more significantly than old genes.
Acknowledgments
We acknowledge Y. Gilad, A. McLysaght, M. Garieri, and F. Santoni for discussions and Joshua Plotkin and two anonymous reviewers for constructive criticisms. This study was supported by Swiss National Science Foundation (SNSF) grant 144082 and European Research Council (ERC) grant 249968 to S.E.A. and by SNSF grant 31003A_130342, ERC grant 260927 POPRNASEQ, and the Louis-Jeantet foundation to E.T.D. K.Y.P. was supported by European Molecular Biology Organization long-term fellowship program ALTF 527-2010 and Novartis grants N3A15 and N14B065. J.S. was supported by the Wellcome Trust (grants 093941/Z/10/Z to J.S. and 090532/Z/09/Z to the Wellcome Trust Centre for Human Genetics). G.A.B. and V.B.S. were supported by the Russian Ministry of Education and Science (grant 11.G34.31.0008, http://eng.mon.gov.ru/) and the Molecular and Cellular Biology Program of the Russian Academy of Sciences (http://molbiol.edu.ru/). P.I. was supported by the SNSF award 31003A-125350 and 31003A-143936 to E.M.Z. The computations were performed at the Vital-IT Center for high-performance computing of the Swiss Institute of Bioinformatics.
Contributor Information
Konstantin Y. Popadin, Email: konstantin.popadin@unige.ch.
Stylianos E. Antonarakis, Email: stylianos.antonarakis@unige.ch.
Supplemental Data
Web Resources
The URL for data presented herein is as follows:
References
- 1.Romero I.G., Ruvinsky I., Gilad Y. Comparative studies of gene expression and the evolution of gene regulation. Nat. Rev. Genet. 2012;13:505–516. doi: 10.1038/nrg3229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wray G.A. The evolutionary significance of cis-regulatory mutations. Nat. Rev. Genet. 2007;8:206–216. doi: 10.1038/nrg2063. [DOI] [PubMed] [Google Scholar]
- 3.Gilad Y., Oshlack A., Smyth G.K., Speed T.P., White K.P. Expression profiling in primates reveals a rapid evolution of human transcription factors. Nature. 2006;440:242–245. doi: 10.1038/nature04559. [DOI] [PubMed] [Google Scholar]
- 4.Fraser H.B. Gene expression drives local adaptation in humans. Genome Res. 2013;23:1089–1096. doi: 10.1101/gr.152710.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kudaravalli S., Veyrieras J.-B., Stranger B.E., Dermitzakis E.T., Pritchard J.K. Gene expression levels are a target of recent natural selection in the human genome. Mol. Biol. Evol. 2009;26:649–658. doi: 10.1093/molbev/msn289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lappalainen T., Montgomery S.B., Nica A.C., Dermitzakis E.T. Epistatic selection between coding and regulatory variation in human evolution and disease. Am. J. Hum. Genet. 2011;89:459–463. doi: 10.1016/j.ajhg.2011.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gilad Y., Oshlack A., Rifkin S.A. Natural selection on gene expression. Trends Genet. 2006;22:456–461. doi: 10.1016/j.tig.2006.06.002. [DOI] [PubMed] [Google Scholar]
- 8.Brawand D., Soumillon M., Necsulea A., Julien P., Csárdi G., Harrigan P., Weier M., Liechti A., Aximu-Petri A., Kircher M. The evolution of gene expression levels in mammalian organs. Nature. 2011;478:343–348. doi: 10.1038/nature10532. [DOI] [PubMed] [Google Scholar]
- 9.Kimura M. Cambridge University Press; Cambridge: 1983. The Neutral Theory of Molecular Evolution. [Google Scholar]
- 10.Ohta T. The Nearly Neutral Theory of Molecular Evolution. Annu. Rev. Ecol. Syst. 1992;23:263–286. [Google Scholar]
- 11.Gutierrez-Arcelus M., Lappalainen T., Montgomery S.B., Buil A., Ongen H., Yurovsky A., Bryois J., Giger T., Romano L., Planchon A. Passive and active DNA methylation and the interplay with genetic variation in gene regulation. Elife. 2013;2:e00523. doi: 10.7554/eLife.00523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu L., Candille S.I., Choi Y., Xie D., Jiang L., Li-Pook-Than J., Tang H., Snyder M. Variation and genetic control of protein abundance in humans. Nature. 2013;499:79–82. doi: 10.1038/nature12223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stranger B.E., Montgomery S.B., Dimas A.S., Parts L., Stegle O., Ingle C.E., Sekowska M., Smith G.D., Evans D., Gutierrez-Arcelus M. Patterns of cis regulatory variation in diverse human populations. PLoS Genet. 