

Abstract
Background
Titanium dioxide nanoparticles (TiO2 NPs) are widely used in the biological sciences. The increasing use of TiO2 NPs increases the risk of humans and the environment being exposed to NPs. We previously showed that toll-like receptors (TLRs) play an important role in the interactions between NPs and cells. Our previous results indicated that TLR4 increased the DNA damage response induced by TiO2 NPs, due to enhanced NP uptake into the cytoplasm, whereas TLR3 expression decreased the DNA damage response induced by TiO2 NPs because of NP retention in the endosome. In this study, we explored the molecular mechanism of the DNA damage response induced by TiO2 NPs using TLR3 or TLR4 transfected cells. We examined the effect of TLR3 or TLR4 over-expression on oxidative stress and the effect of DNA damage induced by TiO2 NPs on gene expression levels.
Results
Our results showed evidence for elevated oxidative stress, including the generation of reactive oxygen species (ROS), with increased hydrogen peroxide levels, decreased glutathione peroxidase, and reduced glutathione and activated caspase-3 levels in cells exposed for 48 h to 10 μg/ml TiO2 NPs. These effects were enhanced by TLR4 and reduced by TLR3 over-expression. Seventeen genes related to DNA double-strand breaks and apoptosis were induced, particularly IP6K3 and ATM.
Conclusion
Our results indicated that TiO2 NPs induced ROS, and the above molecules are implicated in the genotoxicity induced by TiO2 NPs.
Keywords: TiO2NPs, TLRs, DNA damage, ROS, Apoptosis
Background
Nanotechnology is one of the fastest growing sectors of the high-tech economy. Several consumer products currently use nano-materials; these products have personal, commercial, medical, and military uses [1,2]. Engineered nano-materials are suited to a wide range of novel applications in the electronics, healthcare, cosmetics, technologies and engineering industries. The dearth of toxicological data on nano-materials makes it difficult to determine if there is a risk associated with exposure to nano-materials. Thus, there is an urgent need to develop rapid, accurate and efficient testing strategies to assess the health effects of these emerging materials [3].
Titanium dioxide nanoparticles (TiO2 NPs) possess dramatically different physicochemical properties compared to TiO2 fine particles (FPs). TiO2 NPs are widely used in the biomedical and bioengineering fields due to their strong oxidizing properties and chemical inertness [4]. Moreover, TiO2 nano-materials are widely used in industrial and consumer products due to their strong catalytic activity attributed to their small size, which provides a larger surface area per unit mass. These properties of TiO2 NPs may present both unique bioactivity properties and challenges to human health [5-7]. Indeed, the physicochemical properties of TiO2 NPs have been demonstrated to correlate with their toxicological effects [8]. TiO2 nano-materials have attracted much interest in medical fields due to their photo-reactivity [9]. It was previously reported that a TiO2 photo-catalyst can kill bacterial cells in water due to the generation of compounds such as reactive oxygen species (ROS) [10,11]. Furthermore, photo-excited anatase TiO2 nanoparticles effectively induce cytotoxicity in HeLa cancer cells [12]. TiO2 NPs are used increasingly in industrial products, such as toothpastes, sunscreens, cosmetics, pharmaceuticals, and food products [13]. Human exposure may occur during both the manufacturing process and use. The widespread use of TiO2 NPs, and their potential entry into the body through dermal, ingestion, and inhalation routes, suggests that TiO2 NPs pose a potential exposure risk to humans, livestock, and the ecosystem [14-18]. TiO2 NP toxicity may be due to the ease with which these NPs can pass through the cellular membranes and disrupt biological systems [19]. It has been suggested that the small size and corresponding large specific surface area are the major determinants of NP toxicity [20]. It has also been proposed that the large surface area of NPs greatly increases their ability to produce potentially toxic ROS [21].
ROS are reactive molecules and free radicals derived from molecular oxygen. These molecules are produced as byproducts during the mitochondrial electron transport of aerobic respiration or by oxidoreductase enzymes and metal catalyzed oxidation, and have the potential to cause a number of deleterious events. ROS play a role in cellular signaling, including apoptosis, gene expression, and the activation of cell signaling cascades [22]. Oxidative DNA damage induced by ROS and free radicals is important in the pathogenesis of many human diseases, including cancer, muscle degeneration, coronary heart disease and ageing [23]. Moreover, studies have indicated that TiO2 NPs induce photo-damage to DNA in human cells, mouse lymphoma cells, and phage [24-26].
Toll-like receptors (TLRs) play an essential role in the activation of innate immunity by recognizing specific molecular patterns of microbial components. TLRs are transmembrane proteins composed of both an extra-cellular domain (responsible for ligand recognition) and a cytoplasmic domain (required for initiating signaling) [27]. As suggested by their range of ligands and subcellular locations, TLRs recognize a wide range of foreign materials [28,29]. For example, TLRs that localize to the cell surface, such as TLR4, primarily recognize bacterial components. In contrast, TLRs that localize to the endocytic compartments, such as TLR3, mainly recognize viruses. We have previously shown that TLRs are also involved in the cellular response and cellular uptake of TiO2 NPs [30,31]. We have also shown that the exposure of HepG2 cells to TiO2 NPs induces DNA damage responses; this damage was increased by TLR4 over-expression, and decreased by TLR3 over-expression [32]. TLR3, which has a subcellular location distinct from TLR4, reduced the DNA damage response caused by TiO2 NPs. These results suggested that TLRs could be involved in many cellular responses, including genotoxicology. However, the molecular mechanism of DNA damage induced by TiO2 NPs is unknown.
In the present study, we aimed to elucidate the molecular mechanism of DNA damage induced by TiO2 NPs by using PCR array and real-time PCR (RT-PCR). Specifically, we examined the effect of TiO2 NP exposure on gene expression changes in DNA damage signaling pathways involving apoptosis, cell-cycle arrest, and DNA repair. Furthermore, we also confirmed elevated ROS generation by demonstrating increased hydrogen peroxide (H2O2) levels, decreased glutathione peroxidase (GPX) and glutathione (GSH) levels, as well as caspase-3 activation, in cells exposed to TiO2 NPs with or without TLR3 and TLR4 over-expression. We expect our work to advance the understanding of the molecular mechanism of DNA damage induced by TiO2 NPs.
Results
In general, ROS generation results in DNA damage. Our aim was to examine TiO2 NP-induced ROS generation and its association with DNA damage responses. ROS generation was measured in HepG2 cells exposed to TiO2 NPs with and without TLR3 or TLR4 over-expression (Figure 1). The results showed that in TiO2 NP-exposed cells, ROS levels were significantly increased (approximately 1.9 fold) compared with control cells (untreated, untransfected HepG2 cells). Cells exposed to TiO2 NPs and over-expressing TLR4 showed a significant increase in ROS levels compared to untransfected cells exposed to TiO2 NPs, reaching an approximately 2.6 fold increase compared to control. In comparison, ROS levels in HepG2 cells exposed to TiO2 NPs with TLR3 over-expression were slightly (≈1.3 fold) increased compared to control cells, as shown in Figure 1. These results indicate that ROS generation is a factor in the DNA damage response induced by TiO2 NPs.
Figure 1.
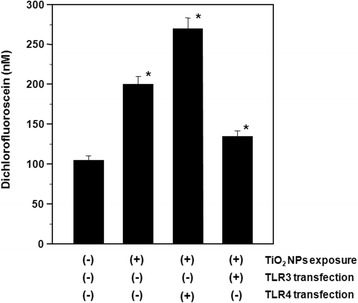
Reactive oxygen species (ROS) generation in TiO 2 NP-exposed HepG2 cells with and without TLR3 or TLR4 transfection. The transfected cells were exposed to 10 μg/ml TiO2 NPs for 48 h. Each plot was produced from at least 3 replicate measurements. All values are presented as mean ± S.D. (n ≥3), (*P <0.05).
Oxidative stress reflects an imbalance between the systemic manifestation of reactive oxygen species and a biological system’s ability to readily detoxify the reactive intermediates. Certain oxidant and antioxidant parameters were evaluated in order to obtain information on the cellular mechanism of oxidative stress in response to TiO2 NP exposure, as well as the effect of TLR3 or TLR4 over-expression. H2O2 is a reactive oxygen metabolic byproduct that serves as a key regulator of a number of oxidative stress-related states. Measurement of this reactive species provides an indication of the modulation of intracellular pathways by oxidative stress during TiO2 NP exposure and TLR3 and TLR4 over-production. In the present study, H2O2 concentrations were elevated 1.9 fold in cells exposed to TiO2 NPs compared to control cells. H2O2 concentration was further increased in cells expressing TLR4 (3.2 fold), whereas cells expressing TLR3 exhibited only a 1.4 fold increase (Figure 2). Our data show that H2O2 is an intermediate in TiO2 NP-induced oxidative stress, regardless of TLR3 or TLR4 over-expression.
Figure 2.
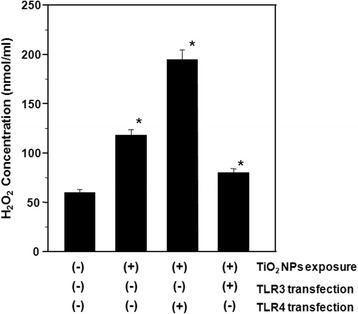
Hydrogen peroxide (H 2 O 2 ) levels in TiO 2 NP-exposed HepG2 cells with and without TLR3 or TLR4 transfection. The transfected cells were exposed to 10 μg/ml TiO2 NPs for 48 h. Each plot was produced from at least 3 replicate measurements. All values are presented as mean ± S.D. (n ≥3), (*P <0.05).
GPX catalyzes the reduction of hydroperoxides, including H2O2, using GSH tripeptide as a hydrogen donor. In order to confirm the elevated H2O2 levels, we measured GPX activity and GSH levels. The results showed that TiO2 NP treatment inhibited GPX enzyme activity in HepG2 cells by 1.6 and 3.6 fold in the absence and presence of TLR4 transfection, respectively, when compared to control cells. Over-expression of TLR3 with TiO2 NP exposure resulted in a 1.3 fold decrease in GPX activity compared to control cells (Figure 3). Similarly, TLR4 over-expression exacerbated TiO2 NP-induced reductions in GSH levels, while TLR3 over-expression appeared to reduce the effects of TiO2 NP exposure (Figure 4). These results showed that reduced GPX detoxification of H2O2 is involved in the oxidative stress response stimulated by TiO2 NPs, regardless of TLR3 or TLR4 over-expression, and confirmed the accumulation of H2O2 due to inhibition of the antioxidants GPX and GSH.
Figure 3.
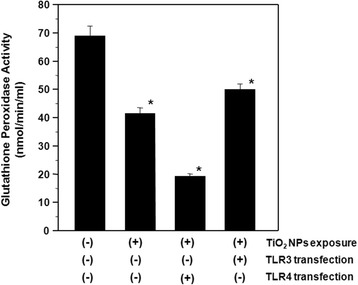
Glutathione peroxidase (GPx) activities in TiO 2 NP-exposed HepG2 cells with and without TLR3 or TLR4 transfection. The transfected cells were exposed to 10 μg/ml TiO2 NPs for 48 h. Each plot was produced from at least 3 replicate measurements. All values are presented as mean ± S.D. (n ≥3), (*P <0.05).
Figure 4.
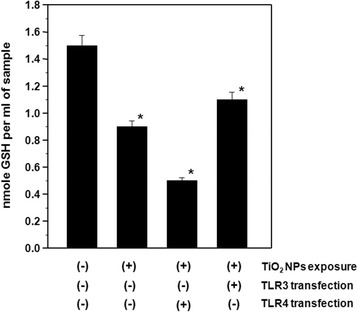
Reduced glutathione (GSH) levels in TiO 2 NP-exposed HepG2 cells with and without TLR3 or TLR4 transfection. The transfected cells were exposed to 10 μg/ml TiO2 NPs for 48 h. Each plot was produced from at least 3 replicate measurements. All values are presented as mean ± S.D. (n ≥3), (*P <0.05).
Caspase-3, an effector cysteine protease involved in apoptosis and necrosis, is activated by H2O2 [33]. Therefore, monitoring caspase-3 activation is important for evaluating apoptotic responses to oxidative stress occurring in HepG2 cells exposed to TiO2 NPs. Our results showed that caspase-3 activity significantly increased in cells exposed to TiO2 NPs, and that TLR4 expression further increased caspase-3 activation. Conversely, TLR3 expression resulted in an almost complete reversal of TiO2 NP-induced caspase-3 activation (Figure 5). Therefore, TiO2 NP-treatment induces apoptosis involving elevated caspase-3 activity in TLR4-expressing and normal HepG2 cells.
Figure 5.
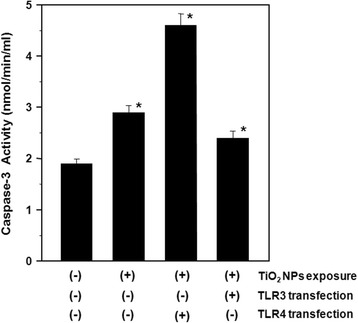
Caspase-3 activities in TiO 2 NP-exposed HepG2 cells with and without TLR3 or TLR4 transfection. The transfected cells were exposed to 10 μg/ml TiO2 NPs for 48 h. Each plot was produced from at least 3 replicate measurements. All values are presented as mean ± S.D. (n ≥3), (*P <0.05).
We used PCR array and RT-PCR to assess the cellular mechanisms operating in response to TiO2 NP exposure in the presence of TLR4 over-expression. The genes up-regulated by greater than 1-fold are listed in Table 1, and consist of genes in the human DNA damage signaling pathways (Table 1). In particular, the expression of the genes for apurinic/apyrimidinic exonuclease 1 (APEX1), ataxia telangiectasia mutated (ATM), growth arrest and DNA-damage-inducible, alpha (GADD45A), inositol hexakisphosphate kinase 3 (IP6K3), methyl-CpG binding domain protein 4 (MBD4), and structural maintenance of chromosomes 1A (SMC1A) were increased by >1.5 fold, with the remainder of the genes in the PCR array exhibiting <1.5 fold changes. In order to confirm the induction of the above-mentioned genes, the mRNA induction levels were determined by real-time PCR. The real-time PCR results confirmed the induction of mRNA expression observed for each of the genes (Figure 6). Indeed, the real-time PCR results indicated that the genes induced to the greatest extent were ATM and IP6K3, which is consistent with double-strand breaks in the DNA that result in DNA fragmentation and apoptosis [34].
Table 1.
Induction of gene mRNA expression in HepG2 cells transfected with TLR4 expression vectors
| Symbols of genes | Description of the genes | Fold regulation |
|---|---|---|
| ABL1 | C-abl oncogen 1, non-receptor tyrosine kinase | 1.56 |
| APEX1 | APEX nuclease (multifunctional DNA repair enzyme) 1 | 1.86 |
| ATM | Ataxia telangiectasia mutated | 1.91 |
| ATR | Ataxia telangiectasia and Rad3 related | 1.42 |
| BRCA1 | Breast cancer 1, early onset | 1.23 |
| CHEK2 | CHK2 checkpoint homolog (S.pombe) | 1.41 |
| CIDEA | Cell death-inducing DFFA-like effector a | 1.32 |
| GADD45A | Growth arrest and DNA-damage inducible, alpha | 1.77 |
| GML | Glycosylphosphatidylinositol anchored molecule like protein | 1.3 |
| IP6K3 | Inositol hexaphosphate kinase 3 | 2.23 |
| MAPK6 | Mitogen-activated protein kinase 6 | 1.34 |
| MBD4 | Methyl-CpG binding domain protein 4 | 1.65 |
| BTG2 | B-cell translocation gene 2 | 1.43 |
| RAD18 | RAD18 homolog (S. cerevisiae) | 1.34 |
| RAD21 | RAD21 homolog (S. pombe) | 1.39 |
| SMC1A | Structural maintenance of chromosomes 1A | 1.62 |
| PNKP | Polynucleotide kinase 3′-phosphate | 1.27 |
Cells exposed to 10 μg/ml TiO2 NPs for 48 h prior to gene expression analysis with PCR array. Fold regulation values greater than 1 indicate positive regulation (up-regulation).
Figure 6.
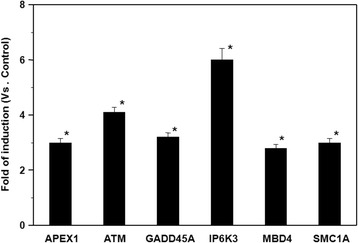
Expression of DNA damage marker mRNAs in TiO 2 NP-exposed HepG2 cells. Cells were exposed to 10 μg/ml TiO2 NPs for 48 h. Results are shown as the mean ± SD, n ≥3 for each marker, (*P <0.05).
To confirm the enhancement of apoptosis in HepG2 cells, we used Hoechst DNA staining to observe nuclear fragmentation as an indication of apoptosis. As shown in Figure 7, morphological changes consistent with cellular apoptosis, including condensation of chromatin and nuclear fragmentation, were observed in the cells exposed to TiO2 NPs. Again, expression of TLR4 (Figure 7C) appeared to enhance the effects of TiO2 NPs (Figure 7B) on apoptosis. Microscopy analysis confirmed that cells exposed to TiO2 NPs and transfected with TLR4 undergo programmed cell death (apoptosis) because of DNA damage. HepG2 cells that did not express TLRs and that were not exposed to TiO2 NPs also underwent apoptosis: by counting the number of apoptotic cells, we determined that 28% of untreated, untransfected cells had fragmented nuclei (Figure 7A), 55% of HepG2 cells exposed to TiO2 NPs were apoptotic (Figure 7B), and 75% of cells over-expressing TLR4 and exposed to TiO2 NPs were apoptotic (Figure 7C).
Figure 7.
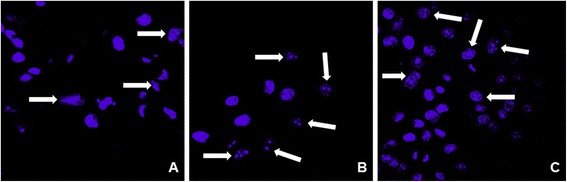
Confocal laser scanning microscopic images of HepG2 cells, with and without TLR4 transfection, treated with TiO 2 NPs. (A) HepG2 cells without transfection and without TiO2 NP exposure, (B) Cells exposed to TiO2 NPs only, (C) Cells transfected with TLR4 expression vector and exposed to 10 μg/ml TiO2 NPs for 48 h. The white arrows show the apoptotic, nuclear fragmented cells. The confocal microscopic images show condensation of chromatin and nuclear fragmentation in HepG2 cells transfected with TLR4 expression vector and exposed to TiO2 NPs.
Discussion
The purpose of this study was to examine the molecular mechanism of DNA damage caused by exposure to TiO2 NPs (10 μg/ml). A high concentration of TiO2 NPs should amplify the effects of the NPs and thus aid examination of their mechanism of action. The interactions of NPs with cells resulted in the generation of ROS, and the resultant oxidative stress may cause DNA fragmentation [35,36]. We found a significant increase in ROS generation in cells exposed to TiO2 NPs, which is consistent with our previous report of DNA damage responses in TiO2 NP-exposed cells [32]. In this paper, the results indicated that TiO2 NPs induced oxidative stress in cells, which can cause oxidative DNA damage and lead to the activation of p53 tumor suppressors and bcl-2 apoptotic factors. Additionally, oxidative stress can affect the mitochondria, the richest source of ROS, in which oxygen is metabolized and converted to O2− by several components of the mitochondrial respiratory chain (Figure 8).
Figure 8.
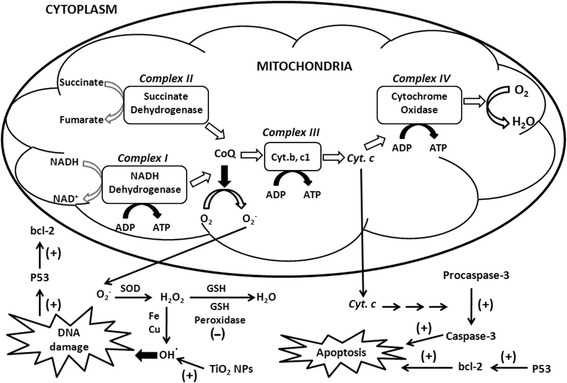
A schematic representation of mitochondrial ROS implicated in DNA damage and apoptosis induced by TiO 2 NP exposure, with and without TLR4 over-expression. The figure shows the site of induction and inhibition of respiratory complexes and oxidative stress molecules involved in ROS generation in HepG2 cells exposed to TiO2 NPs with and without TLR4 over-expression. (+) indicate the sites of activation and (-) show the sites of inhibition.
TLRs recognize and respond to exogenous and endogenous ligands through signaling pathways, leading to inflammatory cascade mediator production, which directs the innate and adaptive immune responses. TLRs interact with microbial components, such as lipopeptides, and non-self nucleic acids [37]. TLR4 localizes to the cell surface and TLR3 localizes in the endosome. We have shown that TLR4 is involved in TiO2 NP-induced inflammatory responses and TiO2 NP-incorporation [38,39]. We also have shown that TLR3 and TLR4 are involved in DNA damage induced by TiO2 NPs, indicating that TLR3 reduces DNA damage while TLR4 exacerbates it [32]. In this paper, our results showed more significant effects in HepG2 cells exposed to TiO2 NPs with TLR4 over-expression due to increased TiO2 NP uptake into the cytoplasm and increased signal transduction involving ROS in TLR4-dependent activation of NF-kB [40], while TLR3 reduced the effects caused by TiO2 NPs.
Various endogenous and exogenous genotoxic insults induce DNA-damage checkpoint signaling. The biological outcomes of checkpoint signaling include the control and coordination of cell-cycle progression, transcription, DNA replication, DNA repair, and apoptosis. DNA lesions trigger the activation of various kinases, which constitute the primary transducers in the signaling cascade. Of utmost importance are the phosphoinositide-3-kinase-related protein kinase (PIKK) family members, ATM, ATR, and DNA-dependent protein kinase. While ATR activation is associated with single-stranded DNA and stalled DNA replication forks, ATM and DNA-dependent protein kinase respond mainly to DNA double-strand breaks (DSBs) [34]. To identify marker genes of DNA damage-related cytotoxic stimulation, PCR array and RT-PCR analysis were performed using a commercial array system. Our results showed the induction of six genes, as follows: (1) APEX1 gene: a multifunctional DNA repair enzyme that plays a central role in the cellular response to oxidative stress. The major function of APEX1 in DNA repair and redox regulation of transcriptional factors is as an apurinic/apyrimidinic (AP) endodeoxyribonuclease in the DNA base excision repair pathway of DNA lesions induced by oxidative and alkylating agents. (2) ATM genes: the protein encoded by this gene belongs to the PI3/PI4-kinase family. This protein is an important cell cycle checkpoint kinase that functions as a regulator of a wide variety of downstream proteins, including tumor suppressor protein p53. It also functions as a serine/threonine protein kinase that activates checkpoint signaling upon DSBs, apoptosis, and genotoxic stresses, thereby acting as a DNA damage sensor. (3) GADD45A gene: the protein encoded by this gene responds to environmental stresses by mediating activation of the p38/JNK pathway via MTK1/MEKK4 kinase. DNA damage-induced transcription of this gene is mediated by both p53-dependent and -independent mechanisms. (4) IP6K3 gene: encodes a protein of the IPK family. IP6Ks regulate numerous biological processes, including chemotaxis, telomere length, and apoptosis [41]. IP6K3 impacts cell death, induces p53-mediated apoptosis, and its over-expression sensitizes cells to diverse apoptotic stimuli. (5) MBD4 gene: the protein encoded by this gene belongs to a family of nuclear proteins related by the presence of a methyl-CpG binding domain (MBD). These proteins are capable of binding specifically to methylated DNA, and possess mismatch-specific DNA N-glycosylase activity involved in DNA repair. They can also remove uracil or 5-fluorouracil in G:U mismatches. (6) Finally, the sixth gene is SMC1A; the encoded protein is thought to be an important part of functional kinetochores. This protein interacts with BRCA1 and is phosphorylated by ATM, indicating a potential role for this protein in DNA repair. These data suggest that these six genes are useful markers for DNA damage signaling pathways in response to TiO2 NP exposure, with the highest induction observed with ATM and IP6K3, as illustrated in Figure 6. Given that ATM and IP6K3 gene induction are involved in DSBs [34], the type of DNA damage induced by TiO2 NP exposure is likely DNA DSBs that cause eventual DNA fragmentation and apoptosis.
Conclusions
Our results showed that exposing HepG2 cells to TiO2 NPs enhances ROS generation and activates caspase-3 and oxidative stress-induced apoptosis. These effects were increased by TLR4 over-expression and decreased by TLR3 over-expression. We conclude that exposure to TiO2 NPs causes oxidative stress, with increased H2O2 and ·OH levels leading to DNA damage and p53 activation, and induces apoptosis by releasing cytochrome c into the cytoplasm and activating caspase-3. Over-expression of TLR3 protects against oxidative stress-induced damage in response to TiO2 NP exposure, but over-production of TLR4 enhances the oxidative stress mediated by TiO2 NPs. TiO2 NPs induce the expression of 17 DNA damage marker genes, especially the ATM and IP6K3 genes. This indicates that the type of DNA damage caused in HepG2 cells is double strand breaks, as well as chromatin condensation, nuclear fragmentation, and apoptosis.
Materials and methods
Cells and cell culture
HepG2 cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM, Nacalai Tesque, Inc., Kyoto, Japan) supplemented with 10% fetal bovine serum (FBS, Biowest, Nuaillé, France), 100 U/mL penicillin, and 100 μg/mL streptomycin (Nacalai Tesque, Inc.) at 37°C in a humidified atmosphere containing 5% CO2.
Plasmids employed
TLR-encoding genes were purchased from InvivoGen (San Diego, CA, USA). The pUNO1-mcs expression vector was used as an “empty” control vector. Since pUNO1-mcs does not contain a therapeutic gene, it can be used in conjunction with other vectors of the pUNO1 family to serve as an experimental control. Overproduction of TLR3 and TLR4 was provided by transfection with pUNO-hTLR3 (which encodes the human TLR3 protein) and pUNO1-hTLR04a (CD284a) (which harbors the human TLR04a (CD284a) encoding open reading frame), respectively. HepG2 cells were seeded in 6-well plates. After overnight incubation, the cells were co-transfected with TLR3 or TLR4 expression vectors and control plasmid (pUNO1-mcs) using Lipofectamine™ LTX Reagent (Invitrogen, Carlsbad, CA, USA) according to the supplier’s protocol. Transfection efficiency of at least 50% was obtained.
Preparation and exposure to TiO2 NPs
The preparation and characterization of TiO2 NPs were described in previous studies [26]. Briefly, nano-TiO2 (AeroxideR P25; Sigma-Aldrich, St Louis, MO, USA) was dispersed in distilled water and autoclaved at 120°C for 20 min. The suspension was cooled to room temperature and then sonicated for 10 min at 200 kHz using a high-frequency ultrasonic sonicator (MidSonic 600, Kaijo Corp., Tokyo, Japan). The resulting nano-TiO2 suspension was designated “TiO2 NPs”. The concentration of TiO2 NPs was determined using a UV–vis spectrophotometer at 370 nm (UV-1600, Shimadzu, Kyoto, Japan). The suspension was adjusted to the desired concentration by the addition of distilled water and stored at 4°C until use. The particle size distribution was measured by dynamic light scattering (Zetasizer Nano-ZS, Malvern Instruments, Malvern, UK). The aggregated particle size of the TiO2 NPs was determined to be 216 ± 70 nm. The size of the aggregated TiO2 NPs remained stable for several weeks under the indicated storage conditions. Prior to addition to the cell cultures, the suspension of TiO2 NPs was diluted with supplemented medium and used as described above. For the reporter gene (transfected cell) assays, the culture medium was replaced (1 day after transfection) with medium containing the TiO2 NPs at the indicated concentration. Specifically, TiO2 NPs were added to the culture medium immediately before the medium was applied to the cells. After 48 h, the cells were harvested and assayed.
DCF assay for oxidative stress determination
The accumulation of intracellular free radicals was quantified using a ROS assay kit (OxiSelect, Cell Biolabs, Inc., San Diego, CA, USA), which employs the cell-permeable fluorogenic probe 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA). DCFH-DA can cross cell membranes and be deacetylated by intracellular esterases to non-fluorescent 2′, 7′-dichlorodihydrofluorescein (DCFH). In the presence of ROS, DCFH is rapidly oxidized to the highly fluorescent DCF, which is readily detectable. The fluorescence intensity is proportional to the ROS levels in the cell cytosol. HepG2 cells were cultured in 96-well black plates; after overnight incubation, the cells were co-transfected with TLR3, TLR4 or control plasmid (pUNO1-mcs). After 24 h, the cells were exposed to TiO2 NPs for 48h, and were then incubated with DCHF-DA for 30 min in the dark. Parallel sets of wells containing freshly cultured cells, which were not treated with NPs or plasmids, and were suspended in the same concentration ratio of DPBS and DMEM, were regarded as negative controls. The fluorescence emission of DCF was monitored at regular intervals at an excitation wavelength of 480 nm and an emission wavelength of 530 nm using a fluorescence plate reader (Twinkle LB 970 Microplate Fluorometer, BERTHOLD TECHNOLOGIES GmbH & Co. KG, Calmbacher, Bad Wildbad Germany). The amount of DCF formed was calculated from a calibration curve constructed using an authentic DCF standard.
Measurement of H2O2
The levels of hydrogen peroxide (H2O2) were measured using a hydrogen peroxide assay kit (ab102500, Abcam, Tokyo, Japan). In the presence of horseradish peroxidase (HRP), the OxiRed Probe reacts with H2O2 to produce a colored product. Following the experiment, the cells were collected in H2O2 assay buffer and then centrifuged for 15 min at 1000 × g. A total of 50 μl of the supernatant was mixed with 50 μl of the reaction mix (assay buffer: 46 μl; OxiRed Probe: 2 μl; HRP: 2 μl) and then incubated at room temperature for 10 min. The optical density at 570 nm was read with a microplate reader (Benckmark Plus microplate spectrophotometer, BioRad, [city?] CA, USA), and the H2O2 concentration was calculated according to a standard concentration curve.
Measurement of GPX
Glutathione peroxidase activity was measured using a glutathione peroxidase assay kit provided by Cayman Chemical Company (Ann Arbor, MI, USA). Cells were washed in phosphate buffer, pH 7.4, collected by centrifugation (2000 × g for 10 min at 4°C), then homogenized in cold assay buffer (50 mM Tris-HCL, pH 7.5, 5 mM EDTA, 1 mM DTT). Following centrifugation at 10,000 × g for 15 min at 4°C, the supernatant was removed for assay. Sample (20 μl of supernatant) was added to the desired well of a 96-well plate, then 100 μL of assay buffer and 50 μl of co-substrate mixture was added. The reaction was initiated by adding 20 μl of cumene hydroperoxide to each reaction well, then mixed by shaking for second. The absorbance was read at 340 nm using a plate reader. At least 5 time points were obtained.
Measurement of GSH
The total glutathione concentration (reduced and oxidized forms) was determined in a microtitre plate assay using a glutathione assay kit (Sigma-Aldrich). After TLR transfection and nanoparticle exposure, HepG2 cells were washed twice with phosphate-buffered saline (PBS), resuspended in a 5% 5-sulfosalicylic acid solution, then centrifuged at 10,000 × g for 10 min. Supernatant (10 μl) was mixed with 150 μl of working solution, incubated for 5 min at room temperature, then 50 μl of the diluted NADPH solution was added. The absorbance of each sample was measured at 412 nm using the plate reader, as was the absorbance of the reagent blank (10 μl of 5% 5-sulfosalicylic acid); the absorbance of the blank was then subtracted from the absorbance of each sample. The final concentration of the components in the reaction mixture was 95 mM potassium phosphate buffer, pH 7.0, containing 0.95 mM ethylenediamine tetra-acetic acid (EDTA), 0.038 mg/ml (48 μM) NADPH, 0.031 mg/ml DTNB, 0.115 units/ml glutathione reductase, and 0.24% 5-sulfosalicylic acid. All measurements were performed in triplicate; the concentration (nmoles) of GSH in the samples was calculated.
Measurement of caspase-3 activity
All reagents for assessing caspase-3 activity were provided in a caspase-3 colormetric assay kit, (Sigma Aldrich). HepG2 cells (1 × 106) were cultured in 6-well plates and treated as described above. At the end of the experiment, the cells were washed and lysed in 100 μl of lysis buffer provide in the kit, then 80 μl of the sample was added to 10 μl of the 10× assay buffer and 10 μl of in a well of a 96-well plate. The reaction mix was incubated for 10 hours at 37°C, then the absorbance was read at 405 nm.
Gene expression analysis: PCR array
For polymerase chain reaction (PCR) array analysis, HepG2 cells (at 6 × 105 cells/ml) with or without TLR4 transfection were seeded in a culture dish containing culture medium with or without TiO2 NPs (suspended at 10 μg/ml). After 48 h exposure to the TiO2 NPs, the cells were detached by mechanical dissociation and total cellular RNA was extracted using an RNeasy kit (Qiagen, MD, USA). An aliquot (1 μg) of the extracted total RNA was reverse transcribed into cDNA with random hexamer primers using a RT2 First Strand kit (SABiosciences/Quiagen MD, USA) and the expression of 89 human DNA damage-related genes involved in signaling pathways were examined using a RT2 Profiler PCR array kit (SABiosciences/Quiagen) according to the manufacturer’s instructions. PCR array analysis was performed using an ABI PRISM 7000 sequence detection system (Applied Biosystems, Singapore).
Real-Time (RT) PCR
For mRNA expression analysis, 6 × 105 HepG2 cells/ml were seeded in cell culture dishes, the cells were transfected with TLR4 expression vector and exposed to a suspension of TiO2 NPs at a final concentration of 10 μg/ml for 48 h, then the cells were detached and subjected to gene expression analysis. The expression of marker genes was determined using quantitative real-time PCR (RT-PCR) as follows. Total RNA and cDNA were synthesized as described for the PCR array. The PCR primers for human APEX1, ATM, GADD45A, IP6K3, MBD4, SMC1A were purchased from SABiosciences/Qiagen. The data were normalized using the housekeeping gene glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as an endogenous control in the same reaction as the gene of interest [42]. The reaction mixture was composed of 12.5 μl of RT2 SYBR Green qPCR Master Mix (SABiosciences;/Qiagen), 1 μl of 10 μM gene-specific RT2 qPCR forward and reverse primers, 2 μl of cDNA, and nuclease-free water to a final volume of 25 μl. The thermocycling conditions were 95°C for 10 min, followed by 40 cycles of 95°C for 15 s and 60°C for 1 min.
Confocal microscopy observation
Confocal laser scanning microscopy was performed using a Zeiss LSM510 microscope (Carl Zeiss, Oberkochen, Germany). HepG2 cells were cultured on cover-slips (13 mm diameter; Matsunami Glass Ind., Ltd., Osaka, Japan). The following day, cultures were transfected (using the Lipofectamine™ LTX Reagent, as described above) with the expression vector encoding TLR4. At 24 h after transfection, the culture medium was replaced with medium containing 10 μg/ml TiO2 NPs. Untransfected cells and cells without NP exposure were used as controls. After 48 h incubation, the cells were washed with PBS and fixed with 4% paraformaldehyde for 5 min. Fixed cells were then stained for nuclei using 1 μg/ml Hoechst33342 (Dojin Chemical, Japan) for 30 min in a 5% CO2 environment. Figures were created using NIH ImageJ software.
Statistical analysis
Data were expressed as the mean ± SD, (n ≥3). All experiments were carried out independently. The data were analyzed using Student’s t test to evaluate the significance of differences between the treated groups and control groups. Statistical significance was accepted at P < 0.05.
Acknowledgements
This work was supported by Interaction Center for Material Nanoarchitectonics (MANA), National Institute for Materials Science (NIMS), Japan.
Abbreviations
- TiO2 NPs
Titanium dioxide nanoparticles
- TLRs
Toll-like receptors
- PCR
Polymerase chain reaction
- ROS
Reactive oxygen species
- DCF
Dichlorofluoroscein
- NADH
Nicotinamide adenine dinucleotide hydrogen
- APEX1
Apurinic/Apyrimidinic exonuclease 1
- ATM
Ataxia telangiectasia mutated
- GADD45A
Growth arrest and DNA-damage-inducible, alpha
- IP6K3
Inositol hexakisphosphate kinase 3
- MBD4
Methyl-CpG binding domain protein 4
- SMC1A
Structural maintenance of chromosomes
- H2O2
Hydrogen peroxide
- GPx
Glutathione peroxidase
- GSH
Reduced glutathione
- RAD
Radiation associated damage “DNA repair gene”
Footnotes
Competing interests
All authors declare that they have no competing of interests.
Authors’ contribution
KS conducted the laboratory work, developed the study parameters and design, and wrote the manuscript. KK assisted with TiO2 NP experiments. AT and EM conceived of and supervised the study, and AT supervised KS and KK. The all authors read and approved the final draft of the manuscript.
Contributor Information
Karim Samy El-Said, Email: SAMYELSAIDALI.Karim@nims.go.jp.
Ehab Mostafa Ali, Email: ehab_1964@hotmail.com.
Koki Kanehira, Email: koki.kanehira@jp.toto.com.
Akiyoshi Taniguchi, Email: taniguchi.akiyoshi@nims.go.jp.
References
- 1.Brumfiel G. Consumer products leap aboard the nano bandwagon. Nature. 2006;440:262. doi: 10.1038/440262b. [DOI] [PubMed] [Google Scholar]
- 2.Griffitt RJ, Weil R, Hyndman KA, Denslow ND, Powers K, Taylor D, Barber DS. Exposure to copper nanoparticles causes gill injury and acute lethality in zebrafish (Danio rerio) Environ Sci Technol. 2007;41:8178–8186. doi: 10.1021/es071235e. [DOI] [PubMed] [Google Scholar]
- 3.Hu X, Cook S, Wang P, Hwang HM. In vitro evaluation of cytotoxicity of engineered metal oxide nanoparticles. Sci Total Environ. 2009;407:3070–3072. doi: 10.1016/j.scitotenv.2009.01.033. [DOI] [PubMed] [Google Scholar]
- 4.Bernard BK, Osheroff MR, Hofman A, Mennear JH. Toxicology and carcinogenesis studies of dietary titanium dioxide-coated mica in male and female Fischer 344 rats. Environ Health. 1989;28:415–426. doi: 10.1080/15287399009531402. [DOI] [PubMed] [Google Scholar]
- 5.Maynard AD, Kuempel ED. Airborne nanostructured particles and occupational health. J Nanopart Res. 2005;6:587–614. doi: 10.1007/s11051-005-6770-9. [DOI] [Google Scholar]
- 6.Tsuji JS, Maynard AD, Howard PC, James JT, Lam CW, Warheit DB, Santamaria AB. Research strategies for safety evaluation of nanomaterials, part IV: risk assessment of nanoparticles. Toxicol Sci. 2006;89:42–50. doi: 10.1093/toxsci/kfi339. [DOI] [PubMed] [Google Scholar]
- 7.Shi H, Magaye R, Castranova V, Zhao J. Titanium dioxide nanoparticles: a review of current toxicological data. Part Fibre Toxicol. 2013;10:15. doi: 10.1186/1743-8977-10-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Iavicoli I, Leso V, Fontana L, Bergamaschi A. Toxicological effects of titanium dioxide nanoparticles: a review of in vitro mammalian studies. Eur Rev Med Pharmacol Sci. 2011;15:481–508. [PubMed] [Google Scholar]
- 9.Sakai H, Ito E, Cai RX, Yoshioka T, Hashimoto K, Fujishima A. Intracellular Ca2+ concentration change of T24 cell under irradiation in the presence of TiO2 ultrafine particles. Biochim Biophys. 1994;1201:259–265. doi: 10.1016/0304-4165(94)90049-3. [DOI] [PubMed] [Google Scholar]
- 10.Sunada K, Kikuchi Y, Hashimoto K, Fujishima A. Bactericidal and detoxification effects of TiO2 thin film photocatalyst. Environ Sci Technol. 1998;32:726–728. doi: 10.1021/es970860o. [DOI] [Google Scholar]
- 11.Maness PC, Smolinski S, Blake DM, Huang Z, Wolfrum EJ, Jacoby WA. Bactericidal activity of photocatalytic TiO2 reaction: toward an understanding of Its killing mechanism. Appl Environ Microbiol. 1999;65(9):4094–4098. doi: 10.1128/aem.65.9.4094-4098.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cai R, Kubota Y, Shiumi T, Sakai H, Hashimoto K, Fujishima A. Induction of cytotoxicity by photoexcited TiO2 particles. Cancer Res. 1992;52:2346–2348. [PubMed] [Google Scholar]
- 13.Wang JJ, Sanderson BJ, Wang H. Cyto- and genotoxicity of ultrafine TiO2 particles in cultured human lymphoblastoid cells. Mutat Res. 2007;628:99–106. doi: 10.1016/j.mrgentox.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 14.Oberdorster G, Maynard A, Donaldson K, Castranova V, Fitzpatrick J, Ausman K, Carter J, Karn B, Kreyling W, Lai D, Olin S, Monteiro-Riviere N, Warheit D, Yang H. Principles for characterizing the potential human health effects from exposure to nanomaterials: elements of a screening strategy. Part Fibre Toxicol. 2005;2:8. doi: 10.1186/1743-8977-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Long MS, Keating CD. Nanoparticle conjugation increases protein partitioning in aqueous two-phase systems. Anal Chem. 2006;78:379–386. doi: 10.1021/ac051882t. [DOI] [PubMed] [Google Scholar]
- 16.Jin CY, Zhu BS, Wang XF, Lu QH. Cytotoxicity of titanium dioxide nanoparticles in mouse fibroblast cells. Chem Res Toxicol. 2008;21:1871–1877. doi: 10.1021/tx800179f. [DOI] [PubMed] [Google Scholar]
- 17.Zhao J, Bowman L, Zhang X, Vallyathan V, Young SH, Castranova V, Ding M. Titanium dioxide (TiO2) nanoparticles induce JB6 cell apoptosis through activation of the caspase-8/Bid and mitochondrial pathways. J Toxicol Environ Health A. 2009;72:1141–1149. doi: 10.1080/15287390903091764. [DOI] [PubMed] [Google Scholar]
- 18.Liu S, Xu L, Zhang T, Ren G, Yang Z. Oxidative stress and apoptosis induced by nanosized titanium dioxide in PC12 cells. Toxicology. 2010;267:172–177. doi: 10.1016/j.tox.2009.11.012. [DOI] [PubMed] [Google Scholar]
- 19.Mishra VK, Mishra SK, Jha A. Application of nanomaterials in mesenchymal stem cell engineering. Dig J Nanomater Biostructures. 2008;3:203–208. [Google Scholar]
- 20.Huang CC, Aronstam RS, Chen DR, Huang YW. Oxidative stress, calcium homeostasis, and altered gene expression in human lung epithelial cells exposed to ZnO nanoparticles. Toxicol Vitro. 2010;24:45–55. doi: 10.1016/j.tiv.2009.09.007. [DOI] [PubMed] [Google Scholar]
- 21.Pujalte I, Passagne I, Brouillaud B, Treguer M, Durand E, Courtes CO, Lazou B. Cytotoxicity and oxidative stress induced by different metallic nanoparticles on human kidney cells. Part Fibre Toxicol. 2011;ᅟ:ᅟ. doi: 10.1186/1743-8977-8-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hancock JT, Desikan R, Neill SJ. Role of reactive oxygen species in cell signaling pathways. Bioch Biomed Aspects Oxidative Modif. 2001;29(2):345–350. doi: 10.1042/0300-5127:0290345. [DOI] [PubMed] [Google Scholar]
- 23.Dong-Hyun L, Timothy R, Gred P. Oxidative DNA damage induced by copper and hydrogen peroxide promotes CG_TT tandem mutations at methylated CpG dinucleotides in nucleotide excision repair-deficient cells. Nucleic Acids Res. 2002;30:3566–3573. doi: 10.1093/nar/gkf478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dunford R, Salinaro A, Cai L, Serpone N, Horikoshi S, Hidaka H, Knowland J. Chemical oxidation and DNA damage catalyzed by inorganic sunscreen ingredients. FEBS Lett. 1997;418:87–90. doi: 10.1016/S0014-5793(97)01356-2. [DOI] [PubMed] [Google Scholar]
- 25.Nakagawa Y, Wakuri S, Sakamoto K, Tanaka N. The photogenotoxicity of titanium dioxide particles. Mutat Res. 1997;394:125–132. doi: 10.1016/S1383-5718(97)00126-5. [DOI] [PubMed] [Google Scholar]
- 26.Kashige N, Kakita Y, Nakashima Y, Miake F, Watanabe K. Mechanism of the photocatalytic inactivation of Lactobacillus casei phage PL-1 by titania thin film. Curr Microbiol. 2001;42:184–189. doi: 10.1007/s002840010201. [DOI] [PubMed] [Google Scholar]
- 27.Huyton T, Rossjohn J, Wilce M. Toll-like receptors: structural pieces of a curve-shaped puzzle. Immunol Cell Biol. 2007;85:406–410. doi: 10.1038/sj.icb.7100089. [DOI] [PubMed] [Google Scholar]
- 28.Trinchieri G, Sher A. Cooperation of Toll-like receptors signals in innate immune defense. Nat Rev Immunol. 2007;7:179–190. doi: 10.1038/nri2038. [DOI] [PubMed] [Google Scholar]
- 29.Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. 2010;11:373–384. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- 30.Chen P, Kanehira K, Sonezaki S, Taniguchi A. Detection of cellular response to titanium dioxide nanoparticles agglomerates by sensor cells using heat shock protein promoter. Biotechnol Bioeng. 2012;109:3112–3118. doi: 10.1002/bit.24583. [DOI] [PubMed] [Google Scholar]
- 31.Chen P, Migita S, Kanehira K, Taniguchi A. Role of toll-like receptors 3, 4 and 7 in cellular uptake and response to titanium dioxide nanoparticles. Sci Tech Adv Mater. 2013;14:ᅟ. doi: 10.1088/1468-6996/14/1/015008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.El-Said KS, Ali EM, Kanehira K, Taniguchi A. Effects of toll-like receptors 3 and 4 induced by titanium dioxide nanoparticles in DNA damage-detecting sensor cells. J Biosens Bioelectron. 2013;4:144. [Google Scholar]
- 33.Piperakis M, Karanastasi G, Lakovidou-Kritsi Z, Piperakis M. The use of comet assay in measuring DNA damage and repair efficiency in child, adult, and old age populations. Cell Biol Toxicol. 2009;25:65–71. doi: 10.1007/s10565-007-9046-6. [DOI] [PubMed] [Google Scholar]
- 34.Durocher D, Jackson SP. DNA-PK, ATM and ATR as sensors of DNA damage: variations on a theme. Curr Opin Cell Biol. 2001;13:225–231. doi: 10.1016/S0955-0674(00)00201-5. [DOI] [PubMed] [Google Scholar]
- 35.Petruska JM, Leslie KO, Mossman BT. Enhanced lipid peroxidation in lung lavage of rats after inhalation of asbestos. Free Radic Biol Med. 1991;11:425–432. doi: 10.1016/0891-5849(91)90160-5. [DOI] [PubMed] [Google Scholar]
- 36.Shukla RK, Kumar A, Pandey AK, Singh SS, Dhawan A. Titanium dioxide nanoparticles induce oxidative stress-mediated apoptosis in human keratinocyte cells. J Biomed Nanotechnol. 2011;7:100–101. doi: 10.1166/jbn.2011.1221. [DOI] [PubMed] [Google Scholar]
- 37.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 38.Mano SS, Kanehira K, Taniguchi A. Comparison of cellular uptake and inflammatory response via toll-like receptor 4 to lipopolysaccharide and titanium dioxide nanoparticles. Mol Sci. 2013;14(7):13154–13170. doi: 10.3390/ijms140713154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mano SS, Kanehira K, Sonezaki S, Taniguchi A. Toll-like receptor 4 is involved in titanium dioxide nanoparticle incorporation into cells. Sci Adv Mater. 2014;6:1562–1568. doi: 10.1166/sam.2014.1821. [DOI] [Google Scholar]
- 40.Asehnoune K, Strassheim D, Mitra S, Kim JY, Abraham E. Involvement of reactive oxygen species in toll-like receptor 4-dependent activation of NF-kB. J Immun. 2004;172:2522–2529. doi: 10.4049/jimmunol.172.4.2522. [DOI] [PubMed] [Google Scholar]
- 41.Nagata E, Luo HR, Saiardi A, Bae BI, Suzuki N, Snyder SH. Inositol hexakisphosphate kinase-2, a physiologic mediator ofcell death. J Biol Chem. 2005;280:1634–1640. doi: 10.1074/jbc.M409416200. [DOI] [PubMed] [Google Scholar]
- 42.Matsumoto A, Harada H, Saito M, Taniguchi A. Induction of enamel matrix protein expression in an ameloblast cell line co-cultures with a mesenchymal cell line in vitro. In Vitro Cell Dev Biol Anim. 2011;47:39–44. doi: 10.1007/s11626-010-9362-7. [DOI] [PubMed] [Google Scholar]