

Abstract
Arrhythmogenic cardiomyopathy (ACM) is a primary myocardial disorder characterized by the early appearance of ventricular arrhythmias often out of proportion to the degree of ventricular remodeling and dysfunction. ACM typically presents in adolescence or early adulthood. It accounts for 10% of sudden cardiac deaths in individuals under the age of 18 years. Although there has been significant progress in recognizing the genetic determinants of ACM, how specific gene mutations cause the disease remains poorly understood. Here, we review insights gained from studying the human disease as well as in vivo and in vitro experimental models. These observations have advanced our understanding of the molecular mechanisms underlying the pathogenesis of ACM and may lead to development of new mechanism-based therapies.
Keywords: Arrhythmogenic cardiomyopathy, sudden cardiac death, plakoglobin, SB216763
Introduction
Arrhythmogenic cardiomyopathy (ACM) is apparently the most arrhythmogenic form of heart disease known to man. Its pathological hallmark is progressive myocardial degeneration with subsequent replacement by scar and fatty tissue.1, 2 Originally described as arrhythmogenic right ventricular cardiomyopathy (ARVC), it is now recognized to include left dominant and biventricular forms. Arrhythmias arise early in the natural history of ACM, often preceding structural remodeling of the myocardium.1, 2 In this sense, ACM is more reminiscent of ion channelopathies than the other non-ischemic cardiomyopathies. However, arrhythmias in ACM do not depend on ion channel mutations but instead arise in structurally normal hearts with normal ion channel genes.
It is estimated that in the United States, ACM affects 1:5000 individuals and accounts for 17% of sudden cardiac death (SCD) cases <35 year of age.3 In certain regions of the world, such as Northern Italy, prevalence reaches 1:1000 and ACM is the number one cause of SCD among young athletes.4 ACM is familial in at least 50% of cases and is predominantly inherited in an autosomal dominant manner although recessive forms have been recognized.5 Currently, diagnosis relies on fulfilling clinical criteria set by an International Task Force, which although relatively specific are not highly sensitive.6 Increasingly, overlap is recognized with other forms of cardiomyopathy segregating in the same family.
Early genetic studies were hampered by the low genetic penetrance, phenotypic variation and age-related progression characterizing the disease.5 Thus, it was not until 2000 that analysis of patients from the Greek island of Naxos with a rare, highly penetrant recessive form of ACM led to the identification of the first disease-causing mutation, a deletion (2057del2) in the gene encoding the desmosomal protein plakoglobin (γ-catenin). So-called Naxos disease is a triad of classical ARVC, woolly hair and palmoplantar keratoderma.7 This discovery provided the initial insight that ACM is a ‘disease of the desmosome’ and paved the way for identification of mutations in other desmosomal genes including those encoding desmoplakin, plakophilin-2, desmocollin-2 and desmoglein-2.5
The disease in children
ACM-related mutations are present from conception, but the clinical phenotype is not manifested until at least adolescence or more typically early adulthood. It is therefore of pivotal importance to recognize ACM in young individuals early, before the onset of risk of fatal arrhythmias.8 Not long after ARVC was established as a new morbid entity,9 Dungan et al. diagnosed the disease in 3/8 children undergoing angiography for sustained monomorphic ventricular tachycardia (VT) of left bundle branch block (LBBB) morphology in the context of seemingly structurally normal hearts.10 Currently, the diagnostic yield of ACM among children who present with this type of arrhythmia remains the same, although the diagnosis of ACM is now facilitated by analysis of the ECG, Holter monitoring, signal-averaged (SA)-ECG, echocardiography, MRI and voltage mapping as well as by mutation screening.8 ECG and SA-ECG findings can identify >50% of children that fulfill the Task Force Criteria. By contrast, histological findings in endomyocardial biopsy samples were diagnostic of the disease in only 16/48 children.8
Although the diagnostic utility of MRI is growing among the adult population with ACM, 11 it remains controversial in pediatric cases. Aviram et al. reviewed MRI findings in 26 children undergoing clinical assessment for ACM and showed a moderate correlation between MRI abnormalities and diagnostic status.12 Fogel et al. reviewed MRI findings in a population of 81 children and concluded that MRI is a low-yield test for ACM diagnosis in this age group.13 Despite the difficulties in disease diagnosis, however, ACM accounts for 10% of SCD cases among individuals below the age of 18.8 Current guidelines suggest that asymptomatic children diagnosed with ACM based on clinical and/or genetic evaluation should avoid competitive sports, modify recreational sports and be evaluated annually8. These restrictions are of paramount importance given that 10% of ACM-related deaths occur before adulthood and exercise may to trigger arrhythmic events.8, 14
Lessons from the human disease
Studies of the molecular pathology of ACM in patient myocardial tissues have revealed consistent patterns of redistribution of key intercalated disk proteins and features of inflammation and apoptosis. Here, we briefly describe these features and discuss their implications in terms of disease mechanisms.
Immunoreactive signal for the desmosomal protein plakoglobin, also known as γ-catenin, is reduced at intercalated disks in the majority of ACM myocardial samples regardless of the underlying pathogenic mutation (Fig 1A).15 In experimental models, this is accompanied by redistribution to intracellular and intranuclear sites. Such redistribution probably also occurs in patients but is more difficult to demonstrate by immunohistochemistry in formalin-fixed, paraffin-embedded tissues. As a member of the catenin family of proteins, β-catenin and plakoglobin (α-catenin) appear to have different roles in normal cell function. Whereas β-catenin fulfills roles both as a structural protein in desmosomes and adherens junctions and as a nuclear transcription co-factor regulating gene expression programs, plakoglobin serves mainly if not entirely as an anchoring molecule confined to cell-cell junctions.16,17 The distribution of β-catenin in junctional and nuclear pools is tightly regulated by the canonical Wnt pathway.16–18 Wnt pathways are involved in fundamental biological processes including normal heart development and differentiation, cardiac hypertrophy, heart failure and aging.18 Experimental evidence suggests that nuclear localization of plakoglobin in ACM dysregulates Wnt/β-catenin signaling raising the possibility that altered signaling through this pathway may be part of the disease mechanism in ACM.19
Figure 1.

(A) Representative confocal immunofluorescence images of control ventricular myocardium and myocardium from two patients with ACM. Specific immunoreactive signal for plakoglobin, Cx43 and Nav1.5 is significantly depressed at intercalated disks in both ACM cases. Control human myocardium shows immunoreactive signal for SAP97 concentrated at intercalated disks and in a sarcomeric pattern. By contrast, a marked reduction in SAP97 signal is seen in the myocardium of both patients with ACM. (B) Representative immunoperoxidase staining for TNFα in ventricular myocardial samples from two patients with ACM. The intensity of immunoreactive signal indicated by the brown reaction product is increased in ACM myocardium compared to control myocardium (x20).
Gap junction remodeling, evidenced by reduced immunoreactive signal at cell-cell junctions for the major ventricular gap junction protein Cx43, also appears to be a consistent feature of ACM (Fig 1A).15 It apparently develops as an early manifestation of disease. Only limited tissue samples are available from ACM patients during the early “concealed” phase of the disease, but in one striking example, marked loss of junctional Cx43 signal was seen in the heart of a 7 year-old child with Naxos disease in whom >14,000 premature ventricular contractions were identified on 24 hr Holter monitoring. This child died of an acute leukemia and at autopsy had what appeared to be a structurally normal heart.20 Although anecdotal, this observation raises the possibility that gap junction remodeling may contribute to arrhythmias prior to the appearance of structural abnormalities.