2012;8:e1002639. doi: 10.1371/journal.pgen.1002639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Popadin K., Gutierrez-Arcelus M., Dermitzakis E.T., Antonarakis S.E. Genetic and epigenetic regulation of human lincRNA gene expression. Am. J. Hum. Genet. 2013;93:1015–1026. doi: 10.1016/j.ajhg.2013.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Necsulea A., Soumillon M., Warnefors M., Liechti A., Daish T., Zeller U., Baker J.C., Grützner F., Kaessmann H. The evolution of lncRNA repertoires and expression patterns in tetrapods. Nature. 2014;505:635–640. doi: 10.1038/nature12943. [DOI] [PubMed] [Google Scholar]
- 16.Zhang Y.E., Vibranovski M.D., Landback P., Marais G.A.B., Long M. Chromosomal redistribution of male-biased genes in mammalian evolution with two bursts of gene gain on the X chromosome. PLoS Biol. 2010;8:13. doi: 10.1371/journal.pbio.1000494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Khan Z., Ford M.J., Cusanovich D.A., Mitrano A., Pritchard J.K., Gilad Y. Primate transcript and protein expression levels evolve under compensatory selection pressures. Science. 2013;342:1100–1104. doi: 10.1126/science.1242379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang B., Horvath S. A general framework for weighted gene co-expression network analysis. Stat. Appl. Genet. Mol. Biol. 2005;4:e17. doi: 10.2202/1544-6115.1128. [DOI] [PubMed] [Google Scholar]
- 19.Langfelder P., Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pai A.A., Cain C.E., Mizrahi-Man O., De Leon S., Lewellen N., Veyrieras J.-B., Degner J.F., Gaffney D.J., Pickrell J.K., Stephens M. The contribution of RNA decay quantitative trait loci to inter-individual variation in steady-state gene expression levels. PLoS Genet. 2012;8:e1003000. doi: 10.1371/journal.pgen.1003000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Obayashi T., Okamura Y., Ito S., Tadaka S., Motoike I.N., Kinoshita K. COXPRESdb: a database of comparative gene coexpression networks of eleven species for mammals. Nucleic Acids Res. 2013;41(Database issue):D1014–D1020. doi: 10.1093/nar/gks1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dang V.T., Kassahn K.S., Marcos A.E., Ragan M.A. Identification of human haploinsufficient genes and their genomic proximity to segmental duplications. Eur. J. Hum. Genet. 2008;16:1350–1357. doi: 10.1038/ejhg.2008.111. [DOI] [PubMed] [Google Scholar]
- 23.Drummond D.A., Raval A., Wilke C.O. A single determinant dominates the rate of yeast protein evolution. Mol. Biol. Evol. 2006;23:327–337. doi: 10.1093/molbev/msj038. [DOI] [PubMed] [Google Scholar]
- 24.Stranger B.E., Nica A.C., Forrest M.S., Dimas A., Bird C.P., Beazley C., Ingle C.E., Dunning M., Flicek P., Koller D. Population genomics of human gene expression. Nat. Genet. 2007;39:1217–1224. doi: 10.1038/ng2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vishnoi A., Kryazhimskiy S., Bazykin G.A., Hannenhalli S., Plotkin J.B. Young proteins experience more variable selection pressures than old proteins. Genome Res. 2010;20:1574–1581. doi: 10.1101/gr.109595.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Albà M.M., Castresana J. Inverse relationship between evolutionary rate and age of mammalian genes. Mol. Biol. Evol. 2005;22:598–606. doi: 10.1093/molbev/msi045. [DOI] [PubMed] [Google Scholar]
- 27.Capra J.A., Stolzer M., Durand D., Pollard K.S. How old is my gene? Trends Genet. 2013;29:659–668. doi: 10.1016/j.tig.2013.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Warnefors M., Eyre-Walker A. The accumulation of gene regulation through time. Genome Biol. Evol. 2011;3:667–673. doi: 10.1093/gbe/evr019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wolf Y.I., Novichkov P.S., Karev G.P., Koonin E.V., Lipman D.J. The universal distribution of evolutionary rates of genes and distinct characteristics of eukaryotic genes of different apparent ages. Proc. Natl. Acad. Sci. USA. 2009;106:7273–7280. doi: 10.1073/pnas.0901808106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dimas A.S., Deutsch S., Stranger B.E., Montgomery S.B., Borel C., Attar-Cohen H., Ingle C., Beazley C., Gutierrez Arcelus M., Sekowska M. Common regulatory variation impacts gene expression in a cell type-dependent manner. Science. 2009;325:1246–1250. doi: 10.1126/science.1174148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fu W., O’Connor T.D., Jun G., Kang H.M., Abecasis G., Leal S.M., Gabriel S., Rieder M.J., Altshuler D., Shendure J., NHLBI Exome Sequencing Project Analysis of 6,515 exomes reveals the recent origin of most human protein-coding variants. Nature. 2013;493:216–220. doi: 10.1038/nature11690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lohmueller K.E., Indap A.R., Schmidt S., Boyko A.R., Hernandez R.D., Hubisz M.J., Sninsky J.J., White T.J., Sunyaev S.R., Nielsen R. Proportionally more deleterious genetic variation in European than in African populations. Nature. 2008;451:994–997. doi: 10.1038/nature06611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim W.K., Marcotte E.M. Age-dependent evolution of the yeast protein interaction network suggests a limited role of gene duplication and divergence. PLoS Comput. Biol. 2008;4:e1000232. doi: 10.1371/journal.pcbi.1000232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Capra J.A., Pollard K.S., Singh M. Novel genes exhibit distinct patterns of function acquisition and network integration. Genome Biol. 2010;11:R127. doi: 10.1186/gb-2010-11-12-r127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang N., Lee I., Marcotte E.M., Hurles M.E. Characterising and predicting haploinsufficiency in the human genome. PLoS Genet. 2010;6:e1001154. doi: 10.1371/journal.pgen.1001154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Grundberg E., Small K.S., Hedman Å.K., Nica A.C., Buil A., Keildson S., Bell J.T., Yang T.-P., Meduri E., Barrett A., Multiple Tissue Human Expression Resource (MuTHER) Consortium Mapping cis- and trans-regulatory effects across multiple tissues in twins. Nat. Genet. 2012;44:1084–1089. doi: 10.1038/ng.2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang S., Liu Y., Jiang N., Chen J., Leach L., Luo Z., Wang M. Genome-wide eQTLs and heritability for gene expression traits in unrelated individuals. BMC Genomics. 2014;15:13. doi: 10.1186/1471-2164-15-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Powell J.E., Henders A.K., McRae A.F., Wright M.J., Martin N.G., Dermitzakis E.T., Montgomery G.W., Visscher P.M. Genetic control of gene expression in whole blood and lymphoblastoid cell lines is largely independent. Genome Res. 2012;22:456–466. doi: 10.1101/gr.126540.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kaessmann H. Origins, evolution, and phenotypic impact of new genes. Genome Res. 2010;20:1313–1326. doi: 10.1101/gr.101386.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Domazet-Lošo T., Tautz D. A phylogenetically based transcriptome age index mirrors ontogenetic divergence patterns. Nature. 2010;468:815–818. doi: 10.1038/nature09632. [DOI] [PubMed] [Google Scholar]
- 41.Cai J.J., Woo P.C.Y., Lau S.K.P., Smith D.K., Yuen K.-Y. Accelerated evolutionary rate may be responsible for the emergence of lineage-specific genes in ascomycota. J. Mol. Evol. 2006;63:1–11. doi: 10.1007/s00239-004-0372-5. [DOI] [PubMed] [Google Scholar]
- 42.Cai J.J., Borenstein E., Petrov D.A. Broker genes in human disease. Genome Biol. Evol. 2010;2:815–825. doi: 10.1093/gbe/evq064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Papp B., Pál C., Hurst L.D. Dosage sensitivity and the evolution of gene families in yeast. Nature. 2003;424:194–197. doi: 10.1038/nature01771. [DOI] [PubMed] [Google Scholar]
- 44.Kondrashov F.A., Kondrashov A.S. Role of selection in fixation of gene duplications. J. Theor. Biol. 2006;239:141–151. doi: 10.1016/j.jtbi.2005.08.033. [DOI] [PubMed] [Google Scholar]
- 45.Conant G.C., Wolfe K.H. Turning a hobby into a job: how duplicated genes find new functions. Nat. Rev. Genet. 2008;9:938–950. doi: 10.1038/nrg2482. [DOI] [PubMed] [Google Scholar]
- 46.Edger P.P., Pires J.C. Gene and genome duplications: the impact of dosage-sensitivity on the fate of nuclear genes. Chromosome Res. 2009;17:699–717. doi: 10.1007/s10577-009-9055-9. [DOI] [PubMed] [Google Scholar]
- 47.Makino T., McLysaght A. Ohnologs in the human genome are dosage balanced and frequently associated with disease. Proc. Natl. Acad. Sci. USA. 2010;107:9270–9274. doi: 10.1073/pnas.0914697107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Makino T., McLysaght A., Kawata M. Genome-wide deserts for copy number variation in vertebrates. Nat. Commun. 2013;4:2283. doi: 10.1038/ncomms3283. [DOI] [PubMed] [Google Scholar]
- 49.Liao B.-Y., Zhang J. Low rates of expression profile divergence in highly expressed genes and tissue-specific genes during mammalian evolution. Mol. Biol. Evol. 2006;23:1119–1128. doi: 10.1093/molbev/msj119. [DOI] [PubMed] [Google Scholar]
- 50.Lemos B., Bettencourt B.R., Meiklejohn C.D., Hartl D.L. Evolution of proteins and gene expression levels are coupled in Drosophila and are independently associated with mRNA abundance, protein length, and number of protein-protein interactions. Mol. Biol. Evol. 2005;22:1345–1354. doi: 10.1093/molbev/msi122. [DOI] [PubMed] [Google Scholar]
- 51.Warnefors M., Kaessmann H. Evolution of the correlation between expression divergence and protein divergence in mammals. Genome Biol. Evol. 2013;5:1324–1335. doi: 10.1093/gbe/evt093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gu X., Zhang Z., Huang W. Rapid evolution of expression and regulatory divergences after yeast gene duplication. Proc. Natl. Acad. Sci. USA. 2005;102:707–712. doi: 10.1073/pnas.0409186102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Leach L.J., Zhang Z., Lu C., Kearsey M.J., Luo Z. The role of cis-regulatory motifs and genetical control of expression in the divergence of yeast duplicate genes. Mol. Biol. Evol. 2007;24:2556–2565. doi: 10.1093/molbev/msm188. [DOI] [PubMed] [Google Scholar]
- 54.Gu X., Su Z. Tissue-driven hypothesis of genomic evolution and sequence-expression correlations. Proc. Natl. Acad. Sci. USA. 2007;104:2779–2784. doi: 10.1073/pnas.0610797104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fraser H.B., Wall D.P., Hirsh A.E. A simple dependence between protein evolution rate and the number of protein-protein interactions. BMC Evol. Biol. 2003;3:11. doi: 10.1186/1471-2148-3-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fraser H.B., Hirsh A.E., Steinmetz L.M., Scharfe C., Feldman M.W. Evolutionary rate in the protein interaction network. Science. 2002;296:750–752. doi: 10.1126/science.1068696. [DOI] [PubMed] [Google Scholar]
- 57.Gout J.-F., Kahn D., Duret L., Paramecium Post-Genomics Consortium The relationship among gene expression, the evolution of gene dosage, and the rate of protein evolution. PLoS Genet. 2010;6:e1000944. doi: 10.1371/journal.pgen.1000944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cooper D.N., Kehrer-Sawatzki H. Exploring the potential relevance of human-specific genes to complex disease. Hum. Genomics. 2011;5:99–107. doi: 10.1186/1479-7364-5-2-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lappalainen T., Sammeth M., Friedländer M.R., ’t Hoen P.A.C., Monlong J., Rivas M.A., Gonzàlez-Porta M., Kurbatova N., Griebel T., Ferreira P.G., Geuvadis Consortium Transcriptome and genome sequencing uncovers functional variation in humans. Nature. 2013;501:506–511. doi: 10.1038/nature12531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nica A.C., Montgomery S.B., Dimas A.S., Stranger B.E., Beazley C., Barroso I., Dermitzakis E.T. Candidate causal regulatory effects by integration of expression QTLs with complex trait genetic associations. PLoS Genet. 2010;6:e1000895. doi: 10.1371/journal.pgen.1000895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nicolae D.L., Gamazon E., Zhang W., Duan S., Dolan M.E., Cox N.J. Trait-associated SNPs are more likely to be eQTLs: annotation to enhance discovery from GWAS. PLoS Genet. 2010;6:e1000888. doi: 10.1371/journal.pgen.1000888. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.