

Abstract
Corin is a membrane-bound protease that regulates blood pressure by activating the natriuretic peptides. CORIN variants have been associated with hypertension and heart disease in African Americans. In this study, we conducted targeted exome sequencing and identified an insertion variant, c.102_103insA, in exon 1 of the CORIN gene. Analysis of two independent cohorts showed that the variant was preferentially present in hypertensive patients (38/795 or 4.78% vs. 4/632 or 0.63% in normal individuals, p = 4.14E-6). The insertion shifted the reading frame, resulting in a corin variant with a truncated cytoplasmic tail. In cell-based studies, the corin variant exhibited poor trafficking in the Golgi, reduced cell surface expression and zymogen activation, and low natriuretic peptide processing activity. Compared with normal individuals with the wild-type allele, individuals with the variant allele had lower levels of plasma corin [0.59 ± 0.07 ng/mL (n = 25) vs. 0.91 ± 0.02 ng/mL (n = 215), p<0.001] and higher levels of plasma N-terminal pro-atrial natriuretic peptide (NT-pro-ANP) [2.39 ± 3.6 nmol/L (n = 21) vs. 0.87 ± 0.6 nmol/L (n = 48), p = 0.005]. These results indicate that the variant altered corin structure and impaired the natriuretic peptide processing activity in vivo. The results highlight corin defects as an important underlying mechanism in hypertension.
Hypertension is a major cardiovascular disease, which increases the risk for stroke, myocardial infarction, and heart failure. Genetic factors play an important role in hypertension1,2,3,4. To date, molecular defects in most hypertensive patients remain unclear2,3, suggesting that more contributing factors are yet to be identified. Atrial and B-type natriuretic peptides (ANP and BNP) are cardiac hormones that regulate sodium homeostasis and blood pressure5,6. In cardiomyocytes, these peptides are synthesized in pro-forms, i.e. pro-ANP and pro-BNP, which are converted to active ANP and BNP by corin, a membrane-bound serine protease7. The importance of corin in regulating blood pressure has been shown in corin knockout mice, which exhibited reduced sodium excretion and salt-sensitive hypertension due to impaired natriuretic peptide processing8,9,10.
Structurally, corin belongs to the type II transmembrane serine protease family, a group of trypsin-like enzymes involved in diverse biological processes11,12. All these membrane-bound proteases consist of an N-terminal cytoplasmic tail, a single-span transmembrane domain and an extracellular region containing various modules and a C-terminal protease domain. The transmembrane domain anchors the proteases on the cell surface, where proteolytic processes take place11,12. It has been shown that cell surface targeting is important for corin zymogen activation and biological activity13,14,15.
Human CORIN gene consists of 22 exons and spans >200 kb in length on chromosome 416, making it one of the largest protease genes in the human genome. In principle, natural variants and mutations are more likely to occur in large genes. To date, CORIN variants have been associated with hypertension in African Americans17,18,19,20. CORIN mutations that reduce corin activity also have been found in patient families with hypertension and preeclampsia21,22,23,24. It remains unknown if additional CORIN variants are present in general populations that may contribute to hypertension.
In this study, we conducted targeted exome sequencing and identified a novel insertion variant in the CORIN gene that was preferentially present in patients with hypertension. Biochemical experiments showed that the variant altered corin cytoplasmic tail sequence and reduced corin cell surface expression, zymogen activation, and pro-ANP processing activity. Our results suggest that naturally occurring CORIN variants may reduce corin activity and contribute to hypertension in patients.
Results
Identification of CORIN Gene Variant
We sequenced CORIN exons and intron-exon boundaries in genomic DNA samples from normal and hypertensive individuals and identified a variant allele with a single adenine insertion at nucleotide position 102 (c.102_103insA, referred to as “insA variant” hereafter) in exon 1 of the CORIN gene (Fig. 1A)16,25. The insertion shifted the reading frame, creating a down-stream stop codon (Fig. 1B).
Figure 1. Genetic variant altering corin cytoplasmic tail.

(A) CORIN gene sequencing data from a WT allele (left) and a variant allele with an insertion (right). (B) Corin domain structure. TM, transmembrane; Fz, frizzled; LDLR, LDL receptor; SR, scavenger receptor. The catalytic sites His (H), Asp (D) and Ser-985 (S) in the protease domain are shown. The activation cleavage site R801-I802 is indicated. The insertion causes frameshift, creating a down-stream stop codon (*) in the cytoplasmic tail. An alternative initiation site, Met-30, is indicated by an arrowhead.
The variant allele occurred more frequently in hypertensive patients than normal individuals. In the first study cohort, 23 of 401 hypertensive patients (5.74%) were heterozygous for this variant allele, whereas 2 of 217 normal individuals (0.92%) were heterozygous for the variant allele (p = 0.0037). We verified this finding in a second study cohort. Among 394 hypertensive patients, 15 or 3.81% were heterozygous for the variant allele, whereas 2 of 415 normal individuals (0.48%) were heterozygous for the variant allele (p = 0.00098). When the data of both cohorts were combined, 38 of 795 hypertensive patients (4.78%) and 4 of 632 normal individuals (0.63%) were heterozygous for the variant allele (p = 4.14E-6). We searched the 1000 genome database (www.1000genomes.org) and found the insA CORIN allele with a minor allele frequency of 0.0037 (8 alleles in 1094 individuals), which is similar to that in our normal controls (0.0032; 4 alleles in 632 individuals; p = 0.81). The results indicate that the variant allele is present in general populations but its occurrence is particularly high in hypertensive patients. In agreement with this, multivariate logistic regression analysis showed consistent association of the insA variant with hypertension in both study cohorts (Supplementary Tables S1 and S2).
Characterization of insA Variant Corin
The insertion of an adenine at nucleotide position 102 shifts the reading frame (Fig. 1B), which is expected to prevent corin expression. We made the plasmid, pcDNAinsA, containing the adenine insertion and transfected it in HEK293 cells. WT corin (positive) and inactive mutants R801A and S985A (negative) were used as controls (Fig. 1B). Surprisingly, Western analysis detected the insA variant expression in transfected HEK293 cells (Fig. 2A).
Figure 2. Expression and activity of the insA variant in HEK293 cells.

(A) Western analysis of corin WT, the insA variant, and mutants R801A and S985A protein levels in transfected HEK293 cells. (B) Pro-ANP processing activity of the insA variant. WT and inactive mutants R801A and S985A were positive and negative controls, respectively. (C) Quantitative data of pro-ANP processing activity were from densitometric analysis. The cropped blots are used in the main figures and full-length blots are included in the supplementary information (Supplementary Fig. S1). Data are representative of at least four independent experiments.
To test if the insA variant corin is biologically active, a pro-ANP processing assay was performed. The variant exhibited pro-ANP processing activity, although the level was markedly reduced compared to that of WT (Fig. 2B). In negative controls (vector, mutants R801A and S985A), little pro-ANP processing activity was observed (Fig. 2B). By densitometric analysis, the variant had 41.3 ± 8.5% activity compared with that of WT (p<0.01) (Fig. 2C).
Zymogen Activation of Corin Variant
One possible explanation for the insA variant expression is that a down-stream methionine was used as the translation initiation site. Indeed, there is a methionine at position 30 that may be used as an alternative initiation site (Fig. 1B). To test this hypothesis, we expressed the variant in HEK293 cells together with a corin isoform that contains methionine 30 (Met-30) but lacks methionine 1 (Met-1)15. By Western analysis, similar expression levels of the insA variant, the Met-30 isoform (Met-30), WT, and mutants R801A and S985A were observed (Fig. 3A). On the Western blots, an ~40-kDa band (corin-p), representing the corin protease domain cleaved at the activation site Arg-801 (Fig. 1B), was detected in WT (Fig. 3A, lane 2). This band also was detected in the catalytic site mutant S985A (Fig. 3A, lane 5), but not in mutant R801A, in which the cleavage site is abolished (Fig. 3A, lane 4). The intensity of this band in the insA variant was markedly reduced (Fig. 3A, lane 3). Similar results were observed in the Met-30 isoform (Fig. 3A, lane 6). By densitometric analysis, the ~40-kDa band in WT represented 12.73 ± 1.97% of the total corin protein in lysate, whereas the band in the insA variant was 4.04 ± 1.02%, significantly lower than that in WT (n = 5, p<0.01) (Fig. 3B). In the Met-30 isoform, this band was 3.99 ± 2.67% of the total corin protein, which was similar to that in the insA variant (Fig. 3B). The data support that the insertion resulted in a variant corin with a shorter cytoplasmic tail and reduced zymogen activation and activity.
Figure 3. Zymogen activation of the insA variant in HEK293 cells.
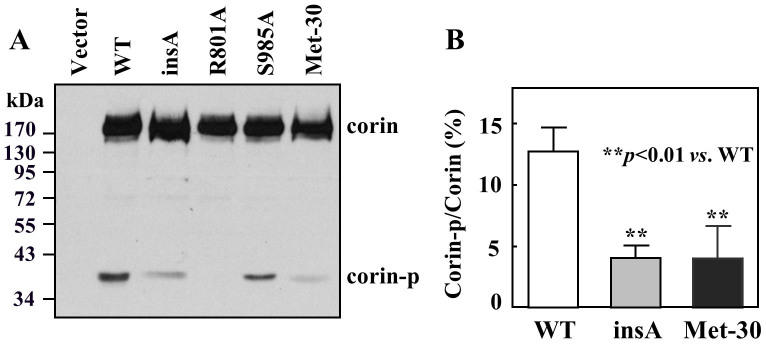
(A) Western analysis of corin proteins in cell lysate. The ~40-kDa band represented the corin protease domain (corin-p) from activation cleavage. This band was absent in mutant R801A, in which the cleavage site was abolished. Data are representative of five independent experiments. (B) Percentage of the activated protease domain fragment (corin-p) vs. corin zymogen protein (corin) was estimated by densitometry. Data are mean ± S.D. from five independent experiments.
Corin Expression on Cell Surface
Corin is a membrane-bound protease and its activation depends on cell surface expression14. Previous studies indicated that cytoplasmic tail sequences may alter corin cell surface expression15. We examined the variant expression in biotin-labeled HEK293 cells. Surface expression of the insA variant was much less abundant than that of WT (5.5 ± 1.5 vs. 31.5 ± 8.4%, n = 3, p<0.01) (Fig. 4A and B). Similarly reduced cell surface expression was observed in the Met-30 isoform (6.7 ± 3.1%, n = 3, p<0.01 vs. WT) (Fig. 4A and B). As controls, the cell surface expression of mutants R801A and S985A was similar to that of WT (Fig. 4A). In cell lysate, total corin levels were comparable in all samples (Fig. 4A, right panel). As expected, GAPDH was absent in labeled cell surface proteins, but present at similar levels in all lysate samples (Fig. 4A, lower panels).
Figure 4. Cell surface expression of corin proteins.

(A) Western analysis of biotin-labeled corin proteins on the cell surface (left panel) and in cell lysate (right panel). GAPDH was used as a control (lower panels). The cropped blots are used in the main figure and full-length blots are included in the supplementary information (Supplementary Fig. S2). (B) Corin bands on Western blots were analyzed by densitometry. Percentage of corin proteins on the cell surface was estimated. Data are mean ± S.D. from three independent experiments. (C) Analysis of cell surface corin proteins by flow cytometry. Data are representative of three independent experiments.
We verified these results by flow cytometry. In HEK293 cells expressing WT, 32.7 ± 8.5% were corin-positive on the surface (Fig. 4C). In contrast, only 7.9 ± 3.7 and 6.9 ± 6.4%, respectively, were corin-positive in cells expressing the insA variant and the Met-30 isoform (n = 3; both p values <0.01 vs. WT).
Subcellular Localization of insA Variant
We next examined the intracellular distribution of the variant by immunostaining. In HEK293 cells expressing WT, predominant corin staining was on the cell surface (Fig. 5A). Such staining was absent in vector-transfected cells. In cells expressing the insA variant, corin staining was more prominent intracellularly. By co-staining with TGN46, a Golgi marker, a significant portion of the insA variant was found in the Golgi (Fig. 5A). In parallel experiments of co-staining with PDI, an ER marker, no significant staining of WT or the variant was found in the ER (Fig. 5B). The data indicate that the insA variant was less efficient in trafficking through the Golgi network.
Figure 5. Subcellular distribution of corin proteins.

(A) HEK293 cells expressing WT and the insA variant were stained for corin (green) and TGN46, a Golgi marker (red). (B) HEK293 cells expressing WT and the insA variant were stained for corin (red) and PDI, an ER marker (green). In both experiments, cell nuclei were stained with DAPI (blue) and vector-transfected cells were used as a negative control. Bar: 5 μm.
Soluble Corin in Cultured Cell Medium
As reported previously26, cell surface corin undergoes proteolytic shedding, generating three fragments of ~180-, ~160-, and ~100-kDa, respectively, in the conditioned medium. The ~180-kDa fragment was from a disintegrin and metalloprotease (ADAM)-mediated cleavage, whereas the ~160- and ~100-kDa fragments were from corin autocleavage26. We examined corin shedding in HEK293 cells. Three bands of the predicted sizes were found in the conditioned medium from cells expressing WT (Fig. 6A). In cells expressing the insA variant and the Met-30 isoform, the ~180-kDa fragment was less abundant and the ~100-kDa fragment was hardly visible (Fig. 6A). By densitometric analysis of the Western blots, the percentage of the ~180-kDa fragment (Fig. 6B) or all three soluble corin fragments (Fig. 6C) was significantly lower than that of WT (n = 4, p values <0.01). In inactive mutants R801A and S985A, only the ~180-kDa ADAM-cleaved fragment was detected, but not the ~160- and ~100-kDa autocleavage fragments, as predicted. The results indicate that the insA variant had reduced shedding, likely due to impaired cell surface targeting and zymogen activation.
Figure 6. Shed corin fragments in conditioned medium and plasma levels of soluble corin and NT-pro-ANP.

(A) Western analysis of corin fragments in the conditioned medium from cells expressing corin WT, the variant and mutants (upper panel). Corin proteins in cell lysate are shown as a control (lower panel). The cropped blots are used in the main figure and full-length blots are included in the supplementary information (Supplementary Fig. S3). Data are representative of four independent experiments. Western blots were analyzed by densitometry. Percentages of the ~180-kDa corin fragment (B) or all there soluble corin fragments (C) were estimated. Data are mean ± S.D. from four independent experiments. (D) Plasma levels of soluble corin in individuals with WT (Control) and the variant (insA) alleles. (E) Plasma levels of NT-pro-ANP in individuals with WT (Control) and the variant (insA) alleles.
Plasma Soluble Corin and NT-pro-ANP Levels
We also measured plasma corin in individuals with WT and the insA variant CORIN alleles. The levels in 215 normal individuals with the WT allele were 0.91 ± 0.02 ng/mL, whereas the levels in 25 individuals with the variant allele were significantly lower (0.59 ± 0.07 ng/mL, p<0.001) (Fig. 6D). When the 25 individuals with the insA variant allele were separated into normotensive and hypertensive groups, the levels in two normotensive individuals (mean ± range: 0.91 ± 0.04 ng/mL) were higher than that in 23 hypertensive individuals (mean ± SD: 0.57 ± 0.07 ng/mL). However, the sample size in the normotensive group was too small for statistical analysis. In a subset of samples from individuals with the WT allele, plasma corin levels were lower in hypertensive individuals (0.62 ± 0.09 ng/mL, n = 60, p<0.01) than that in normotensive individuals (0.86 ± 0.43 ng/mL, n = 60).
We next measured plasma levels of NT-pro-ANP in individuals with WT and the insA variant alleles (Fig. 6E). Plasma NT-pro-ANP levels in 48 normal individuals with the WT allele were 0.87 ± 0.6 nmol/L, whereas the levels in 21 individuals with the variant allele were 2.39 ± 3.6 nmol/L, significantly higher than that in the normal control (p = 0.005).
Discussion
In this study, we performed targeted exome sequencing and identified an insertion variant, c.102_103insA, in the CORIN gene. The occurrence of this variant allele in the normal controls from two independent cohorts (4/632 or 0.63%) is similar to that in the 1000 genome database (8/1094 or 0.73%, p = 0.81). This allele, however, is much more prevalent in hypertensive patients (23/401 or 5.74% in the first cohort and 15/394 or 3.81% in the second cohort, both p values <0.001 vs. normal controls in respective cohorts), suggesting that the variant allele may contribute to hypertension in the patients. Previously, a corin minor allele, T555I/Q568P, is linked to hypertension and heart disease17,18,19. The T555I/Q568P variant allele appears unique in African Americans, as it is mostly absent in other ethnic groups17. Here we have identified a novel corin variant allele that is associated with hypertension in different ethnic populations.
The insertion of a single adenine at nucleotide position 102 shifts the reading frame, causing a down-stream stop codon. In principle, the insertion may result in a null allele. In functional studies, however, we found that the insA variant was expressed at a similar level to that of WT in HEK293 cells, suggesting that the variant may use Met-30 as an alternative initiation site to produce a corin protein with a shorter cytoplasmic tail. On Western blots, the variant and WT corin migrated similarly, indicating that Western blots are not sensitive enough to detect such a small size difference in corin cytoplasmic tails. This hypothesis was supported by a similar migration pattern in the Met-30 isoform, which contains Met-30 but lacks Met-1 (Fig. 3A).
Previously, we found that different cytoplasmic tail sequences may alter corin expression and activity on the cell surface15. In our functional studies, the insA variant exhibited low pro-ANP processing activity (Fig. 2), which was likely due to reduced cell surface expression (Fig. 4) and impaired zymogen activation (Fig. 3). By immunostaining, we found that the variant was partially retained in the Golgi, reducing its cell surface targeting (Fig. 5). Since corin zymogen activation occurs on the cell surface but not intracellularly14, these results may explain the reduced zymogen activation and activity of the variant. Earlier, a protein motif, DDNN, which precedes Met-30 in human corin cytoplasmic tail (Fig. 1B), was found to be important for corin cell surface targeting15. Consistent with these data, the insA variant lacking the DDNN motif exhibited reduced cell surface expression and zymogen activation in our experiments.
As a mechanism to regulate corin activity on the cell surface, corin is shed by autocleavage and ADAM-mediated proteolysis26. Similar shedding has been reported in other membrane-bound proteases27,28,29,30. In this study, we found reduced corin shedding fragments in HEK293 cells expressing the insA variant, consistent with the reduced cell surface expression and zymogen activation of the variant. In agreement with this finding, plasma corin levels were significantly lower in individuals with the variant allele than with WT allele, suggesting that the variant may have poor cell surface expression, impaired zymogen activation, and reduced shedding in vivo.
Low levels of plasma corin antigen and activity have been reported in patients with hypertension and heart disease31,32,33. Low levels of serum corin also were found to be a predictor for poor outcomes in patients with coronary heart disease34. In this study, low plasma corin levels were found in hypertensive patients with the variant allele. Consistently, plasma levels of NT-pro-ANP were much higher in these individuals. These data indicate that the activity of the insA variant is lower in vivo, which may impair natriuretic peptide processing in the individuals with the variant alleles.
The ANP signaling pathway is important for salt-water balance and normal blood pressure. Genetic variants and mutations in the genes coding for ANP or its receptor have been found in patients with hypertension and heart disease35,36,37,38,39,40,41. Corin acts as the pro-ANP convertase at the top of the ANP pathway7,24,42,43. Here we report a novel CORIN variant allele that is present preferentially in hypertensive patients. In functional studies, the variant exhibited poor intracellular trafficking, reduced cell surface expression, and impaired zymogen activation, which may explain low plasma corin and higher plasma NT-pro-ANP levels in the patients with the variant allele. Together, these results indicate that corin defects that impair the ANP pathway may represent an important mechanism underlying hypertension.
Methods
Blood Samples
The ethics committee of the First Affiliated Hospital of Soochow University approved this study. Written informed consent was obtained from all study participants. All experiments were performed in accordance with the approved guidelines and regulations. Blood samples were collected from normal individuals and hypertensive patients. Normal individuals had systolic blood pressure <140 mmHg and diastolic blood pressure <90 mmHg with no history of hypertension, heart disease and other diseases. Patients with hypertension had either systolic blood pressure >140 mmHg and/or diastolic blood pressure >90 mmHg, or having a history of hypertension and taking anti-hypertensive drugs. The first study cohort consisted of 217 normal individuals and 401 patients with hypertension. The second cohort consisted of 415 normal individuals and 394 hypertensive patients. The basic characteristics of the normal controls and hypertensive patients of these two cohorts are shown in Supplementary Tables S3 and S4, respectively.
Plasma Soluble Corin and N-terminal pro-ANP
Plasma corin levels were measured by ELISA (R&D Systems)44,45. Briefly, plasma samples were added to 96-well plates coated with an anti-corin antibody. After 2 h at room temperature, the plates were washed with phosphate-buffered saline. A biotin-labeled anti-corin secondary antibody was added for 2 h. After washing, horseradish peroxidase-conjugated streptavidin was added. After incubation and washing, a horseradish peroxidase substrate (3,3,5,5-tetramethylbenzidine) was added. The reaction was stopped by adding H2SO4 and the optical density in wells was recorded by a spectrometer at 450 nm. A ELISA kit (Biomedica Group) was used to measure plasma N-terminal pro-ANP (NT-pro-ANP), i.e. pro-ANP 1-9846. Experimental procedures were similar to those of the corin ELISA, as indicated by the manufacturer's instructions.
DNA Sequencing
Genomic DNA was isolated from white blood cells using a Qiagen kit. The samples were used in PCR to amplify CORIN gene exons and intron-exons boundaries16. Amplified PCR products were sequenced. Nucleotide polymorphisms found were verified by independent PCR followed by subcloning the PCR fragments and DNA sequencing.
Plasmid Constructs
Plasmid pcDNA3.1 (Invitrogen) was used to express human corin wild-type (WT) and mutants14. Plasmids expressing corin activation cleavage site mutant R801A and active site mutant S985A were described previously14,15. The corin proteins encoded by the plasmids had a C-terminal V5 tag for detection by Western blotting using an anti-V5 antibody (Invitrogen). Plasmid expressing corin insA variant was made using WT corin plasmid as a template. By site-directed mutagenesis, a single base of adenine was inserted at nucleotide position 10225. The resulting plasmid, pcDNAinsA, and all other plasmids were verified by DNA sequencing.
Cell Transfection and Western Blotting
Human embryonic kidney 293 (HEK293) cells were cultured in Dulbecco's Modified Eagle's Medium with 10% fetal bovine serum at 37°C. Plasmids were transfected into HEK293 cells using TurboFect reagents (Thermo Scientific). Conditioned medium was collected after 48 h and the transfected cells were lysed with a lysis buffer14. Procedures for immunoprecipitation and Western analysis of corin proteins in the conditioned medium and cell lysate were described previously14.
Pro-ANP Processing Assay
Corin pro-ANP processing activity was examined by a cell-based assay14. Briefly, corin WT and mutants were expressed in HEK293 cells. Conditioned medium containing human pro-ANP from stable cells was added to the cells at 37°C. After 1 h, the conditioned medium was collected. Pro-ANP processing was analyzed by immunoprecipitation and Western blotting14,15. In these experiments, WT corin was used as a positive control and inactive mutants R801A and S985A were used as negative controls.
Corin Expression on Cell Surface
To examine corin cell surface expression, HEK293 cells expressing corin proteins were labeled with membrane impermeable Sulfo-NHS-SS-biotin (Thermo Scientific). The surface-labeled cells were lysed with a lysis buffer14. Biotin-labeled proteins were precipitated with avidin-agarose beads and analyzed by Western blotting. The optical density of corin bands on the blots was measured by densitometry, and the percentage of the activated corin fragments and corin fragments on the surface or conditioned medium was calculated, as described previously15.
Flow Cytometry
HEK293 cells expressing corin on the cell surface were analyzed by flow cytometry using an anti-V5 antibody, followed by an FITC-conjugated secondary antibody, as described previously15. Life-cell gating was performed using pyridinium iodide (Sigma). Data were acquired on Calibur flow cytometer (BD Biosciences) and analyzed by FlowJo v7.6.5 software (Tree Star).
Immunostaining
Corin-expressing HEK293 cells on coverslips were fixed with 4% paraformaldehyde or cold acetone and co-stained with a mouse anti-V5 antibody and a rabbit anti-TGN46 antibody (Sigma), or a rabbit anti-corin antibody and a mouse anti-protein disulfide isomerase (PDI) antibody (BD Biosciences). Secondary antibodies were conjugated with Alexa Fluor 488 (green) or 594 (red) (Invitrogen). DAPI (4′, 6-diamidino-2-phenylindole dihydrochloride) was used to stain cell nuclei. Immunostaining images were examined under a confocal microscope (Olympus).
Statistical Analysis
Data were analyzed using the Prism 5 (GraphPad) and SPSS18.0 software, and presented as means ± S.D. Comparisons between two groups were analyzed by Student's t-test. Multi-group comparisons were analyzed by ANOVA. Comparisons between two populations were analyzed by chi-squared test. Multivariate logistic regression analysis was done using SPSS 18.0. software. A p value of <0.05 was considered to be statistically significant.
Author Contributions
Y.Z., H.L., N.D. and Q.W. conceived and designed the experiments. Y.Z., H.L., C.W., M.L. and T.Z. performed the experiments. Y.Z., H.L., N.D. and Q.W. analyzed the data. J.Z., J.Y. and Z.L. contributed reagents/materials/analysis tools. A.W. and Y.-H.Z. provided DNA samples of the second cohort study. Y.Z., H.L., N.D. and Q.W. wrote the paper. All authors reviewed the manuscript. Y.Z. and H.L. contributed equally to this work.
Supplementary Material
Supplementary info
Acknowledgments
We thank Dr. Yueping Shen (Soochow University) and Dr. Jiang He (Tulane University) for helpful discussions and suggestions. This work was supported in part by grants from the National Natural Science Foundation (81301844, 81370718 and 81170247), the Key Project of Chinese Ministry of Education (213016A), the Priority Academic Program Development of Jiangsu Higher Education Institutions, and the Danish-Chinese Center for Proteases and Cancer (31161130356). This work was also supported in part by the NIH grant R01 HL089298.
References
- Cowley A. W. Jr The genetic dissection of essential hypertension. Nat Rev Genet 7, 829–840 (2006). [DOI] [PubMed] [Google Scholar]
- Lifton R. P., Gharavi A. G. & Geller D. S. Molecular mechanisms of human hypertension. Cell 104, 545–556 (2001). [DOI] [PubMed] [Google Scholar]
- Oparil S., Zaman M. A. & Calhoun D. A. Pathogenesis of hypertension. Ann Intern Med 139, 761–776 (2003). [DOI] [PubMed] [Google Scholar]
- Sanada H., Jones J. E. & Jose P. A. Genetics of salt-sensitive hypertension. Curr Hypertens Rep 13, 55–66 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishimoto I., Tokudome T., Nakao K. & Kangawa K. Natriuretic peptide system: an overview of studies using genetically engineered animal models. Febs J 278, 1830–1841 (2011). [DOI] [PubMed] [Google Scholar]
- Levin E. R., Gardner D. G. & Samson W. K. Natriuretic peptides. N Engl J Med 339, 321–328 (1998). [DOI] [PubMed] [Google Scholar]
- Wu Q., Xu-Cai Y. O., Chen S. & Wang W. Corin: new insights into the natriuretic peptide system. Kidney Int 75, 142–146 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan J. C. et al. Hypertension in mice lacking the proatrial natriuretic peptide convertase corin. Proc Natl Acad Sci U S A 102, 785–790 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W. et al. Salt-sensitive hypertension and cardiac hypertrophy in transgenic mice expressing a corin variant identified in blacks. Hypertension 60, 1352–1358 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W. et al. Impaired sodium excretion and salt-sensitive hypertension in corin-deficient mice. Kidney Int 82, 26–33 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antalis T. M., Bugge T. H. & Wu Q. Membrane-anchored serine proteases in health and disease. Prog Mol Biol Transl Sci 99, 1–50 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo R. & Bugge T. H. Membrane-anchored serine proteases in vertebrate cell and developmental biology. Annu Rev Cell Dev Biol 27, 213–235 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gladysheva I. P., Robinson B. R., Houng A. K., Kovats T. & King S. M. Corin is co-expressed with pro-ANP and localized on the cardiomyocyte surface in both zymogen and catalytically active forms. J Mol Cell Cardiol 44, 131–142 (2008). [DOI] [PubMed] [Google Scholar]
- Liao X., Wang W., Chen S. & Wu Q. Role of glycosylation in corin zymogen activation. J Biol Chem 282, 27728–27735 (2007). [DOI] [PubMed] [Google Scholar]
- Qi X., Jiang J., Zhu M. & Wu Q. Human corin isoforms with different cytoplasmic tails that alter cell surface targeting. J Biol Chem 286, 20963–20969 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan J. et al. Genomic structures of the human and murine corin genes and functional GATA elements in their promoters. J Biol Chem 277, 38390–38398 (2002). [DOI] [PubMed] [Google Scholar]
- Dries D. L. et al. Corin gene minor allele defined by 2 missense mutations is common in blacks and associated with high blood pressure and hypertension. Circulation 112, 2403–2410 (2005). [DOI] [PubMed] [Google Scholar]
- Rame J. E. et al. Corin I555(P568) allele is associated with enhanced cardiac hypertrophic response to increased systemic afterload. Hypertension 49, 857–864 (2007). [DOI] [PubMed] [Google Scholar]
- Rame J. E. et al. Dysfunctional corin I555(P568) allele is associated with impaired brain natriuretic peptide processing and adverse outcomes in blacks with systolic heart failure: results from the Genetic Risk Assessment in Heart Failure substudy. Circ Heart Fail 2, 541–548 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W. et al. Corin variant associated with hypertension and cardiac hypertrophy exhibits impaired zymogen activation and natriuretic peptide processing activity. Circ Res 103, 502–508 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Y. et al. Role of corin in trophoblast invasion and uterine spiral artery remodelling in pregnancy. Nature 484, 246–250 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong N. et al. Corin mutation R539C from hypertensive patients impairs zymogen activation and generates an inactive alternative ectodomain fragment. J Biol Chem 288, 7867–7874 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong N. et al. Corin mutations K317E and S472G from preeclamptic patients alter zymogen activation and cell surface targeting. J Biol Chem 289, 17909–17916 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y. & Wu Q. Corin in natriuretic peptide processing and hypertension. Curr Hypertens Rep 16, 415 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan W., Sheng N., Seto M., Morser J. & Wu Q. Corin, a mosaic transmembrane serine protease encoded by a novel cDNA from human heart. J Biol Chem 274, 14926–14935 (1999). [DOI] [PubMed] [Google Scholar]
- Jiang J. et al. Ectodomain shedding and autocleavage of the cardiac membrane protease corin. J Biol Chem 286, 10066–10072 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadorn B., Steiner N., Sumida C. & Peters T. J. Intestinal enterokinase. Mechanisms of tts "secretion" into the lumen of the small intestine. Lancet 1, 165–166. (1971). [DOI] [PubMed] [Google Scholar]
- Larsen B. R. et al. Hepatocyte growth factor activator inhibitor-2 prevents shedding of matriptase. Exp Cell Res 319, 918–929 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C. Y. et al. Zymogen activation, inhibition, and ectodomain shedding of matriptase. Front Biosci 13, 621–635 (2008). [DOI] [PubMed] [Google Scholar]
- Wu Q. Type II transmembrane serine proteases. Curr Top Dev Biol 54, 167–206 (2003). [DOI] [PubMed] [Google Scholar]
- Dong N., Chen S., Wang W., Zhou Y. & Wu Q. Corin in clinical laboratory diagnostics. Clin Chim Acta 413, 378–383 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong N. et al. Plasma soluble corin in patients with heart failure. Circ Heart Fail 3, 207–211 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibebuogu U. N., Gladysheva I. P., Houng A. K. & Reed G. L. Decompensated heart failure is associated with reduced corin levels and decreased cleavage of pro-atrial natriuretic peptide. Circ Heart Fail 4, 114–120 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peleg A., Ghanim D., Vered S. & Hasin Y. Serum corin is reduced and predicts adverse outcome in non-ST-elevation acute coronary syndrome. Eur Heart J Acute Cardiovasc Care 2, 159–165 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbato E. et al. Influence of rs5065 atrial natriuretic peptide gene variant on coronary artery disease. J Am Coll Cardiol 59, 1763–1770 (2012). [DOI] [PubMed] [Google Scholar]
- Cannone V. et al. Atrial natriuretic peptide genetic variant rs5065 and risk for cardiovascular disease in the general community: a 9-year follow-up study. Hypertension 62, 860–865 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgson-Zingman D. M. et al. Atrial natriuretic peptide frameshift mutation in familial atrial fibrillation. N Engl J Med 359, 158–165 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch A. I. et al. Pharmacogenetic association of the NPPA T2238C genetic variant with cardiovascular disease outcomes in patients with hypertension. Jama 299, 296–307 (2008). [DOI] [PubMed] [Google Scholar]
- Lynch A. I., Claas S. A. & Arnett D. K. A review of the role of atrial natriuretic peptide gene polymorphisms in hypertension and its sequelae. Curr Hypertens Rep 11, 35–42 (2009). [DOI] [PubMed] [Google Scholar]
- Nakayama T. et al. Functional deletion mutation of the 5′-flanking region of type A human natriuretic peptide receptor gene and its association with essential hypertension and left ventricular hypertrophy in the Japanese. Circ Res 86, 841–845 (2000). [DOI] [PubMed] [Google Scholar]
- Sciarretta S. et al. C2238 atrial natriuretic peptide molecular variant is associated with endothelial damage and dysfunction through natriuretic peptide receptor C signaling. Circ Res 112, 1355–1364 (2013). [DOI] [PubMed] [Google Scholar]
- Armaly Z., Assady S. & Abassi Z. Corin: a new player in the regulation of salt-water balance and blood pressure. Curr Opin Nephrol Hypertens 22, 713–722 (2013). [DOI] [PubMed] [Google Scholar]
- Ichiki T., Huntley B. K. & Burnett J. C. Jr BNP molecular forms and processing by the cardiac serine protease corin. Adv Clin Chem 61, 1–31 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong N. et al. Effects of anticoagulants on human plasma soluble corin levels measured by ELISA. Clin Chim Acta 411, 1998–2003 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peleg A., Jaffe A. S. & Hasin Y. Enzyme-linked immunoabsorbent assay for detection of human serine protease corin in blood. Clin Chim Acta 409, 85–89 (2009). [DOI] [PubMed] [Google Scholar]
- Hoffmann U. et al. Increased plasma levels of NT-proANP and NT-proBNP as markers of cardiac dysfunction in septic patients. Clin Lab 51, 373–379 (2005). [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary info