

Abstract
Long-QT syndromes (LQTSs) have been described in all ages and are a significant cause of cardiovascular mortality, especially in structurally normal hearts. Abnormalities in transmembrane ion conduction channels and structural proteins produce these clinical syndromes, labeled LQT1-LQT12; however, genotype-positive patients still represent only about 70% of LQTSs. Future research will determine the etiology of the remaining cases, further risk-stratify the known genetic defects, improve current treatment options for these syndromes, and uncover novel therapies.
Introduction
In 1957, Jervell and Lange-Nielsen described a family with congenital deafness, prolongation of the QT interval on surface electrocardiogram (ECG), and sudden death in 3 of the 4 children. A few years later, Romano and Ward independently described a similar familial cardiac arrhythmia syndrome, without deafness (Romano et al. 1963, Ward 1964). An entire field of investigation has grown from Jervell and Lange-Nielsen’s initial description, with 50 years of remarkable progress since their original suggestion of asystole as the etiology of the sudden death.
Long-QT syndromes (LQTSs) cause a significant proportion of premature sudden cardiac death, especially in patients with structurally normal hearts. Estimates suggest that LQTS deaths occur on the order of 5000 deaths per year in the United States (Vincent 1998).
Long-QT syndrome can be subclassified into congenital and acquired forms. Thus far, the genes that underpin the congenital forms of LQTS have been found in cardiac ion channels, membrane-associated proteins, and trafficking proteins in electrically active cells. Twelve genetic forms of LQTS have been described, given the uninspired names LQT1-LQT12, based on the order of discovery. The autosomal recessive Jervell-Lange-Nielsen syndrome, the autosomal dominant Romano-Ward syndrome, as well as other related diagnoses, including Andersen-Tawil syndrome, Timothy syndrome and some cases of sudden infant death syndrome, have been linked with LQTS. Drugs or other acquired exposures may unmask previously unsuspected congenital concealed mutations or prolong the QT interval even in the absence of known genetic abnormalities (Czosek and Berul 2008, Kannankeril 2008).
Diagnosis
Prolongation of the QT interval is the hallmark ECG feature of LQTS. Normally, the QT shortens with increasing heart rate and prolongs with lower heart rates; thus, a rate correction is typically performed with the use of Bazett’s formula. The corrected QT interval is defined as the QT interval divided by square root of the preceding RR interval: QTc = QT/√RR (Figure 1). Although calculating Bazett’s formula is straight-forward, controversy exists about how to measure its primary variable, the QT interval. QT intervals vary throughout a standard 12-lead ECG. Published normal measurements are often based on lead II; however, some studies use the clearest QT measurements on each ECG or the longest measurement that can be calculated from a given ECG (Al-Khatib et al. 2003). With signal averaging technology, summation techniques have been used as well (Rijnbeek et al. 2001).
Figure 1.
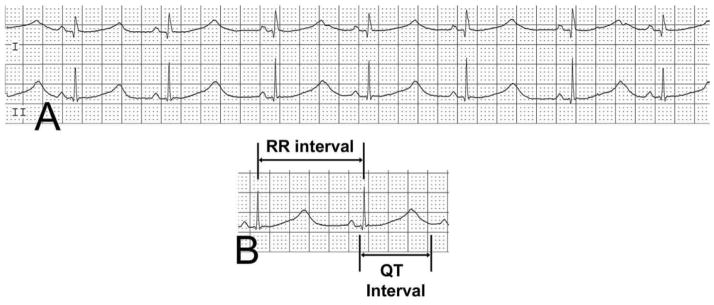
Surface ECG showing a prolonged QTc of 687. Measurement of the QTc is calculated as the duration of the QT interval, divided by the square root of the preceding RR interval.
The mean QTc interval should be calculated over 3 to 5 consecutive beats. Averaging the results over serial ECGs on different dates can produce a consensus measurement of any one patient’s true QTc. A U wave, if present, is typically excluded from the calculation unless it is merged with the T wave and is at least 50% of the T wave’s amplitude. This measurement tradition is based on convention rather than scientific rationale. In special cases, such as atrial fibrillation or marked sinus arrhythmia, where the QT interval and heart rate can vary dramatically throughout a single ECG, either a larger number of beats can be averaged or the extremes of QT interval on the ECG record can be measured and averaged (Al-Khatib et al. 2003). In patients with a wide QRS complex, use of the corrected JT interval (measured from the J point at the termination of the QRS complex to the termination of the T wave) has been suggested (Berul et al. 1994). Other authors have suggested a using a broader definition of a prolonged QTc in the setting of a wide QRS, such as any QTc greater than 500 milliseconds (Al-Khatib et al. 2003).
Mathematically, Bazett’s formula is imperfect. It overcorrects at high heart rates and under-corrects at low heart rates. Some studies have attempted to model a best-fit regression curve on known patient data. The most prominent of the patient-based modeling efforts has been the model proposed from studying the Framingham heart data [QTc = QT + 0.154 × (1 RR)] (Sagie et al. 1992). In drug trials and other research protocols, alternative methods have been used, including using cube root derivations and individual regression analysis on a patient-by-patient basis (Funck-Brentano and Jaillon 1993, Hnatkova and Malik 1999, Malik 2001). However, the simplicity of Bazett’s formula and the large amount of research that has already drawn conclusions based on its measurement have made it the dominant formula for correction of the QT interval in clinical medicine.
Population-based studies, such as a Dutch study of 1912 healthy children, show normal QTc intervals up to 462 milliseconds (Rijnbeek et al. 2001). This is not a firm cutoff. The International Long-QT Syndrome registry has classified patients as electrocardiographically affected (QTc >470 milliseconds), borderline (QTc 440–469 milliseconds), or electrocardiographically unaffected (QTc <440 milliseconds) (Goldenberg et al. 2008a). Even in affected families, the QTc in members who carry the affected allele and those that do not carry the affected allele overlap, with some affected members having a QTc less than 440 ms (Kaufman et al. 2001, Vincent et al. 1992). As a single tool, the QTc is an imperfect method of diagnosing LQTS.
Other features of the ECG can provide additive information. The repolarization abnormalities of LQTS can manifest as T- wave alternans, where alternating positive and negative inscriptions of the T wave are recorded on the ECG (macroscopic T-wave alternans). Microscopic T-wave alternans also occurs in LQTS and refers to the phenomenon of alterations in the shape and magnitude of the T wave, suggesting repolarization instability at the cellular level, giving rise to areas of local tissue repolarization abnormalities.
Long-QT syndrome creates an abnormal, heterogeneous pattern of repolarization in cardiac tissue. This heterogeneous repolarization increases the variability in QT duration throughout the ECG. QT dispersion was previously used as a simplistic, noninvasive measurement of this variability. It is calculated by subtracting the minimum QT from the maximum QT on a single ECG. QT dispersion has been shown to be increased in patients with LQTS; however, this method is not in common clinical use owing to the significant overlap in QT dispersion in affected and unaffected patients and lack of scientific validation (Shah et al. 1997).
Sinus bradycardia is also observed in some LQTS patients, particularly in children. In addition, infants with fast baseline heart rates and correspondingly short RR intervals can occasionally have QTc intervals sufficiently long that ongoing ventricular repolarization blocks conduction of the subsequent p wave to the ventricle, producing 2:1 heart block (Figure 2).
Figure 2.
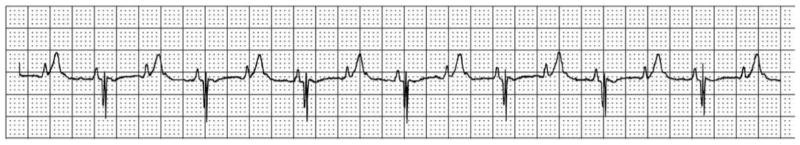
Surface ECG rhythm strip showing a QTc of 675 and 2:1 atrioventricular block.
Twenty-four-hour recordings of ECG activity (Holter monitors) may establish longer records of variation in heart rate, QT interval, T-wave abnormalities, and may document spontaneous ectopy. During exercise, failure of the normal shortening of the QT interval with increasing heart rate may be observed in patients with LQTS and the U wave may increase in prominence (Vincent et al. 1991). Exercise testing may also unveil concealed T-wave alternans during exertion or recovery. Provocative testing with epinephrine or isoproterenol may uncover patients with suspected, but unproven, LQTS. Healthy subjects will shorten their QT interval relative to heart rate (shorter QTc) as the infusion increases (Berul et al. 1998, Vincent et al. 1991). Vyas et al. (2006) treated 125 patients with a 25-minute infusion protocol and found that patients with LQT1 paradoxically prolonged their QTc in the setting of epinephrine testing. Patients with LQTS may prolong their QTc during facial immersion, whereas the QTc in patients without LQTS remains unchanged (Katagiri-Kawade et al. 1995).
Risk Factors
There is wide phenotypic variability in the clinical course of LQTS, related in part to age, sex, genotype, mutation location, symptoms and treatment status. Two major resources have contributed many of the available data on LQTS risk factors: the International Long QT Registry and the Pediatric and Congenital Electrophysiology Society. Registry data suggest that among children younger than 13 years, girls had a slightly longer corrected QT interval (QTc) than boys; however, boys experienced cardiac events at a markedly higher rate (5% vs 1%). Boys with a QTc of greater than 500 milliseconds were at highest risk, with a hazard ratio as high as 12 times the risk for age-matched girls (Goldenberg et al. 2008b, Locati et al. 1998). Prior syncope was associated with a higher risk of cardiac events for all patients. Remote syncope (syncope more than 2 years before evaluation) was associated with a hazard ratio of 2.67 for boys and 12.04 for girls over baseline risk. A history of recent syncope (within 2 years) more than doubled the hazard ratio in both groups (Goldenberg et al. 2008b). Among adolescents (13–20 years old), a markedly increased QTc and recent syncope remained significant risk factors, although the sex difference was no longer statistically significant (Hobbs et al. 2006). Across multiple age and sex groups, as the QTc increased, the risk of cardiac events increased as well (Moss et al. 1991b, Zareba et al. 1998). In several studies, a QTc greater than 500 milliseconds predicted a marked elevation in risk and is a clinically useful cutoff for high-risk QTc duration (Goldenberg et al. 2008b, Priori et al. 2003, Sauer et al. 2007).
Women who had LQTS and were pregnant constituted a notable subgroup. A lower cardiac event rate was observed during pregnancy, but in the 9-month postpartum period, there was an increased risk, with a hazard ratio of 2.7 vs the baseline pre-pregnancy risk. Women with the LQT2 genotype had the greatest increase in risk. Fortunately, β-blocker therapy reduced cardiac events in the postpartum period, with the hazard ratio for treated women only 0.37 times that of untreated women (Seth et al. 2007).
Genetics
To date, 12 genes have been linked to LQTS; research is ongoing to identify more and to gain understanding into genetic implications and genotype-phenotype correlations. Comprehensive genetic screening can lead to a specific diagnosis in approximately 70% of patients with LQTS (Figure 3) (Splawski et al. 2000, Tester et al. 2005). LQT1, LQT2, and LQT3 together represent most of the genotype-positive patients. In patients with a genetic diagnosis, approximately 45% have LQT1, 34% have LQT2, and 10% have LQT3 (Tester et al. 2005).
Figure 3.
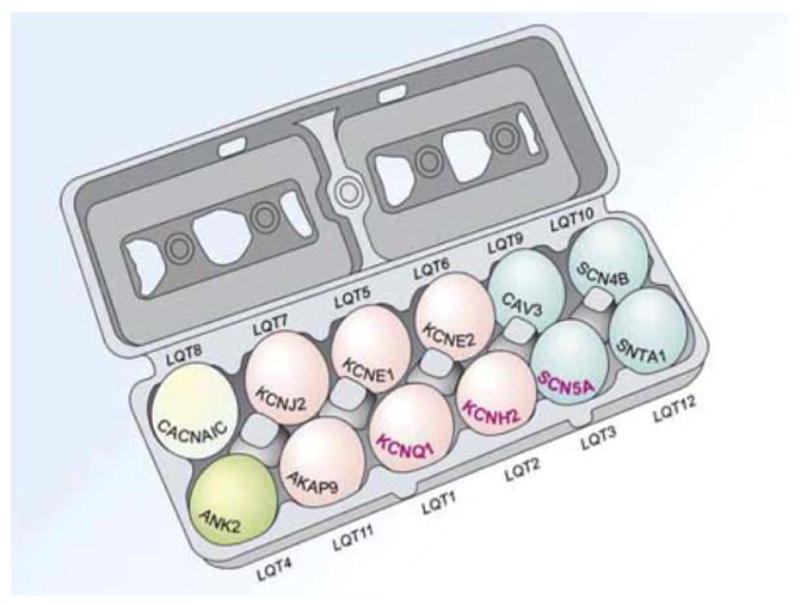
Cartoon diagram of the 12 known LQTS genes and the LQT syndromes associated with mutations in each gene (LQT1-LQT12). The 3 most common (LQT1-3) are in bold. Eggs are color-coded by the functionality of each gene and its respective protein: pink, potassium channel; blue, sodium channel; green, calcium channel; ivory, structural protein.
The effects of genotype on risk have been studied by several groups, primarily by looking at an end point of “cardiac events.” Although this end point has been described slightly differently in different studies, it has typically referred to syncope, aborted cardiac arrest, and sudden death. In a registry study, Zareba et al. studied 246 genotype positive patients and found that LQT1 had the highest risk of a first cardiac event. By the age of 15, 53% of subjects in the LQT1 group had experienced syncope, aborted cardiac arrest, or death vs 29% in the LQT2 group and 6% in the LQT3 group. However, a study by Priori et al. of 580 genotype-positive patients showed that untreated LQT1 patients had the lowest risk of a first cardiac event before 40 years of age and, in 2008, Goldenberg et al. reported that in children from 1 to 12 years of age there was no additional risk of cardiac events by genotype once clinical factors were accounted for (Goldenberg et al. 2008b, Priori et al. 2003). By adulthood, the registry data suggest that LQT1 has an intermediate risk between LQT2 and LQT3, with LQT2 having the highest risk of any cardiac events (Sauer et al. 2007).
Although the LQT3 genotype has not been associated with the highest risk of cardiac events, those events that do occur were more likely to be fatal. Zareba et al. (1998) showed that 20% of cardiac events in patients with LQT3 were likely to be fatal vs only 4% of events in LQT1 and LQT2.
There is clearly not a consensus regarding risk stratification by genotype. When LQTS was first identified, patients primarily presented with a markedly long QT interval on ECG or symptoms. Consequently, the prevalence of all cardiac events was fairly high in this population. As LQTS has become more thoroughly characterized and the genetic component of the syndrome has been more fully appreciated, registries and clinical studies have broadened to include a wide variety of LQT phenotypes, from clinically affected probands to family members who are asymptomatic but genotypically affected. It is unsurprising to find that as groups with heterogeneous clinical features are compared, there are some differences in genotypic risk factors. Furthermore, the information we have is primarily limited to LQT1-LQT3. Nine additional genes have been identified and the clinical risk of these genetic disorders is still largely unknown. Although the clinical phenotypes in Anderson-Tawil syndrome (LQT7) and Timothy syndrome (LQT8) have been well described, further work is still required on their risk of sudden death from LQTS.
The known LQTS genetic defects were named in order of discovery but are presented here as they are associated with their respective proteins. Most defects lie in ion channels and modulate either the inward rectifying potassium current or the inward sodium current of the heart.
Potassium Channels
There are 2 major inward rectifying potassium current channels: a slowly activating channel (IKs) and a rapidly activating channel (IKr). Each of these channels is composed of its own α and β subunits. Four α subunits assemble with minK β subunits to create the IKs channel. Similarly, 4 α subunits assemble with MiRP1 β-subunits to create the IKr channel.
The KCNQ1 gene (previously published as KVLQT1), encodes the α subunit of the IKs channel. Alterations in this gene produce the LQT1 genotype and may cause the Jervell-Lange-Nielsen syndrome. Many individual mutations have been described. Crotti et al. (2007) studied 78 patients with a specific mutation, KCNQ1/A341V, and found that the presence of this mutation predicts high clinical severity. In addition to its cardiac defects, KCNQ1 is also found to be expressed in mouse inner ear, which correlates with the deafness in the Jervell-Lange-Nielsen syndrome (Neyroud et al. 1997).
Clinically, the LQT1 population has been found to have a shorter mean QTc than patients with LQT2 and LQT3 (466, 490, and 496 milliseconds, respectively) (Priori et al. 2003). In a study of 371 patients with LQT1, 62% of events were associated with exercise. LQT1 also accounts for most cases of cardiac events associated with swimming. However, LQT1 is not the only cause of swimming-related arrhythmias. Cardiac events while swimming have also been associated with LQT2 and with defects in the RYR2-encoded ryanodine receptor, a calcium release channel that is the responsible molecular mechanism for most cases of catecholaminergic polymorphic ventricular tachycardia.
The KCNE1 gene encodes a β-subunit protein, minK that associates with the product of KCNQ1 to create the IKs cannel. Mutations in KCNE1 cause LQT5, which is a rare mutation, occurring in fewer than 1% of all genotyped patients. The conclusion that LQT1 and LQT5 genes both contribute to the IKs channel is supported by the clinical observations that homozygous mutations in KCNE1 can independently contribute to the development of Jervell-Lange-Nielsen syndrome (Schulzse-Bahr et al. 1997).
The other major potassium channel implicated in LQTS is the IKr channel. The α subunit of this channel is encoded by KCNH2, also known as HERG—the human “ether-a-go-go” related gene. Mutations in this gene cause LQT2, the second most common form of genotype-positive LQTS. Most drugs that cause acquired LQTS do so by blocking IKr (Hancox and James 2008). Exercise, emotion, and noise (“arousal events”) have been noted to cause cardiac symptoms more frequently in LQT1 and LQT2 than in LQT3. Cardiac events with auditory arousal such as alarm clocks, horns, or telephones are most typically seen in LQT2. LQT2 may also predispose patients to pause-dependent mechanism of ventricular tachycardia (VT). Based on a series of 50 patients, 68% of LQT2 patients had pause-dependent torsades de pointes vs no LQT1 patients with a similar trigger (Tan et al. 2006).
The minK-related protein (MiRP1) is the corresponding β subunit for the IKr channel. MiRP1 is encoded by the KCNE2 gene; mutations in KCNE2 cause LQT6. Although mutations in MiRP1 have been noted in drug-associated LQTS as well, patients with LQT6 are rare (<1%) and the clinical consequences of LQT6 have been incompletely described.
Defects in other potassium channels have been reported in individual patients, suggesting that they only rarely account for clinical cases; however, they may continue to provide insight into the pathophysiology of the disease. Defects in the potassium channel gene KCNJ2 cause LQT7 (Andersen-Tawil syndrome) and defects in the potassium channel gene encoded by ACAP9 cause LQT11. Genotype-specific risk factors have not yet been described for these less common mutations.
Sodium Channels
Loss-of-function mutations in the rectifying current modulated by potassium channels cause LQTS by causing instability during repolarization. Gain-of-function mutations in the sodium channels have also been described to cause LQTS. By permitting persistent, noninactivating inward sodium current throughout repolarization, defects in the sodium channel cause prolongation of the cardiac action potential. LQT3 is caused by defects in the voltage-dependent sodium channel gene SCN5A. Men with LQT3 appear to be at somewhat higher risk, although the numbers remain modest (Schwartz et al. 2001). Patients with LQT3 have the highest risk of events when at rest or asleep and a relatively low risk of events during arousal. β-Adrenergic blocker therapy, which has been shown to be effective therapy in LQTS overall, may potentially be less efficacious in this subgroup, although the data are not robust. Some studies suggest that propranolol may be effective owing to its nonspecific ion channel blocking effects rather than β-blockade. Genotype-specific treatment may improve clinical response. As an example, mexiletine has been shown to affect the gating properties of sodium channels with SCN5A (LQT3) mutations and may contribute to pharmacotherapy in patients with these mutations (Ruan et al. 2007).
Three other sodium channel genes have been described very recently. CAV3 (LQT9), SCN4B (LQT10), and SNTA1 (LQT12) encode proteins associated with sodium channels (Medeiros-Domingo et al. 2007, Tester and Ackerman 2008, Ueda et al. 2008, Vatta et al. 2006). To date, very few patients have been reported with abnormalities of these genes.
Other Proteins
It is notable that LQTS mutations do not fall exclusively within the purview of sodium and potassium ion channels. Mutation in ankyrin-B causes LQT4, which is a minor contributor to genotype- positive patients (3%) (Sherman et al. 2005). Ankyrin-B is a structural protein that binds with a sodium/potassium ATPase, a sodium/calcium exchanger, and inositol-1,4,5-triphosphate receptors. Abnormalities of ankyrin-B reduce targeting of proteins to the transverse tubules and lower overall protein level. Alterations in calcium ion signaling, linked to ankyrin-B, have been shown to result in extrasystoles and may be the trigger for arrhythmias in LQT4 (Mohler and Bennett 2005, Yong et al. 2003). A single calcium channel (CACNA1C, LQT8) gene has been described as well. Failure of the calcium channel to close may cause depolarizing calcium currents to persist throughout repolarization.
Treatment
The arrhythmia most commonly associated with LQTS is torsades de pointes; however, other ventricular arrhythmias, including monomorphic VT, polymorphic VT, and ventricular extrasystoles, have been described as well. Other arrhythmias, such as sinus node dysfunction, atrial fibrillation, and atrioventricular block exist, but treatment modalities primarily focus on prevention and therapy of the life-threatening consequences of VT.
Several large studies from the Pediatric and Congenital Electrophysiology Society and the International Long QT Registry, supported by a number of smaller studies, show that lifelong β-blocker therapy is effective at reducing fatal cardiac events (Garson et al. 1993, Sauer et al. 2007). Depending on the study population, β-blocker therapy is associated with a 42% to 78% reduction of aborted cardiac arrest or sudden cardiac death (Goldenberg et al. 2008b, Schwartz et al. 1991, Seth et al. 2007). Compliance with medications is never perfect and, as mentioned above, some studies have suggested that although risk of cardiac events is decreased in patients with LQT1 or LQT2, there may be little reduction of risk in patients with LQT3 (Gow 2001).
Other antiarrhythmic medications, including labetolol, verapamil, nicorandil, mexiletine, and lidocaine, have been explored in individual patients and in animal models but have not been tested in large clinical trials (Table 1). These additional antiarrhythmic medications may be of particular interest moving forward and they may be the first step toward gene-specific pharmacotherapy.
Table 1.
Medical and procedural treatment options in LQTS
| Medical treatment | Notes |
|---|---|
| β-Adrenergic blockers | Documented to reduce risk of cardiac events |
| Labetolol | Described in limited clinical situations. Has both α- and β-adrenergic blockage actions, less specific antisympathetic effects |
| Verapamil | Hypothesized to decrease calcium channel– mediated heterogeneity of repolarization |
| Nicorandil | Potassium channel opener decreases heterogeneity of repolarization and shortens action potential duration in LQT1 and LQT2 (and other K+-channel mutation mediated LQTS) |
| Mexiletine | Sodium channel blockade thought to decrease gain-of function mutation in LQT3 |
| Lidocaine | Blocks inactivated sodium channels, thought to decrease sodium leak current in LQT3 |
| Procedural treatment | Notes |
|---|---|
| ICD placement | Recommended for patients with aborted cardiac arrest or VT on medical treatment, other indications (see text) |
| Pacemaker placement | Recommended for rate smoothing in pause-related VT |
| Left cardiac sympathetic denervation | Decreases frequency of cardiac events. May decrease frequency of ICD defibrillation |
| Electrophysiologic study/catheter ablation | Case reports of successful ablation of arrhythmogenic foci. Not in common diagnostic or therapeutic use. |
VT = Ventricular tachycardia, ICD = Implantable cardioverter-defibrillator
Triggered activity may induce torsades de pointes (Pelleg et al. 1995, Vos et al. 1995). An electrical pause can precipitate heterogeneity in repolarization and cause the subsequent QT interval to prolong. If a premature beat occurs during the vulnerable portion of the repolarization period, VT can ensue. Pacemakers prohibit long pauses by setting a lower rate limit. When feasible, atrial-based devices with rate-smoothing algorithms that shorten the QT interval may be beneficial. Patients with LQT2 and LQT3, who may be especially susceptible to pause-dependent phenomena, may particularly benefit from this strategy. The data for pacemakers are limited to studies on small numbers of patients (Eldar et al. 1987, Moss et al. 1991a). The largest series to date concludes that combined pacemaker and β-blocker therapy may be beneficial but notes a cardiac event rate of 24% in this group and suggests that backup implantable cardiac defibrillators (ICDs) may be indicated (Dorostkar et al. 1999). The most recent consensus guidelines state that “permanent pacing is indicated for sustained pause-dependent VT, with or without QT prolongation” (Epstein et al. 2008).
Given the propensity toward ventricular tachyarrhythmias in LQTS, the placement of ICDs has become standard of care for a subgroup of patients considered to be at higher potential risk (Figure 4). Current guidelines suggest ICD implantation for selected patients with recurrent syncope despite drug therapy, sustained ventricular arrhythmias, or sudden cardiac arrest. In addition, the guidelines recommend consideration of ICDs for primary prevention in patients with a strong family history of sudden cardiac death or where compliance with medications is a strong concern (Epstein et al. 2008).
Figure 4.

(A) ICD ventricular intracardiac electrogram and a marker channel. The electrogram begins with sinus rhythm at a ventricular rate of 100 beats per minute with sudden initiation of a polymorphic ventricular rhythm at a rate of approximately 215 beats per minute. The marker channel assigns the electrical deflections as ventricular sensing (VS) and ventricular fibrillation detection (FS). (B) Ventricular intracardiac electrogram recording demonstrating successful ICD shock. The electrogram shows polymorphic ventricular tachycardia with a rate of 300 beats per minute. The marker channel assigns the electrical deflections as ventricular sensing (VS) and refractory (VR). The device discharges (CD) and the rhythm returns to a ventricular rate of approximately 110 beats per minute.
Implantable cardiac defibrillators have known risks, including lead fracture, dislodgement, infection, inappropriate discharge, and electrical storm. Studies in adults and children have suggested that there are at least short-term psychiatric consequences of ICD placement in some patients (DeMaso et al. 2004, Sears and Conti 2002) Devices may require replacement several times during life and younger patients are more likely to require reintervention. Careful consideration on a case-by-case basis is required.
Left cardiac sympathetic denervation is a process in which the lower half of the left stellate ganglion as well as the thoracic ganglia of T2 through T4 is ablated (Moss and McDonald 1971). Decreasing adrenergic stimulation from the left cardiac sympathetic network is hypothesized to suppress ectopic beats that may trigger arrhythmias. The procedure remains relevant in the post-ICD era as a useful adjunct to other therapies. It may decrease VT events and thus decrease the frequency of ICD dis charges, which may be painful or sufficiently frequent as to be debilitating. In a study of 174 patients after left cardiac sympathetic denervation, frequency of cardiac events per patient decreased by 91%. The procedure carries relatively low risks of Horner syndrome and surgical morbidity. A minimally invasive approach using videoscope-assisted thorascopic surgery for left cardiac sympathetic denervation is feasible (Atallah et al. 2008).
Invasive electrophysiologic ablation has been shown in selected cases to be able to suppress ventricular ectopy that may trigger VT but has not been reproducible and is currently not a routine part of LQTS evaluation or therapy (Stephenson and Berul 2007).
Future Directions
Genetic testing has transformed the study of LQTS, creating a cellular model for testing hypotheses. It opens the door to genetic risk stratification in the genotype-positive population. Hundreds of mutations have been described in a dozen genes so far; however, nearly 30% of the tested population remains without a genetic diagnosis, suggesting that there are further insights still to come in the field. Elucidation of these remaining genetic causes is a major frontier of further research, be they in proteins, pathway trafficking, or in regulators of DNA and RNA expression. It may be that many of the remaining genes are unique to individual family pedigrees (ie, private mutations).
As knowledge of the LQT genotypes expand, the ability to risk-stratify probands and their families will become more robust. Counseling will improve and the ability to assign therapy may become more tailored. Pharmacogenomic genotype-specific drug treatment is already being investigated. Long-QT syndrome has well-described mutations in single proteins and may eventually be a candidate for directed genomic therapy.
Overall, the LQTS has been a prototype genetic model for mechanistic understanding of inherited arrhythmias, growing from a single case study to a well-described heterogeneous syndrome that now constitutes a host of genetic diagnoses with a growing armory of treatment options.
Acknowledgments
This work was supported by the National Institutes of Health under award number: T32HL007572.
Footnotes
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
References
- Al-Khatib SM, LaPointe NM, Kramer JM, Califf RM. What clinicians should know about the QT interval. JAMA. 2003;289:2120–2127. doi: 10.1001/jama.289.16.2120. [DOI] [PubMed] [Google Scholar]
- Atallah J, Fynn-Thompson F, Cecchin F, et al. Video-assisted thoracoscopic cardiac denervation: a potential novel therapeutic option for children with intractable ventricular arrhythmias. Ann Thorac Surg. 2008;86:1620–1625. doi: 10.1016/j.athoracsur.2008.07.006. [DOI] [PubMed] [Google Scholar]
- Berul CI, Sweeten TL, Dubin, et al. Use of the rate-corrected JT interval for prediction of repolarization abnormalities in children. Am J Cardiol. 1994;74:1254–1257. doi: 10.1016/0002-9149(94)90558-4. [DOI] [PubMed] [Google Scholar]
- Berul CI, Sweeten TL, Hill SL, Vetter VL. Provocative testing in children with suspect congenital long QT syndrome. Ann Noninvasive Electrocardiol. 1998;3:3–11. [Google Scholar]
- Crotti L, Spazzolini C, Schwartz PJ, et al. The common long-QT syndrome mutation KCNQ1/A341V causes unusually severe clinical manifestations in patients with different ethnic backgrounds: toward a mutation-specific risk stratification. Circulation. 2007;116:2366–2375. doi: 10.1161/CIRCULATIONAHA.107.726950. [DOI] [PubMed] [Google Scholar]
- Czosek RJ, Berul CI. Congenital long-QT syndrome concealed by hypercalcemia in Williams syndrome. J Cardiovasc Electrophysiol. 2008 doi: 10.1111/j.1540-8167.2008.01263.x. [DOI] [PubMed] [Google Scholar]
- DeMaso DR, Lauretti A, Spieth L, et al. Psychosocial factors and quality of life in children and adolescents with implantable cardioverter-defibrillators. Am J Cardiol. 2004;93:582–587. doi: 10.1016/j.amjcard.2003.11.022. [DOI] [PubMed] [Google Scholar]
- Dorostkar PC, Eldar M, Belhassen B, Scheinman MM. Long-term follow-up of patients with long-QT syndrome treated with beta-blockers and continuous pacing. Circulation. 1999;100:2431–2436. doi: 10.1161/01.cir.100.24.2431. [DOI] [PubMed] [Google Scholar]
- Eldar M, Griffin JC, Abbott JA, et al. Permanent cardiac pacing in patients with the long QTsyndrome. J Am Coll Cardiol. 1987;10:600–607. doi: 10.1016/s0735-1097(87)80203-6. [DOI] [PubMed] [Google Scholar]
- Epstein AE, DiMarco JP, Ellenbogen KA, et al. ACC/AHA/HRS 2008 Guidelines for Device-Based Therapy of Cardiac Rhythm Abnormalities: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the ACC/AHA/NASPE 2002 Guideline Update for Implantation of Cardiac Pacemakers and Antiarrhythmia Devices): developed in collaboration with the American Association for Thoracic Surgery and Society of Thoracic Surgeons. Circulation. 2008;117:e350–e408. doi: 10.1161/CIRCUALTIONAHA.108.189742. [DOI] [PubMed] [Google Scholar]
- Funck-Brentano C, Jaillon P. Rate-corrected QT interval: techniques and limitations. Am J Cardiol. 1993;72:17B–22B. doi: 10.1016/0002-9149(93)90035-b. [DOI] [PubMed] [Google Scholar]
- Garson A, Jr, Dick M, 2nd, Fournier A, et al. The long QTsyndrome in children. An international study of 287 patients. Circulation. 1993;87:1866–1872. doi: 10.1161/01.cir.87.6.1866. [DOI] [PubMed] [Google Scholar]
- Goldenberg I, Moss AJ, Bradley J, et al. Long-QT syndrome after age 40. Circulation. 2008;117:2192–2201. doi: 10.1161/CIRCULATIONAHA.107.729368. [DOI] [PubMed] [Google Scholar]
- Goldenberg I, Moss AJ, Peterson DR, et al. Risk factors for aborted cardiac arrest and sudden cardiac death in children with the congenital long-QT syndrome. Circulation. 2008;117:2184–2191. doi: 10.1161/CIRCULATIONAHA.107.701243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gow RM. Effectiveness and limitations of beta-blocker therapy in congenital long QT syndrome. Circulation. 2001;103:E24. doi: 10.1161/01.cir.103.4.e24. [DOI] [PubMed] [Google Scholar]
- Hancox JC, James AF. Refining insights into high-affinity drug binding to the human ether-a-go-go–related gene potassium channel. Mol Pharmacol. 2008;73:1592–1595. doi: 10.1124/mol.108.047563. [DOI] [PubMed] [Google Scholar]
- Hnatkova K, Malik M. “Optimum” formulae for heart rate correction of the QT interval. Pacing Clin Electrophysiol. 1999;22:1683–1687. doi: 10.1111/j.1540-8159.1999.tb00390.x. [DOI] [PubMed] [Google Scholar]
- Hobbs JB, Peterson DR, Moss AJ, et al. Risk of aborted cardiac arrest or sudden cardiac death during adolescence in the long-QT syndrome. JAMA. 2006;296:1249–1254. doi: 10.1001/jama.296.10.1249. [DOI] [PubMed] [Google Scholar]
- Kannankeril PJ. Understanding drug-induced torsades de pointes: a genetic stance. Expert Opin Drug Saf. 2008;7:231–239. doi: 10.1517/14740338.7.3.231. [DOI] [PubMed] [Google Scholar]
- Katagiri-Kawade M, Ohe T, Arakaki Y, et al. Abnormal response to exercise, face immersion, and isoproterenol in children with the long-QT syndrome. Pacing Clin Electrophysiol. 1995;18:2128–2134. doi: 10.1111/j.1540-8159.1995.tb04637.x. [DOI] [PubMed] [Google Scholar]
- Kaufman ES, Priori SG, Napolitano C, et al. Electrocardiographic prediction of abnormal genotype in congenital long QT syndrome: experience in 101 related family members. J Cardiovasc Electrophysiol. 2001;12:455–461. doi: 10.1046/j.1540-8167.2001.00455.x. [DOI] [PubMed] [Google Scholar]
- Locati EH, Zareba W, Moss AJ, et al. Age- and sex-related differences in clinical manifestations in patients with congenital long-QT syndrome: findings from the International LQTS Registry. Circulation. 1998;97:2237–2244. doi: 10.1161/01.cir.97.22.2237. [DOI] [PubMed] [Google Scholar]
- Malik M. Problems of heart rate correction in assessment of drug-induced QT interval prolongation. J Cardiovasc Electrophysiol. 2001;12:411–420. doi: 10.1046/j.1540-8167.2001.00411.x. [DOI] [PubMed] [Google Scholar]
- Medeiros-Domingo A, Kaku T, Tester DJ, et al. SCN4B-encoded sodium channel beta4 subunit in congenital long-QT syndrome. Circulation. 2007;116:134–142. doi: 10.1161/CIRCULATIONAHA.106.659086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohler PJ, Bennett V. Ankyrin-based cardiac arrhythmias: a new class of channelopathies due to loss of cellular targeting. Curr Opin Cardiol. 2005;20:189–193. doi: 10.1097/01.hco.0000160372.95116.3e. [DOI] [PubMed] [Google Scholar]
- Moss AJ, Liu JE, Gottlieb S, et al. Efficacy of permanent pacing in the management of high-risk patients with long QT syndrome. Circulation. 1991;84:1524–1529. doi: 10.1161/01.cir.84.4.1524. [DOI] [PubMed] [Google Scholar]
- Moss AJ, McDonald J. Unilateral cervicothoracic sympathetic ganglionectomy for the treatment of long QT interval syndrome. N Engl J Med. 1971;285:903–904. doi: 10.1056/NEJM197110142851607. [DOI] [PubMed] [Google Scholar]
- Moss AJ, Schwartz PJ, Crampton RS, et al. The long QT syndrome. Prospective longitudinal study of 328 families. Circulation. 1991;84:1136–1144. doi: 10.1161/01.cir.84.3.1136. [DOI] [PubMed] [Google Scholar]
- Neyroud N, Tesson F, Denjoy I, et al. A novel mutation in the potassium channel gene KVLQT1 causes the Jervell and Lange-Nielsen cardioauditory syndrome. Nat Genet. 1997;15:186–189. doi: 10.1038/ng0297-186. [DOI] [PubMed] [Google Scholar]
- Pelleg A, Hurt CM, Xu J. Reproducible induction of EADs and torsade de pointes. Circulation. 1995;92:1666–1667. [PubMed] [Google Scholar]
- Priori SG, Schwartz PJ, Napolitano C, et al. Risk stratification in the long-QT syndrome. N Engl J Med. 2003;348:1866–1874. doi: 10.1056/NEJMoa022147. [DOI] [PubMed] [Google Scholar]
- Rijnbeek PR, Witsenburg M, Schrama E, et al. New normal limits for the paediatric electrocardiogram. Eur Heart J. 2001;22:702–711. doi: 10.1053/euhj.2000.2399. [DOI] [PubMed] [Google Scholar]
- Romano C, Gemme G, Pongiglione R. Aritmie cardiache rare dell’età pediatrica. ii. Accessi sincopali per fibrillazione ventricolare parossistica (presentazione del primo caso della letteratura pediatrica italiana) Clin Pediatr (Bologna) 1963;45:656–683. [PubMed] [Google Scholar]
- Ruan Y, Liu N, Bloise R, et al. Gating properties of SCN5A mutations and the response tomexiletine in long-QT syndrome type 3 patients. Circulation. 2007;116:1137–1144. doi: 10.1161/CIRCULATIONAHA.107.707877. [DOI] [PubMed] [Google Scholar]
- Sagie A, Larson MG, Goldberg RJ, et al. An improved method for adjusting the QT interval for heart rate (the Framingham Heart Study) Am J Cardiol. 1992;70:797–801. doi: 10.1016/0002-9149(92)90562-d. [DOI] [PubMed] [Google Scholar]
- Sauer AJ, Moss AJ, McNitt S, et al. Long QT syndrome in adults. J Am Coll Cardiol. 2007;49:329–337. doi: 10.1016/j.jacc.2006.08.057. [DOI] [PubMed] [Google Scholar]
- Schulze-Bahr E, Wang Q, Wedekind H, et al. KCNE1 mutations cause Jervell and Lange-Nielsen syndrome. Nat Genet. 1997;17:267–268. doi: 10.1038/ng1197-267. [DOI] [PubMed] [Google Scholar]
- Schwartz PJ, Priori SG, Spazzolini C, et al. Genotype-phenotype correlation in the long-QT syndrome: gene-specific triggers for life-threatening arrhythmias. Circulation. 2001;103:89–95. doi: 10.1161/01.cir.103.1.89. [DOI] [PubMed] [Google Scholar]
- Schwartz PJ, Zaza A, Locati E, Moss AJ. Stress and sudden death. The case of the long QT syndrome. Circulation. 1991;83:II71–II80. [PubMed] [Google Scholar]
- Sears SF, Jr, Conti JB. Quality of life and psychological functioning of ICD patients. Heart. 2002;87:488–493. doi: 10.1136/heart.87.5.488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seth R, Moss AJ, McNitt S, et al. Long QT syndrome and pregnancy. J Am Coll Cardiol. 2007;49:1092–1098. doi: 10.1016/j.jacc.2006.09.054. [DOI] [PubMed] [Google Scholar]
- Shah MJ, Wieand TS, Rhodes LA, et al. QT and JT dispersion in children with long QT syndrome. J Cardiovasc Electrophysiol. 1997;8:642–648. doi: 10.1111/j.1540-8167.1997.tb01827.x. [DOI] [PubMed] [Google Scholar]
- Sherman J, Tester DJ, Ackerman MJ. Targeted mutational analysis of ankyrin-B in 541 consecutive, unrelated patients referred for long QT syndrome genetic testing and 200 healthy subjects. Heart Rhythm. 2005;2:1218–1223. doi: 10.1016/j.hrthm.2005.07.026. [DOI] [PubMed] [Google Scholar]
- Splawski I, Shen J, Timothy KW, et al. Spectrum of mutations in long-QT syndrome genes. KVLQT1, HERG, SCN5A, KCNE1, and KCNE2. Circulation. 2000;102:1178–1185. doi: 10.1161/01.cir.102.10.1178. [DOI] [PubMed] [Google Scholar]
- Stephenson EA, Berul CI. Electrophysiological interventions for inherited arrhythmia syndromes. Circulation. 2007;116:1062–1080. doi: 10.1161/CIRCULATIONAHA.106.655779. [DOI] [PubMed] [Google Scholar]
- Tan HL, Bardai A, Shimizu W, et al. Genotype-specific onset of arrhythmias in congenital long-QT syndrome: possible therapy implications. Circulation. 2006;114:2096–2103. doi: 10.1161/CIRCULATIONAHA.106.642694. [DOI] [PubMed] [Google Scholar]
- Tester DJ, Ackerman MJ. Novel gene and mutation discovery in congenital long QT syndrome: let’s keep looking where the street lamp standeth. Heart Rhythm. 2008;5:1282–1284. doi: 10.1016/j.hrthm.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tester DJ, Will ML, Haglund CM, Ackerman MJ. Compendium of cardiac channel mutations in 541 consecutive unrelated patients referred for long QT syndrome genetic testing. Heart Rhythm. 2005;2:507–517. doi: 10.1016/j.hrthm.2005.01.020. [DOI] [PubMed] [Google Scholar]
- Ueda K, Valdivia C, Medeiros-Domingo A, et al. Syntrophin mutation associated with long QT syndrome through activation of the nNOS-SCN5A macromolecular complex. Proc Natl Acad Sci U S A. 2008;105:9355–9360. doi: 10.1073/pnas.0801294105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vatta M, Ackerman MJ, Ye B, et al. Mutant caveolin-3 induces persistent late sodium current and is associated with long-QT syndrome. Circulation. 2006;114:2104–2112. doi: 10.1161/CIRCULATIONAHA.106.635268. [DOI] [PubMed] [Google Scholar]
- Vincent GM. The molecular genetics of the long QT syndrome: genes causing fainting and sudden death. Annu Rev Med. 1998;49:263–274. doi: 10.1146/annurev.med.49.1.263. [DOI] [PubMed] [Google Scholar]
- Vincent GM, Jaiswal D, Timothy KW. Effects of exercise on heart rate, QT, QTc and QT/QS2 in the Romano-Ward inherited long QT syndrome. Am J Cardiol. 1991;68:498–503. doi: 10.1016/0002-9149(91)90785-j. [DOI] [PubMed] [Google Scholar]
- Vincent GM, Timothy KW, Leppert M, Keating M. The spectrum of symptoms and QT intervals in carriers of the gene for the long-QT syndrome. N Engl J Med. 1992;327:846–852. doi: 10.1056/NEJM199209173271204. [DOI] [PubMed] [Google Scholar]
- Vos MA, Verduyn SC, Gorgels AP, et al. Reproducible induction of early after depolarizations and torsade de pointes arrhythmias by D-sotalol and pacing in dogs with chronic atrioventricular block. Circulation. 1995;91:864–872. doi: 10.1161/01.cir.91.3.864. [DOI] [PubMed] [Google Scholar]
- Vyas H, Hejlik J, Ackerman MJ. Epinephrine QT stress testing in the evaluation of congenital long-QT syndrome: diagnostic accuracy of the paradoxical QT response. Circulation. 2006;113:1385–1392. doi: 10.1161/CIRCULATIONAHA.105.600445. [DOI] [PubMed] [Google Scholar]
- Ward OC. A new familial cardiac syndrome in children. J Ir Med Assoc. 1964;54:103–106. [PubMed] [Google Scholar]
- Yong S, Tian X, Wang Q. LQT4 gene: the “missing” ankyrin. Mol Interv. 2003;3:131–136. doi: 10.1124/mi.3.3.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zareba W, Moss AJ, Schwartz PJ, et al. Influence of genotype on the clinical course of the long-QT syndrome. International Long-QT Syndrome Registry Research Group. N Engl J Med. 1998;339:960–965. doi: 10.1056/NEJM199810013391404. [DOI] [PubMed] [Google Scholar]