Pathologists have long recognized myocardial inflammation as a feature of ACM, often associated with disease flares associated with increased arrhythmic activity.21 It is unclear, however, whether inflammation plays a primary role in ACM pathogenesis, or represents a secondary response to myocyte injury. Mononuclear inflammatory infiltrates are commonly seen in the hearts of ACM patients and in some cases can be especially prominent and suggest myocarditis as a primary diagnosis.21 Even in the absence of inflammatory cells, cardiac myocytes in ACM express pro-inflammatory cytokines (Fig 1B), some of which, including interleukin-17 (IL-17) and tumor necrosis factor alpha (TNFα), can promote the rapid redistribution of plakoglobin from junctional to intracellular sites in vitro, thus establishing a potential mechanistic link between inflammation and redistribution of intercalated disk proteins.22 Increased circulating levels of pro-inflammatory cytokines have also been found in patients with ACM.22
Reduced densities of the cardiac Na+ current and the inward rectifying K+ current, IK1, have been observed in experimental models of ACM 23, 24 which, combined with reduced gap junctional coupling, could contribute to the highly arrhythmogenic phenotype. Only limited characterization of cellular electrophysiology has been performed in patients with ACM and this involves analysis of cardiac myocytes from patient-derived induced pluripotent stem cells. Such cells from a patient with ACM due to a mutation in plakophilin2 show reduced Na+ current density25 and abnormal Ca2+ dynamics.26 It has also been reported that immunoreactive signal for Nav1.5 (Fig 1A), the major protein subunit responsible for the cardiac sodium current, is reduced at intercalated disks in patients with ACM.27 These observations raise the possibility that defective localization of ion channel proteins could contribute to reduced INa and IK1 densities in ACM. In this regard, the PDZ domain protein synapse-associated-protein 97 (SAP97) is known to participate in the regulation of Nav1.5 and Kir2.1, the major protein subunits responsible for the cardiac Na+ current and the inward rectifying potassium current IK1, respectively.28 Indeed, abnormal distribution of immunoreactive signal for SAP97 has been described in experimental models of ACM and in patient myocardial samples (Fig 1A).24
Lessons from zebrafish
To gain further insights into disease mechanisms and to facilitate high-throughput chemical screening for drug discovery, we created a transgenic zebrafish model of ACM with cardiac myocyte-specific expression of human plakoglobin bearing the 2057del2 mutation.24 Transgenic fish exhibited bradycardia, reduced stroke volume and reduced cardiac output by only 48 hours post-fertilization. As young adults, they demonstrated cardiomegaly, peripheral edema and cachexia, and experienced substantial mortality from arrhythmias or heart failure.24 Ventricular myocytes from fish expressing 2057del2 plakoglobin exhibited changes in action potential morphology including marked reduction in the maximum rate-of-rise of phase 0 and reduced resting membrane potential. There were corresponding reductions in the peak INa and IK1 densities but not in the delayed rectifier potassium current IKr. Unlike Nav1.5 and Kir2.1, trafficking of Kv11.1, the major protein subunit underlying IKr, is not regulated by SAP97.29
To identify potential tool compounds, we screened a library of bioactive compounds for modifiers of the disease phenotype in zebrafish.24 One compound in particular, SB216763, prevented development of bradycardia and contractility defects and significantly increased survival in the mutant fish. This small molecule also rapidly reversed the highly abnormal action potential phenotype and corrected the marked decreases in INa and IK1 exhibited by mutant fish myocytes.24
SB216763 has been annotated as an ATP-competitive inhibitor of glycogen synthase kinase 3β (GSK-3β).30 It is, therefore, an activator of the canonical Wnt/β-catenin signaling pathway.18 When this pathway is quiescent, cytosolic β-catenin becomes phosphorylated by GSK-3β and targeted for degradation. In response to Wnt ligands, however, GSK-3β is prevented from phosphorylating β-catenin which can enter the nucleus where it regulates gene expression.18 The observation that an apparent Wnt pathway activator can rescue the features of the ACM phenotype in fish, is consistent with previous studies in mouse models suggesting an association between ACM and suppression of this signaling pathway.19
Lessons from in vitro models of ACM
Neonatal rat ventricular myocytes (NRVM) transfected to express plakoglobin bearing the 2057del2 mutation exhibit many of the same features identified in patients with ACM.24 For example, myocytes expressing mutant plakoglobin showed intracellular and intranuclear accumulation of plakoglobin as well as gap junction remodeling within 24 hours of transfection (Fig 2A). These marked changes in protein localization occur with no apparent change in the total cellular content of plakoglobin and Cx43, thus implicating abnormal protein distribution rather than abnormal protein synthesis or degradation as a mechanism underlying ACM pathogenesis. Transfected NRVMs also show decreased immunoreactive signal for Nav1.5 (Fig 2B), and abnormal localization of SAP97 (Fig 2A), and exhibit increased rates of apoptosis (Fig 2C), which are further increased in response to mechanical stress.24 NRVMs expressing 2057del2 plakoglobin secrete numerous pro-inflammatory cytokines into the culture medium including IL-17, TNFα and macrophage inflammatory protein-1α, all of which have previously been found at high circulating levels in patients with ACM (Fig 2D).22, 24 And like fish myocytes expressing mutant plakoglobin, transfected NRVMs exhibit marked alterations in action potential morphology.24
Figure 2.
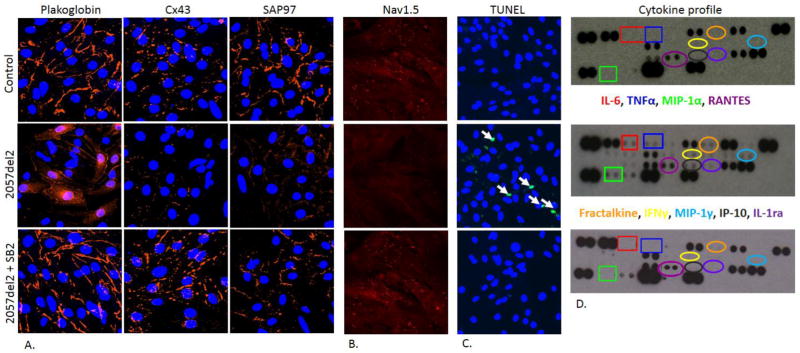
(A) Representative confocal immunofluorescence images showing plakoglobin, Cx43 and SAP97 immunoreactive signal in control neonatal rat ventricular myocytes (NRVMs), cells transfected with 2057del2 plakoglobin and 2057del2-plakoglobin expressing cells exposed to SB216763 (SB2) for 24 hours. Cell nuclei were visualized with 4′,6-diamidino-2-phenylindole (DAPI) staining (blue). (B) Specific immunoreactive signal for Nav1.5 appears to be distributed both intra-cellularly and at the cell surface in control NRVMs. A marked reduction in Nav1.5 signal is seen in myocytes expressing mutant plakoglobin, and this was rapidly reversed in cells exposed to SB2 for 24 hours. (C) Increased apoptosis in NRVM expressing 2057del2 plakoglobin as shown by the number of nuclear with positive terminal deoxynucleotidyl transferase dUTP end-labeling (TUNEL) (green). All nuclei were visualized with 4′,6-diamidino-2-phenylindole (DAPI) staining (blue). White arrows point to apoptotic nuclei. (D) Cytokine expression profiles showing increased secretion of interleukin-6 (IL-6), tumor necrosis factor-α (TNFα), macrophage inflammatory protein 1α (MIP-1α), the chemokine RANTES, fractalkine, interferon γ (IFNγ), MIP-1γ, IP10 and IL-1 receptor α (IL-1α) in NRVMs expressing 2057del2 plakoglobin compared to control cells. The cytokine profile of mutant cells was normalized following treatment with SB2 for 48 hours.
To investigate whether SB216763 exerts salutary effects in a mammalian model of ACM, we treated NRVMs expressing mutant plakoglobin with the drug and determined whether the disease features previously demonstrated in these cells were reversed. After only 24 hours of exposure to SB216763, the distribution of plakoglobin and Cx43 appeared to be restored to normal. SB216763 also virtually eliminated apoptosis (Fig 2C) and normalized cytokine levels in media recovered from transfected cultures (Fig 2D). And as observed in fish, exposure of 2057del2 NRVMs to SB216763 for only 24 hours reversed action potential remodeling.24 Finally, SB216763 restored the normal localization of Nav1.5 and SAP97 in mutant plakoglobin-expressing myocytes (Fig 2A and 2B) without apparent changes in total protein expression levels. These observations provide independent evidence that changes in cellular electrophysiology in ACM are related to defective forward trafficking of key ion channel proteins rather to insufficient ion channel protein production. They also suggest that a common mechanism, sensitive to the mitigating effects of a single small molecule, is responsible for both the myocyte injury and the arrhythmia phenotype characterizing ACM.24
To further investigate the role of SAP97 in ACM, we knocked down its expression in normal NRVMs using short hairpin RNA (shRNA) and examined the distribution of key intercalated disk proteins.24 Knock-down of SAP97 prevented junctional localization of Nav1.5, as reported.28 However, it also reduced junctional immunoreactive signal for plakoglobin, but not for other cell-cell adhesion proteins including plakophilin-2 and desmoplakin. This unexpected observation suggests a central role for SAP97 in localization of plakoglobin in cell-cell junctions. It also potentially links abnormal trafficking mediated by SAP97 to Wnt signaling abnormalities in ACM.24
Normal NRVMs respond to even brief intervals of cyclical uniaxial stretch by up-regulating immunoreactive signal for various cell-cell junction proteins, including plakoglobin and Cx43.31 By contrast, NRVMs expressing an ACM-causing mutation, fail to up-regulate junctional signal for these proteins in response to stretch and instead show greatly increased levels of apoptosis.24 These results are consistent with clinical studies suggesting that increased mechanical stress in the form of endurance exercise increases the risk of arrhythmias and accelerates disease progression in patients with ACM.14 Yet, normal responses to stress are restored once mutant NRVMs are exposed to SB216763 for 24 hours.24
Finally, to investigate the effects of SB216763 on human cells, we performed experiments on induced pluripotent stem cell (iPSC)-derived cardiac myocytes generated from peripheral blood mononuclear cells obtained from ACM patients bearing two different mutations in the gene encoding plakophilin-2 (Q617X and 2013delC).24 iPSC-derived cardiac myocytes from both probands showed significantly reduced immunoreactive signal for plakoglobin, Cx43, Nav1.5 and SAP97 at cell-cell junctions compared to myocytes derived from their unaffected, mutation-negative respective siblings. Distribution of all four proteins was, however, restored in both myocyte lines following a 24hr exposure to SB216763, validating our observations in fish and NRVM models of ACM.24
Conclusions
Though relatively uncommon, ACM is a highly arrhythmogenic form of human heart disease and a significant cause of SCD in the young, and particularly in athletes. Features of the molecular pathology of ACM in patients suggest that defective trafficking of key proteins to the intercalated disk may play a role in disease pathogenesis. Additional studies in zebrafish and mammalian models of ACM further support such a mechanism. And, high-throughput chemical screening in a fish model has identified a small molecule that appears to be remarkably effective in preventing or reversing selected features of the molecular pathology identified in patients with ACM. There is still much to discover about the pathogenesis of this disease. Future studies may determine whether SB216763 or a related compound will be of value in treating patients with ACM and possibly other heart diseases associated with a high risk of sudden death.
Acknowledgments
This work was supported by NIH grants R01 HL102361, R01 HL109264, and RCHL100110; a Scientist Development Grant from the American Heart Association (12SDG8800009) and a Harvard Stem Cell Institute Award.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Basso C, Corrado D, Marcus FI, Nava A, Thiene G. Arrhythmogenic right ventricular cardiomyopathy. Lancet. 2009;373:1289–1300. doi: 10.1016/S0140-6736(09)60256-7. [DOI] [PubMed] [Google Scholar]
- 2.Saffitz JE. The pathobiology of arrhythmogenic cardiomyopathy. Annu Rev Pathol. 2011;28:299–321. doi: 10.1146/annurev-pathol-011110-130151. [DOI] [PubMed] [Google Scholar]
- 3.Peters S, Trümmel M, Meyners W. Prevalence of right ventricular dysplasia-cardiomyopathy in a non-referral hospital. Int J Cardiol. 2008;97:499–501. doi: 10.1016/j.ijcard.2003.10.037. [DOI] [PubMed] [Google Scholar]
- 4.Basso C, Corrado D, Thiene G. Cardiovascular causes of sudden death in young individuals including athletes. Cardiol Rev. 1999;7:127–135. doi: 10.1097/00045415-199905000-00009. [DOI] [PubMed] [Google Scholar]
- 5.Sen-Chowdhry S, Syrris P, McKenna WJ. Genetics of right ventricular cardiomyopathy. J Cardiovasc Electrophysiol. 2005;16:927–935. doi: 10.1111/j.1540-8167.2005.40842.x. [DOI] [PubMed] [Google Scholar]
- 6.Marcus FI, McKenna WJ, Sherrill D, et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the task force criteria. Circulation. 2010;121:1533–1541. doi: 10.1161/CIRCULATIONAHA.108.840827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McKoy G, Protonotarios N, Crosby A, et al. Identification of a deletion in plakoglobin in arrhythmogenic right ventricular cardiomyopathy with palmoplantar keratoderma and woolly hair (Naxos disease) Lancet. 2000;335:2119–2124. doi: 10.1016/S0140-6736(00)02379-5. [DOI] [PubMed] [Google Scholar]
- 8.Hamilton RM, Fidler L. Right ventricular cardiomyopathy in the young: an emerging challenge. Heart Rhythm. 2009;6:571–575. doi: 10.1016/j.hrthm.2009.01.026. [DOI] [PubMed] [Google Scholar]
- 9.Halphen C, Beaufils P, Azancot I, Baudouy P, Manne B, Slama R. Recurrent ventricular tachycardia due to right ventricular dysplasia. Association with left ventricular anomalies. Arch Mal Coeur Vaiss. 1981;74:1113–1118. [PubMed] [Google Scholar]
- 10.Dungan WT, Garson A, Jr, Gillette PC. Arrhythmogenic right ventricular dysplasia: a cause of ventricular tachycardia in children with apparently normal hearts. Am Heart J. 1981;102:745–750. doi: 10.1016/0002-8703(81)90101-0. [DOI] [PubMed] [Google Scholar]
- 11.Deac M, Alpendurada F, Fanaie F, et al. Prognostic value of cardiovascular magnetic resonance in patients with suspected arrhythmogenic right ventricular cardiomyopathy. Int J Cardiol. 2013;168:3514–3521. doi: 10.1016/j.ijcard.2013.04.208. [DOI] [PubMed] [Google Scholar]
- 12.Aviram G, Fishman JE, Young ML, Redha E, Biliciler-Denktas G, Rodriguez MM. MR evaluation of arrhythmogenic right ventricular cardiomyopathy in pediatric patients. AJR Am J Roentgenol. 2003;180:1135–1141. doi: 10.2214/ajr.180.4.1801135. [DOI] [PubMed] [Google Scholar]
- 13.Fogel MA, Weinberg PM, Harris M, Rhodes L. Usefulness of magnetic resonance imaging for the diagnosis of right ventricular dysplasia in children. Am J Cardiol. 2006;98:1232–1237. doi: 10.1016/j.amjcard.2005.11.045. [DOI] [PubMed] [Google Scholar]
- 14.James CA, Bhonsale A, Tichnell C, et al. Exercise increases age-related penetrance and arrhythmic risk in arrhythmogenic right ventricular dysplasia/cardiomyopathy-associated desmosomal mutation carriers. J Am Coll Cardiol. 2013;62:1290–1297. doi: 10.1016/j.jacc.2013.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Asimaki A, Tandri H, Huang H, et al. A new diagnostic test for arrhythmogenic right ventricular cardiomyopathy. N Engl J Med. 2009;360:1075–1084. doi: 10.1056/NEJMoa0808138. [DOI] [PubMed] [Google Scholar]
