

Abstract
Genome-wide association studies (GWAS) are a powerful tool for understanding the genetic underpinnings of human disease. In this article, we briefly review the role and findings of GWAS in common neurological diseases, including Stroke, Alzheimer’s disease, Parkinson’s disease, epilepsy, multiple sclerosis, migraine, amyotrophic lateral sclerosis, frontotemporal lobar degeneration, restless legs syndrome, intracranial aneurysm, human prion diseases and moyamoya disease. We then discuss the present and future implications of these findings with regards to disease prediction, uncovering basic biology, and the development of potential therapeutic agents.
Keywords: Neurology, neurological diseases, genetics, genome-wide association studies (GWAS), stroke, alzheimer’s disease, parkinson’s disease, epilepsy, polymorphism
Introduction
Many neurological disorders are said to be complex, meaning they are oligogenic and multifactorial. That is, they are the end result of the complex effects of several genes interacting with the environment. To identify their genetic background is just as important as finding the environmental factors, in view of their high population prevalence, severe course, serious impact on patients’ disability and progressive mental and physical de-adaptation.
One of the earliest ventures into understanding the genetic basis of these complex diseases was through investigating candidate genes with plausible biological function. The association between the disease and a specific allele of a single nucleotide polymorphism (SNP) within the functional candidate genes is analyzed between patients and controls. Most of these studies included relatively small numbers of patients and controls. Therefore, results have been conflicting or have not been replicated. Another main disadvantage of the hypothesis-based approach of candidate gene studies is that genes involved in the pathogenesis of a disease through unknown pathways are overlooked, while, the hypothesis-free approach by genome-wide association studies (GWAS) allow researchers to overcome this drawback because in these studies nearly all common variants in the entire genome can be tested for association with a disease (1,2). Additionally, in contrast to linkage studies, GWAS utilizes readily available case-control datasets rather than multiplex family based sets, permitting collection of much larger datasets (Figure 1).
Figure 1.
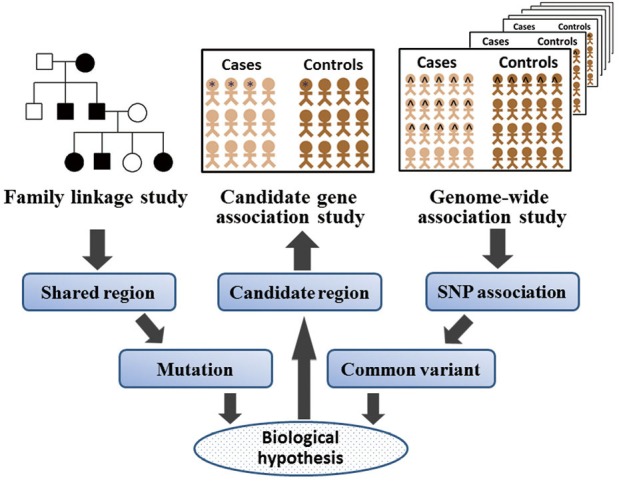
Three major genetic approaches used in complex diseases. Many approaches have been used to understand the genetic basis of the complex diseases. A brief summary and comparison of three major genetic approaches was shown, that includes the classic linkage study, candidate-gene association study, and genome-wide association study. SNP, single nucleotide polymorphism.
Over the past few years, tremendous progress has been made in understanding the genetic basis of complex diseases and hundreds of genetic loci have been identified through the use of GWAS. In this article, we briefly review the role and findings of GWAS in some common neurological diseases. We will then discuss the present and future implications of these findings with regards to disease prediction, uncovering basic biology, and the development of potential therapy.
Stroke
Stroke, as a heterogeneous multifactorial disorder, is the world’s third leading cause of death and a major cause of adult chronic disability. Although epidemiological data from twin and family studies provide substantial evidence for a genetic basis for stroke, the contribution of genetic factors identified so far is small. More recently, the GWAS approach is just beginning to affect the study of stroke.
As we all known, over 80% of strokes are ischaemic, as opposed to haemorrhagic. Ischaemic stroke (IS) itself includes several subtypes with differing pathophysiological mechanisms, the most common of which are large-vessel disease stroke, small-vessel disease stroke, and cardioembolic stroke. Most early stroke studies utilizing the GWAS examined whether associations detected by applying the GWAS to other cardiovascular diseases are also risk factors in stroke. Two loci (PITX2 and ZFHX3) associated with atrial fibrillation have both been shown to be risk factors for cardioembolic stroke (3,4). And a locus on chromosome 9p21 originally associated with coronary artery disease was shown to be a risk factor for large-vessel stroke (5). Indeed, it has demonstrated that alleles of 9p21 locus were associated with a broad variety of vascular diseases (6,7). What is worth more noting is an important finding from GWAS, that is different genetic risk factors appear to predispose to specific stroke subtypes. In Japanese populations, a variant in the protein kinase C family (PRKCH) was associated with small-vessel stroke (8). This association was further replicated in patients with subcortical silent brain infarction (9). Moreover, SNPs in CELSR1 gene have been identified as IS risk factors in Japanese individuals (10), while this has not yet been confirmed in the non-Japanese population. A meta-analysis of prospective population based cohort studies reported an association with the 12p13 region, thought to be with the NINJ2 gene, although this result was not replicated in a larger case-control sample (11,12). Recently, the Wellcome Trust Case Control Consortium 2 (WTCCC2) IS GWAS reported a novel association on chromosome 7p21 within the HDAC9 gene, which associated only with large-vessel ischaemic stroke (13). More importantly, through a largest meta-analysis of available IS GWAS datasets, four previously described associations were further confirmed and all these associations were specific to stroke subtypes; PITX2 and ZFHX3 were specific to cardioembolic stroke, whereas HDAC9 and the 9p21 locus were specific to large-vessel strokes (14).
Anyway, GWAS to date have identified robust associations with stroke. Most associations occur with specific stroke subtypes, emphasizing the importance of careful subtyping in genetic epidemiology studies. However, we must know that conventional environmental risk factors play an important role in stroke pathogenesis. Future studies should fully take into account the gene-environment interactions.
Alzheimer’s disease (AD)
AD is a complex multifactorial neurodegenerative disease that is the leading cause of dementia in the elderly. Genetic susceptibility at multiple loci and interactions among these genes influence the risk of developing AD; recent estimates of heritability range from 58-79%. For many years amyloid precursor protein (APP) and the presenilin genes 1 and 2 (PSEN1, PSEN2) have been the only unequivocally established susceptibility genes for early-onset familial AD, and apolipoprotein E (APOE) the only confirmed susceptibility gene for common late-onset AD (LOAD). Since 2009, large GWAS changed the face of the complex genetics of AD. At least nine novel risk loci (CR1, BIN1, CLU, PICALM, CD2AP, CD33, EPHA1, ABCA7 and MS4A cluster) were uncovered, which along with APOE4 contribute a high proportion of genetic risk for LOAD (15-19). Interestingly, these new identified susceptibility genes are not random, but show patterns of putative functional relation, which increase understanding of the molecular underpinnings of AD by showing prominent roles of the lipid-processing pathway (APOE, CLU, and ABCA7), the immune system (CLU, CR1, ABCA7, CD33, and EPHA1), and the synaptic-cell-functioning pathways (PICALM, BIN1, CD33, and CD2AP) (20-22). Moreover, a recent study has demonstrated that multiple AD-associated genes with evidence for positive selection encode proteins that physically interact within an independently defined protein-protein interaction network, suggesting such loci variants may have functional consequences and be involved in executing a shared molecular function (23). Besides, the combination of information about SNPs at different AD risk genes led to significantly associated genotype patterns that affected episodic memory performance (24). In short, a single locus direct effect is after all small. Further research investigating potential functional gene variants and their mechanistic role in AD is very needed.
Parkinson’s disease (PD)
PD is a chronic neurodegenerative disorder that leads to motor and cognitive disability. Although the causes of PD are thought to be primarily environmental, recent studies suggest that a number of genes influence susceptibility. To date, several GWAS for PD have been reported (25-32) and four meta-analyses have been performed (33-36). All GWAS indicate a strong association to the SNCA gene and most studies also confirm an association with the MAPT gene (25-31). Early in 2011, the International Parkinson’s Disease Genomics Consortium (IPDGC) performed a meta-analysis of datasets from five PD GWAS across the United States and Europe in order to identify novel risk loci for PD (33). Besides six previously identified (MAPT, SNCA, HLA-DRB5, BST1, GAK and LRRK2), five novel loci (ACMSD, STK39, MCCC1/LAMP3, SYT11 and CCDC62/HIP1R) were identified. A second meta-analysis by the IPDGC in collaboration with the Wellcome Trust Case Control Consortium 2 revealed another five PD risk loci (PARK16, STX1B, FGF20, STBD1 and GPNMB) (34). The third and most comprehensive meta-analysis included data from seven million polymorphisms originating either from GWAS datasets or from smaller scale PD association studies (35). Many previously reported risk loci were confirmed and evidence for a new risk variant in the ITGA8 gene was found (35). Along with the deepening in PD meta-analysis, another PD susceptibility locus RIT2 was also identified (36). To sum up, GWAS identified a number of susceptibility loci in sporadic PD, meta-analyses greatly expanded the list of proposed associations, and further studies replicated and confirmed most of these well-established loci in independent populations (37). By applying the genome-wide pathway analysis, three candidate SNPs, two genes (MAPT and HSD3B7), and 21 pathways involving protein domain specific binding and neurogenesis have also been identified (38). Further efforts to pinpoint functional variants and understand the biological implications of each risk locus are warranted.
Epilepsy
Epilepsy is one of the most common neurological disorders characterized by recurrent unprovoked seizures due to neuronal hyperexcitability and abnormal synchronization. Despite advances in epilepsy research, the heterogeneous and complex molecular mechanisms involved in epileptogenesis remain elusive. Genetic factors play a predominant role in 40% of epilepsies, however, early candidate gene-based studies in epilepsy proved ineffective in identifying genetic risk factors. And GWAS of epilepsy patients have been largely negative (39), with the exception of several putative susceptibility loci discovered in Han Chinese focal epilepsy and European Caucasian genetic generalized epilepsy (GGE) patients (40,41). In the combined analysis of the 2-stage Han Chinese GWAS of symptomatic epilepsy (40), the strongest signals were observed with two highly correlated SNPs, rs2292096 and rs6660197, on 1q32.1 in the CAMSAP1L1 gene, which encodes a cytoskeletal protein. More recently, the EPICURE study published a first large GWAS in GGE and reported the allelic associations at 2p16.1 (rs13026414) and 17q21.3 (rs72823592) (41). Further replication studies in independent populations are necessary to validate these novel association findings. Meanwhile, we should be aware that these susceptibility loci appear likely to account for only a small fraction of the heritability of epilepsy, fuelling the effort to search for alternative genetic contributors is quite essential.
Multiple sclerosis (MS)
MS is a chronic neuroinflammatory autoimmune disease believed to arise from complex interactions of both environmental and genetic factors. The importance of genetic in MS is best illustrated by observations of strong familial clustering and a significantly increased risk in first-degree relatives. Meanwhile, some studies in monozygotic and dizygotic twins also indicate a strong genetic component. To date, alleles of the human leukocyte antigen (HLA) are known to make the single strongest contribution to MS susceptibility (42). Additionally, many loci of more modest effect have been identified in recent GWAS. In 2007, the first MS GWAS identified the first non-HLA regions with genome-wide significance. IL7Ra and IL2Ra were found to be significantly associated with MS (43). Simultaneously, the IL7Ra gene was confirmed to be associated in other MS cohorts (44,45). In the years that followed, a new series of GWAS and meta-analyses were performed in different MS cohorts, steadily adding more regions to the list of confirmed MS-associated loci (46-55). What is worth noting is a larger-scale collaborative GWAS reported in 2011, which replicated almost all of the previously suggested associations and identified at least 29 novel susceptibility loci (56). More importantly, a recent analysis shown that approximately 30% of the genetic variation in liability to MS may be directly explained by variants represented by current GWAS arrays (57). And further genome-wide pathway analysis in identification of candidate SNPs, pathways, and hypothetical biological mechanisms (58) will continue to be conducted.
It is also remarkable that the vast majority of the confirmed MS-associated loci are located close or inside genes encoding immune system-related molecules, strongly supporting the hypothesis that MS is primarily an immune-mediated disease. Moreover, one-third of the identified loci were reportedly associated with at least one other autoimmune disease, strengthening the notion that common disease mechanism(s) may underlie most if not all autoimmune diseases (59,60). It is very interesting that a core of genes is shared among multiple autoimmune diseases, and some are disease specific. Future investigation of the shared susceptibility genes might be of great value.
Migraine
Migraine is an episodic and disabling neurological disorder and has a strong genetic basis, with heritability estimates of 40-57%. The two most prevalent forms are migraine without aura (MO) and migraine with aura (MA). Many linkage studies and candidate gene studies have suggested causative genes in MO and MA, but few have been replicated. Recent attempts using GWAS have yielded four SNPs, located on chromosome 8q22.1, 2q37.1, 12q13.3 and 1p36.32, that are significantly associated with MA and/or MO (61,62). Meanwhile, some meta-analysis replicated and confirmed these findings in independent populations (63,64). In another recent GWAS, three additional SNPs located at 1q22 and 3p24 have shown convincing association with MO as well (65). However, all the aforementioned associations conferred only a small to moderate change in risk for migraine. Ongoing large international collaborations to identify additional gene variants for migraine might be very needed in the future. Incidentally, Cox et al. recently did a pedigree-based GWAS in an isolated population of Norfolk Island with a high prevalence of migraine and several novel variants in migraine susceptibility were identified (66). Although the associations might be specific only for the isolated population, we still believe this finding will bring some insights and inspiration to future research.
Amyotrophic lateral sclerosis (ALS)
ALS is a fatal disease characterized by motor neuron degeneration, which leads to muscle atrophy and paralysis. Typically, this progressive muscle wasting results in death from respiratory failure within 2-4 years after onset of symptoms. The majority of ALS cases are sporadic; approximately 5% of cases show a family history of disease (67). Although several causative genes have been identified in familial ALS, the genetic contributors to sporadic ALS have been more difficult to identify. GWAS in sporadic ALS have implicated a number of genes or regions (DPP6, ITPR2, UNC13A, FGGY, ELP3, KIFAP3, 9p21.2) (68-76), but replication of these findings in independent populations has proven difficult (77-81). A possible exception is the 9p21.2 region, which was recently replicated by two large independent studies and seems to be important in both familial and sporadic ALS (82,83). Additionally, the genetic risk factor rs12608932 in UNC13A has been identified as a modifier of survival among ALS patients, which might provide promising therapeutic targets to slow disease progression (84,85). Moreover, environmental exposures also appear to play a role in ALS incidence. Following reports of an increased incidence of ALS in U.S. veterans, interestingly, a GWAS was designed to identify genetic factors that may contribute to ALS in a sample of U.S. veterans (86), while no SNPs reached genome-wide significance for either ALS outcome or survival. Further investigation of the nature of the relationship between military service and ALS merits are still needed.
Frontotemporal lobar degeneration (FTLD)
FTLD is one of the common causes of presenile dementia worldwide, and familial forms occur in 30%-50% of all frontotemporal dementia (FTD) patients. It manifests, clinically, with behavioural changes and language impairment and is pathologically associated with tau- or ubiquitin-positive inclusions detected in neurons and glial cells of the frontal and temporal lobes in the brain. Up to 50% of familial cases show association with chromosome 17: mutations in the microtubule-associated protein tau (MAPT) and progranulin (GRN) are considered as the main genetic cause of FTD. Rare defects in VCP, CHMP2B, TDP-43 and FUS genes have been found in a small number of families (87).
Recently, a first GWAS in FTLD identified common genetic variability at the TMEM106B gene on chromosome 7p21.3 as a potential important risk modifying factor for FTLD with pathologic inclusions of TAR DNA-binding protein (FTLD-TDP), the most common pathological subtype in FTLD (88). To gather additional evidence for the implication of TMEM106B in FTLD risk, multiple replication studies in geographically distinct populations were set up (89,90). It has demonstrated that the most significant association of TMEM106B polymorphisms with risk of FTLD-TDP was observed in patients with GRN mutations. Subsequent studies suggested an inverse correlation between TMEM106B expression and GRN levels in patient serum (90). Accordingly, a biochemical and cell biological basis for the understanding of the pathological role of TMEM106B in FTLD was also presented (91). Additional studies to further unravel the molecular role of TMEM106B in disease onset and progression are still needed.
It is particularly worth mentioning that FTLD may be found alone or accompanied by corticobasal degeneration, parkinsonism, or ALS. As many as 50% of ALS patients have cognitive and behavioral deficits and about half of FTLD cases have motor neuron impairment. Along this line, Vass et al. investigated whether this FTLD-TDP associated TMEM106B risk genotypes could also contribute to the risk and disease presentation of ALS. Although no association between the TMEM106B genotypes and ALS was demonstrated, the protective allele at TMEM106B did appear to be associated with preserved cognition in ALS patients (92). This finding, although preliminary, are of interest in light of the TDP-43 proteinopathy spectrum that binds FTLD and ALS. In addition, gene linkage to chromosome 9p has already been established in familial FTLD-ALS. And recently, a genetic locus associated with FTLD and ALS was identified as a hexanucleotide repeat located on chromosome 9, open reading frame 72 (C9ORF72) (74,83,93), which causes both familial and sporadic types of these two disease and can be believed as the most common cause of familial FTD-ALS. As the pathophysiological mechanisms by which this mutation leads to FTD and ALS begin to be understood, we have reason to believe new targets for disease-modifying therapies will likely be unveiled (94).
Restless legs syndrome (RLS)
RLS is a sensorimotor disorder in which affected individuals suffer from uncomfortable sensations and an urge to move their lower limbs; it occurs mainly in resting situations during the evening or at night. The high familial aggregation of RLS suggests a genetic component. Through GWAS analysis, RLS was identified an association with variants in gene MEIS1, BTBD9, and MAP2K5/LBXCOR1 on chromosomes 2p, 6p and 15q (95,96). These findings were further confirmed in several studies (97,98). Recently, another GWAS identified two additional RLS susceptibility loci within an intergenic region on chromosome 2p14 and 16q12.1 (99). Opportunities now exist to study the functionality of the RLS risk susceptibility loci and the potential molecular pathways involved. Some illustrative examples include the study reporting that the MEIS1 risk variant influences iron metabolism (100) and BTBD9 regulates brain dopamine levels and controls iron homeostasis through the iron regulatory protein-2 (101), adding to growing body of evidence that dopaminergic neurotransmission and iron dysregulation might contribute to the pathogenesis of RLS.
Intracranial aneurysm (IA)
The rupture of IA is the most common cause of subarachnoid hemorrhage associated with high morbidity and mortality, while its pathogenesis is still unknown. Because catastrophic hemorrhage is commonly the first sign of disease, early identification is very essential. Two GWAS in European and Japanese cohorts have successfully identified common variants located on chromosomes 8q11, 9p21, 10q24, 13q13 and 18q11 that are associated with IA (102,103). Some independent GWAS in the Japanese population was also conducted (104,105). A recent larger-scale GWAS replicated and validated rs10757272 on CDKN2BAS at chromosome 9p21.3 to be significantly associated with IA, meanwhile, a novel functional SNP, rs6842241, near EDNRA at chromosome 4q31.22 was found (105), which might affect the expression of EDNRA and subsequently result in the IA susceptibility. In addition, a meta-analysis also yielded genome-wide significance for SNP in CDKN2BAS on chromosome 9p (rs6475606) and supported previously reported SNP in SOX17 on chromosome 8q (rs1072737) contributing to IA susceptibility (106).
Among the loci above, 9p21 locus seems most intriguing because the SNPs identified by the GWAS of IA are in linkage disequilibrium with SNPs associated with several other arterial diseases (coronary artery disease, abdominal aortic aneurysm, and peripheral arterial disease) (7), and this risk locus overlaps an annotated noncoding RNA, called ANRIL (or CDKN2BAS) (107). More importantly, 9p21 variation is found associated with the site distribution of IA, especially IA of the posterior communicating artery and the posterior circulation than in the anterior circulation, which are clinically important (108).
Human prion diseases
Prion diseases are fatal neurodegenerative diseases of humans and animals caused by the misfolding and aggregation of prion protein (PrP). Mammalian prion diseases are under strong genetic control but few risk factors are known apart from the PrP gene locus (PRNP). In order to providing other (non-PRNP) genetic modifiers for human prion disease, many studies were conducted based on candidates derived from close functional links to PrP, screening human genes orthologous to mouse candidates, and also human GWAS (109). From a GWAS in variant Creutzfeldt-Jakob disease (vCJD), it has been demonstrated that SNPs upstream of the RARB and STMN2 loci were candidates for vCJD association (110). Interestingly, markers on human chromosome 8 in this human vCJD study share conservation of synteny with the region of cattle chromosome 14, where an association was reported with classical bovine spongiform encephalopathy (111). Moreover, another large GWAS in human prion diseases provide evidence for several non-PRNP genetic risk factors as well, although none of these loci achieved genome-wide significance in meta-analyses between regions or aetiologies. It is unsurprising that PRNP genotypes were found strongly associated with risk in all categories and regions of human prion diseases (112). Further genetic analyses are required to provide convincing evidence for association at these non-PRNP loci. Meanwhile, the mechanism by which the existing loci exert their influence on nearby genes and potential molecular pathways involved need to be investigated.
Moyamoya disease (MMD)
Moyamoya, a poorly understood arteriopathy derives its name from the characteristic angiographic pattern. The name means ‘hazy puffs of smoke’ describing the small vessel collateral system that develops in response to hyperplastic stenosis and occlusion of the distal internal carotid and proximal vessels of the circle of Willis. MMD occurs worldwide, but its prevalence is highest in East Asian countries and 15% of MMD cases have a family history (113). Genetic analyses in familial MMD and GWAS represent promising strategies for underling its pathogenesis. A recent GWAS was performed in Japanese MMD patients, resulting in a strong association of chromosome 17q25-ter with MMD risk (114). And candidate locus on 17q25 was revealed associated with MMD before. What’s more, a single haplotype consisting of seven SNPs at the RNF213 locus was tightly associated with MMD and mutational analysis of RNF213 revealed a founder mutation greatly increases MMD risk (114). Therefore, RNF213 can be identified as a susceptibility gene for MMD and its possible role in vascular development (115) should be discussed further.
Conclusions and future directions
Significant progress has been made in recent years in unraveling the genetic roots of complex diseases. GWAS have played a major role in this effort as they have allowed for the discovery of novel genetic associations that are not based on prior knowledge. The studies reviewed above provide a synopsis of the major advances in several topics related to neurological diseases.
There are a number of key concepts that can be gleaned from these studies. Firstly, larger sample sizes are needed to detect variants with small effect sizes. Therefore, a meta-analysis of available GWAS datasets and further GWAS in different populations are of great value. Despite large collaborative efforts, so far, GWAS findings explain only a small proportion of the heritability of complex diseases, which makes genetic risk prediction tests currently unfeasible for these diseases. It is evident that a proportion of the genetic predisposition will be explained by highly penetrant rare variants, while these rare variants will not be captured with GWAS. Further research, including new approaches to detect rare variants using next generation sequencing and structural variants, including copy number variants such as insertions and deletions, might improve genetic risk prediction for these diseases.
What’s more, reviewing some compelling loci based on current data from GWAS, genes that have been linked to more than one disease entity are emphasized. It is very interesting that a core of genes is shared among multiple diseases in a general category. Such overlapping findings could point to several common genetic and mechanistic denominators, especially in terms of neurodegeneration, vascular disease or autoimmune diseases. Unveiling the identity of these shared genetic factors will not only improve our understanding of the underlying pathophysiology, but also may lead to new avenues for preventing and treating these devastating diseases.
It is particularly worth noting that GWAS is not a method to conclude something. “Significantly associated” does not mean that the association identified by GWAS is significant in biology, medicine, or actual life. It’s just a method to map the gene and presents candidates of the causal variant. To identify and prove the causality, we need to find the best associated variations by fine mapping around the smoking gun GWAS signal, followed by functional biological studies to understand the underlying biology for them. Further efforts are necessary to make full use of its potential power to medicine. Therefore, identification of the significantly associated SNP is just ‘end of the beginning’ to get to the final goal: treatment of patients with common diseases. However, at present, after identification of ‘marker’ SNPs by GWASs, we have no golden road to find ‘causal’ variants. We have no customized method to identify functional variants. By studying the meaning of allelic differences of causal variants, we can clarify the molecular pathogenesis of the disease. With the knowledge of the pathogenesis, we can walk directly to develop innovative treatments and discover new drugs. So, there is still a long and winding road before us.
Although current examples of immediate clinical impact from GWAS studies are limited, we strongly believe that GWAS have begun to revolutionize our understanding of the genetic underpinnings of human disease. It’s just the beginning and it gives us wonderful start points. Future work should prioritize the studies designed to ascertain rarer SNPs with possibly larger functional effects, analysis of potential pathway and further study of the modification of clinical phenotype by genetic polymorphism. We believe that starting with GWAS results will increase the likelihood of successful biological and therapeutic discoveries in neurology in the years to come.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (81000544, 81171209), the Shandong Provincial Natural Science Foundation, China (ZR2010HQ004, ZR2011HZ001), the Medicine and Health Science Technology Development Project of Shandong Province (2011WSA02018, 2011WSA02020), and the Shandong Provincial Outstanding Medical Academic Professional Program.
Disclosure: The authors declare no conflict of interest.
References
- 1.Hindorff LA, Sethupathy P, Junkins HA, et al. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci U S A 2009;106:9362-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.A Catalog of Published Genome-Wide Association Studies. Available online: http://www.genome.gov/26525384
- 3.Gretarsdottir S, Thorleifsson G, Manolescu A, et al. Risk variants for atrial fibrillation on chromosome 4q25 associate with ischemic stroke. Ann Neurol 2008;64:402-9. [DOI] [PubMed] [Google Scholar]
- 4.Gudbjartsson DF, Holm H, Gretarsdottir S, et al. A sequence variant in ZFHX3 on 16q22 associates with atrial fibrillation and ischemic stroke. Nat Genet 2009;41:876-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gschwendtner A, Bevan S, Cole JW, et al. Sequence variants on chromosome 9p21.3 confer risk for atherosclerotic stroke. Ann Neurol 2009;65:531-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Holdt LM, Teupser D. Recent studies of the human chromosome 9p21 locus, which is associated with atherosclerosis in human populations. Arterioscler Thromb Vasc Biol 2012;32:196-206. [DOI] [PubMed] [Google Scholar]
- 7.Helgadottir A, Thorleifsson G, Magnusson KP, et al. The same sequence variant on 9p21 associates with myocardial infarction, abdominal aortic aneurysm and intracranial aneurysm. Nat Genet 2008;40:217-24. [DOI] [PubMed] [Google Scholar]
- 8.Kubo M, Hata J, Ninomiya T, et al. A nonsynonymous SNP in PRKCH (protein kinase C eta) increases the risk of cerebral infarction. Nat Genet 2007;39:212-7. [DOI] [PubMed] [Google Scholar]
- 9.Serizawa M, Nabika T, Ochiai Y, et al. Association between PRKCH gene polymorphisms and subcortical silent brain infarction. Atherosclerosis 2008;199:340-5. [DOI] [PubMed] [Google Scholar]
- 10.Yamada Y, Fuku N, Tanaka M, et al. Identification of CELSR1 as a susceptibility gene for ischemic stroke in Japanese individuals by a genome-wide association study. Atherosclerosis 2009;207:144-9. [DOI] [PubMed] [Google Scholar]
- 11.Ikram MA, Seshadri S, Bis JC, et al. Genomewide association studies of stroke. N Engl J Med 2009;360:1718-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.International Stroke Genetics Consortium , Wellcome Trust Case-Control Consortium 2. Failure to validate association between 12p13 variants and ischemic stroke. N Engl J Med 2010;362:1547-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bellenguez C, Bevan S, Gschwendtner A, et al. Genome-wide association study identifies a variant in HDAC9 associated with large vessel ischemic stroke. Nat Genet 2012;44:328-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Traylor M, Farrall M, Holliday EG, et al. Genetic risk factors for ischaemic stroke and its subtypes (the METASTROKE collaboration): a meta-analysis of genome-wide association studies. Lancet Neurol 2012;11:951-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Harold D, Abraham R, Hollingworth P, et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat Genet 2009;41:1088-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hollingworth P, Harold D, Sims R, et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat Genet 2011;43:429-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lambert JC, Heath S, Even G, et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat Genet 2009;41:1094-9. [DOI] [PubMed] [Google Scholar]
- 18.Naj AC, Jun G, Beecham GW, et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat Genet 2011;43:436-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Seshadri S, Fitzpatrick AL, Ikram MA, et al. Genome-wide analysis of genetic loci associated with Alzheimer disease. JAMA 2010;303:1832-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Morgan K.The three new pathways leading to Alzheimer’s disease. Neuropathol Appl Neurobiol 2011;37:353-7. [DOI] [PubMed] [Google Scholar]
- 21.Carter C. Alzheimer’s Disease: APP, Gamma Secretase, APOE, CLU, CR1, PICALM, ABCA7, BIN1, CD2AP, CD33, EPHA1, and MS4A2, and Their Relationships with Herpes Simplex, C. Pneumoniae, Other Suspect Pathogens, and the Immune System. Int J Alzheimers Dis 2011;2011:501862. [DOI] [PMC free article] [PubMed]
- 22.Schellenberg GD, Montine TJ. The genetics and neuropathology of Alzheimer’s disease. Acta Neuropathol 2012;124:305-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Raj T, Shulman JM, Keenan BT, et al. Alzheimer disease susceptibility loci: evidence for a protein network under natural selection. Am J Hum Genet 2012;90:720-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barral S, Bird T, Goate A, et al. Genotype patterns at PICALM, CR1, BIN1, CLU, and APOE genes are associated with episodic memory. Neurology 2012;78:1464-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Simón-Sánchez J, Schulte C, Bras JM, et al. Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat Genet 2009;41:1308-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Do CB, Tung JY, Dorfman E, et al. Web-based genome-wide association study identifies two novel loci and a substantial genetic component for Parkinson’s disease. PLoS Genet 2011;7:e1002141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Satake W, Nakabayashi Y, Mizuta I, et al. Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nat Genet 2009;41:1303-7. [DOI] [PubMed] [Google Scholar]
- 28.Edwards TL, Scott WK, Almonte C, et al. Genome-wide association study confirms SNPs in SNCA and the MAPT region as common risk factors for Parkinson disease. Ann Hum Genet 2010;74:97-109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Saad M, Lesage S, Saint-Pierre A, et al. Genome-wide association study confirms BST1 and suggests a locus on 12q24 as the risk loci for Parkinson’s disease in the European population. Hum Mol Genet 2011;20:615-27. [DOI] [PubMed] [Google Scholar]
- 30.Spencer CC, Plagnol V, Strange A, et al. Dissection of the genetics of Parkinson’s disease identifies an additional association 5’ of SNCA and multiple associated haplotypes at 17q21. Hum Mol Genet 2011;20:345-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hamza TH, Zabetian CP, Tenesa A, et al. Common genetic variation in the HLA region is associated with late-onset sporadic Parkinson’s disease. Nat Genet 2010;42:781-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu X, Cheng R, Verbitsky M, et al. Genome-wide association study identifies candidate genes for Parkinson’s disease in an Ashkenazi Jewish population. BMC Med Genet 2011;12:104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nalls MA, Plagnol V, Hernandez DG, et al. Imputation of sequence variants for identification of genetic risks for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet 2011;377:641-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.International Parkinson’s Disease Genomics Consortium (IPDGC) , Wellcome Trust Case Control Consortium 2 (WTCCC2). A two-stage meta-analysis identifies several new loci for Parkinson’s disease. PLoS Genet 2011;7:e1002142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lill CM, Roehr JT, McQueen MB, et al. Comprehensive research synopsis and systematic meta-analyses in Parkinson’s disease genetics: The PDGene database. PLoS Genet 2012;8:e1002548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pankratz N, Beecham GW, DeStefano AL, et al. Meta-analysis of Parkinson’s disease: identification of a novel locus, RIT2. Ann Neurol 2012;71:370-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pihlstrøm L, Axelsson G, Bjørnarå KA, et al. Supportive evidence for 11 loci from genome-wide association studies in Parkinson’s disease. Neurobiol Aging 2013;34:1708.e7-13. [DOI] [PubMed]
- 38.Song GG, Lee YH. Pathway analysis of genome-wide association studies for Parkinson’s disease. Mol Biol Rep 2013;40:2599-607. [DOI] [PubMed] [Google Scholar]
- 39.Kasperaviciūte D, Catarino CB, Heinzen EL, et al. Common genetic variation and susceptibility to partial epilepsies: a genome-wide association study. Brain 2010;133:2136-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Guo Y, Baum LW, Sham PC, et al. Two-stage genome-wide association study identifies variants in CAMSAP1L1 as susceptibility loci for epilepsy in Chinese. Hum Mol Genet 2012;21:1184-9. [DOI] [PubMed] [Google Scholar]
- 41.Steffens M, Leu C, Ruppert AK, et al. Genome-wide association analysis of genetic generalized epilepsies implicates susceptibility loci at 1q43, 2p16.1, 2q22.3 and 17q21.32. Hum Mol Genet 2012;21:5359-72. [DOI] [PubMed] [Google Scholar]
- 42.Lincoln MR, Montpetit A, Cader MZ, et al. A predominant role for the HLA class II region in the association of the MHC region with multiple sclerosis. Nat Genet 2005;37:1108-12. [DOI] [PubMed] [Google Scholar]
- 43.Hafler DA, Compston A, Sawcer S, et al. Risk alleles for multiple sclerosis identified by a genomewide study. N Engl J Med 2007;357:851-62. [DOI] [PubMed] [Google Scholar]
- 44.Gregory SG, Schmidt S, Seth P, et al. Interleukin 7 receptor alpha chain (IL7R) shows allelic and functional association with multiple sclerosis. Nat Genet 2007;39:1083-91. [DOI] [PubMed] [Google Scholar]
- 45.Lundmark F, Duvefelt K, Iacobaeus E, et al. Variation in interleukin 7 receptor alpha chain (IL7R) influences risk of multiple sclerosis. Nat Genet 2007;39:1108-13. [DOI] [PubMed] [Google Scholar]
- 46.Australia and New Zealand Multiple Sclerosis Genetics Consortium (ANZgene) . Genome-wide association study identifies new multiple sclerosis susceptibility loci on chromosomes 12 and 20. Nat Genet 2009;41:824-8. [DOI] [PubMed] [Google Scholar]
- 47.Jakkula E, Leppa V, Sulonen AM, et al. Genome-wide association study in a high-risk isolate for multiple sclerosis reveals associated variants in STAT3 gene. Am J Hum Genet 2010;86:285-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sanna S, Pitzalis M, Zoledziewska M, et al. Variants within the immunoregulatory CBLB gene are associated with multiple sclerosis. Nat Genet 2010;42:495-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nischwitz S, Cepok S, Kroner A, et al. Evidence for VAV2 and ZNF433 as susceptibility genes for multiple sclerosis. J Neuroimmunol 2010;227:162-6. [DOI] [PubMed] [Google Scholar]
- 50.International Multiple Sclerosis Genetics Consortium . Genome-wide association study of severity in multiple sclerosis. Genes Immun 2011;12:615-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.De Jager PL, Jia X, Wang J, et al. Meta-analysis of genome scans and replication identify CD6, IRF8 and TNFRSF1A as new multiple sclerosis susceptibility loci. Nat Genet 2009;41:776-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Baranzini SE, Wang J, Gibson RA, et al. Genome-wide association analysis of susceptibility and clinical phenotype in multiple sclerosis. Hum Mol Genet 2009;18:767-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Comabella M, Craig DW, Camina-Tato M, et al. Identification of a novel risk locus for multiple sclerosis at 13q31.3 by a pooled genome-wide scan of 500,000 single nucleotide polymorphisms. PLoS One 2008;3:e3490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Patsopoulos NA, Esposito F, Reischl J, et al. Genome-wide meta-analysis identifies novel multiple sclerosis susceptibility loci. Ann Neurol 2011;70:897-912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Matesanz F, Gonzalez-Perez A, Lucas M, et al. Genome-wide association study of multiple sclerosis confirms a novel locus at 5p13.1. PLoS One 2012;7:e36140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sawcer S, Hellenthal G, Pirinen M, et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature 2011;476:214-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Watson CT, Disanto G, Breden F, et al. Estimating the proportion of variation in susceptibility to multiple sclerosis captured by common SNPs. Sci Rep 2012;2:770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Song GG, Choi SJ, Ji JD, et al. Genome-wide pathway analysis of a genome-wide association study on multiple sclerosis. Mol Biol Rep 2013;40:2557-64. [DOI] [PubMed] [Google Scholar]
- 59.Baranzini SE. The genetics of autoimmune diseases: a networked perspective. Curr Opin Immunol 2009;21:596-605. [DOI] [PubMed] [Google Scholar]
- 60.Cotsapas C, Voight BF, Rossin E, et al. Pervasive sharing of genetic effects in autoimmune disease. PLoS Genet 2011;7:e1002254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Anttila V, Stefansson H, Kallela M, et al. Genome-wide association study of migraine implicates a common susceptibility variant on 8q22.1. Nat Genet 2010;42:869-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chasman DI, Schurks M, Anttila V, et al. Genome-wide association study reveals three susceptibility loci for common migraine in the general population. Nat Genet 2011;43:695-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Esserlind AL, Christensen AF, Le H, et al. Replication and meta-analysis of common variants identifies a genome-wide significant locus in migraine. Eur J Neurol 2013;20:765-72. [DOI] [PubMed] [Google Scholar]
- 64.Ligthart L, de Vries B, Smith AV, et al. Meta-analysis of genome-wide association for migraine in six population-based European cohorts. Eur J Hum Genet 2011;19:901-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Freilinger T, Anttila V, de Vries B, et al. Genome-wide association analysis identifies susceptibility loci for migraine without aura. Nat Genet 2012;44:777-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cox HC, Lea RA, Bellis C, et al. A genome-wide analysis of ‘Bounty’ descendants implicates several novel variants in migraine susceptibility. Neurogenetics 2012;13:261-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Byrne S, Walsh C, Lynch C, et al. Rate of familial amyotrophic lateral sclerosis: a systematic review and meta-analysis. J Neurol Neurosurg Psychiatry 2011;82:623-7. [DOI] [PubMed] [Google Scholar]
- 68.Dunckley T, Huentelman MJ, Craig DW, et al. Whole-genome analysis of sporadic amyotrophic lateral sclerosis. N Engl J Med 2007;357:775-88. [DOI] [PubMed] [Google Scholar]
- 69.van Es MA, Van Vught PW, Blauw HM, et al. ITPR2 as a susceptibility gene in sporadic amyotrophic lateral sclerosis: a genome-wide association study. Lancet Neurol 2007;6:869-77. [DOI] [PubMed] [Google Scholar]
- 70.Cronin S, Berger S, Ding J, et al. A genome-wide association study of sporadic ALS in a homogenous Irish population. Hum Mol Genet 2008;17:768-74. [DOI] [PubMed] [Google Scholar]
- 71.van Es MA, van Vught PW, Blauw HM, et al. Genetic variation in DPP6 is associated with susceptibility to amyotrophic lateral sclerosis. Nat Genet 2008;40:29-31. [DOI] [PubMed] [Google Scholar]
- 72.Landers JE, Melki J, Meininger V, et al. Reduced expression of the Kinesin-Associated Protein 3 (KIFAP3) gene increases survival in sporadic amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A 2009;106:9004-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.van Es MA, Veldink JH, Saris CG, et al. Genome-wide association study identifies 19p13.3 (UNC13A) and 9p21.2 as susceptibility loci for sporadic amyotrophic lateral sclerosis. Nat Genet 2009;41:1083-7. [DOI] [PubMed] [Google Scholar]
- 74.Laaksovirta H, Peuralinna T, Schymick JC, et al. Chromosome 9p21 in amyotrophic lateral sclerosis in Finland: a genome-wide association study. Lancet Neurol 2010;9:978-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Shatunov A, Mok K, Newhouse S, et al. Chromosome 9p21 in sporadic amyotrophic lateral sclerosis in the UK and seven other countries: a genome-wide association study. Lancet Neurol 2010;9:986-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Simpson CL, Lemmens R, Miskiewicz K, et al. Variants of the elongator protein 3 (ELP3) gene are associated with motor neuron degeneration. Hum Mol Genet 2009;18:472-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cronin S, Tomik B, Bradley DG, et al. Screening for replication of genome-wide SNP associations in sporadic ALS. Eur J Hum Genet 2009;17:213-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Schymick JC, Scholz SW, Fung HC, et al. Genome-wide genotyping in amyotrophic lateral sclerosis and neurologically normal controls: first stage analysis and public release of data. Lancet Neurol 2007;6:322-8. [DOI] [PubMed] [Google Scholar]
- 79.Chiò A, Schymick JC, Restagno G, et al. A two-stage genome-wide association study of sporadic amyotrophic lateral sclerosis. Hum Mol Genet 2009;18:1524-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Van Es MA, Van Vught PW, Veldink JH, et al. Analysis of FGGY as a risk factor for sporadic amyotrophic lateral sclerosis. Amyotroph Lateral Scler 2009;10:441-7. [DOI] [PubMed] [Google Scholar]
- 81.Fernández-Santiago R, Sharma M, Berg D, et al. No evidence of association of FLJ10986 and ITPR2 with ALS in a large German cohort. Neurobiol Aging 2011;32:551.e1-4. [DOI] [PubMed]
- 82.Renton AE, Majounie E, Waite A, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011;72:257-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.DeJesus-Hernandez M, Mackenzie IR, Boeve BF, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011;72:245-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Diekstra FP, van Vught PW, van Rheenen W, et al. UNC13A is a modifier of survival in amyotrophic lateral sclerosis. Neurobiol Aging 2012;33:630.e3-8. [DOI] [PubMed]
- 85.Chiò A, Mora G, Restagno G, et al. UNC13A influences survival in Italian amyotrophic lateral sclerosis patients: a population-based study. Neurobiol Aging 2013;34:357.e1-5. [DOI] [PMC free article] [PubMed]
- 86.Kwee LC, Liu Y, Haynes C, et al. A high-density genome-wide association screen of sporadic ALS in US veterans. PLoS One 2012;7:e32768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ferrari R, Hardy J, Momeni P.Frontotemporal dementia: from Mendelian genetics towards genome wide association studies. J Mol Neurosci 2011;45:500-15. [DOI] [PubMed] [Google Scholar]
- 88.Van Deerlin VM, Sleiman PM, Martinez-Lage M, et al. Common variants at 7p21 are associated with frontotemporal lobar degeneration with TDP-43 inclusions. Nat Genet 2010;42:234-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.van der Zee J, Van Langenhove T, Kleinberger G, et al. TMEM106B is associated with frontotemporal lobar degeneration in a clinically diagnosed patient cohort. Brain 2011;134:808-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Finch N, Carrasquillo MM, Baker M, et al. TMEM106B regulates progranulin levels and the penetrance of FTLD in GRN mutation carriers. Neurology 2011;76:467-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lang CM, Fellerer K, Schwenk BM, et al. Membrane orientation and subcellular localization of transmembrane protein 106B (TMEM106B), a major risk factor for frontotemporal lobar degeneration. J Biol Chem 2012;287:19355-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Vass R, Ashbridge E, Geser F, et al. Risk genotypes at TMEM106B are associated with cognitive impairment in amyotrophic lateral sclerosis. Acta Neuropathol 2011;121:373-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Mok K, Traynor BJ, Schymick J, et al. Chromosome 9 ALS and FTD locus is probably derived from a single founder. Neurobiol Aging 2012;33:209.e3-8. [DOI] [PMC free article] [PubMed]
- 94.Sha SJ, Boxer A. Treatment implications of C9ORF72. Alzheimers Res Ther 2012;4:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Winkelmann J, Schormair B, Lichtner P, et al. Genome-wide association study of restless legs syndrome identifies common variants in three genomic regions. Nat Genet 2007;39:1000-6. [DOI] [PubMed] [Google Scholar]
- 96.Stefansson H, Rye DB, Hicks A, et al. A genetic risk factor for periodic limb movements in sleep. N Engl J Med 2007;357:639-47. [DOI] [PubMed] [Google Scholar]
- 97.Yang Q, Li L, Chen Q, et al. Association studies of variants in MEIS1, BTBD9, and MAP2K5/SKOR1 with restless legs syndrome in a US population. Sleep Med 2011;12:800-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kemlink D, Polo O, Frauscher B, et al. Replication of restless legs syndrome loci in three European populations. J Med Genet 2009;46:315-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Winkelmann J, Czamara D, Schormair B, et al. Genome-wide association study identifies novel restless legs syndrome susceptibility loci on 2p14 and 16q12.1. PLoS Genet 2011;7:e1002171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Catoire H, Dion PA, Xiong L, et al. Restless legs syndrome-associated MEIS1 risk variant influences iron homeostasis. Ann Neurol 2011;70:170-5. [DOI] [PubMed] [Google Scholar]
- 101.Freeman A, Pranski E, Miller RD, et al. Sleep fragmentation and motor restlessness in a Drosophila model of Restless Legs Syndrome. Curr Biol 2012;22:1142-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Bilguvar K, Yasuno K, Niemela M, et al. Susceptibility loci for intracranial aneurysm in European and Japanese populations. Nat Genet 2008;40:1472-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Yasuno K, Bilguvar K, Bijlenga P, et al. Genome-wide association study of intracranial aneurysm identifies three new risk loci. Nat Genet 2010;42:420-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Akiyama K, Narita A, Nakaoka H, et al. Genome-wide association study to identify genetic variants present in Japanese patients harboring intracranial aneurysms. J Hum Genet 2010;55:656-61. [DOI] [PubMed] [Google Scholar]
- 105.Low SK, Takahashi A, Cha PC, et al. Genome-wide association study for intracranial aneurysm in the Japanese population identifies three candidate susceptible loci and a functional genetic variant at EDNRA. Hum Mol Genet 2012;21:2102-10. [DOI] [PubMed] [Google Scholar]
- 106.Foroud T, Koller DL, Lai D, et al. Genome-wide association study of intracranial aneurysms confirms role of Anril and SOX17 in disease risk. Stroke 2012;43:2846-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Pasmant E, Laurendeau I, Heron D, et al. Characterization of a germ-line deletion, including the entire INK4/ARF locus, in a melanoma-neural system tumor family: identification of ANRIL, an antisense noncoding RNA whose expression coclusters with ARF. Cancer Res 2007;67:3963-9. [DOI] [PubMed] [Google Scholar]
- 108.Nakaoka H, Takahashi T, Akiyama K, et al. Differential effects of chromosome 9p21 variation on subphenotypes of intracranial aneurysm: site distribution. Stroke 2010;41:1593-8. [DOI] [PubMed] [Google Scholar]
- 109.Lukic A, Mead S.Genome wide association studies and prion disease. Prion 2011;5:154-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Mead S, Poulter M, Uphill J, et al. Genetic risk factors for variant Creutzfeldt-Jakob disease: a genome-wide association study. Lancet Neurol 2009;8:57-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Murdoch BM, Murdoch GK, Settles M, et al. Genome-wide scan identifies loci associated with classical BSE occurrence. PLoS One 2011;6:e26819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Mead S, Uphill J, Beck J, et al. Genome-wide association study in multiple human prion diseases suggests genetic risk factors additional to PRNP. Hum Mol Genet 2012;21:1897-906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kuroda S, Houkin K.Moyamoya disease: current concepts and future perspectives. Lancet Neurol 2008;7:1056-66. [DOI] [PubMed] [Google Scholar]
- 114.Kamada F, Aoki Y, Narisawa A, et al. A genome-wide association study identifies RNF213 as the first Moyamoya disease gene. J Hum Genet 2011;56:34-40. [DOI] [PubMed] [Google Scholar]
- 115.Liu W, Morito D, Takashima S, et al. Identification of RNF213 as a susceptibility gene for moyamoya disease and its possible role in vascular development. PLoS One 2011;6:e22542. [DOI] [PMC free article] [PubMed] [Google Scholar]