

Abstract
Coagulation is a dynamic process and the understanding of the blood coagulation system has evolved over the recent years in anaesthetic practice. Although the traditional classification of the coagulation system into extrinsic and intrinsic pathway is still valid, the newer insights into coagulation provide more authentic description of the same. Normal coagulation pathway represents a balance between the pro coagulant pathway that is responsible for clot formation and the mechanisms that inhibit the same beyond the injury site. Imbalance of the coagulation system may occur in the perioperative period or during critical illness, which may be secondary to numerous factors leading to a tendency of either thrombosis or bleeding. A systematic search of literature on PubMed with MeSH terms ‘coagulation system, haemostasis and anaesthesia revealed twenty eight related clinical trials and review articles in last 10 years. Since the balance of the coagulation system may tilt towards bleeding and thrombosis in many situations, it is mandatory for the clinicians to understand physiologic basis of haemostasis in order to diagnose and manage the abnormalities of the coagulation process and to interpret the diagnostic tests done for the same.
Keywords: Anaesthesia, Coagulation system, haemostasis
INTRODUCTION
The concept of blood coagulation dates back to 1960's when Davie, Ratnoff and Macfarlane described the “waterfall” and “cascade” theories outlining the fundamental principle of cascade of proenzymes leading to activation of downstream enzymes.[1] Haemostasis, defined as arrest of bleeding, comes from Greek, haeme meaning blood and stasis meaning to stop.[2] This thrombohaemmorhagic balance is maintained in the body by complicated interactions between coagulation and the fibrinolytic system as well as platelets and vessel wall.
Usually, the coagulation process is under the inhibitory control of several inhibitors that limit the clot formation, thus avoiding the thrombus propagation. This delicate balance is interrupted whenever the procoagulant activity of the coagulation factors is increased, or the activity of naturally occurring inhibitors is decreased.[3] Some of the thrombogenic and antithrombogenic components are listed in Table 1.
Table 1.
Thrombogenic and antithrombogenic components in the body

It is important for a perioperative physician to understand the intricacies of two systems (more so in a preexisting haematological disorder) that go side by side in maintaining the circulating blood in a fluidic state.
Pathological situations requiring surgery or anaesthesia or any other invasive procedure trigger the haemostatic system. This balance is also disturbed by trauma, cytokines or infectious agents. Thus, the perioperative period is at high risk for both prohaemorrhagic and prothrombotic abnormalities. Hypoxia, hypothermia, metabolic acidosis and extracorporeal circulation may also further aggravate the situation.[4]
Coagulopathy, may also be encountered by the intensivist due to physiological disturbances, disturbances in the primary haemostasis, abnormalities of blood, plasma or due to disseminated intravascular coagulation (DIC).[5]
This review aims to simplify the understanding of the coagulation system as a whole as well as discuss various abnormalities of the same, which may have an impact in the perioperative period and ICU. They can be classified as those that affect the primary haemostasis, the coagulation pathways and the fibrinolytic system.
PRIMARY HAEMOSTASIS
Primary haemostasis results from complex interactions between platelets, vessel wall and adhesive proteins leading to the formation of initial ‘platelet plug’. The endothelial cells lining the vascular wall exhibit the antithrombotic properties due to multiple factors viz: negatively charged heparin-like glycosaaminoglycans, neutral phospholipids, synthesis and secretion of platelet inhibitors, coagulation inhibitors and fibrinolysis activators. In contrast, subendothelial layer is highly thrombogenic and contains collagen, Von Willebrand factor (vWF) and other proteins like laminin, thrombospondin and vitronectin that are involved in platelet adhesion. Any vascular insult results in arteriolar vasospasm, mediated by reflex neurogenic mechanisms and release of local mediators like endothelin and platelet-derived thromboxane A2 (TxA2).[6,7,8]
Platelets are disc shaped, anucleate cellular fragments derived from megakaryocytes. They have a pivotal role in haemostasis by forming the initial haemostatic plug that provides a surface for the assembly of activated coagulation factors leading to the formation of fibrin stabilized platelet aggregates and subsequent clot retraction. Platelets have two types of granules:
α granules-contain P-selectin, fibrinogen, fibronectin, factor V, factor VIII, platelet factor IV, platelet-derived growth factor and tumour growth factor-α (TGF-α)[9]
δ granules or Dense granules-contain adenosine triphosphate (ATP), adenosine diphosphate (ADP), calcium (Ca), serotonin, histamine and epinephrine.[9]
Normally platelets do not adhere to intact vascular endothelium. Subsequent to the vascular injury, platelets adhere to collagen and vWF in the subendothelial tissue and undergo a morphological change by assuming irregular surface, forming numerous pseudopods thus drastically increasing their surface area.[10] The formation of the platelet plug involves a series of steps:
Platelet adhesion
After vascular injury vWf acts as a bridge between endothelial collagen and platelet surface receptors GpIb and promotes platelet adhesion.[9] The platelet glycoprotein complex I (GP-Ib) is the principal receptor for vWF.
Platelet secretion
After adhesion, degranulation from both types of granules takes place with the release of various factors. Release of calcium occurs here. Calcium binds to the phospholipids that appear secondary to the platelet activation and provides a surface for assembly of various coagulation factors.
Platelet aggregation
Thromboxane A2 produced by activated platelets provide stimulus for further platelet aggregation. TxA2 along with ADP enlarge this platelet aggregate leading to the formation of the platelet plug, which seals off vascular injury temporarily. ADP binding also causes a conformational change in GpIIb/IIIa receptors presents on the platelet surface causing deposition of fibrinogen. Thrombin generation also catalyses the conversion of this fibrinogen to fibrin which adds to the stability of the platelet plug and is now known as secondary haemostasis.[9]
Prostacyclin inhibits platelet aggregation (platelet anti aggregating effect) and the balance between TxA2 and prostacyclin leads to localized platelet aggregation thus preventing extension of the clot thereby maintaining the vessel lumen patency.[6,11]
COAGULATION DISORDERS INVOLVING PRIMARY HEMOSTASIS
Defects of primary haemostasis may be due to abnormalities of the vessel wall or qualitative/quantitative defects of platelets that may cause bleeding in varying severity.
Thrombocytopenia may be observed secondary to numerous causes listed in Table 2. The inherited platelet disorders are uncommon and if present, are, usually, diagnosed in childhood and these patients need to be given platelet concentrates before and after surgery.
Table 2.
Disorders of primary haemostasis

High platelet counts may occur in the perioperative period after major bleeding, splenectomy, major reconstruction surgery or may just represent the inflammatory response.
Certain inherited bleeding disorders with platelet glycoprotein deficiency as seen in Glanzmann thromboasthenia (deficiency of IIb/IIIa) or Bernard-Soulier syndrome (deficiency of platelet glycoprotein Ib) may be seen secondary to viral infections and are associated with immediate failure of haemostasis.[12,13]
Massive blood transfusion entails the replacement of one whole blood volume within a period of 24 h with stored blood that is deficient in both functional platelets and coagulation factors. Transfused red cells dilute patient's native coagulation reserve also. All these effects are exacerbated by infusion of fluids especially colloids. Hence, massive blood transfusion may lead to dilutional coagulopathy.
COAGULATION PATHWAYS
The coagulation proteins are the core components of the coagulation system that lead to a complex interplay of reactions resulting in the conversion of soluble fibrinogen to insoluble fibrin strands.
CLOTTING FACTORS (COAGULATION PROTEINS)
Majority of clotting factors are precursors of proteolytic enzymes known as zymogens that circulate in an inactive form. The activation of each zymogen is depicted by suffixing letter “a” to the Roman numeral identifying that particular zymogen. Most of the procoagulants and anticoagulants are produced by liver except factor III, IV and VIII. These proteins undergo a post translational modification (vitamin K dependent ϒ carboxylation of glutamic acid residues) which enables them to bind calcium and other divalent cations and participate in clotting cascade.[14] Deficiency of vitamin K or administration of vitamin K antagonists (warfarin) lead to anticoagulation.
Nomenclature of coagulation proteins is rather complex [Table 3]. The first 4 of the 12 originally identified factors are referred to by their common names, i.e., fibrinogen, prothrombin, tissue factor (TF), and calcium and are not assigned any Roman numerals. FVI no longer exists. The more recently discovered clotting factors (e.g. prekallikrein and high-molecular-weight kininogen) have not been assigned Roman numerals. Some factors have more than one name. Factors V and VIII are also referred to as the labile factors because their coagulant activity is not durable in stored blood.
Table 3.
Nomenclature of the coagulation proteins/clotting factors
Prothrombin is a plasma protein formed by liver (MW 68700). It is an unstable protein, splitting into smaller proteins one of which is thrombin (MW33700). Thrombin generated from prothrombin also has pro-inflammatory effects which are exerted by the activation of protease activating receptors present on monocytes, lymphocytes, endothelium and dendritic cells.[15]
Von Willebrand factor is a glycoprotein present in blood plasma and produced constitutively as ultra-large vWf in endothelium, megakaryocytes, and subendothelial connective tissue. It mediates platelet adhesion to subendothelial surface. It also acts as a carrier protein for coagulant activity of Factor VIII and is referred there as VIII: C.[16,17]
Fibrinogen is an essential coagulation protein produced by liver (MW340 kDa) and is the precursor of fibrin that ultimately defines the strength of clot.[18,19]
Factor III or TF is a membrane bound procoagulant glycoprotein (MW47-kDa) present in the subendothelial tissue and fibroblasts and is not exposed to blood until disruption of the vessel wall.[14] It is the primary initiator of coagulation in vivo. TF is localised predominantly to the tunica media and tunica adventitia of blood vessels and a smaller quantity as circulating TF on monocytes. Tissue factor may be activated by physical injury (activation of Vessel wall TF), by direct vascular injury or functional injury (activation of circulating TF), by hypoxia, sepsis, malignancy, inflammation, etc.[20,21]
Clotting factors can also be classified into three groups [Table 4].
Table 4.
Classification of coagulation factors

Fibrinogen Family
Vitamin K dependent proteins
Contact family.
Hypoxia up regulates the expression of P selectin present in the α granules of platelets on the endothelium leading to recruitment of monocytes containing TF, thus initiating coagulation.[22,23] With the exposure of TF to factor VII/VIIIa in the blood, it allows for the formation of TF-VIIIa complex and thus initiate the coagulation cascade.
NATURALLY OCCURRING ANTICOAGULANTS IN THE BODY
The anticoagulant system exerts a regulatory role over the procoagulant activity in blood thus localizing the thrombus formation.[13] The main anticoagulant mechanisms naturally present in the body include the following:
Antithrombin
Antithrombin (AT), previously known as AT III is the main inhibitor of thrombin. It is a serine protease inhibitor, which binds and inactivates thrombin, factor IXa, Xa, XIa and XIIa. The enzymatic activity of AT is enhanced in the presence of heparin. However, the plasma concentration of heparin is low and does not contribute significantly to the in vivo activation of AT. AT is activated by binding of heparin sulphate present on endothelial cell surface. AT binds coagulation factors in a ratio of 1:1 and this complex is removed by reticuloendothelial cells. Other thrombin inhibitors are heparin cofactor II, α2 macroglobulin and α1-antitrypsin.[24,25]
Tissue factor plasminogen inhibitor
It is a polypeptide produced by endothelial cells. It acts as a natural inhibitor of the extrinsic pathway by inhibiting TF-VIIa complex.[25,26] Protein S enhances the interaction of factor Xa in the presence of calcium and phospholipids.[27]
Protein C pathway
The propagation phase of the coagulation is inhibited by the Protein C pathway that primarily consist of four key elements:
Protein C is a serine protease with potent anticoagulant, profibrinolytic and anti-inflammatory properties. It is activated by thrombin to form activated protein C (APC) and acts by inhibiting activated factors V and VIII (with Protein S and phospholipids acting as cofactors)
Thrombomodulin - A transmembrane receptor on the endothelial cells, it prevents the formation of the clot in the undamaged endothelium by binding to the thrombin
Endothelial protein C receptor is another transmembrane receptor that helps in the activation of Protein C
Protein S is a vitamin K-dependent glycoprotein, synthesised by endothelial cells and hepatocytes. It exists in plasma as both free (40%) and bound (60%) forms (bound to C4b-binding protein). The anticoagulant activity is by virtue of free form while the bound form acts as an inhibitor of the complement system and is up regulated in the inflammatory states, which reduce the Protein S levels thus resulting in procoagulant state. It functions as a cofactor to APC in the inactivation of FVa and FVIIIa. It also causes direct reversible inhibition of the prothrombinase (FVa–FXa) complex.[28]
Protein Z dependent protease inhibitor/protein Z (PZI)
It is a recently described component of the anticoagulant system that is produced in the liver. It inhibits Factor Xa in reaction requiring PZ and calcium.[29]
COAGULATION CASCADE
It has been traditionally classified into intrinsic and extrinsic pathways, both of which converge on factor X activation. The classical theory of blood coagulation is particularly useful for understanding the In vitro coagulation tests, but fails to incorporate the central role of cell-based surfaces in In vivo coagulation process.[4] Interestingly contact activation critical for In vivo haemostasis does not get support from following observations. Persons lacking FXII, prekallikrein, or high-molecular-weight kininogen do not bleed abnormally. Second, patients with only trace quantities of FXI can withstand major trauma without unusual bleeding, and those who completely lack factor XI (haemophilia C) exhibit mild haemorrhagic disorder. Deficiencies of FVIII and FIX (both intrinsic pathway factors) lead to haemophilia A and B, respectively, however the classic description of two pathways of coagulation leave it unclear as to why either type of haemophiliac cannot not simply clot blood via the unaffected pathway.
To answer all this, the modern time-based structuring of blood coagulation provides more authentic description of the coagulation process. It is now appreciated that the classic theories may provide only a reasonable model of in vitro coagulation tests (i.e., aPTT and PT).
Extrinsic pathway
It is considered as the first step in plasma mediated haemostasis. It is activated by TF, which is expressed in the subendothelial tissue.[7] Under normal physiological conditions, normal vascular endothelium minimises contact between TF and plasma procoagulants, but vascular insult expose TF which binds with factor VIIa and calcium to promote the conversion of factor X to Xa.[30]
Intrinsic pathway
It is a parallel pathway for thrombin activation by factor XII. It begins with factor XII, HMW kininogen, prekallekerin and factor XI, which results in activation of factor XI. Activated factor XI further activates factor IX, which then acts with its cofactor (factor VIII) to form tenase complex on a phospholipid surface to activate factor X.[15,31]
Common pathway
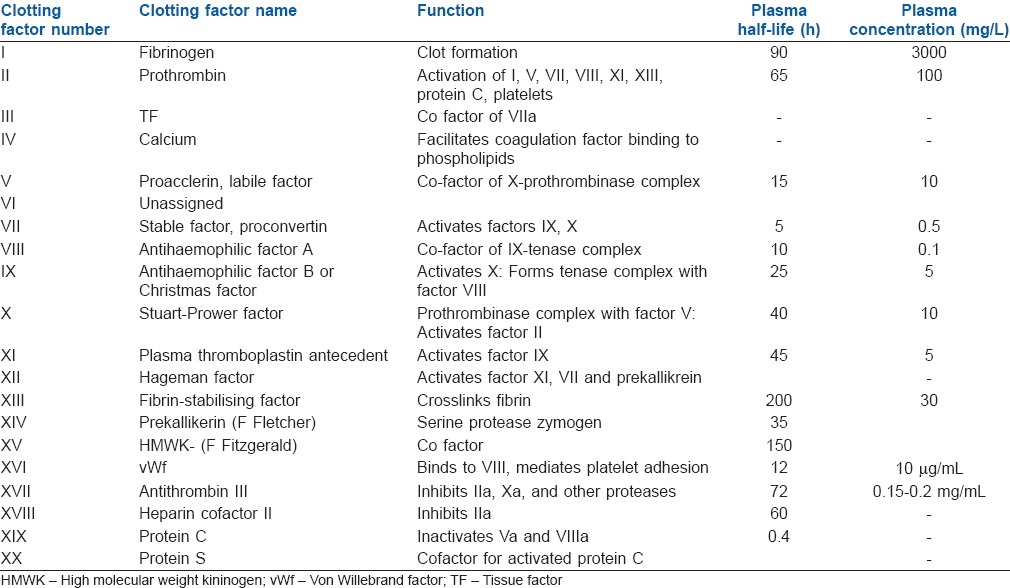
Activated factor X along with its cofactor (factor V), tissue phospholipids, platelet phospholipids and calcium forms the prothrombinase complex which converts prothrombin to thrombin. This thrombin further cleaves circulating fibrinogen to insoluble fibrin and activates factor XIII, which covalently crosslinks fibrin polymers incorporated in the platelet plug. This creates a fibrin network which stabilises the clot and forms a definitive secondary haemostatic plug[15,31] [Figure 1].
Figure 1.
Earlier concept of coagulation
CURRENT CONCEPT OF COAGULATION
Current evidence supports the understanding that intrinsic pathway is not a parallel pathway but indeed it augments thrombin generation primarily initiated by the extrinsic pathway[8] Newer model describes coagulation with following steps:
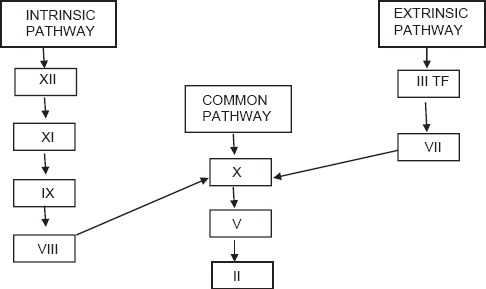
Initiation
It occurs by expression of TF in damaged vessel which binds factor VIIa to activate factor IX and factor X. This activation of factor IX by TF-VIIa complex serves as the bridge between classical extrinsic and intrinsic pathways. Factor Xa then binds to factor II to form thrombin (factor IIa). Thrombin generation through this reaction is not robust and can be effectively terminated by TF pathway inhibitor [Figure 2].
Figure 2.
Current concept of coagulation (initiation phase)
Amplification
Since the amount of thrombin generated is not sufficient, therefore numerous positive feedback loops are present that bind thrombin with platelets. Thrombin that is generated in the initiation phase further activates factor V and factor VIII, which serves as a cofactor in prothrombinase complex and accelerates the activation of Factor II by F Xa and of F Xa by F IXa, respectively.
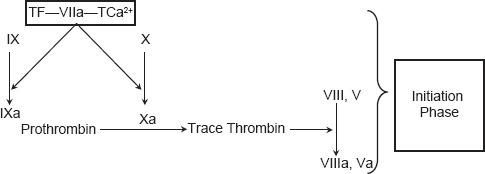
Propagation
The accumulated enzyme complexes (tenase complex and prothrombinase complex) on platelet surface support robust amounts of thrombin generation and platelet activation. This ensures continuous generation of thrombin and subsequently fibrin to form a sufficiently large clot [Figure 3].
Figure 3.
Current concepts of coagulation (propagation phase)
Stabilization
Thrombin generation leads to activation of factor XIII (fibrin stabilizing factor) which covalently links fibrin polymers and provides strength and stability to fibrin incorporated in platelet plug. In addition, thrombin activates thrombin activatable fibrinolysis inhibitor (TAFI) that protects the clot from fibrinolysis.[4,7]
DISORDERS OF COAGULATION
A balance between clotting and bleeding is always maintained in the body under normal physiology. However any pathological scenario will tilt this balance to either haemorrhagic or thrombotic complications. Hence as a corollary disorders of haemostasis can be categorised into those that lead to abnormal bleeding and those that lead to abnormal clotting [Table 5].
Table 5.
Classification of disorders of coagulation
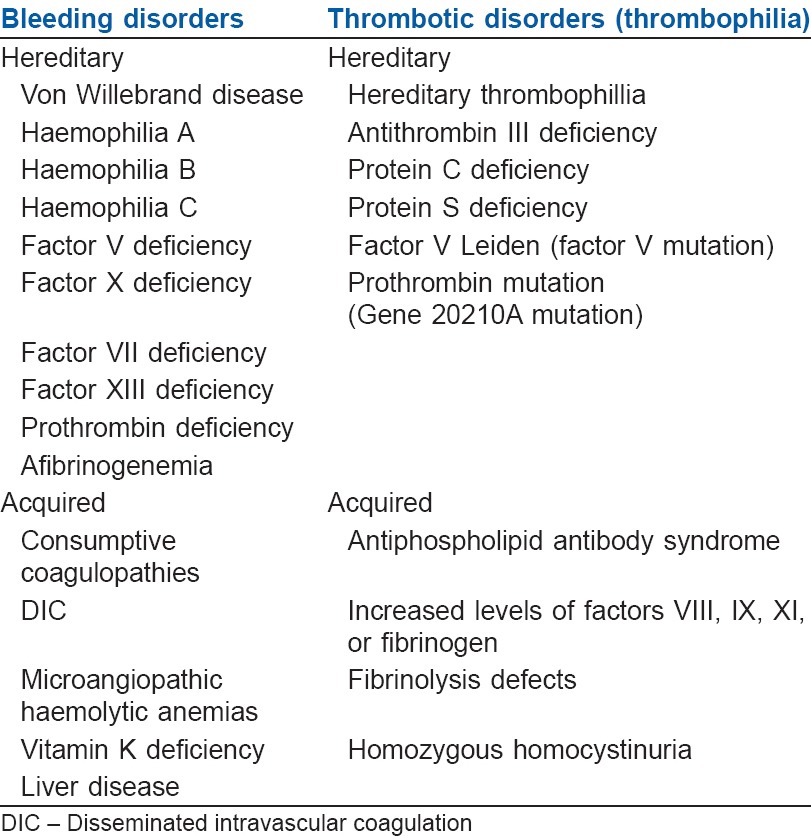
BLEEDING DISORDERS
Haemophilias
Haemophilia A, the most common form of haemophilia is associated with the deficiency of factor VIII and B (Christmas disease) is secondary to deficiency of factor IX. C is only found in 1% of the population and is due to deficiency of factor XI. Haemophilia's are X linked recessive disorders and occur in males. The severity of the bleeding tendency is directly related to the levels of the coagulation factors. The levels of factor VIII need to be assessed in the preoperative period and human or recombinant factor VIII concentrates are transfused to keep the factor VIII levels 100% in the perioperative period.[32]
Von Willebrand disease is the most common autosomal dominant inherited disorder of coagulation due to abnormality in the production of vWF, which may be qualitative or quantitative. vWF acts as a carrier molecule for factor VIII coagulant protein and the deficiency of former has a profound effect on the stability of latter. The clinical presentation may be variable and complete deficiency, though rare leads to severe bleeding. The diagnosis is usually made by prolonged bleeding time in the presence of adequate platelets. Antifibrinolytic agent and tranexamic acid may be used for minor bleeding. Desmopressin D arginine vasopressin administered in the preoperative period is expected to raise the concentration of factor VIII in patients with quantitative disorders.[32]
Fibrinogen deficiency
Total or partial is an extremely rare inherited bleeding disorder. Afibrinogenemia is rather well tolerated and may manifest as subcutaneous haematoma or umbilical haematoma at birth. The clinical findings are variable in childhood and adults.[33] DIC is the most common consumptive coagulopathy that maybe observed secondary to numerous causes. It often presents as diffuse bleeding associated with consumption of coagulation factors and thrombocytopenia secondary to widespread small vessel thrombosis. Common causes include sepsis/infections, obstetric causes, incompatible blood transfusion, shock, trauma and embolism.
Liver disease
Majority of clotting factors are synthesized in liver therefore severe liver disease is associated with coagulopathy. Since liver is also involved in the clearance of activated clotting factors and fibrinolytic products, it may predispose to DIC. Management of bleeding secondary to liver disease is based on the laboratory values of various coagulation tests.
Hypothermia is also associated with anticoagulatory effects, which are more pronounced in the presence of acidosis. The effects may result from platelet dysfunction in mild hypothermia (below 35°C) to decreased synthesis of clotting enzymes and plasminogen activator inhibitors when temperatures is <33°C.[34]
The availability of newer oral anticoagulants, targeting either thrombin (dabigatran etexilate) or factor Xa (rivaroxaban or apixaban) exhibit rapid onset/offset and minimal drug interactions with more predictable pharmacokinetics thus eliminating the need for frequent coagulation monitoring. All these features give newer oral anticoagulants a major pharmacological benefits over vitamin K antagonists.[35]
THROMBOTIC DISORDERS
Plasma concentration of certain coagulation factors (factor V, VII, VIII, IX, fibrinogen) increase progressively with age.[36] Same is true for vWF, a key protein in platelet vessel wall interaction.[37] High incidence of cardiovascular events seen in elderly may be due to increased levels of plasma fibrinogen, which enhance the bridging of the platelets via glycoprotein IIb-IIIa receptor and act as a direct substrate for clot and/or by increasing the blood viscosity.[3]
The constitutive or acquired disorders of thrombosis are termed as thrombophillia.
There are number of factors that are associated with the hypercoaguable states. In addition to the genetic and hereditary disorders that predispose to thrombosis, several risk factors such as smoking, obesity, pregnancy, immobility, malignancy, surgery, females on oral contraceptives may also contribute to its development.[38] Disorders of ATIII deficiency, reduced protein C and Protein S are inherited in autosomal dominant fashion and are associated with increased risk of thrombosis. Acquired Protein C and Protien S deficiency may be observed in vitamin K deficiency, warfarin therapy, pregnancy, liver cirrhosis and sepsis.[39] Numbers of observational studies have shown decreased levels of APC in critically ill-patients that may have a direct correlation with the mortality.[40]
The risk of thromboembolism in the perioperative period is well recognized. Therefore, patients with herditary thrombophillia should be given thromboprophylaxis.
During pregnancy stasis due to obstruction of inferior vene cava by gravid uterus along with increase in the majority of clotting factors, fibrinogen and vWF is observed. Activity of Protein S decreases with simultaneous resistance of protein C. In addition, fibrinolytic system is also impaired thus contributing to a hypercoaguable state that makes the parturient more prone to thromboembolism.[2]
During surgery and trauma, prolonged immobility promotes stasis which results in local hypoxia. Physical disruption leads to exposure of TF thus triggering thrombosis.[37] Furthermore during the first hours of surgery, there is increase in TF, tissue plasminogen activator (tPA) and vWF, leading to hypercoaguable state thus promoting venous thrombosis. Even a venepuncture cause vascular wall injury thus, predisposing to thrombus formation. Since lower limb is associated with stasis and immobilization during surgery, venepuncture preferably should be avoided in the lower limb.
FIBRINOLYTIC SYSTEM
Fibrinolytic system is a parallel system which is activated along with activation of coagulation cascade and serves to limit the size of clot. Fibrinolysis is an enzymatic process that dissolves the fibrin clot into fibrin degradation products (FDPs) by plasmin originating from fibrin bound plasminogen in liver. This reaction is catalysed by tPA or urokinase plasminogen activator (u-PA) released from vascular endothelium. The release of t-PA is stimulated by tissue occlusion, thrombin, epinephrine, vasopressin and strenuous exercise.
Plasmin activity is tightly regulated by its inhibitor (α-2 antiplasmin) thus preventing widespread fibrinolysis[41] [Figure 4]. In vivo activity of the fibrinolytic system is assessed clinically by measuring the FDP's. D dimers are produced by digestion of cross linked fibrin and are specific indicators of fibrinolysis used in the assessment and diagnosis of pulmonary embolism, DIC or deep vein thrombosis.[13]
Figure 4.
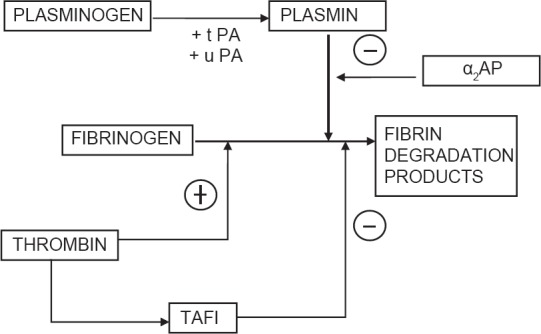
Regulation of the fibrinolytic system
Since plasmin has the potential to degrade fibrinogen leading to deleterious consequences, the fibrinolytic activity is limited by following factors:
Plasminogen activator inhibitor - It is the main physiological inhibitor of fibriolysis and acts by inhibiting t-PA and u-PA irreversibly
TAFI - It is a plasma proenzyme synthesized by liver and activated by thrombin. It decreases the affinity of plasminogen to fibrin and augments the action of anti-trypsin in inhibiting plasmin
Plasmin inhibitors - α2 antiplasmin and α2Macroglobulin are the glycoproteins that exert action by virtue of plasmin inhibition.[25]
DISORDERS OF FIBRINOLYSIS
Congenital disorders pertaining to fibrinolytic system are rare. Although the hyperfibrinolytic state is associated with increased tendency to bleed, deficiency of the same predisposes to thromboembolism.[42] Excessive activation of fibrinolysis may be observed during cardiopulmonary bypass, hence antifibrinolytics have a beneficial role in the prevention of same. Acquired hyperfibrinolysis may be encountered in trauma, liver cirrhosis, amniotic fluid embolism, multiple myeloma, snake bite and conditions associated with massive activation of t-PA, which can lead to DIC and haemorrhage.[25]
SUMMARY
Haemostasis is a complex physiological process, maintaining the fluidity of blood and is regulated by delicate balance existing between thrombogenic and anti thrombogenic mechanisms present in the body. Imbalance between the two components predisposes a patient to either bleed or present with thrombosis. The physiology of the same therefore, needs to be understood to predict the pathological and clinical consequences of the same before implementing any pharmacological interventions.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared
REFERENCES
- 1.Achneck HE, Sileshi B, Parikh A, Milano CA, Welsby IJ, Lawson JH. Pathophysiology of bleeding and clotting in the cardiac surgery patient: from vascular endothelium to circulatory assist device surface. Circulation. 2010;122:2068–77. doi: 10.1161/CIRCULATIONAHA.110.936773. [DOI] [PubMed] [Google Scholar]
- 2.Thornton P, Douglas J. Coagulation in pregnancy. Best Pract Res Clin Obstet Gynaecol. 2010;24:339–52. doi: 10.1016/j.bpobgyn.2009.11.010. [DOI] [PubMed] [Google Scholar]
- 3.Previtali E, Bucciarelli P, Passamonti SM, Martinelli I. Risk factors for venous and arterial thrombosis. Blood Transfus. 2011;9:120–38. doi: 10.2450/2010.0066-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bombeli T, Spahn DR. Updates in perioperative coagulation: Physiology and management of thromboembolism and haemorrhage. Br J Anaesth. 2004;93:275–87. doi: 10.1093/bja/aeh174. [DOI] [PubMed] [Google Scholar]
- 5.Meybohm P, Zacharowski K, Weber CF. Point-of-care coagulation management in intensive care medicine. Crit Care. 2013;17:218. doi: 10.1186/cc12527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cines DB, Pollak ES, Buck CA, Loscalzo J, Zimmerman GA, McEver RP, et al. Endothelial cells in physiology and in the pathophysiology of vascular disorders. Blood. 1998;91:3527–61. [PubMed] [Google Scholar]
- 7.Lasne D, Jude B, Susen S. From normal to pathological hemostasis. Can J Anesth. 2006;53:S2–11. doi: 10.1007/BF03022247. [DOI] [PubMed] [Google Scholar]
- 8.Triplett DA. Coagulation and bleeding disorders: Review and update. Clin Chem. 2000;46:1260–9. [PubMed] [Google Scholar]
- 9.Heemskerk JW, Bevers EM, Lindhout T. Platelet activation and blood coagulation. Thromb Haemost. 2002;88:186–93. [PubMed] [Google Scholar]
- 10.Andrews RK, Berndt MC. Platelet physiology and thrombosis. Thromb Res. 2004;114:447–53. doi: 10.1016/j.thromres.2004.07.020. [DOI] [PubMed] [Google Scholar]
- 11.Ashby B, Daniel JL, Smith JB. Mechanisms of platelet activation and inhibition. Hematol Oncol Clin North Am. 1990;4:1–26. [PubMed] [Google Scholar]
- 12.Yenicesu I, Yetgin S, Ozyürek E, Aslan D. Virus-associated immune thrombocytopenic purpura in childhood. Pediatr Hematol Oncol. 2002;19:433–7. doi: 10.1080/08880010290097233. [DOI] [PubMed] [Google Scholar]
- 13.Colvin BT. Physiology of haemostasis. Vox Sang. 2004;87(Suppl 1):43–6. doi: 10.1111/j.1741-6892.2004.00428.x. [DOI] [PubMed] [Google Scholar]
- 14.Monroe DM, 3rd, Hoffman M, Roberts HR. Williams Hematology. 8th ed. New York NY: McGraw-Hill Professional Publishing; 2010. Molecular biology and biochemistry of the coagulation factors and pathways of hemostasis; pp. 614–6. [Google Scholar]
- 15.Hall JE. Guyton and Hall Textbook of Medical Physiology: Enhanced E-Book. 11th ed. Philadelphia: Elsevier Health Sciences; 2010. Hemostasis and blood coagulation; pp. 457–9. [Google Scholar]
- 16.Sadler JE. Biochemistry and genetics of von Willebrand factor. Annu Rev Biochem. 1998;67:395–424. doi: 10.1146/annurev.biochem.67.1.395. [DOI] [PubMed] [Google Scholar]
- 17.Barash PG, Cullen BF, Stoelting RK, editors. 5th ed. Philadelphia: Lippincott Williams and Wilkins; 2006. Clinical Anesthesia; pp. 224–6. [Google Scholar]
- 18.Doolittle RF, Spraggon G, Everse SJ. Three-dimensional structural studies on fragments of fibrinogen and fibrin. Curr Opin Struct Biol. 1998;8:792–8. doi: 10.1016/s0959-440x(98)80100-0. [DOI] [PubMed] [Google Scholar]
- 19.Kamath S, Lip GY. Fibrinogen: Biochemistry, epidemiology and determinants. QJM. 2003;96:711–29. doi: 10.1093/qjmed/hcg129. [DOI] [PubMed] [Google Scholar]
- 20.Mackman N, Tilley RE, Key NS. Role of the extrinsic pathway of blood coagulation in hemostasis and thrombosis. Arterioscler Thromb Vasc Biol. 2007;27:1687–93. doi: 10.1161/ATVBAHA.107.141911. [DOI] [PubMed] [Google Scholar]
- 21.Manly DA, Boles J, Mackman N. Role of tissue factor in venous thrombosis. Annu Rev Physiol. 2011;73:515–25. doi: 10.1146/annurev-physiol-042210-121137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Myers DD, Hawley AE, Farris DM, Wrobleski SK, Thanaporn P, Schaub RG, et al. P-selectin and leukocyte microparticles are associated with venous thrombogenesis. J Vasc Surg. 2003;38:1075–89. doi: 10.1016/s0741-5214(03)01033-4. [DOI] [PubMed] [Google Scholar]
- 23.Closse C, Seigneur M, Renard M, Pruvost A, Dumain P, Belloc F, et al. Influence of hypoxia and hypoxia-reoxygenation on endothelial P-selectin expression. Thromb Res. 1997;85:159–64. doi: 10.1016/s0049-3848(96)00233-2. [DOI] [PubMed] [Google Scholar]
- 24.Opal SM, Kessler CM, Roemisch J, Knaub S. Antithrombin, heparin, and heparan sulfate. Crit Care Med. 2002;30:S325–31. doi: 10.1097/00003246-200205001-00024. [DOI] [PubMed] [Google Scholar]
- 25.Ejiofor JA. Anticlotting mechanisms I: Physiology and pathology. Contin Educ Anesth Crit Care Pain. 2013;13:87–92. [Google Scholar]
- 26.Price GC, Thompson SA, Kam PC. Tissue factor and tissue factor pathway inhibitor. Anaesthesia. 2004;59:483–92. doi: 10.1111/j.1365-2044.2004.03679.x. [DOI] [PubMed] [Google Scholar]
- 27.Dahm AE, Sandset PM, Rosendaal FR. The association between protein S levels and anticoagulant activity of tissue factor pathway inhibitor type 1. J Thromb Haemost. 2008;6:393–5. doi: 10.1111/j.1538-7836.2008.02859.x. [DOI] [PubMed] [Google Scholar]
- 28.Rigby AC, Grant MA. Protein S: A conduit between anticoagulation and inflammation. Crit Care Med. 2004;32:S336–41. doi: 10.1097/01.ccm.0000126360.00450.f8. [DOI] [PubMed] [Google Scholar]
- 29.Corral J, González-Conejero R, Hernández-Espinosa D, Vicente V. Protein Z/Z-dependent protease inhibitor (PZ/ZPI) anticoagulant system and thrombosis. Br J Haematol. 2007;137:99–108. doi: 10.1111/j.1365-2141.2007.06548.x. [DOI] [PubMed] [Google Scholar]
- 30.Owens AP, 3rd, Mackman N. Tissue factor and thrombosis: The clot starts here. Thromb Haemost. 2010;104:432–9. doi: 10.1160/TH09-11-0771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kumar V, Abbas AK, Fausto N, Aster JC. Robbins and Cotran Pathologic Basis of Disease. 8th ed. Philadelphia, PA: Saunders Elsevier; 2010. Hemodynamic disorders, thromboembolic disease and shock; pp. 118–20. [Google Scholar]
- 32.Martlew VJ. Peri-operative management of patients with coagulation disorders. Br J Anaesth. 2000;85:446–55. doi: 10.1093/bja/85.3.446. [DOI] [PubMed] [Google Scholar]
- 33.Roberts HR, Stinchcombe TE, Gabriel DA. The dysfibrinogenaemias. Br J Haematol. 2001;114:249–57. doi: 10.1046/j.1365-2141.2001.02892.x. [DOI] [PubMed] [Google Scholar]
- 34.Polderman HK. Hypothermia and coagulation. Crit Care. 2012;16:A20. [Google Scholar]
- 35.Bauer KA. Pros and cons of new oral anticoagulants. Hematology Am Soc Hematol Educ Program 2013. 2013:464–70. doi: 10.1182/asheducation-2013.1.464. [DOI] [PubMed] [Google Scholar]
- 36.Franchini M. Hemostasis and aging. Crit Rev Oncol Hematol. 2006;60:144–51. doi: 10.1016/j.critrevonc.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 37.Coppola R, Mari D, Lattuada A, Franceschi C. Von Willebrand factor in Italian centenarians. Haematologica. 2003;88:39–43. [PubMed] [Google Scholar]
- 38.Pinjala RK, Reddy LR, Nihar RP, Praveen GV, Sandeep M. Thrombophilia - How far and how much to investigate? Indian J Surg. 2012;74:157–62. doi: 10.1007/s12262-011-0407-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Martinelli I, Bucciarelli P, Mannucci PM. Thrombotic risk factors: Basic pathophysiology. Crit Care Med. 2010;38:S3–9. doi: 10.1097/CCM.0b013e3181c9cbd9. [DOI] [PubMed] [Google Scholar]
- 40.Jagneaux T, Taylor DE, Kantrow SP. Coagulation in sepsis. Am J Med Sci. 2004;328:196–204. doi: 10.1097/00000441-200410000-00002. [DOI] [PubMed] [Google Scholar]
- 41.Cesarman-Maus G, Hajjar KA. Molecular mechanisms of fibrinolysis. Br J Haematol. 2005;129:307–21. doi: 10.1111/j.1365-2141.2005.05444.x. [DOI] [PubMed] [Google Scholar]
- 42.Schuster V, Hügle B, Tefs K. Plasminogen deficiency. J Thromb Haemost. 2007;5:2315–22. doi: 10.1111/j.1538-7836.2007.02776.x. [DOI] [PubMed] [Google Scholar]