- 16.Ben-Ze’ev A, Geiger B. Differential molecular interactions of beta-catenin and plakoglobin in adhesion, signaling and cancer. Curr Opin Cell Biol. 1998;10:629–639. doi: 10.1016/s0955-0674(98)80039-2. [DOI] [PubMed] [Google Scholar]
- 17.Zhurinsky J, Shtutman M, Ben-Ze’ev A. Plakoglobin and beta-catenin: protein interactions, regulation and biological roles. J Cell Sci. 2000;113:3127–3139. doi: 10.1242/jcs.113.18.3127. [DOI] [PubMed] [Google Scholar]
- 18.Rao TP, Kuhl M. An updated overview on Wnt signaling pathways. A prelude for more. Circ Res. 2010;106:1798–1806. doi: 10.1161/CIRCRESAHA.110.219840. [DOI] [PubMed] [Google Scholar]
- 19.Garcia-Gras E, Lombardi R, Giocondo MJ, et al. Suppression of canonical Wnt/beta catenin signaling by nuclear plakoglobin recapitulates phenotype of arrhythmogenic right ventricular cardiomyopathy. J Clin Invest. 2006;116:2012–2021. doi: 10.1172/JCI27751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kaplan SR, Gard JJ, Protonotarios N, et al. Remodeling of myocyte gap junctions in arrhythmogenic right ventricular cardiomyopathy due to a deletion in plakoglobin (Naxos disease) Heart Rhythm. 2004;1:3–11. doi: 10.1016/j.hrthm.2004.01.001. [DOI] [PubMed] [Google Scholar]
- 21.Bassso C, Thiene G, Corrado D, Angelini A, Nava A, Valente M. Arrhythmogenic right ventricular cardiomyopathy: dysplasia, dystrophy or myocarditis? Circulation. 1996;94:983–991. doi: 10.1161/01.cir.94.5.983. [DOI] [PubMed] [Google Scholar]
- 22.Asimaki A, Tandri H, Duffy ER, et al. Altered desmosomal proteins in granulomatous myocarditis and potential pathogenic links to arrhythmogenic right ventricular cardiomyopathy. Circ Arrhythm Electrophysiol. 2011;4:743–752. doi: 10.1161/CIRCEP.111.964890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sato PY, Musa H, Coombs W, et al. Loss of plakophilin-2 expression leads to decreased sodium current and slower conduction velocity in cultured cardiac myocytes. Circ Res. 2009;105:523–526. doi: 10.1161/CIRCRESAHA.109.201418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Asimaki A, Kapoor S, Plovie E, et al. Identification of a new modulator of the intercalated disc in a zebrafish model of arrhythmogenic cardiomyopathy. Sci Transl Med. 2014;6:240ra74. doi: 10.1126/scitranslmed.3008008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cerrone M, Lin X, Zhang M, et al. Missense mutations in plakophilin-2 cause sodium current deficit and associate with a Brugada syndrome phenotype. Circulation. 2014;129:1092–1103. doi: 10.1161/CIRCULATIONAHA.113.003077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim C, Wong J, Wen J, et al. Studying arrhythmogenic right ventricular dysplasia with patient-specific iPSCs. Nature. 2013;494:105–110. doi: 10.1038/nature11799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Noorman M, Hakim S, Kessler E, et al. Remodeling of the cardiac sodium channel, connexin43 and plakoglobin at the intercalated disk in patients with arrhythmogenic cardiomyopathy. Heart Rhythm. 2013;10:412–419. doi: 10.1016/j.hrthm.2012.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Milstein ML, Musa H, Balbuena DP, et al. Dynamic reciprocity of sodium and potassium channel expression in a macromolecular complex controls cardiac excitability and arrhythmia. Proc Natl Acad Sci USA. 2012;109:E2134–E2143. doi: 10.1073/pnas.1109370109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gutman GA, Chandy KG, Grissmer S, et al. Nomenclature and molecular relationships of voltage-gated potassium channels. Pharmacol Rev. 2005;57:473–508. doi: 10.1124/pr.57.4.10. [DOI] [PubMed] [Google Scholar]
- 30.Eldar-Finkelman H, Martinez A. GSK-3 inhibitors: preclinical and clinical focus on CNS. Front Mol Neurosci. 2011;4:32. doi: 10.3389/fnmol.2011.00032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yamada K, Green KG, Samarel AM, Saffitz JE. Distinct pathways regulate expression of cardiac electrical and mechanical junction proteins in response to stretch. Circ Res. 2005;97:346–353. doi: 10.1161/01.RES.0000178788.76568.8a. [DOI] [PubMed] [Google Scholar]