

Abstract
Genetic studies of type 1 diabetes (T1D) have been advanced by comparative analysis of multiple susceptible and resistant rat strains with a permissive class II MHC haplotype, RT1u. LEW.1WR1 (but not resistant LEW.1W or WF) rats are susceptible to T1D induced by a TLR3 agonist (poly I:C) followed by infection with parvovirus. We have mapped genetic loci for virus-induced T1D susceptibility, identifying a major susceptibility locus (Iddm37) near the MHC. Iddm37 homologues on mouse and human chromosomes are also diabetes-linked. We report that a major effect gene within Iddm37 is diubiquitin (Ubd). Gene expression profiling of pancreatic lymph nodes in susceptible and resistant rats during disease induction showed differences in Ubd transcript abundance. The LEW.1WR1 Ubd promoter allele leads to higher inducible levels of UBD than that of LEW.1W or WF. Using zinc finger nucleases (ZFN), we deleted a segment of the LEW.1WR1 Ubd gene and eliminated its expression. UBD-deficient rats show substantially reduced diabetes after viral infection. Complementation studies show that there may be another diabetes gene in addition to Ubd in the Iddm37 interval. These data prove that Ubd is a diabetes susceptibility gene, providing insight into the interplay of multiple genes and environmental factors in T1D susceptibility.
Keywords: Autoimmune diabetes, susceptibility genes, virus-induced, rat model
Introduction
Type 1 diabetes (T1D) afflicts a million Americans 1 causing numerous complications and shortening life. It is a polygenic, T cell-mediated disease that results from the interaction of multiple gene variants 2 and environmental factors 3. There are to date no approved methods for preventing or abrogating the disease. T1D susceptibility has strong genetic linkage to the major histocompatibility complex (MHC) in all tested species. Genome wide association studies (GWAS) have identified more than 50 non-MHC T1D candidates, many of which are classified in gene ontology analyses as members of “immune system process” 4. These loci each have only a modest effect (odds ratio <1.5), though the concatenation of risk alleles may amplify risk 5. Many GWAS candidate genes are just that – candidates, with the informative SNPs located nearby in non-coding regions with no clear function 6. It is not always possible to deduce from human allelic variation in GWAS regions the actual genomic change that influences risk or the level at which the environment interacts with the genome.
T1D in humans remains quite perplexing in other ways. T1D is becoming more common, for unknown reasons, and it is suggested that virus exposure elicits T1D or, paradoxically, protects against it. Evidence that human viral infections are protective against autoimmunity (i.e. hygiene hypothesis) is correlative 7, 8. Significant evidence has mounted linking enteroviral infections to the initiation of T1D 9 and to progression from islet autoimmunity to overt T1D 10, 11.
Animal models are invaluable in sorting out the mechanism (a recent example is 12) by which GWAS genes could influence complex diseases such as autoimmune diabetes. A recent example is the discovery that the GWAS gene encoding the transcription factor BACH2 plays a critical role in mouse regulatory T cell (Treg) function12, known to be compromised in NOD mice13. They are potentially invaluable for analyzing the interaction of genes and environmental perturbants, especially viral infection. Both NOD mice and various rat strains have been used for this purpose. The NOD mouse model has not been helpful in this regard 14 15, 16 because most viral infections prevent T1D in this animal. In contrast, we have documented that the growing evidence for environmental triggers of disease can be modeled in a number of rat strains, allowing for the dissection of specific gene-environment collaborations in initiating diabetes 17, 18. Rats have been a good (albeit negative) predictor of human clinical T1D trial outcomes (reviewed in 17, 18). The need for new clinical strategies is pressing. With the exception of generalized immunosuppression 19, which is unacceptably toxic 20, no preventive intervention has achieved clinical success 21.
In order to map T1D genes and their relation to environmental factors, we have used rat models of the disease. The LEW.1WR1 rat develops spontaneous diabetes at a low rate but is also very susceptible to the induction of diabetes using a small, non-diabetogenic priming dose of the TLR3 agonist polyinosinic:polycytidylic acid (poly I:C) followed by infection with Kilham rat virus (KRV); it has been shown that KRV does not directly infect the pancreas and its role in the breakdown of self-tolerance is not completely understood 22, 23. With this treatment, rats develop symptoms of diabetes 10-18 days after infection.
As is true for humans and the NOD mouse, T1D in rats is strongly associated with a permissive class II MHC haplotype. In the rat, the MHC locus is designated RT1 and the permissive class II haplotype is designated RT1-B/DU 24. The RT1-B locus is homologous to human HLA-DQ and RT1-D to HLA-DR. In the rat the two class two loci are in strong linkage disequilibrium, and this is reflected in the “B/D” nomenclature. In addition, a previous genome wide linkage study 25 was used to identify regions of the rat genome that control T1D due to virus exposure. In this linkage study, (LEW.1WR1 × WF)F2 animals were used to map two major quantitative trait loci (QTL), both derived from LEW.1WR1, that promote diabetes susceptibility in rats. In addition to an established susceptibility locus (Iddm14)26–28, a new locus (Iddm37) near the MHC on chromosome 4 was found to be a major determinant of T1D disease susceptibility virus-induced T1D 25. Interestingly, Iddm37 homologues on mouse 29 and human chromosomes 30, 31 are also linked to T1D.
In the present study, we took advantage of congenic strains that are closely related to LEW.1WR1 and informative for the Iddm37 interval. The LEW.1WR1 rat was generated from two LEW MHC-congenics, LEW.1W and LEW.1A (Figure 1). Based on their genotypes, we predicted that both parental strain rats would be resistant to induction of diabetes by KRV+poly I:C but for different reasons. LEW.1W has the permissive class II MHC (RT1-B/D u) but not the permissive Iddm37, whereas LEW.1A has the permissive Iddm37 but a non-permissive class II region (RT1-B/Da)32, 33. LEW.1WR1 rats bear RT1-D/Bu (MHC class II) and a recombination distal to the Iddm37 interval and are susceptible.
Figure 1.
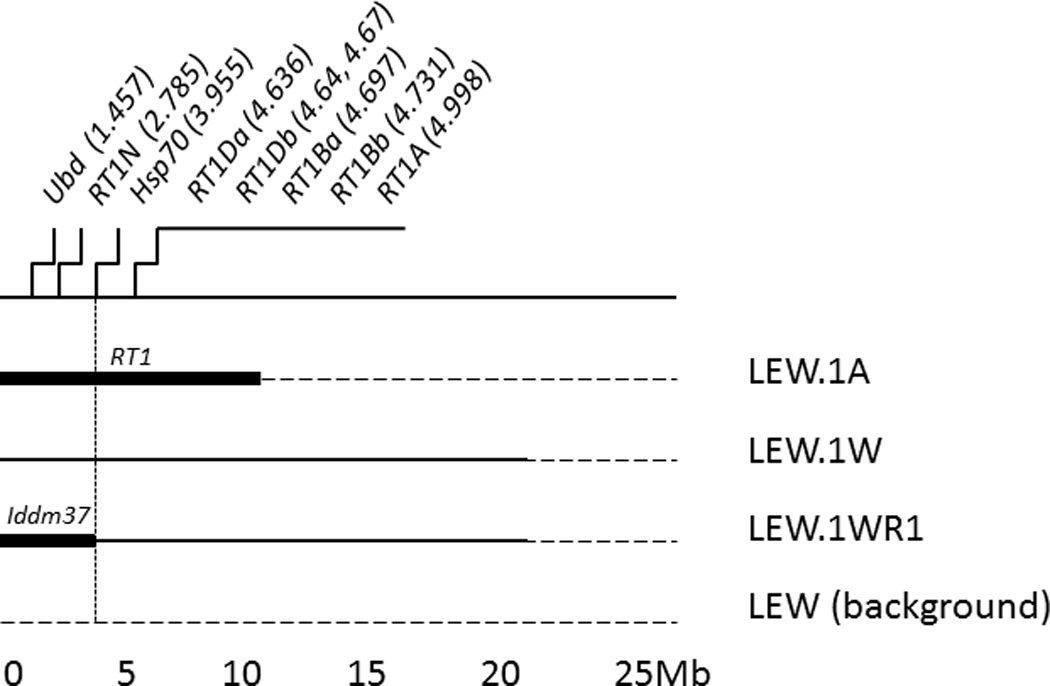
The centromeric end of rat Chromosome 20. We determined informative alleles by determining single nucleotide polymorphism (SNP) genotypes of the four congenic strains (Blankenhorn et al., 2009), and this cartoon (not to scale) shows the proximal 25 megabases of Chromosome 20 and the approximate borders of intervals retained in each congenic rat strain. LEW.1WR1 is a recombinant descendant of LEW.1A × LEW.1W.
We confirmed this hypothesis and proceeded to positional gene identification for Iddm37 using the LEW.1WR1 and LEW.1W strains. To do so, we performed gene expression studies of the genes in the interval, and identified two genes for further study. The strain distribution pattern of sequence polymorphisms and expression patterns of one of these genes, diubiquitin (Ubd), closely resembled the strain distribution pattern of virus-induced diabetes in rats 24. The Ubd gene was then deleted on both the LEW.1WR1 and the LEW.1W backgrounds, which resulted in modestly but significantly reduced diabetes incidence, as would be expected of a single gene in the polygenetic context of T1D. To our knowledge, this is the first genetic deletion using ZFN technology of any autoimmune candidate gene in rats.
Results and Discussion
Susceptibility to KRV-induced T1D in congenic rat strains
We first assessed susceptibility of the three LEW congenic rat strains to virus-induced diabetes. As shown in Figure 2, we documented diabetes susceptibility in LEW.1WR1 rat and diabetes resistance in LEW.1A rats, which bear the non-permissive RT1-B/Da haplotype. This experiment also confirmed our prediction that LEW.1W congenic rats, bearing a permissive RT1-B/Du but a resistant haplotype for the Iddm37 interval, are T1D-resistant in response to viral infection.
Figure 2.

Kaplan-Meyer survival plot of virus-induced T1D in LEW RT1-congenic rats. The rat strains show highly significantly different susceptibility to T1D (χ2=28.99, p<0.0001). Days PI, days post-induction by poly I:C + KRV. For these studies, a total of 18 LEW.1WR1, 12 LEW.1W, and 6 LEW.1A rats were induced for T1D when they reached 28-30 days of age.
Microarray
There are 370 known genes and gene elements in the Iddm37 region (Rat genome v4, http://genome.ucsc.edu/) and we examined the expression differences for all of them using the Affymetrix RG230 2.0 GeneChip. For this experiment, we treated one group of nine LEW.1WR1 rats and two groups of diabetes resistant rats (LEW.1W and WF.Iddm4, nine of each) to induce diabetes. The WF.Iddm4 is a congenic diabetes-resistant substrain of the WF rat into which a segment of BBDR rat chromosome 4 was introduced34. It is susceptible to diabetes induced by poly I:C but not rat virus 34. Using these animals, we then examined global transcript levels in pancreatic lymph nodes (PLN) across three early time points in disease progression (day 0, after poly I:C; day 3 after KRV: and day 5 after KRV). Comparisons were made between LEW.1WR1 and each of the other rat strains. We accepted a false discovery rate (FDR) of 0.20, to account for the biological diversity among the three replicates at each time.
Nine genes within the Iddm37 interval were differentially regulated in the same direction in comparisons of LEW.1WR1 vs. LEW.1W, and LEW.1WR1 vs. WF.Iddm4, as would be expected for an Iddm37 candidate gene. Confirmatory quantitative RT-PCR was conducted for all nine of these genes (Table 1). We selected for further study the two most significantly different genes that were concordant and different across all time points: UBD and RT1-N1 (Table 1). RT1-N1 is an MHC class Ib gene (http://rgd.mcw.edu/rgdweb/report/gene/main.html?id=3498). Notably, there was an excess of LEW.1WR1 Ubd transcripts, and an excess of LEW.1W RT1-N1 transcripts on day 0, even before the KRV inoculation.
Table 1.
Fold-change of expression (LEW.1WR1:other rat strain) is shown for nine genes with significant expression in PLN on the microarray (chip). Highlighted in bold type are genes that are significantly elevated in LEW.1WR1, and in blue are those that are significantly decreased in expression. Quantitative RT-PCR was performed on the same triplicate samples from PLN, to determine which of the microarray differences between the rats for expression of these genes is genuine. Only Ubd and RT1-N1/N2 showed significantly different ratios by both assays. RT-PCR is necessary to rule out differences due to polymorphism of the probe and target. Differences in gene expression levels were assessed by determining the false discovery rate (FDR) and use of a nonparametric rank product test 58 for the chip, and a t-test for the RT-PCR values.
| Day 0 (poly I:C only) | Day 3 | Day 5 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| LEW.1WR1 vs LEW.1W |
LEW.1WR1 vs WF.Iddm4 |
LEW.1WR1 vs LEW.1W |
LEW.1WR1 vs WF.Iddm4 |
LEW.1WR1 vs LEW.1W |
LEW.1WR1 vs WF.Iddm4 |
|||||||||
| gene | NAME | position (MB) | RT-PCR | Chip | RT-PCR | Chip | RT-PCR | Chip | RT-PCR | Chip | RT-PCR | Chip | RT-PCR | Chip |
| Ubd | ubiquitin D | 1.48 | 2.7‡ | 2.5†‡ | 3.3‡ | 2.3†‡ | 2.5‡ | 1.4‡ | 3.3* | 3.4†‡ | 3.3‡ | 2.5†‡ | 4.1‡ | 5.2†‡ |
| RT1-N1/RT1-N2 | RT1-N1 class Ib | 2.79 | −10.4£ | −5.5†‡ | −9.2* | −4.5†‡ | −8.8* | −7.2†‡ | −11.5‡ | −5.6†‡ | −14.5£ | −4.8†‡ | −19.6£ | −5.2†‡ |
| Nrm | Nurim | 3.04 | 1.3 | 1.5‡ | 1.6‡ | 1.4‡ | 1.9 | 1.2‡ | 1.1 | 1.6‡ | 1.5 | 2.3†‡ | 1 | 2.4†‡ |
| Lta | Lymphotoxin alpha | 3.66 | −1.6 | −1.1 | −1.3 | −1.2 | 1.8 | 1.2‡ | 1 | 1.1 | 1.7 | 2.4†‡ | −1.5 | 1.6‡ |
| Ltb | Lymphotoxin beta | 3.67 | −1.1 | −1.2 | −1.4 | −1.1 | −2.1 | 1.1 | 1.1 | 1.2 | 1.0 | 2.0†‡ | −1.5 | 1.7‡ |
| Tnf | Tnf | 3.67 | 1.2 | 1.1 | 1.3 | 1.4‡ | −1.1 | −1.2 | 1.3 | 1.0 | 1.0 | 1.1 | 1.4 | 2.3†‡ |
| Lst1 | leukocyte specific transcript 1 | 3.69 | 1.2 | 1.3 | 1.2 | 1.1 | 1.2 | −1.3 | −1.2 | 1.1 | −1.2 | 1.3 | 1.0 | 2.2†‡ |
| Vars | valyl-tRNA synthetase | 3.87 | −1.1 | 1.2 | 1.1 | 1.2 | 1.1 | −1.1 | 1.2 | 1.4‡ | 1 | 1.7‡ | −1.1 | 2.1†‡ |
| Neu1 | Sialidase 1 | 3.99 | −1.3 | 1.4‡ | 1.2 | 1.6‡ | 2.5 | 1.2 | 1.9 | 1.5‡ | 1.2 | 1.9‡ | −1.4 | 2.5†‡ |
Symbols:
At FDR<20%
p<0.05
p<0.01
p<0.001.
Iddm37 candidate gene strain distribution patterns (SDP)
To determine which of the two genes was the more likely candidate for Iddm37, it was necessary to characterize their alleles and haplotypes in a number of rat strains (i.e., to determine their SDP). To obtain an SDP for the RT1-N1 gene, we first identified polymorphic variants between LEW.1W and LEW.1WR1 by sequencing, which identified one synonymous SNP (at nucleotide 2,785,930 on chromosome 20) between LEW.1W and LEW.1WR1 in exon 2 and a second SNP (nucleotide 2,785,102) in the promoter region that distinguishes LEW.1W and WF from LEW.1WR1. We also identified a polymorphic repetitive element 115 bp from the TAA stop site. Strains that carry the RT1a or RT1l haplotype (LEW.1WR1, LEW.1A, DA, and LEW) at the RT1-N1 locus have a 35bp insertion at this repetitive element, whereas rat strains carrying the RT1u haplotype (WF, LEW.1W, and BBDR) were characterized by the absence of this additional 35bp (Table 2).
Table 2. Strain distribution patterns (SDP) of genotypes, KRV-induced T1D, and gene expression in related rats.
Rats in bold font are susceptible to T1D induced by KRV + Poly I:C. RT1-N1 haplotypes were determined by comparison of alleles at two SNPs and one microsatellite region. LEW.1WR1, LEW.1A and LEW are designated RT1-N1A. Rat strains carrying the RT1u haplotype (WF, LEW.1W, and BBDR) carry RT1-N1-U. Ubd haplotypes were declared on the basis of the presence or absence of a SINE element and two non-synonymous SNPs in exon 2. The UbdL haplotype has no SINE, and G and A at SNP1 and SNP2, respecitively; UbdBB haplotype is SINE absent, A,A; and UbdA haplotype bears the SINE element, and has A and C at SNPs 1 and 2. Gene expression was ascertained by qRT-PCR on spleen mRNA samples.
| Strain | LEW.1WR1 | BBDR | LEW.1W | WF | LEW.1AR1 | LEW.1A | LEWIS |
|---|---|---|---|---|---|---|---|
| RT1 Haplotype (A - B/D) | U-U | U-U | U-U | U-U | A-U | A-A1 | L-L1 |
| KRV-T1D | Susceptible | Susceptible | Resistant | Resistant | Resistant2 | Resistant | Resistant |
| UBD-SINE | Absent | Absent | Present | Present | Present | Absent | Present |
| Ubd mRNA expression | High | High | Low | Low | Low | High | Low |
| Ubd exon haplotype | A | BB | L | L | L | A | L |
| RT1-N1 haplotype | A | U | U | U | U | A | A |
| RT1-N1 expression | Low | High | High | High | High | Low | nd |
RT1 haplotypes that are non-permissive for T1D induction. Other strains (LEW.1WR1, LEW.1W, and LEW.1A) were tested in the present report or in previous publications24.
unpublished data.
Nd, not determined.
We also used sequencing to characterize the Ubd haplotypes. In our previous report, we presented two haplotypes for the coding region of Ubd 25. We now report an additional Ubd haplotype for the BBDR rat and several other strains that have been classified for T1D susceptibility 24 (Supplemental Figure 1). Sequencing the promoters and 5’UTR regions of Ubd from LEW.1W, LEW, WF, and LEW.1WR1 haplotypes revealed two major allelic variants among these strains. There is a polymorphic microsatellite region at –775 relative to the transcriptional start site, and, in all the resistant strains, an insertion of 59 nucleotides just upstream of the 5’UTR. This 59-nt insertion is a short interspersed element (B2-SINE) as described by the UCSC database (rat genome v4, genome.ucsc.edu).
The polymorphisms of Ubd – including the SINE element -- were compiled to make an SDP of Ubd haplotypes for comparison to the SDP for RT1-N1 sequence polymorphisms and the SDP of diabetes susceptibility (Table 2). The results show that RT1-N1 is a less probable candidate gene than Ubd, because BBDR rats do not have the same RT1-N1 allele as LEW.1WR1, yet both are susceptible to virus-induced T1D. The Ubd SINE element, on the other hand, has an identical SDP for T1D in these rat strains. All the RT1u rat strains that are diabetes resistant have low Ubd expression, and Ubd is highly expressed (and cytokine inducible25) in both KRV-T1D susceptible rat strains, whereas RT1-N1 expression is discordant. In addition, no functionally relevant polymorphisms were found in the RT1-N1 sequence between LEW.1W and LEW.1WR1, strengthening the notion that Ubd is the gene that more likely underlies the Iddm37 susceptibility locus.
Ubd promoter polymorphisms control allele-specific Ubd expression
The gene expression data showing reduced Ubd gene expression in LEW.1W and WF.Iddm4 rats were not surprising, as we have previously demonstrated that expression of UBD in draining LN of poly I:C+KRV inoculated animals is four-fold higher in LEW.1WR1 rats than in WF rats; this difference is even more dramatic in splenocytes 25. In that study, we proved that this was not due to a deficiency of required inflammatory cytokines, which were expressed at similar levels in both rats, indicating that rats of both strains had comparable cytokine responses to poly I:C and to the KRV infection.
It was important to link the phenotype (low Ubd mRNA expression) to the allelic polymorphism seen in LEW.1W. We analyzed the SINE insertion and the region surrounding it using a program to highlight transcription factor binding sites (Transfac, http://www.gene-regulation.com/index2.html); the result suggested that insertion of the SINE element could add new transcription factor binding sites, and/or distance native Ubd promoter elements from the transcriptional start site. The contribution of the microsatellite polymorphism in the upstream region of the Ubd promoter, however, was unknown. To distinguish which of the two polymorphic promoter elements is responsible for the UBD expression difference, luciferase reporter vectors were constructed (Supplemental figure 2) containing each variant region in the promoter from either the LEW.1WR1 or the LEW.1W rat, and measured luciferase expression. When tested in vitro in Huh7 cells, the level of luciferase in cells transfected with the SINE element-containing promoter from LEW.1W was significantly lower compared to the level of luciferase driven by the Ubd promoter from LEW.1WR1 (Figure 3). The region containing the microsatellite polymorphism did not drive expression of the reporter (not shown). The transfected cells were also grown in the presence of TNF-α, IFN-γ or IFN-β, all of which have been demonstrated to induce Ubd transcription in vitro 35, 36. The results show that the LEW.1WR1 Ubd promoter is highly cytokine-inducible, and the presence of the SINE element inhibits cytokine induction of Ubd in LEW.1W (Figure 3).
Figure 3.

The Ubd allele from LEW.1WR1 but not LEW.1W promotes both high constitutive as well as induced UBD expression. In closed circles are results from reporter plasmids containing the LEW.1W promoter, in open circles are LEW.1WR1 results. Mean ± standard deviation are shown per condition per strain, performed once (LEW.1A strain) or twice (for LEW.1W and LEW.1WR1 strains).
UBD-KO rats
These results suggested but did not prove that expression of Ubd, the top candidate for Iddm37, is a modifier of diabetes susceptibility. To test this hypothesis, we targeted Ubd for genetic deletion in LEW.1WR1 rats. Nucleotide sequencing of tail DNA for heterozygosity at the Ubd locus identified a number of candidate founder rats. One founder (LEW.1WR1-UbdemUmass, or UBD-KO) had a 65-base pair deletion of intron 1/exon2 that eliminates a splice acceptor site (Supplemental Figure 3). Progeny from this founder were crossed to produce heterozygous and homozygous UBD-KO rats for comparison to the wild-type (WT) rats on the LEW.1WR1 background. Rats bearing a homozygous deletion of Ubd were viable and expressed no Ubd transcripts in their spleens (data not shown).
Absence of UBD expression reduces susceptibility to virus-induced T1D in LEW.1WR1 rats
We first compared diabetes susceptibility of LEW.1WR1 rats to that of LEW.1WR1 rats with either heterozygous or homozygous deletion of Ubd. As shown in Figure 4, overall diabetes frequency in wild-type LEW.1WR1 rats was highest of the three strains, LEW.1WR1-UBD-KO heterozygotes were less susceptible, and the homozygous LEW.1WR1-UBD-KO rats were the least susceptible. The diabetes-free survival proportions are significantly increased in the heterozygotes (13%) and homozygotes (26%) (χ2 test for trend, p=0.016, Figure 4B). Latency to onset in rats that became diabetic was similar in the three groups, and for this reason, the Kaplan Meyer plot (Figure 4A) of the three groups showed a trend that did not reach statistical significance (p<0.2).
Figure 4.

Deletion of Ubd affects diabetes incidence on the LEW.1WR1 background
(A) Kaplan-Meyer survival plot of survival to virus-induced T1D in LEW.1WR1 rats with homozygous WT/WT, Ubd-KO/Ubd-KO, or heterozygous Ubd-KO/WT genotypes. The rat strains show a trend toward different susceptibility to T1D (χ2=3.235, p=0.198).
(B) Comparison of diabetes-free survival among the same three rat strains. The difference due to Ubd-genotype is significant ((χ2=5.83, p<0.016). For these studies, 31 LEW.1WR1-UBD/KO, 13 heterozygous LEW.1WR1KO/WT, and 17 (WT) LEW.1WR1 littermate rats were studied as reached 28-30 days of age.
Genetic complementation enhances susceptibility of resistant LEW.1W rats to virus-induced T1D
One advantage to using the knockout strategy is that the resulting genetically deficient animals can be bred to a strain bearing a putative defective allele to determine if the deleted allele is identical to the defective one. We hypothesize that LEW.1W carries a defective allele of the Ubd gene, because we have shown that it cannot be up-regulated normally by cytokines and thus is likely to be hypofunctional in the setting of diabetes. To test this hypothesis, we bred LEW.1W rats to heterozygous LEW.1WR1-UBD-KO rats. If Iddm37 is Ubd, then the Ubd-KO-bearing chromosome from the heterozygous parent will not be able to ‘complement’ the defective LEW.1W Iddm37 allele, and the hybrid (LEW.1W/LEW.1WR1-UBD-KO) rat will remain resistant to diabetes. If Iddm37 is not Ubd, the “authentic” Iddm37 gene on the UBD-KO chromosome (derived from LEW.1WR1) will bear a susceptible complementing allele for Iddm37, and the hybrid will be as susceptible as the normal F1 hybrid between LEW.1W and LEW1.WR1. The other, wild type (WT) chromosome from the LEW.1WR1-UBD-KO heterozygous parent serves as an internal control for the experiment, providing a confirming comparison for the known T1D susceptibility of Iddm37 heterozygotes 25.
The results of the complementation experiment were revealing. The UBD-KO showed a significant effect on the LEW.1W background (Kaplan Meier statistic, p=0.013; Figure 5A). The hybrid rat bearing a deletion of Ubd on one chromosome and a defective allele from LEW.1W on the other (LEW.1W/LEW.1WR1-UBD-KO) shows greatly increased diabetes-free survival rats (60%) compared to their WT control, LEW.1W/LEW.1WR1 (25%). This compound heterozygote (LEW.1W/LEW.1WR1UBD-KO) was not as resistant as the LEW.1W/LEW.1W homozygote (100% diabetes-free survival), meaning that the UBD-KO partially complemented the defective allele in LEW.1W. The fact that the LEW.1W defect was partially but not fully complemented by the LEW.1WR1 chromosome carrying the Ubd knockout allele means that Ubd plus an additional unidentified gene in the Iddm37 interval both contribute to diabetes. As in the LEW.1WR1 crosses, there is a highly significant difference in incidence in LEW.1W crosses (Figure 5B, χ2 test for trend, p=0.0023), but no significant difference in the latency of those rats that become diabetic. Together, the data from the two crosses support a model where the polymorphic haplotypes of Ubd significantly influence the incidence of KRV+ poly I:C induced diabetes in the rat.
Figure 5.

Deletion of Ubd affects diabetes incidence on the LEW.1W background
(A) Kaplan-Meyer survival plot of survival to virus-induced T1D in LEW.1W rats with homozygous LEW.1W/LEW1W, heterozygous LEW.1WR1Ubd-KO/LEW.1W, or heterozygous LEW.1WR1/LEW.1W genotypes at the Ubd locus. The rat strains show a significantly different susceptibility to T1D (χ2=8.711, p=0.0128).
(B) Comparison of diabetes-free survival among the same three rat strains. The difference due to Ubd genotype is significant (χ2=9.289, p=0.0023). For these studies, 6 LEW.1W, 11 heterozygous LEW.1WR1Ubd-KO/LEW.1W, and 12 heterozygous LEW.1WR1/LEW.1W littermate rats were studied as litters reached 28-30 days of age.
Discussion
Diabetes-susceptible rat strains have very high fidelity to the clinical pathology seen in human autoimmune diabetes. This is especially true for testing the gene-environment interactions that induce the disease. Such interactions are difficult to discern the NOD mouse model of T1D because all environmental perturbations including viral infections that have been studied uniformly prevent the disease17, 37, 38 which cannot be performed in diabetes-susceptible mice. The recently developed technologies that allow targeting of genes for deletion in the rat 39–42 have made this species exceptionally useful for the dissection of the genetic requirements for diabetes susceptibility. In this report, we have taken the diabetes QTL Iddm37 from a map position on the proximal arm of Chr20 that encompasses about 4 megabases 25 to a lead candidate gene, Ubd. Deletion of the Ubd gene substantially and significantly reduced the incidence, but not the latency, of T1D in UBD-KO rats as compared to their respective WT controls in two large cohorts of rats. This is to our knowledge the first successful genetic knockout of a rat autoimmunity gene.
In identifying this gene, we have uncovered a new pathway relevant to autoimmune disease susceptibility. UBD is much more highly expressed in the LEW.1WR1 rat than in the resistant strains. The SINE element in the UBD promoter in resistant strains likely plays a role as a transcriptional repressor of UBD. It has been shown that SINE elements can influence the expression of nearby genes 43, 44 although the repressive mechanism in the Ubd promoter is unknown. The link between higher UBD expression and higher disease incidence suggests that UBD could be required for the recognition or processing of viral or self-antigens, or for activating T cells that recognize them. Although UBD, a diubiquitin with ubiquitin-like properties, is likely expressed in pancreas, in antigen-presenting cells, B cells, and T cells in rats, humans, and mice 25, 45–47, its functional importance in these tissues has not yet been defined. The human homolog of Ubd is called FAT10, for HLA-F-adjacent transcript number 10. It is known that dendritic cell (DC) maturation induces FAT10 in human cells 48, 49. In addition, CD40L-CD40 ligation, TLR agonists such as LPS or poly I:C, and some cytokines cause up-regulation of FAT10 in DC 48. FAT10:protein conjugates undergo rapid proteasome-dependent degradation in DCs 50, supporting a potential role for UBD in antigen presentation. FAT10/UBD is also associated with resistance to apoptosis in lymphocytes 35, 46.
A clue to the role of UBD in rat T1D comes from a closer inspection of the SDP of diabetes in rats. KDP rats are spontaneously diabetic, due to a mutation that inactivates the Casitas B-lineage lymphoma b (Cblb) gene 51. Disabling mutations of Cblb cause hyper-activation of lymphocytes in both rats and mice 51, and Cblb may also be involved in the diabetogenic pathways required for human diabetes 52. Inspection of genotypes of Ubd in rats shows that KDP rats have the same B2-SINE element in their Ubd promoter as do LEW.1W rats (unpublished observations), yet, unlike all other RT1u rats with this repetitive element, they are diabetes-prone. This may indicate that Cblb is downstream of Ubd in a diabetogenic pathway that otherwise requires a functional Ubd gene.
It is of interest that our complementation data suggest further that there is a second gene in addition to Ubd in the Iddm37 interval. When we analyze the entire set of rat UBD-KO crosses tested for T1D susceptibility, there is a highly significant trend associated with both the alleles of Ubd or the absence of Ubd and with a second, as yet unidentified gene (“gene 2”) (Table 3, χ2 test for trend, p<0.0001). If Ubd were the sole gene underlying the effect of Iddm37, the complementation experiment would have shown complete resistance in the LEW.1WR1-KO/LEW.1W hybrid rat, instead of the observed 60% resistance (line 4 of Table 3). This second gene is being sought in our congenic animals. These data indicate additivity of multiple genes, each of relatively small effect size, underlying diabetes. This finding will in future studies allow us to analyze in detail T1D susceptibility as a function of the number of susceptible alleles as we have done for Iddm37 (Table 3). This combinatorial analysis of genetic elements in the rat is similar to the cumulative genetic risk analysis for human T1D developed by Winkler et al. who were able to predict diabetes risk in the BABY DIAB cohort based on non-HLA-risk allele scores 5. The rat models we now have will allow us to generate this kind of risk for individual genotypes and apply them to different pathways leading to T1D: viral infection, Treg deficiency, and other stressors. This should eventually allow us to enrich the repertoire of candidate interventions for halting the progress to T1D in children at risk.
Table 3. Two gene model of Iddm37.
Incidence of KRV + poly I:C induced diabetes rats of all Iddm37 classes in this study.
| Ubd | Gene 2 | Strain | Iddm37S copies | %T1D |
|---|---|---|---|---|
| S/S | S/S | LEW.1WR1 | 4S | 100% |
| KO/S | S/S | LEW.1WR1/LEW.1WR1-KO | 3S | 87% |
| KO/KO | S/S | LEW.1WR1-KO/LEW.1WR1-KO | 2S | 75% |
| S/R | S/R | LEW.1WR1/LEW.1W | 2S | 75% |
| KO/R | S/R | LEW.1WR1-KO/LEW.1W | 1S | 40% |
| R/R | R/R | LEW.1W/LEW.1W | 0S | 0% |
Abbreviations: S, susceptible allele; KO, deleted allele; R, resistant allele.
χ2 test for trend: p<0.0001.
As one example, the identification of Ubd as a diabetes susceptibility gene should reanimate studies of the role of FAT10/Ubd in children at risk for T1D by virtue of HLA haplotype and family history. The Eisenbarth group has reported linkage to the region containing UBD in their study of Type 1 Diabetes Genetics Consortium (T1DGC) cases and controls 30, 31, 53. This human research, coupled with our analyses of the relative necessity of Ubd for diabetes induced by virus in rats, leads us to propose that Ubd is a good candidate gene for T1D, but quite possibly only in the setting of an environmental perturbant. Our studies clearly demonstrate the interaction of genetic requirements of the disease and environmental factors, and comparable analyses may be necessary to achieve a more complete understanding of the origins of T1D in human populations.
Methods
Rats
Inbred LEW.1WR1, LEW.1W, and LEW.1A rats were obtained from BRM, Inc. (Worcester, MA). As depicted in Figure 1, LEW.1WR1 (RT1-Au, B/Du, Ca) is an MHC recombinant congenic strain derived from LEW.1W (RT1u) and LEW.1A (RT1a) rats 54. RT1-A and RT1-C are Class I and Class 1b MHC genes in the rat, respectively. All of these share the diabetes susceptibility allele of Iddm14 (Tcrb-V13S1A1)55 (Iddm14 was formerly designated Iddm4). Congenic WF.Iddm4 rats (RT1u) were developed and maintained at the University of Massachusetts Medical School; they express the diabetes-susceptible allele of Iddm14 but are resistant to KRV + poly I:C-induced diabetes 55. Rats of both sexes were used in roughly equal numbers and were approximately 4 weeks old at the time of experimentation. Animals were housed in viral antibody free conditions, confirmed monthly to be serologically free of rat pathogens 55 and maintained in accordance with institutional (University of Massachusetts School of Medicine Institutional Animal Care and Use Committee) and national guidelines 56
Diabetes Induction
Rats were injected with poly I:C (1 µg/g body weight) three times (on days -3, -2, -1) and then inoculated with 107 plaque forming units (PFU) of KRV. This dose of poly I:C is not itself diabetogenic, but increases the penetrance of virus-induced diabetes in susceptible rats from ~40% to up to 100%. Rats were monitored for glycosuria three times weekly; diabetes in glycosuric rats was diagnosed on the basis of blood glucose concentration >250 mg/dl on consecutive days using a hand held glucose meter (One Touch Ultra). Animals were killed when diabetes was diagnosed or on day 40 after viral infection. Liver, pancreatic draining lymph nodes (PLN), spleens and pancreata were harvested for further analysis.
Genotyping
Genomic DNA was isolated from tail and liver samples using GenElute Mammalian Genomic DNA miniprep kit (Sigma) and analyzed as in our previous publications 27, 28, 57.
Sequencing
Genes of interest were amplified from genomic DNA of both diabetes susceptible and resistant rats using Hi-Fidelity Taq polymerase. The PCR products were purified and sequenced by Genewiz (South Plainfield, NJ). The sequences were analyzed by 4peaks software (http://nucleobytes.com/index.php/4peaks) and aligned by CLUSTALW (http://align.genome.jp/)
Quantitative Real-time PCR (qRT-PCR)
Rats were treated to induce diabetes as above. Pancreas, PLN and spleen were harvested for RNA isolation (Ultraspec, Biotecx and RNeasy RNA kit, Qiagen) on day 0, 3, and 5. cDNA was prepared from total RNA using the ABI High capacity cDNA RT Kit (Life Technologies Corporation). PCR was carried out using Applied Biosystems SYBR Green PCR mix and the Perkin Elmer/Applied Biosystems Division 7900HT Sequence Detector, using the same samples for which global gene expression analyses were done (below).
GeneChip Analyses
Total RNA was isolated from PLN of poly I:C+ KRV-treated susceptible and resistant rats treated to induce diabetes as above on day 0, 3, and 5. RNA was DNAse treated and assessed for quality. For comparison of global gene expression differences, RNA (~100nanograms) was amplified/labeled (Affymetrix two-cycle cDNA synthesis kit, Affymetrix, Santa Clara, CA, USA) and then hybridized to the Affymetrix RG230 2.0 array, which interrogates > 30,000 transcripts and variants, in accordance to the manufacturers' protocol. After hybridization, arrays were washed and stained with Affymetrix fluidics protocol FS450_0001 and scanned with a 7G Affymetrix GeneChip Scanner. Image data were analyzed with Affymetrix Expression Console™ 1.1.2 software and normalized with Robust Multichip Analysis (www.bioconductor.org) to determine signal log ratios. The statistical significance of differentially induced transcription was assessed false discovery rates (FDR) using Partek Genomics Suite 6.5 (Partek, Saint Louis, MO) and use of a nonparametric rank product test 58
Promoter assays
PCR fragments containing the Ubd promoter and the 5’ microsatellite region were amplified from LEW.1WR1 and LEW.1W genomic DNA by Platinum Taq DNA Polymerase High Fidelity (Invitrogen, Grand Island, NY). The PCR products were purified using the QIAquick PCR purification kit (Qiagen, Valencia, CA) and ligated into the luciferase reporter vector, pGL3-basic (Promega, Madison, WI). Competent E. coli (strain DH5α) were transformed with the pGL3 constructs, plated, and colonies screened for insert incorporation by PCR. The constructs were sequenced by Genewiz to confirm that the vector contained the correct insert. For transfection, 0.2 µg pGL3 constructs were transfected into human hepatocarcinoma (Huh7) cells using Lipofectamine 2000 (Invitrogen, Grand Island, NY) and cultured in DMEM supplemented with 10% FBS. To evaluate the effect of cytokine induction, the transfected cells were grown in complete media supplemented post-transfection with one of the following human recombinant cytokines (Peprotech, Rocky Hill, NJ): TNF-α (10 ng/µl), IFN-α (5 pg/ul) or IFN-β (5 pg/ul). In addition, 50 ng of a β-gal expression vector, pCMV-glo-gal, was co-transfected to assess transfection efficiency. After 48 hours the luciferase activity of the pGL3 plasmid was assessed using the Bright-Glo Luciferase assay system (Promega, Madison, WI), and compared to the β-gal activity of the pCMV-glo-gal plasmid, which was evaluated using the βgal-Galacto-Star system (Life Technologies, Grand Island, NY). The activity of the test plasmid was normalized against the control and this was used as a measure of the promoter activity.
Generation of UBD-deficient (UBD-KO) LEW.1WR1 rats
Two zinc finger nucleases (ZFNs) targeting the 5’ coding region in the second exon of Ubd were designed by and purchased from Sigma Aldrich, ST. Louis MO. Sigma-provided quality control tests indicated a cutting efficiency of ~12%. Injections of fertilized single cell embryos from superovulated LEW.1WR1 rats and transfer to pseudopregnant females were performed by the Transgenic Animal Core Facility at the University of Massachusetts Medical School using standard procedures. To prepare high quality DNA template for in vitro transcription, each of the two paired targeting ZFN-encoding expression plasmids were transformed into DH5a and plasmid DNA was isolated using the GenElute HP endotoxin Free Plasmid Maxiprep kit (Sigma). The purified ZFN-encoding expression plasmids were linearized with XbaI (New England Biolabs), extracted with phenol/chloroform/isoamyl alcohol (Sigma) and precipitated with isopropanol (Fisher Scientific). Messenger RNA was in vitro transcribed, capped, and polyadenylated using the MessageMAX T7 ARCA-Capped Message Transcription Kit and Poly(A) polymerase Tailing Kit (Epicentre). The subsequent Poly(A) tailed ZFN mRNA was column purified with the MEGAclear kit (Ambion) before resuspension in RNAse free water. The mRNA was quantified using a NanoDrop-1000 (Fisher Scientific) and assessed for quality with an Agilent bioanalyzer (Agilent). The two paired ZFN mRNAs were combined in a concentration of 400 µg/ml in RNAse free water according to Sigma guidelines. For pronuclear injection, the paired mRNAs were diluted to a final concentration of 10 ng/µl in 1mM Tris HCl, 0.1mM EDTA and kept on ice during the microinjection procedure. One hundred fertilized 1 cell stage LEW.1WR1 embryos were micro-injected with ZFN mRNA into the male pronucleus under standard conditions. The surviving embryos were implanted into pseudopregnant Sprague Dawley female rats and allowed to develop to full term. To assess for mutants, Ubd gene-specific primers were used on genomic DNA to amplify the region by PCR. We also subcloned these amplicons into pUC19 for sequence confirmation by Genewiz. Once mutant rats were identified, heterozygous LEW.1WR1 founder rats were bred to produce homozygous gene knockout animals (Supplemental Figure 1 shows the deleted target region for the UBD-KO).
Supplementary Material
Acknowledgements
This work was supported by the American Diabetes Association (grants 7-08-RA-106 to JPM, 7-09-BS-18 to EPB, and 7-12-BS-075 to MJH); the National Institutes of Health (grants R21AI088480 to EPB, R01AI078713 to MJH).
Footnotes
Online supplemental material
There are three supplemental figures and one supplemental table.
Conflict of interest
The authors declare no conflict of interest.
References
- 1.van Belle TL, Coppieters KT, von Herrath MG. Type 1 diabetes: etiology, immunology, and therapeutic strategies. Physiol Rev. 2011;91(1):79–118. doi: 10.1152/physrev.00003.2010. [DOI] [PubMed] [Google Scholar]
- 2.Polychronakos C, Li Q. Understanding type 1 diabetes through genetics: advances and prospects. Nat Rev Genet. 2011;12(11):781–792. doi: 10.1038/nrg3069. [DOI] [PubMed] [Google Scholar]
- 3.Trucco M. Gene-environment interaction in type 1 diabetes mellitus. Endocrinol Nutr. 2009;56 Suppl 4:56–59. [PubMed] [Google Scholar]
- 4.Concannon P, Rich SS, Nepom GT. Genetics of type 1A diabetes. The New England journal of medicine. 2009;360(16):1646–1654. doi: 10.1056/NEJMra0808284. [DOI] [PubMed] [Google Scholar]
- 5.Winkler C, Krumsiek J, Lempainen J, Achenbach P, Grallert H, Giannopoulou E, et al. A strategy for combining minor genetic susceptibility genes to improve prediction of disease in type 1 diabetes. Genes and immunity. 2012;13(7):549–555. doi: 10.1038/gene.2012.36. [DOI] [PubMed] [Google Scholar]
- 6.Butter F, Davison L, Viturawong T, Scheibe M, Vermeulen M, Todd JA, et al. Proteome-wide analysis of disease-associated SNPs that show allele-specific transcription factor binding. PLoS Genet. 2012;8(9):e1002982. doi: 10.1371/journal.pgen.1002982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bach JF. The effect of infections on susceptibility to autoimmune and allergic diseases. N Engl J Med. 2002;347(12):911–920. doi: 10.1056/NEJMra020100. [DOI] [PubMed] [Google Scholar]
- 8.Ghazarian L, Diana J, Simoni Y, Beaudoin L, Lehuen A. Prevention or acceleration of type 1 diabetes by viruses. Cell Mol Life Sci. 2013;70(2):239–255. doi: 10.1007/s00018-012-1042-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Oikarinen S, Martiskainen M, Tauriainen S, Huhtala H, Ilonen J, Veijola R, et al. Enterovirus RNA in blood is linked to the development of type 1 diabetes. Diabetes. 2011;60(1):276–279. doi: 10.2337/db10-0186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stene LC, Oikarinen S, Hyoty H, Barriga KJ, Norris JM, Klingensmith G, et al. Enterovirus infection and progression from islet autoimmunity to type 1 diabetes: the Diabetes and Autoimmunity Study in the Young (DAISY) Diabetes. 2010;59(12):3174–3180. doi: 10.2337/db10-0866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Richardson SJ, Willcox A, Bone AJ, Foulis AK, Morgan NG. The prevalence of enteroviral capsid protein vp1 immunostaining in pancreatic islets in human type 1 diabetes. Diabetologia. 2009;52(6):1143–1151. doi: 10.1007/s00125-009-1276-0. [DOI] [PubMed] [Google Scholar]
- 12.Roychoudhuri R, Hirahara K, Mousavi K, Clever D, Klebanoff CA, Bonelli M, et al. BACH2 represses effector programs to stabilize T(reg)-mediated immune homeostasis. Nature. 2013;498(7455):506–510. doi: 10.1038/nature12199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.D'Alise AM, Ergun A, Hill JA, Mathis D, Benoist C. A cluster of coregulated genes determines TGF-beta-induced regulatory T-cell (Treg) dysfunction in NOD mice. Proc Natl Acad Sci U S A. 2011;108(21):8737–8742. doi: 10.1073/pnas.1105364108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang L, Gianani R, Nakayama M, Liu E, Kobayashi M, Baschal E, et al. Type 1 diabetes: chronic progressive autoimmune disease. Novartis Foundation symposium. 2008;292:85–94. doi: 10.1002/9780470697405.ch7. discussion 94-8, 122-9, 202-3. [DOI] [PubMed] [Google Scholar]
- 15.Baschal EE, Aly TA, Jasinski JM, Steck AK, Johnson KN, Noble JA, et al. The frequent and conserved DR3-B8-A1 extended haplotype confers less diabetes risk than other DR3 haplotypes. Diabetes, obesity & metabolism. 2009;11 Suppl 1:25–30. doi: 10.1111/j.1463-1326.2008.01000.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aumeunier A, Grela F, Ramadan A, Pham Van L, Bardel E, Gomez Alcala A, et al. Systemic Toll-like receptor stimulation suppresses experimental allergic asthma and autoimmune diabetes in NOD mice. PLoS One. 2010;5(7):e11484. doi: 10.1371/journal.pone.0011484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mordes J, Poussier P, Rossini AA, Blankenhorn EP, Greiner DL. Rat models of type 1 diabetes: Genetics, environment, and autoimmunity. In: Shafrir E, editor. Animal Models of Diabetes: Frontiers in Research. 2007. pp. 1–39. [Google Scholar]
- 18.Mordes J, Serreze DV, Greiner DL, Rossini AA. Animal models of autoimmune diabetes mellitus. In: LeRoith D, Taylor SI, Olefsky JM, editors. Diabetes mellitus. A fundamental and clinical text. 2004. pp. 591–610. [Google Scholar]
- 19.Chase HP, Butler-Simon N, Garg SK, Hayward A, Klingensmith GJ, Hamman RF, et al. Cyclosporine A for the treatment of new-onset insulin-dependent diabetes mellitus. Pediatrics. 1990;85(3):241–245. [PubMed] [Google Scholar]
- 20.Parving HH, Tarnow L, Nielsen FS, Rossing P, Mandrup-Poulsen T, Osterby R, et al. Cyclosporine nephrotoxicity in type 1 diabetic patients. A 7-year follow-up study. Diabetes Care. 1999;22(3):478–483. doi: 10.2337/diacare.22.3.478. [DOI] [PubMed] [Google Scholar]
- 21.Wherrett DK, Daneman D. Prevention of type 1 diabetes. Pediatr Clin North Am. 2011;58(5):1257–1270. xi. doi: 10.1016/j.pcl.2011.07.010. [DOI] [PubMed] [Google Scholar]
- 22.Mordes JP, Bortell R, Blankenhorn EP, Rossini AA, Greiner DL. Rat models of type 1 diabetes: genetics, environment, and autoimmunity. ILAR J. 2004;45(3):278–291. doi: 10.1093/ilar.45.3.278. [DOI] [PubMed] [Google Scholar]
- 23.Mordes JP, Guberski DL, Leif JH, Woda BA, Flanagan JF, Greiner DL, et al. LEW.1WR1 rats develop autoimmune diabetes spontaneously and in response to environmental perturbation. Diabetes. 2005;54(9):2727–2733. doi: 10.2337/diabetes.54.9.2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ellerman KE, Like AA. Susceptibility to diabetes is widely distributed in normal class IIu haplotype rats. Diabetologia. 2000;43(7):890–898. doi: 10.1007/s001250051466. [DOI] [PubMed] [Google Scholar]
- 25.Blankenhorn EP, Cort L, Greiner DL, Guberski DL, Mordes JP. Virus-induced autoimmune diabetes in the LEW.1WR1 rat requires Iddm14 and a genetic locus proximal to the major histocompatibility complex. Diabetes. 2009;58(12):2930–2938. doi: 10.2337/db09-0387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Martin AM, Maxson MN, Leif J, Mordes JP, Greiner DL, Blankenhorn EP. Diabetes-prone and diabetes-resistant BB rats share a common major diabetes susceptibility locus, iddm4: additional evidence for a"universal autoimmunity locus" on rat chromosome 4. Diabetes. 1999;48(11):2138–2144. doi: 10.2337/diabetes.48.11.2138. [DOI] [PubMed] [Google Scholar]
- 27.Martin AM, Blankenhorn EP, Maxson MN, Zhao M, Leif J, Mordes JP, et al. Non-major histocompatibility complex-linked diabetes susceptibility loci on chromosomes 4 and 13 in a backcross of the DP-BB/Wor rat to the WF rat. Diabetes. 1999;48(1):50–58. doi: 10.2337/diabetes.48.1.50. [DOI] [PubMed] [Google Scholar]
- 28.Blankenhorn EP, Rodemich L, Martin-Fernandez C, Leif J, Greiner DL, Mordes JP. The rat diabetes susceptibility locus Iddm4 and at least one additional gene are required for autoimmune diabetes induced by viral infection. Diabetes. 2005;54(4):1233–1237. doi: 10.2337/diabetes.54.4.1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Deruytter N, Boulard O, Garchon HJ. Mapping non-class II H2-linked loci for type 1 diabetes in nonobese diabetic mice. Diabetes. 2004;53(12):3323–3327. doi: 10.2337/diabetes.53.12.3323. [DOI] [PubMed] [Google Scholar]
- 30.Aly TA, Ide A, Jahromi MM, Barker JM, Fernando MS, Babu SR, et al. Extreme genetic risk for type 1A diabetes. Proc Natl Acad Sci U S A. 2006;103(38):14074–14079. doi: 10.1073/pnas.0606349103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Aly TA, Baschal EE, Jahromi MM, Fernando MS, Babu SR, Fingerlin TE, et al. Analysis of single nucleotide polymorphisms identifies major type 1A diabetes locus telomeric of the major histocompatibility complex. Diabetes. 2008;57(3):770–776. doi: 10.2337/db07-0900. [DOI] [PubMed] [Google Scholar]
- 32.Awata T, Guberski DL, Like AA. Genetics of the BB rat: association of autoimmune disorders (diabetes, insulitis, and thyroiditis) with lymphopenia and major histocompatibility complex class II. Endocrinology. 1995;136(12):5731–5735. doi: 10.1210/endo.136.12.7588330. [DOI] [PubMed] [Google Scholar]
- 33.Kohoutava M, Gunther E, Stark O. Genetic definition of a further gene region and identification of at least three different histocompatibility genes in the rat major histocompatibility system. Immunogenetics. 1980;11(5):483–490. doi: 10.1007/BF01567816. [DOI] [PubMed] [Google Scholar]
- 34.Blankenhorn EP, Descipio C, Rodemich L, Cort L, Leif JH, Greiner DL, et al. Refinement of the Iddm4 diabetes susceptibility locus reveals TCRVbeta4 as a candidate gene. Ann N Y Acad Sci. 2007;1103:128–131. doi: 10.1196/annals.1394.020. [DOI] [PubMed] [Google Scholar]
- 35.Hipp MS, Kalveram B, Raasi S, Groettrup M, Schmidtke G. FAT10, a ubiquitin-independent signal for proteasomal degradation. Mol Cell Biol. 2005;25(9):3483–3491. doi: 10.1128/MCB.25.9.3483-3491.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lukasiak S, Schiller C, Oehlschlaeger P, Schmidtke G, Krause P, Legler DF, et al. Proinflammatory cytokines cause FAT10 upregulation in cancers of liver and colon. Oncogene. 2008;27(46):6068–6074. doi: 10.1038/onc.2008.201. [DOI] [PubMed] [Google Scholar]
- 37.Atkinson MA, Leiter EH. The NOD mouse model of type 1 diabetes: as good as it gets? Nat Med. 1999;5(6):601–604. doi: 10.1038/9442. [DOI] [PubMed] [Google Scholar]
- 38.Mordes J, Serreze DV, Greiner DL, Rossini AA. Animal models of autoimmune diabetes mellitus. In: LeRoith D, Taylor SI, Olefsky JM, editors. Diabetes mellitus. A fundamental and clinical text. 2004. pp. 591–610. [Google Scholar]
- 39.Geurts AM, Cost GJ, Freyvert Y, Zeitler B, Miller JC, Choi VM, et al. Knockout rats via embryo microinjection of zinc-finger nucleases. Science. 2009;325(5939):433. doi: 10.1126/science.1172447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Geurts AM, Cost GJ, Remy S, Cui X, Tesson L, Usal C, et al. Generation of gene-specific mutated rats using zinc-finger nucleases. Methods Mol Biol. 2010;597:211–225. doi: 10.1007/978-1-60327-389-3_15. [DOI] [PubMed] [Google Scholar]
- 41.Geurts AM, Moreno C. Zinc-finger nucleases: new strategies to target the rat genome. Clin Sci (Lond) 2010;119(8):303–311. doi: 10.1042/CS20100201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jacob HJ, Lazar J, Dwinell MR, Moreno C, Geurts AM. Gene targeting in the rat: advances and opportunities. Trends Genet. 2010;26(12):510–518. doi: 10.1016/j.tig.2010.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chernova T, Higginson FM, Davies R, Smith AG. B2 SINE retrotransposon causes polymorphic expression of mouse 5-aminolevulinic acid synthase 1 gene. Biochem Biophys Res Commun. 2008;377(2):515–520. doi: 10.1016/j.bbrc.2008.10.020. [DOI] [PubMed] [Google Scholar]
- 44.Mariner PD, Walters RD, Espinoza CA, Drullinger LF, Wagner SD, Kugel JF, et al. Human Alu RNA is a modular transacting repressor of mRNA transcription during heat shock. Mol Cell. 2008;29(4):499–509. doi: 10.1016/j.molcel.2007.12.013. [DOI] [PubMed] [Google Scholar]
- 45.Bates EE, Ravel O, Dieu MC, Ho S, Guret C, Bridon JM, et al. Identification and analysis of a novel member of the ubiquitin family expressed in dendritic cells and mature B cells. Eur J Immunol. 1997;27(10):2471–2477. doi: 10.1002/eji.1830271002. [DOI] [PubMed] [Google Scholar]
- 46.Canaan A, Yu X, Booth CJ, Lian J, