

Abstract
Thoracic aortic aneurysm and dissection (TAAD) is an autosomal-dominant disorder with major life-threatening complications. The disease displays great genetic heterogeneity with some forms allelic to Marfan and Loeys-Dietz syndrome, and an important number of cases still remain unexplained at the molecular level. Through whole-exome sequencing of affected members in a large TAAD-affected family, we identified the c.472C>T (p.Arg158∗) nonsense mutation in MFAP5 encoding the extracellular matrix component MAGP-2. This protein interacts with elastin fibers and the microfibrillar network. Mutation screening of 403 additional probands identified an additional missense mutation of MFAP5 (c.62G>T [p.Trp21Leu]) segregating with the disease in a second family. Functional analyses performed on both affected individual’s cells and in vitro models showed that these two mutations caused pure or partial haploinsufficiency. Thus, alteration of MAGP-2, a component of microfibrils and elastic fibers, appears as an initiating mechanism of inherited TAAD.
Main Text
Thoracic aortic aneurysms and dissection (TAAD) disorder is a major cause of sudden death. TAAD is essentially a multifactorial disease but 20% of cases are inherited in an autosomal-dominant manner and are found in distinct syndromes or represent the only clinical feature of the disease.1 Research of the past 20 years has shown that there is a strong molecular overlap between these different clinical forms of TAAD, mutations in the same genes being found to be responsible for syndromic or isolated TAAD.2 The first syndrome to be associated with TAAD was Marfan syndrome (MFS [MIM 154700/ORPHA558]). In the majority of cases, MFS is due to mutations in FBN1 (MIM 134797) encoding fibrillin-1, the major component of isolated microfibrillar aggregates and the microfibrillar component of elastic fibers in the extracellular matrix (ECM).3 In some cases, MFS might also be due to mutations in TGFBR2 (MIM 190182), thus demonstrating involvement of alterations in TGF-β signaling as an initiating mechanism of TAAD.4 This was further emphasized by the description of Loeys-Dietz syndrome (LDS [MIM 609192/ORPHA60030]) with mutations in TGFBR1 (MIM 190181)/TGFBR2 and the description of aneurysms-osteoarthritis syndrome (AOS [MIM 613795/ORPHA284984]) with mutations in SMAD3 (MIM 190220).5–7 Genetic mutations causing isolated TAAD are found in all previous genes as well as TGFB2 (MIM 190220)8,9 and in genes encoding proteins of the vascular smooth muscle cell (SMC) contractile apparatus: ACTA2 (MIM 102620), MYH11 (MIM 160745), MYLK (MIM 600922), and PRKG1 (MIM 176894).10–13 Although defects in several genes lead to altered TGF-β signaling or SMC contraction, defects in a single ECM protein (i.e., fribrillin-1) have been known until now to account for TAAD. Finally, TAAD shows significant genetic heterogeneity but 80% remain unexplained at the molecular level.2
To identify additional mutations causing TAAD, we performed whole-exome sequencing in TAAD-affected families in which no causal mutation had been identified in genes previously associated with TAAD. Probands and families were recruited through the National Reference Center for Marfan syndrome and related disorders and through related centers nationwide. All affected individuals were screened for diagnosis of possible syndromic (notably MFS) or nonsyndromic TAAD through a multidisciplinary clinic including systematic slit-lamp examination and extensive imaging, aortic echography, and molecular analysis. Blood samples were obtained from affected individuals in agreement with the French Bioethic laws (IRB: CCP Ile de France XI, authorization #11 008; informed, written consent was obtained) and genomic DNA prepared as previously reported.4
Whole-exome sequencing was first performed with DNA from four affected persons (circled individuals in Figure 1A) from TAA-9801 (whole-exome sequencing performed at Integragen S.A.). The female proband (II:14) presented aortic root dilatation discovered during investigation of lone paroxysmal atrial fibrillation at the age of 50 years (aortic root diameter of 46 mm), which was associated with mitral valve prolapse and slight skeletal features (pectus excavatum and arachnodactyly). Family history indicated that her 83-year-old mother (I:1) had a history of atrial fibrillation and presented with Alzheimer disease. Interestingly, a maternal aunt died suddenly at 62 years old and also had a history of atrial fibrillation. Additionally, coronary artery bypass surgery was performed in a maternal uncle at age 60 years, and there was family history of stroke at 55 years, sudden death before the age of 50 years, and heart disease at the age of 55 years reported in three brothers of the maternal grandmother. Clinical familial screening allowed recognition of aortic dilatation (aortic root diameter of 43 mm) in the son of the proband (III:11), without any extra-aortic features, and a sister of the proband (II:10) presented aortic root dilatation (46 mm) and skeletal features (pectus carinatum, arachnodactyly, highly arched palate). Of note, she also suffered lone atrial fibrillation, as did her sister, and complained of migraine; aortic dilatation (aortic root diameter at 50 mm) was also observed in the brother of the proband (II:15), with mild skeletal features (i.e., pectus excavatum). The aortic diameter was within normal limits (Z = 1.8) in his son (III:12) at the age of 16 (who also presented pectus excavatum) and slightly enlarged (Z = 2.3) in his second son at age of 12 (III:13). Lastly, a nephew of the proband (III:1) was screened at the age of 36 years and had an aortic root diameter at the upper limit of normal values (38 mm). Detailed clinical features are presented in Table S1 available online.
Figure 1.
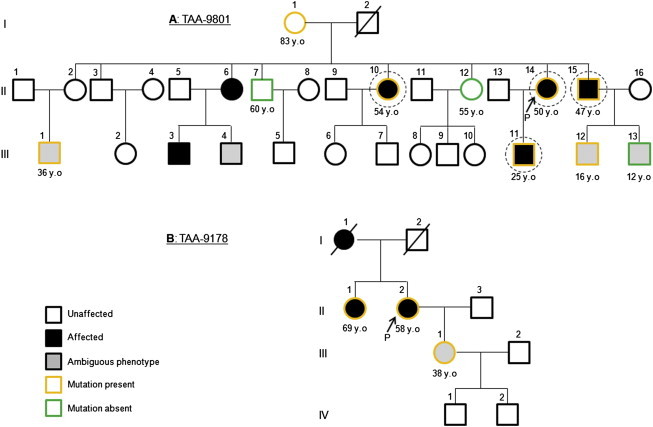
Pedigrees of Families TAA-9801 and TAA-9178
Black arrows indicate probands. DNA from circled individuals was used for whole-exome sequencing. Orange symbols indicate family members heterozygous for mutations in MFAP5: c.472C>T (p.Arg158∗) in TAA-9801 (A) and c.62G>T (p.Trp21Leu) in TAA-9178 (B). The age at diagnosis is specified when available.
DNA was subjected to the exome-capture procedure with the Agilent SureSelect Human All Exon kit (V4, 50 Mb) according to the manufacturer’s protocols. Paired-end sequencing was performed on an Illumina HiSeq 2000, and the short reads (75 bp) were aligned to the Human Genome (UCSC hg19) by CASAVA 1.8. Image analysis and base calling were performed with Illumina Real Time Analysis (RTA) Pipeline v.1.14 with default parameters. The alignment algorithm used was ELANDv.2. Variation annotation was performed with an in-house pipeline of Integragen S.A. Sequencing results were filtered for a depth ≥10× and for novel heterozygous variants shared between these four affected relatives and absent from dbSNP, 1000 Genomes, HapMap, Exome Variant Server, and in-house whole-exome data set of 176 additional exomes. Nonsense, missense, insertion/deletion, and splice variants were carefully examined.
The filtering strategy identified 17 shared novel variants including 14 missense and 3 nonsense variants (Table S3). Nonsense mutations were detected in FER1L5 (c.4111C>T), GATA6 (MIM 601656) (c.457C>A), and MFAP5 (MIM 601103) (c.472C>T). FER1L5 (encoding the Fer-1-Like 5 [C. elegans gene]) is involved in myoblast fusion. GATA6 encodes a zinc finger transcription factor that plays an important role in cell differentiation and organogenesis during vertebrate development. GATA6 haploinsufficiency has been reported as a frequent cause of diabetes with pancreatic agenesis and congenital heart defects.14,15 Pancreatic agenesis and gut and heart malformations in subjects heterozygous for the GATA6 variation were checked and excluded after careful examination of scanner imaging in TAA-9801 individuals, meaning that haploinsufficiency for GATA6 does not lead systematically to the described organogenesis defects. Bidirectional Sanger DNA sequencing of GATA6 mutation (RefSeq accession number NM_005257.5) was performed in other available DNA from members of TAA-9801. The segregation of this variant in TAA-9801 did not corroborate with TAAD (data not shown).
The third candidate, MFAP5 (Microfibrillar Associated Protein 5), encodes MAGP-2 (Microfibril-Associated Glycoprotein 2), a component of the ECM interacting with fibrillin-1 and present in isolated or elastin-associated microfibrils.16 The nonsense MFAP5 mutation (RefSeq NM_003480.3) was sequenced in TAA-9801. Good familial segregation was observed (Figure 1A). Therefore, other mutational events in MFAP5 were looked for in a population of 225 familial probands and 178 simplex TAAD-affected individuals of French origin and 267 TAAD-affected individuals from the Texas collection by systematic Sanger DNA sequencing of all exons (sequencing primers listed in Table S2). One additional MFAP5 variant (c.62G>T) was identified in the proband of TAA-9178 and two affected relatives (Figure 1B). The female proband presented aortic dissection type A at the age of 58 years with no skeletal, ophthalmological, cutaneous, or pulmonary features. Investigation of her 69-year-old sister (II:1) imaged thoracic aortic aneurysm (aortic root diameter at 41 mm) in the context of hypertension with left ventricular hypertrophy. She presented no extra aortic features, but had a history of breast cancer, glaucoma, and hypothyroidism. The proband’s 38-year-old daughter (III:1) had an aortic root diameter at the upper limit of normal values (35 mm), no skeletal features, and no medical problem beyond migraines. Detailed clinical features are presented in Table S1. The variation leads to a substitution in the N-terminal domain of MAGP-2 affecting a very conserved tryptophan (p.Trp21Leu) (Figure 2). This substitution is located close to the putative peptide-signal cleavage site and predicted to be possibly damaging by Polyphen-2 and MutationTaster and as pathogenic by UMD-Predictor.17
Figure 2.

Structure of MFAP5 and MAGP-2
MFAP5 consists of ten exons (squares) with nine coding exons (blue squares). Mutations detected in this study appear under the gene schematic. MFAP5 encodes MAGP-2 for which the protein structure is depicted above the genomic structure. Scissors indicate the putative cleavage sites for the signal peptide (SP) peptidase and for proprotein convertases (RLRR). Putative site for cell attachment (RGD) and N- and O-Glycosylations (N-Glyc and O-Glyc) are also indicated. The electrophoregrams of each mutation obtained from Sanger sequencing appear below the genomic structure. The affected codons are underlined in black. Substitution c.62G>T (left) affects an evolutionary conserved tryptophan among mouse, rat, dog, cat, and chicken. Multiple sequence alignment was performed with CLUSTAL Omega. Comparative sequencing of mutation c.472C>T from genomic DNA and transcript of MFAP5 (cDNA) are presented (right). cDNA from individual TAA-9801 II:15 was obtained from mRNA harvested from primary fibroblasts grown with or without emetine to block the NMD RNA surveillance pathway.
The effect of mutations at the mRNA and protein levels was investigated in subject dermal fibroblasts explanted from three affected members from families TAA-9801 (II:10 and II:15) and TAA-9178 (II:2). Because the c.472C>T variation was located in the last exon of MFAP5, it was not expected to cause nonmediated decay (NMD). This was confirmed by the sequencing of the MFAP5 transcript in fibroblasts from two family relatives who both showed similar levels of mutant and wild-type transcripts in fibroblasts grown with and without emetine (Figure 2, sequencing primers listed in Table S2). Immunoblot analysis was performed with fibroblasts from heterozygous persons with antibodies directed against N-terminal (sc-134437 from Santa Cruz Biotechnology) and C-terminal (ab47826 obtained from Abcam) parts of MAGP-2. Results suggested that although the mutant transcript is expressed, the truncated protein present in cellular lysates is probably degraded by an endoplasmic-reticulum-associated degradation pathway because no extracellular signal was detected for the truncated protein (Figures 3A and 3B). This result showed that c.472C>T leads to pure haploinsufficiency in heterozygous persons (Figure 3C). Immunoblots on fibroblasts from heterozygous persons with the c.62G>T mutation (TAA-9178) were also performed. Presence of MAGP-2 at the expected molecular weight was observed (Figures 3A and 3B). However, intracellular levels of the protein appeared to be lower in c.62G>T-carrying fibroblasts.
Figure 3.

Immunoblot Analysis of MAGP-2 in Fibroblasts from Affected Individuals
Proteins were extracted from cellular lysates (A) and extracellular media (B) of fibroblasts from affected subjects carrying the nonsense (TAA-9801 family) or missense (TAA-9178) mutations and healthy control subjects. Intracellular soluble proteins were extracted with TRIzol reagent (Life Technologies) according to the manufacturer’s instructions. This method avoids contamination by insoluble cell-associated matrix such as matrix-associated MAGP-2. Extracellular proteins were extracted from extracellular media after centrifugation with the AMICON 10 kDa filtration columns (Millipore). Immunoblots were realized via two antibodies recognizing either N- or C-terminal domains of MAGP-2 as mutations affect N-terminal (p.Trp21Leu) or C-terminal (p.Arg158∗) domains or horseradish peroxidase-conjugated β-tubulin (1:5,000 dilution, ab21058 from Abcam). Purified recombinant human MAGP-2 expressed in HEK cells (TP303242 from Origene Technologies) was used as control on each blot. Densitometric analysis was performed with Scion Image software (Scion Corporation). Immunoblots showed the presence in cellular lysates of both WT and truncated MAGP-2 in individuals heterozygous for the nonsense c.472C>T variant. (C) Quantification of immunoblots (N-term) showed that the band corresponding to WT MAGP-2 represents less than 50% of the total MAGP-2 signal detected (normalized with β-tubulin; means ± standard error of the mean). The band corresponding to the truncated protein was not found in extracellular medium.
Site-directed mutagenesis and transfection assays were performed to fully investigate specific effects of each MFAP5 mutation identified as cells from heterozygous subjects constitutively expressing both WT and mutated alleles. The c.472C>T and c.62G>T MFAP5 mutations were introduced separately in plasmids containing the wild-type (WT) coding sequence of MFAP5 with the QuikChange II Site-Directed Mutagenesis Kit, according to manufacturer’s instruction (Agilent Technologies). The coding sequence of MFAP5 is cloned into pCMV6-XL5 vector suitable for mammalian cell overexpression assays (Origene Technologies). After selecting clones, plasmids were purified with the Plasmid Maxi kit (QIAGEN). The presence of the c.472C>T or the c.62G>T mutations and the absence of other coding variations were checked by bidirectional Sanger sequencing via primers spanning the coding sequence of MFAP5 (available upon request). Results confirmed that c.472C>T abrogates MAGP-2 production (Figures 4A and 4B). Conversely, the signal from p.Trp21Leu MAGP-2 was present but significantly lower than that of WT MAGP-2 in cellular lysates (p = 0.03) (Figures 4C and 4E). An important fraction of mutated proteins might be recognized and degraded by the unfolded protein response. The extracellular signal obtained with both antibodies for WT and mutant MAGP-2 constructs was consistently diffuse. However, careful observation of immunoblots revealed for p.Trp21Leu MAGP-2 a shift toward higher molecular weight bands (Figure 4D). So, it is clear that p.Trp21Leu alters posttranslational modifications and/or extracellular processing of MAGP-2. Interestingly, it has been shown that mouse MAGP-2 is subject to a cleavage by proprotein convertases (PC) and that alteration of this cleavage reduces matrix-associated MAGP-2.18,19 Such proteolytic processing seems to have a physiological importance as observed with other ECM proteins including fibrillin-1.20,21 The cleavage site being conserved in human (putative consensus site located between amino acids 152 and 153), p.Trp21Leu might alter the recognition of the cleavage site by proprotein convertases leading to an enrichment in full-length proteins versus cleaved MAGP-2. Thus, alteration of this process by p.Trp21Leu might affect interactions of MAGP-2 with other ECM proteins or deposition in microfibrils.
Figure 4.

Immunoblot Analysis of WT, p.Arg158∗, and p.Trp21Leu MAGP-2 in Transfected HEK
(A and B) Cellular lysates (A) and extracellular media (B) of transfected HEK with WT or c.472C>T (p.Arg158∗) coding vectors were analyzed. Transient transfection assays of constructs were performed in HEK293 H cells (HEK, GIBCO Life Technologies) because they do not express endogenous MFAP5. Cells were transiently cotransfected in serum-free DMEM (GIBCO Life Technologies) with 1 μg of a vector encoding WT or mutated c.472C>T (p.Arg158∗) MAGP-2 and 1 μg of an empty vector (pcDNA3.1). The Fugene 6 transfection agent was used (Promega). After transfection, HEK cells were incubated for 24 hr. The extracellular medium was harvested and cells were washed three times with PBS prior to RNA/protein extractions. Transfection efficiency was checked and showed proper expression of the MFAP5 transcripts for WT and mutant constructs (Figure S1). Protein extraction and IBs were performed as described in Figure 3. WT MAGP-2 was detected in both cellular lysates and extracellular media as expected. p.Arg158∗ truncated MAGP-2 protein was detectable in cellular lysates and totally absent from the extracellular medium.
(C and D) Cellular lysates (C) and extracellular media (D) of transfected HEK with WT or c.62G>T (p.Trp21Leu) coding vectors were analyzed. Immunoblots and checking of transfection efficacy were realized as previously described. Immunoblotting of the N-terminal part of MAGP-2 was not performed because the mutation affects an amino acid located in the epitope.
(E) Quantification of immunoblots confirmed a significant decrease of intracellular presence of p.Trp21Leu MAGP-2. All values are expressed as means ± standard error of the mean. Statistical differences between WT and p.Trp21Leu MAGP-2 were analyzed by a Student’s t test. The absence of p.Arg158∗ MAGP-2 precluded statistical analysis of WT versus p.Arg158∗. Differences were considered statistically significant at values of p < 0.05. Data shown for each construct were pooled from three independent transfection assays.
In parallel, aortic tissue was collected at the time of surgery from the TAA-9178 proband (II:2) (Bichat University Hospital). Histological analyses showed typical features of TAAD: staining with Masson’s trichrome and alcian blue revealed a disorganization of the media with loss of SMCs and proteoglycan accumulation, respectively (Figure 5A). In parallel, a possible effect of MFAP5 loss-of-function mutations on the TGF-β signaling pathway was investigated. Indeed, it is well established that the microfibrillar network acts as a reservoir for latent TGF-β. Assessment of TGF-β1 and nuclear phosphorylated SMAD2/3 showed greatly enhanced TGF-β signaling in the aorta from the individual with MFAP5 mutation (Figure 5B). Paradoxically, upregulation of TGF-β signaling is a major hallmark of syndromic and isolated TAAD aneurysmal aortas regardless of their etiologies.22 Thus, in reducing the TGF-β1 reservoir in ECM, MFAP5 loss of function induces upregulation of the TGF-β/BMP signaling pathway.
Figure 5.

Immunohistology of Aortic Wall from a Dissection Case in Family TAA-9178
Aneurysmal ascending aorta was collected during aortic surgery for the proband from the TAA-9178 family (Bichat University Hospital) (II:2, heterozygous for the c.62G>T MFAP5 mutation). Normal thoracic aorta was obtained from a normal organ transplant donor with the authorization of the French Biomedicine Agency. Aneurysmal tissue was sampled in the outer curvature, the most dilated part of the ascending aorta. Aortic specimens were fixed in Bouin’s solution, and serial sections (6 μm thickness) were obtained from each specimen.
(A) Masson’s trichrome and Alcian blue revealed a disorganization of the tunica media with a loss of smooth muscle cells (SMCs) and proteoglycan accumulation, respectively, compared to healthy aorta.
(B) Immunohistofluorescent staining for the same subject and control samples was performed with antibodies to nuclear phosphorylated SMAD2/3 (phosphorylated Ser 423/425 SMAD2/3, sc-11769 from Santa Cruz Biotechnology) and mouse monoclonal antibody to TGF-β1 (sc-65378 from Santa Cruz Biotechnology). Alexa Fluor 555-conjugated secondary antibodies to mouse and rabbit (Invitrogen, Life Technologies) were used. Because pathology samples had been fixed in Bouin’s solution, SMC nuclei could not be Dapi-stained. Elastic fibers appear in green (TGF-β1) and yellow (p-SMAD2/3). Staining showed enhanced TGF-β signaling in patient compared to healthy aorta.
Finally, deleterious variations in MFAP2 (MIM 156790) (paralog of MFAP5 in mouse and also in human, and encoding MAGP-1) were looked for in the collection of French TAAD-affected individuals. All exons were systematically screened and no variation that could account for the disease was identified (sequencing primers listed in Table S2). Although MGAP-1 and MAGP-2 are described as paralogs, they support distinct functions in vivo that can explain the lack of MFAP2 mutations in TAAD-affected subjects.16 In particular, Gibson et al.16 described distinct patterns of tissue localization between MAGP-1 and MAGP-2 in bovine: whereas MAGP-2 was not detected in the medial layer of fetal thoracic aorta, in much of the peritubular matrix of fetal and mature kidney, and in the mature ocular zonule in contrast to MAGP-1, MAGP-2 was specifically associated with fibrillin-containing microfibrils in nuchal ligament, dermis, adventitia of aorta, glomerular mesangium, and perimysium. Interestingly, homozygous Mfap5 knockout mice display only hematopoietic features characterized by neutropenia.23 However, an age-dependant aortic dilation is observed in double-knockout Mfap5−/− and Mfap2−/− mice. Because all TAAD-affected individuals were systematically screened for mutations both in MFAP2 and MFAP5, the existence in human of a digenism as that seen in mouse can be excluded.
Here we report that mutations in MFAP5 cause familial forms of thoracic aneurysms. Aortic dilations observed in both TAA-9801 and TAA-9178 families affect the aortic root, similar to what has been observed previously with mutations in TAAD-associated genes. The occurrence of other phenotypic features (i.e., other MFS, LDS, or AOS features) was quite infrequent in these two families, suggesting that loss of function of MFAP5 leads to nonsyndromic TAAD. Interestingly and in addition to aortic dilation, familial history of lone paroxysmal atrial fibrillation (PAF) was reported in the TAA-9801 family. The proband was indeed diagnosed during an exploration for lone PAF. This unusual presentation of aortic aneurysms might explain why only two TAAD-affected families with MFAP5 mutations were detected after screening of individuals in the French and Texan populations.
MFAP5 encodes a structural component of the ECM localized in fibrillin-containing microfibrils, either elastin associated or elastin free.16 Multiple experimental findings showed that MAGP-2 plays an important role in the physiology of the ECM through interactions with different microfibrillar components such as fibrillin-1, fibrillin-2, fibulin-1, and tropoelastine.16,24,25 More recently it has been shown that mouse MAGP-2 can bind reversibly different TGF-β/BMP superfamily members such as TGF-β1, TGF-β2, and BMP2 possibly through an acidic region in the N terminus of the protein.23 It must be noted that mutations in genes encoding several partners of MAGP-2, in particular FBN1 and TGFB2, are associated with thoracic aortic aneurysm.8,26 These data confirm a close link between MAGP-2 and pathways involved in pathogenesis of TAAD. Thus, as in FBN1 mutations, loss of function of MFAP5 might affect the structural function of the vessel by directly altering ECM organization and/or deposition of elastic fibers leading to fibers disruption and to dilation of the aorta.
In conclusion, we describe MFAP5 mutations involved in the physiopathology of TAAD. In this disease attention had shifted away from the matrix due to the identification of mutations in genes involved in TGF-β signaling and SMC contraction. Our results highlight the interest of identifying TAAD mutations in genes encoding other structural proteins of the ECM.
Acknowledgments
The authors are extremely grateful to the families involved in this study and to the medical professionals who aided in the collection of clinical data from the families. We thank Jean-Baptiste Michel and Marie-Paule Jacob for fruitful discussions. We also thank Liliane Louedec, Zoubida Karim, and Narjesse Karboul for technical assistance. The following sources provided funding for this study: Projet Hospitalier de Recherche Clinique (PHRC, AOM 10108), Agence Nationale de Recherche (ANR, GDPM2), Fédération Française de Cardiologie, Société Française de Cardiologie, and DHU-FIRE (Assistance Publique-Hôpitaux de Paris).
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes, http://browser.1000genomes.org
Clustal Omega, http://www.ebi.ac.uk/Tools/msa/clustalo/
MutationTaster, http://www.mutationtaster.org/
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
PolyPhen-2, http://www.genetics.bwh.harvard.edu/pph2/
UCSC Genome Browser, http://genome.ucsc.edu
UMD-Predictor, http://umd-predictor.eu/
References
- 1.Milewicz D.M., Guo D.C., Tran-Fadulu V., Lafont A.L., Papke C.L., Inamoto S., Kwartler C.S., Pannu H. Genetic basis of thoracic aortic aneurysms and dissections: focus on smooth muscle cell contractile dysfunction. Annu. Rev. Genomics Hum. Genet. 2008;9:283–302. doi: 10.1146/annurev.genom.8.080706.092303. [DOI] [PubMed] [Google Scholar]
- 2.Jondeau G., Boileau C. Genetics of thoracic aortic aneurysms. Curr. Atheroscler. Rep. 2012;14:219–226. doi: 10.1007/s11883-012-0241-4. [DOI] [PubMed] [Google Scholar]
- 3.Faivre L., Collod-Beroud G., Loeys B.L., Child A., Binquet C., Gautier E., Callewaert B., Arbustini E., Mayer K., Arslan-Kirchner M. Effect of mutation type and location on clinical outcome in 1,013 probands with Marfan syndrome or related phenotypes and FBN1 mutations: an international study. Am. J. Hum. Genet. 2007;81:454–466. doi: 10.1086/520125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mizuguchi T., Collod-Beroud G., Akiyama T., Abifadel M., Harada N., Morisaki T., Allard D., Varret M., Claustres M., Morisaki H. Heterozygous TGFBR2 mutations in Marfan syndrome. Nat. Genet. 2004;36:855–860. doi: 10.1038/ng1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Loeys B.L., Schwarze U., Holm T., Callewaert B.L., Thomas G.H., Pannu H., De Backer J.F., Oswald G.L., Symoens S., Manouvrier S. Aneurysm syndromes caused by mutations in the TGF-beta receptor. N. Engl. J. Med. 2006;355:788–798. doi: 10.1056/NEJMoa055695. [DOI] [PubMed] [Google Scholar]
- 6.van de Laar I.M., Oldenburg R.A., Pals G., Roos-Hesselink J.W., de Graaf B.M., Verhagen J.M., Hoedemaekers Y.M., Willemsen R., Severijnen L.A., Venselaar H. Mutations in SMAD3 cause a syndromic form of aortic aneurysms and dissections with early-onset osteoarthritis. Nat. Genet. 2011;43:121–126. doi: 10.1038/ng.744. [DOI] [PubMed] [Google Scholar]
- 7.Aubart M., Gobert D., Aubart-Cohen F., Detaint D., Hanna N., d’Indya H., Lequintrec J.S., Renard P., Vigneron A.M., Dieudé P. Early-onset osteoarthritis, Charcot-Marie-Tooth like neuropathy, autoimmune features, multiple arterial aneurysms and dissections: an unrecognized and life threatening condition. PLoS ONE. 2014;9:e96387. doi: 10.1371/journal.pone.0096387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boileau C., Guo D.C., Hanna N., Regalado E.S., Detaint D., Gong L., Varret M., Prakash S.K., Li A.H., d’Indy H., National Heart, Lung, and Blood Institute (NHLBI) Go Exome Sequencing Project TGFB2 mutations cause familial thoracic aortic aneurysms and dissections associated with mild systemic features of Marfan syndrome. Nat. Genet. 2012;44:916–921. doi: 10.1038/ng.2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lindsay M.E., Schepers D., Bolar N.A., Doyle J.J., Gallo E., Fert-Bober J., Kempers M.J., Fishman E.K., Chen Y., Myers L. Loss-of-function mutations in TGFB2 cause a syndromic presentation of thoracic aortic aneurysm. Nat. Genet. 2012;44:922–927. doi: 10.1038/ng.2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guo D.C., Pannu H., Tran-Fadulu V., Papke C.L., Yu R.K., Avidan N., Bourgeois S., Estrera A.L., Safi H.J., Sparks E. Mutations in smooth muscle alpha-actin (ACTA2) lead to thoracic aortic aneurysms and dissections. Nat. Genet. 2007;39:1488–1493. doi: 10.1038/ng.2007.6. [DOI] [PubMed] [Google Scholar]
- 11.Zhu L., Vranckx R., Khau Van Kien P., Lalande A., Boisset N., Mathieu F., Wegman M., Glancy L., Gasc J.M., Brunotte F. Mutations in myosin heavy chain 11 cause a syndrome associating thoracic aortic aneurysm/aortic dissection and patent ductus arteriosus. Nat. Genet. 2006;38:343–349. doi: 10.1038/ng1721. [DOI] [PubMed] [Google Scholar]
- 12.Wang L., Guo D.C., Cao J., Gong L., Kamm K.E., Regalado E., Li L., Shete S., He W.Q., Zhu M.S. Mutations in myosin light chain kinase cause familial aortic dissections. Am. J. Hum. Genet. 2010;87:701–707. doi: 10.1016/j.ajhg.2010.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guo D.C., Regalado E., Casteel D.E., Santos-Cortez R.L., Gong L., Kim J.J., Dyack S., Horne S.G., Chang G., Jondeau G., GenTAC Registry Consortium. National Heart, Lung, and Blood Institute Grand Opportunity Exome Sequencing Project Recurrent gain-of-function mutation in PRKG1 causes thoracic aortic aneurysms and acute aortic dissections. Am. J. Hum. Genet. 2013;93:398–404. doi: 10.1016/j.ajhg.2013.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lango Allen H., Flanagan S.E., Shaw-Smith C., De Franco E., Akerman I., Caswell R., Ferrer J., Hattersley A.T., Ellard S., International Pancreatic Agenesis Consortium GATA6 haploinsufficiency causes pancreatic agenesis in humans. Nat. Genet. 2012;44:20–22. doi: 10.1038/ng.1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kodo K., Nishizawa T., Furutani M., Arai S., Yamamura E., Joo K., Takahashi T., Matsuoka R., Yamagishi H. GATA6 mutations cause human cardiac outflow tract defects by disrupting semaphorin-plexin signaling. Proc. Natl. Acad. Sci. USA. 2009;106:13933–13938. doi: 10.1073/pnas.0904744106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gibson M.A., Finnis M.L., Kumaratilake J.S., Cleary E.G. Microfibril-associated glycoprotein-2 (MAGP-2) is specifically associated with fibrillin-containing microfibrils but exhibits more restricted patterns of tissue localization and developmental expression than its structural relative MAGP-1. J. Histochem. Cytochem. 1998;46:871–886. doi: 10.1177/002215549804600802. [DOI] [PubMed] [Google Scholar]
- 17.Frédéric M.Y., Lalande M., Boileau C., Hamroun D., Claustres M., Béroud C., Collod-Béroud G. UMD-predictor, a new prediction tool for nucleotide substitution pathogenicity — application to four genes: FBN1, FBN2, TGFBR1, and TGFBR2. Hum. Mutat. 2009;30:952–959. doi: 10.1002/humu.20970. [DOI] [PubMed] [Google Scholar]
- 18.Donovan L.J., Cha S.E., Yale A.R., Dreikorn S., Miyamoto A. Identification of a functional proprotein convertase cleavage site in microfibril-associated glycoprotein 2. Matrix Biol. 2013;32:117–122. doi: 10.1016/j.matbio.2012.11.009. [DOI] [PubMed] [Google Scholar]
- 19.Miyamoto A., Donovan L.J., Perez E., Connett B., Cervantes R., Lai K., Withers G., Hogrebe G. Binding of MAGP2 to microfibrils is regulated by proprotein convertase cleavage. Matrix Biol. 2014 doi: 10.1016/j.matbio.2014.08.003. Published online August 19, 2014. [DOI] [PubMed] [Google Scholar]
- 20.Raghunath M., Putnam E.A., Ritty T., Hamstra D., Park E.S., Tschödrich-Rotter M., Peters R., Rehemtulla A., Milewicz D.M. Carboxy-terminal conversion of profibrillin to fibrillin at a basic site by PACE/furin-like activity required for incorporation in the matrix. J. Cell Sci. 1999;112:1093–1100. doi: 10.1242/jcs.112.7.1093. [DOI] [PubMed] [Google Scholar]
- 21.Wallis D.D., Putnam E.A., Cretoiu J.S., Carmical S.G., Cao S.N., Thomas G., Milewicz D.M. Profibrillin-1 maturation by human dermal fibroblasts: proteolytic processing and molecular chaperones. J. Cell. Biochem. 2003;90:641–652. doi: 10.1002/jcb.10657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gillis E., Van Laer L., Loeys B.L. Genetics of thoracic aortic aneurysm: at the crossroad of transforming growth factor-β signaling and vascular smooth muscle cell contractility. Circ. Res. 2013;113:327–340. doi: 10.1161/CIRCRESAHA.113.300675. [DOI] [PubMed] [Google Scholar]
- 23.Combs M.D., Knutsen R.H., Broekelmann T.J., Toennies H.M., Brett T.J., Miller C.A., Kober D.L., Craft C.S., Atkinson J.J., Shipley J.M. Microfibril-associated glycoprotein 2 (MAGP2) loss of function has pleiotropic effects in vivo. J. Biol. Chem. 2013;288:28869–28880. doi: 10.1074/jbc.M113.497727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Penner A.S., Rock M.J., Kielty C.M., Shipley J.M. Microfibril-associated glycoprotein-2 interacts with fibrillin-1 and fibrillin-2 suggesting a role for MAGP-2 in elastic fiber assembly. J. Biol. Chem. 2002;277:35044–35049. doi: 10.1074/jbc.M206363200. [DOI] [PubMed] [Google Scholar]
- 25.Lemaire R., Bayle J., Mecham R.P., Lafyatis R. Microfibril-associated MAGP-2 stimulates elastic fiber assembly. J. Biol. Chem. 2007;282:800–808. doi: 10.1074/jbc.M609692200. [DOI] [PubMed] [Google Scholar]
- 26.Détaint D., Faivre L., Collod-Beroud G., Child A.H., Loeys B.L., Binquet C., Gautier E., Arbustini E., Mayer K., Arslan-Kirchner M. Cardiovascular manifestations in men and women carrying a FBN1 mutation. Eur. Heart J. 2010;31:2223–2229. doi: 10.1093/eurheartj/ehq258. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.