

Abstract
A concise thiolation of C(sp3)–H bond of cycloalkanes with diaryl disulfides in the presence of oxidant of di-tert-butylperoxide (DTBP) has been developed. This reaction without using any of metal catalyst, tolerates varieties of disulfides and cycloalkanes substrates, giving good to excellent chemical yields, which provides a useful approach to cycloalkyl aryl sulfides from unactivated cycloalkanes.
Keywords: metal-free, thiolation, oxidative, C(sp3)–H activation, cycloalkane
The formation of C–S bonds represents an active research area in general organic chemistry, material science as well as biological and pharmaceutical chemistry.[1] In the past decade, a number of methodologies have been developed in this field. In particular, transition-metal-catalyzed cross-coupling reactions of thiols with aryl halides provided the most general strategies for constructing C–S bonds.[2] Recently, metal-catalyzed C–S bonds formation through C(sp2)–H bond activation has become an alternative and intriguing methods for preparation of sulfides due to its high atom-economy and efficiency.[3] However, the cleavage of C(sp3)–H bond leading to the C–S bond formation is less studied. Very recently, Xiang and co-workers reported a novel thiolation of C(sp3)–H bond adjacent to a nitrogen or oxygen atom (Scheme 1a and 1b).[4] To date, the thiolation of C(sp3)–H bond of unactivated alkanes still have not been developed.
Scheme 1.
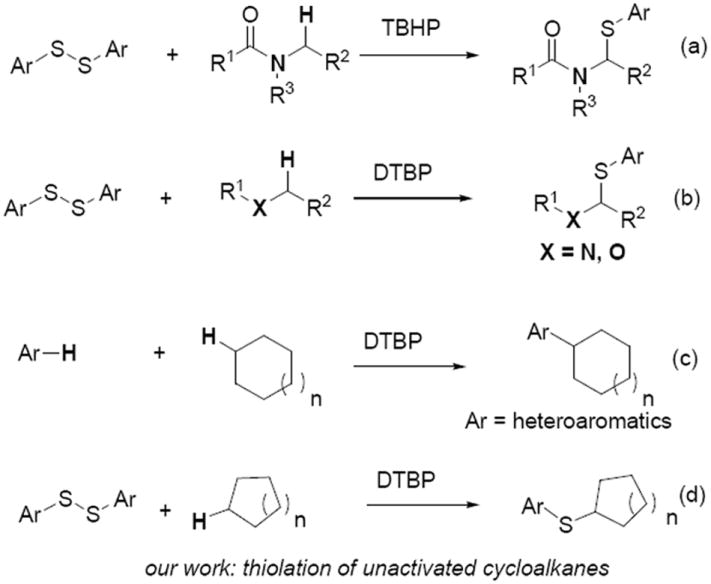
C(sp3)–H bond functionalization.
Direct C(sp3)–H transformation of alkanes, which is a great challenge due to their low reactivity and the lack of a coordination site for the metal catalyst, has attracted many generations of organic chemists in recent years.[5] However, C(sp3)–H bond activation of cycloalkanes to form C–C,[6] C–O,[7] C–N[8] bonds still has not been well reported because C(sp3)–H bond activation of cycloalkanes is more difficult than C(sp3)–H bond activation adjacent to heteroatoms, double bonds, phenyl or electron withdrawing groups. Li and others have done elegant work in this field using a transition-metal catalyst (such as Ru, Sc, Fe, etc.) both for activating the C(sp3)–H bond of cycloalkanes and subsequent coupling to form C–Y (Y = O, N, C) bonds.[9] Recently, our group also developed a Fe-catalyzed decarboxylative alkenylation of cycloalkanes via a radical process.[10] The metal-free C(sp3)–H bond functionalization progress for C(sp2)–C(sp3) bond formation of heteroaromatics and cycloalkanes promoted by DTBP have also been reported (Scheme 1c).[11,12] To the best of our knowledge, no reports for the construction of C–S bond through C(sp3)–H bond functionalization of cycloalkanes are described. Herein, we would report the first realization on the thiolation of C(sp3)–H bond of cycloalkanes through DTBP-mediated oxidative C(sp3)–H bond functionalization without the aid of transition-metal catalyst (Scheme 1d).
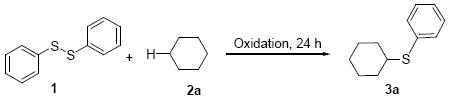
Initially, we conducted our investigation by reacting 1,2-diphenyldisulfane 1 (0.5 mmol) with cyclohexane 2a (2 mL) in the presence 4.0 equiv of tert-butylhydroperoxide (TBHP) at 120 °C for 24 h. The reaction could happen, and afforded the expected product of cyclohexyl(phenyl)sulfane 3a in a poor yield (23%, Table 1, entry 1). Replacing TBHP with DTBP, the yield dramatically increased to 88% without the aid of any metal catalyst or other additives (Table 1, entry 2). The use of other oxidants such as DDQ, K2S2O8, H2O2 (30% aqueous solution) or TBPB did not provide better results (Table 1, entries 3–6). A decreased loading of DTBP to 2.0 equiv or a lower temperature to 100 °C, the lower yield would be obtained in 75% and 69%, respectively (Table 1, entries 7 and 8). No significant effect in yield of 3a was found with 6.0 equiv of DTBP or a higher temperature (Table 1, entries 9 and 10). Furthermore, the addition of metal-catalyst, Cu(OAc)2 (10 mol %), did not result any improvement on the yield (79%, Table 1, entry 11). Further optimization of the conditions showed that the reaction could not proceed without the use of oxidant DTBP (Table 1, entry 12).
Table 1.
Optimization of reaction condition [a]
| |||
|---|---|---|---|
|
| |||
| Entry | Oxidant (equiv) | T (°C) | Yield (%)[b] |
| 1 | TBHP (4.0) | 120 | 23[c] |
| 2 | DTBP (4.0) | 120 | 88 |
| 3 | DDQ (4.0) | 120 | N.D. |
| 4 | K2S2O8 (4.0) | 120 | N.D. |
| 5 | H2O2 (4.0) | 120 | Trace[d] |
| 6 | TBPB (4.0) | 120 | 18 |
| 7 | DTBP (2.0) | 120 | 75 |
| 8 | DTBP (4.0) | 100 | 69 |
| 9 | DTBP (6.0) | 120 | 86 |
| 10 | DTBP (4.0) | 140 | 87 |
| 11 | DTBP (4.0) | 120 | 79[e] |
| 12 | – | 120 | N.D. |
Catalytic conditions: 1 (0.5 mmol), cyclohexane (2 mL), oxidant, 24 h.
Isolated yields based on 1.
70% in water solution.
30% aqueous solution.
Cu(OAc)2 (0.05 mmol) was added.
With the optimized reaction conditions in hand, this approach was then applied to the coupling of cyclohexane to a variety of diaryl disulfides (Scheme 2). As shown in Scheme 2, the process has a broad scope and high compatibility with functional groups, such as methyl, methoxy, halo and nitro substituent groups. Ortho-, meta- and para-substituted diaryl disulfides were well tolerated, and the reactions gave the corresponding products with good to excellent yields of 71–92% (3a-h). The lower yields observed in the case of ortho-substituted diaryl disulfides compared to its para- or meta- analogues, is possibly due to the steric hindrance (3d-f). The reaction with p-methyl and p-methoxy substituted diaryl disulfides afforded the expected product with excellent yields (3b and 3c). However, when p-fluoro and p-nitro substituted diaryl disulfides were used as the coupling partner the product yields dropped (85% for 3d and 75% for 3h). These results imply that the electronegativity of the substituents in the diaryl disulfides has an obvious effect on the chemical yields. In addition, the disubstituted diaryl disulfide is also well tolerated in this reaction to give the target product with a slightly lower yield of 80% (3i). Notably, the heterocyclic disulfides thienyl or pyridinyl disulfide can work well in the reaction to provide the corresponding alkyl heteroaryl sulfide (3j-k). It is worth to note that the reaction with complex heterocyclic disulfides 2,2’-dithiobis(benzothiazole) and 5,5’-dithiobis(1-phenyl-1H-tetrazole) also proceed successfully under the optimized condition (3l-m). Disappointingly, 1,2-dibenzyldisulfane was not a suitable substrate for the current thiolation system (3n).
Scheme 2.
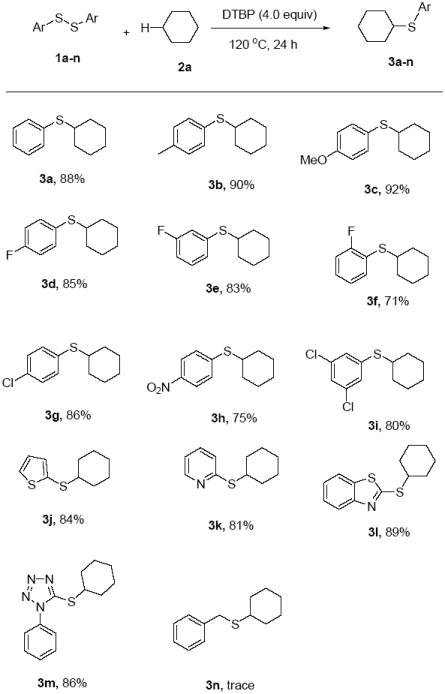
Thiolation of cyclohexane with disulfides. Reaction conditions: 1 (0.5 mmol), cyclohexane (2 mL), DTBP (4.0 equiv), 120 °C, 24 h. Isolated yields based on 1.
Subsequently, other cycloalkanes, including cyclopentane, cycloheptane and cyclooctane were employed as substrates for this reaction to further examination of the reaction scope (Scheme 3). Fortunately, they work well in the system under the optimized conditions, and can react with different diaryl disulfides 1, giving the corresponding products 4a–j in 67–89% chemical yield. Comparing with the results shown in Scheme 2 for cyclohexane as a substrate, the reaction with cyclopentane showed a lower efficiency, and obvious lower yields were found (67–82%, 4a–g). However, for the cases of larger cycloalkanes, such as cycloheptane and cyclooctane, comparative chemical yields were obtained (4h–j).
Scheme 3.
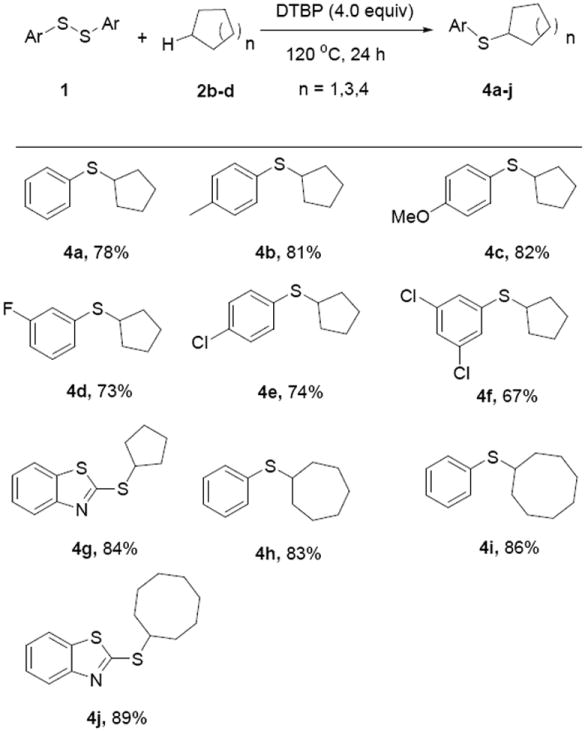
Thiolation of cyclopentane with disulfides. Reaction conditions: 1 (0.5 mmol), cycloalkanes (2 mL), DTBP (4.0 equiv), 120 °C, 24 h. Isolated yields based on 1.
Then intramolecular and intermolecular competition experiments were carried out. Firstly, heteroaryl aryl disulfide 1o was used for the investigation of the intramolecular competition reaction (Scheme 4). We found both of these two ArS• could react with cyclohexane well, giving the cross-coupling products 3l and 3b in 85% and 86% chemical yields respectively.
Scheme 4.
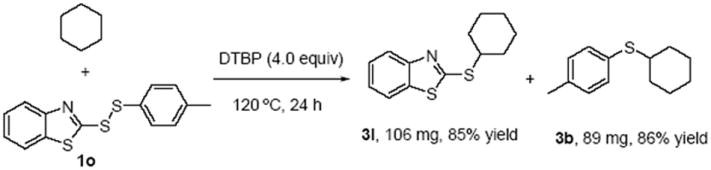
Investigation of the intramolecular competition reaction. Reaction conditions: 1o (0.5 mmol), cyclohexane (2 mL), DTBP (4.0 equiv), 120 °C, 24 h.
Then, the examination of the intramolecular competition reaction was performed with the using of disulfides 1l and 1b as substrates at the same time (Scheme 5). The reaction with both of these two disulfides proceeded very well, and good chemical yields were obtained (3l and 3b in 82% and 84% respectively).
Scheme 5.
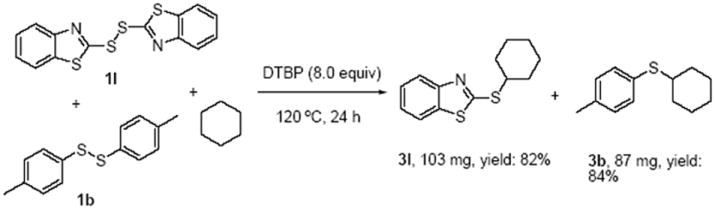
Investigation of the intermolecular competition reaction. Reaction conditions: 1l (0.25 mmol), 1b (0.25 mol), cyclohexane (2 mL), DTBP (8.0 equiv), 120 °C, 24 h.
To gain insight into the reaction mechanism, several control experiments were carried out (Scheme 6). Addition of the radical-trapping reagents 2,2,6,6 tetramethylpiperidine N-oxide (TEMPO) or azobisisobutyronitrile (AIBN) can completely inhibit the reaction and almost no desired product was observed. These results indicate that the transformation may proceed via a radical course.
Scheme 6.
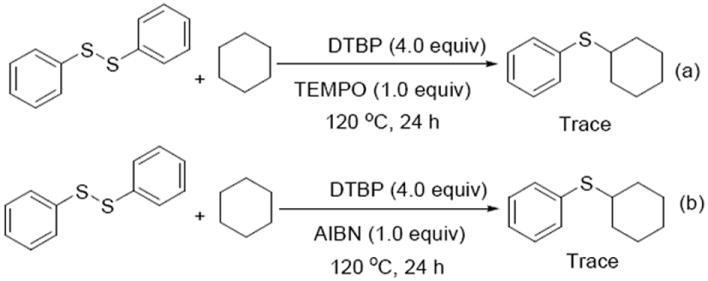
Insights into the mechanism.
Based on the above results and literature reports,[4, 10, 13] a possible mechanism for the cycloalkane thiolation reaction is illustrated in Scheme 7, which includes a key radical oxidative coupling step. At the beginning, homolysis of DTBP give tert-butoxy radical intermediate A under the condition of heating. Then, cyclohexane radical intermediate B is generated via reaction of intermediate A and cyclohexane 2a through a C–H bond cleavage. The following step is the reaction between intermediate B and 1,2-diphenyldisulfane 1, affording the final target product 3a, along with the formation of PhS• free radical intermediate C. Finally, the free radical C couples with cyclohexane readical B, giving another molecular of product 3a.
Scheme 7.
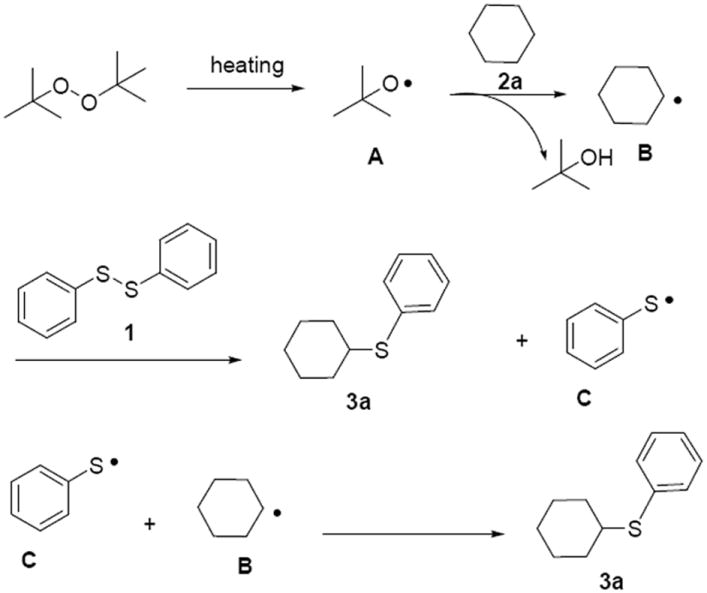
A possible reaction mechanism.
In conclusion, we have presented a novel and highly efficient method for C–S cross-coupling through direct C(sp3)–H bond functionalization of cycloalkanes with diaryl sulfides using DTBP as the oxidant without use of any metal catalyst. Varieties of substituted diphenyl disulfides and heterocyclic disulfides could be tolerated and coupled with cycloalkanes, giving the cycloalkyl aryl sulfides in good to excellent yields. Moreover, this synthetic strategy for direct C–S bond formation might be very valuable and attractive in radical chemistry.
Experimental Section
General procedure for thiolation of cycloalkanes
A sealable reaction tube equipped with a magnetic stirrer bar was charged with diaryl disulfides (0.5 mmol), DTBP (di-tert-butyl peroxide, 2.0 mmol, 377 μL), and cycloalkanes (2.0 mL). The rubber septum was then replaced by a Teflon–coated screw cap, and the reaction vessel placed in an oil bath at 120 °C. After stirring at this temperature for 24 h, it was cooled to room temperature and diluted with ethyl acetate, washed with water, dried over MgSO4. After the solvent was removed under reduced pressure, the residue was purified by column chromatography on silica gel (petroleum) to afford the direct cross–coupling product, cycloalkyl aryl sulfides. In this reaction, 0.5 mol of diaryl disulfides can afford 1 mol of PhS• free radical as shown by Scheme 7, which finally can give 1 mol of cross-coupling product. So the yields and amounts of products are given based on this.
Heating of organic peroxide could be potentially dangerous.
Acknowledgments
We gratefully acknowledge the financial support from the National Natural Science Foundation of China (No. 21102071) and the Fundamental Research Funds for the Central Universities (No. 1107020522 and No. 1082020502). The Jiangsu 333 program (for Pan) and Changzhou Jin-Feng-Huang program (for Han) are also acknowledged.
References
- 1.a) Dua RK, Taylor EW, Phillips RS. J Am Chem Soc. 1993;115:1264–1270. [Google Scholar]; b) Rayner CM. Contemp Org Synth. 1996;3:499–533. [Google Scholar]; c) Hartwig JF. Acc Chem Res. 1998;31:852–860. [Google Scholar]; d) Baird CP, Rayner CM. J Chem Soc Perkin Trans. 1998;1:1973–2003. [Google Scholar]; e) Procter DJ. J Chem Soc Perkin Trans. 1999;1:641–668. [Google Scholar]; f) Ley SV, Thomas AW. Angew Chem Int Ed. 2003;42:5400–5449. doi: 10.1002/anie.200300594. [DOI] [PubMed] [Google Scholar]; g) Gangjee A, Zeng Y, Talreja T, McGuire JJ, Kisliuk RL, Queener SF. J Med Chem. 2007;50:3046–3053. doi: 10.1021/jm070165j. [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Beletskaya IP, Ananikov VP. Chem Rev. 2011;111:1596–1636. doi: 10.1021/cr100347k. [DOI] [PubMed] [Google Scholar]
- 2.a) Li GY, Zheng G, Noonan AF. J Org Chem. 2001;66:8677–8681. doi: 10.1021/jo010764c. [DOI] [PubMed] [Google Scholar]; b) Itoh T, Mase T. Org Lett. 2004;6:4587–4590. doi: 10.1021/ol047996t. [DOI] [PubMed] [Google Scholar]; c) Fernández-Rodríguez MA, Shen Q, Hartwig JF. J Am Chem Soc. 2006;128:2180–2181. doi: 10.1021/ja0580340. [DOI] [PubMed] [Google Scholar]; d) Rout L, Sen TK, Punniyamurthy T. Angew Chem Int Ed. 2007;46:5583–5586. doi: 10.1002/anie.200701282. [DOI] [PubMed] [Google Scholar]; e) Ranu BC, Saha A, Jana R. Adv Synth Catal. 2007;349:2690–2696. [Google Scholar]; f) Zhang Y, Ngeow KC, Ying JY. Org Lett. 2007;9:3495–3498. doi: 10.1021/ol071248x. [DOI] [PubMed] [Google Scholar]; g) Rout L, Saha P, Jammi S, Punniyamurthy T. Eur J Org Chem. 2008:640–643. [Google Scholar]; h) Reddy VP, Swapna K, Kumar AV, Rao KR. J Org Chem. 2009;74:3189–3191. doi: 10.1021/jo802731j. [DOI] [PubMed] [Google Scholar]; i) Chen CK, Chen YW, Lin CH, Lin HP, Lee CF. Chem Commun. 2010;46:282–284. doi: 10.1039/b918117b. [DOI] [PubMed] [Google Scholar]; j) Kovács S, Novák Z. Org Biomol Chem. 2011;9:711–716. doi: 10.1039/c0ob00397b. [DOI] [PubMed] [Google Scholar]; k) Xu HJ, Liang YF, Zhou XF, Feng YS. Org Biomol Chem. 2012;10:2562–2568. doi: 10.1039/c2ob06795a. [DOI] [PubMed] [Google Scholar]; l) Bastug G, Nolan SP. J Org Chem. 2013;78:9303–9308. doi: 10.1021/jo401492n. [DOI] [PubMed] [Google Scholar]
- 3.a) Chu L, Yue X, Qing FL. Org Lett. 2010;12:1644–1647. doi: 10.1021/ol100449c. [DOI] [PubMed] [Google Scholar]; b) Zhang S, Qian P, Zhang M, Hu M, Cheng J. J Org Chem. 2010;75:6732–6735. doi: 10.1021/jo1014849. [DOI] [PubMed] [Google Scholar]; c) Wu Q, Zhao D, Qin X, Lan J, You J. Chem Commun. 2011;47:9188–9190. doi: 10.1039/c1cc13633j. [DOI] [PubMed] [Google Scholar]; d) Ranjit S, Lee R, Heryadi D, Shen C, Wu JE, Zhang P, Huang KW, Liu X. J Org Chem. 2011;76:8999–9007. doi: 10.1021/jo2017444. [DOI] [PubMed] [Google Scholar]; e) Zhou AX, Liu XY, Yang K, Zhao SC, Liang YM. Org Biomol Chem. 2011;9:5456–5462. doi: 10.1039/c1ob05395g. [DOI] [PubMed] [Google Scholar]; f) Zou LH, Reball J, Mottweiler J, Bolm C. Chem Commun. 2012;48:11307–11309. doi: 10.1039/c2cc36711d. [DOI] [PubMed] [Google Scholar]; g) He Z, Luo F, Li Y, Zhu G. Tetrahedron Lett. 2013;54:5907–5910. [Google Scholar]; h) Rosario AR, Casola KK, Oliveira CES, Zeni G. Adv Synth Catal. 2013;355:2960–2966. [Google Scholar]
- 4.Tang RY, Xie YX, Xie YL, Xiang JN, Li JH. Chem Commun. 2011;47:12867–12869. doi: 10.1039/c1cc15397h. [DOI] [PubMed] [Google Scholar]
- 5.For selected examples of C(sp3)–H activation, see: Chatani N, Asaumi T, Yorimitsu S, Ikeda T, Kakiuchi F, Murai S. J Am Chem Soc. 2001;123:10935–10941. doi: 10.1021/ja011540e.; DeBoef B, Pastine SJ, Sames D. J Am Chem Soc. 2004;126:6556–6557. doi: 10.1021/ja049111e.; Song CX, Cai GX, Farrell TR, Jiang ZP, Li H, Gan LB, Shi ZJ. Chem Commun. 2009:6002–6004. doi: 10.1039/b911031c.; Harris JF., Jr J Org Chem. 1981;46:268–271.; Mitroka S, Zimmeck S, Troya D, Tanko JM. J Am Chem Soc. 2010;132:2907–2913. doi: 10.1021/ja903856t.; Antonchick AP, Burgmann L. Angew Chem Int Ed. 2013;52:3267–3271. doi: 10.1002/anie.201209584.; Ryu I, Tani A, Fukuyama T, Ravelli D, Fagnoni M, Albini A. Angew Chem Int Ed. 2011;50:1869–1872. doi: 10.1002/anie.201004854.; Chuprakov S, Malik JA, Zibinsky M, Fokin VV. J Am Chem Soc. 2011;133:10352–10355. doi: 10.1021/ja202969z.; Ravelli D, Montanaro S, Zema M, Fagnoni M, Albini A. Adv Synth Catal. 2011;353:3295–3300.; Samanta R, Matcha K, Antonchick AP. Eur J Org Chem. 2013:5769–5804..
- 6.Yonehara F, Kido Y, Morita S, Yamaguchi M. J Am Chem Soc. 2001;123:11310–11311. doi: 10.1021/ja0164172. [DOI] [PubMed] [Google Scholar]
- 7.a) Iwahama T, Syojyo K, Sakaguchi S, Ishii Y. Org Process Res Dev. 1998;2:255–260. [Google Scholar]; b) Prat I, Gómez L, Canta M, Ribas X, Costas M. Chem Eur J. 2013;19:1908–1913. doi: 10.1002/chem.201203281. [DOI] [PubMed] [Google Scholar]; c) White MC. Science. 2012;335:807–809. doi: 10.1126/science.1207661. [DOI] [PubMed] [Google Scholar]
- 8.a) Deng G, Chen W, Li CJ. Adv Synth Catal. 2009;351:353–356. [Google Scholar]; b) Gephart RT, Huang DL, Aguila MJB, Schmidt G, Shahu A, Warren TH. Angew Chem Int Ed. 2012;51:6488–6492. doi: 10.1002/anie.201201921. [DOI] [PubMed] [Google Scholar]; c) Michaudel Q, Thevenet D, Baran PS. J Am Chem Soc. 2012;134:2547–2550. doi: 10.1021/ja212020b. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Liu Y, Guan X, Wong ELM, Liu P, Huang JS, Che CM. J Am Chem Soc. 2013;135:7194–7204. doi: 10.1021/ja3122526. [DOI] [PubMed] [Google Scholar]; e) Liang C, Collet F, Robert-Peillard F, Müller P, Dodd RH, Dauban P. J Am Chem Soc. 2008;130:343–350. doi: 10.1021/ja076519d. [DOI] [PubMed] [Google Scholar]; f) Fiori KW, Du Bois J. J Am Chem Soc. 2007;129:562–568. doi: 10.1021/ja0650450. [DOI] [PubMed] [Google Scholar]
- 9.a) Zhang Y, Li CJ. Eur J Org Chem. 2007:4654–4657. [Google Scholar]; b) Deng G, Zhao L, Li CJ. Angew Chem Int Ed. 2008;47:6278–6282. doi: 10.1002/anie.200801544. [DOI] [PubMed] [Google Scholar]; c) Deng G, Li CJ. Org Lett. 2009;11:1171–1174. doi: 10.1021/ol900070x. [DOI] [PubMed] [Google Scholar]; d) Newhouse T, Baran PS. Angew Chem Int Ed. 2011;50:3362–3374. doi: 10.1002/anie.201006368. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Guo X, Li CJ. Org Lett. 2011;13:4977–4979. doi: 10.1021/ol202081c. [DOI] [PubMed] [Google Scholar]
- 10.Zhao J, Fang H, Han J, Pan Y. Beilstein J Org Chem. 2013;9:1718–1723. doi: 10.3762/bjoc.9.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Deng G, Ueda K, Yanagisawa S, Itami K, Li CJ. Chem Eur J. 2009;15:333–337. doi: 10.1002/chem.200801893. [DOI] [PubMed] [Google Scholar]
- 12.Xia R, Niu HY, Qu GR, Guo HM. Org Lett. 2012;14:5546–5549. doi: 10.1021/ol302640e. [DOI] [PubMed] [Google Scholar]
- 13.a) Zhao J, Zhou W, Han J, Li G, Pan Y. Tetrahedron Lett. 2013;54:6507–6510. [Google Scholar]; b) Li D, Wang M, Liu J, Zhao Q, Wang L. Chem Commun. 2013;49:3640–3642. doi: 10.1039/c3cc41188e. [DOI] [PubMed] [Google Scholar]; c) Zhou MB, Wang CY, Song RJ, Liu Y, Wei WT, Li JH. Chem Commun. 2013;49:10817–10819. doi: 10.1039/c3cc45861j. [DOI] [PubMed] [Google Scholar]; d) Gu L, Jin C, Guo J, Zhang L, Wang W. Chem Commun. 2013;49:10968–10970. doi: 10.1039/c3cc46375c. [DOI] [PubMed] [Google Scholar]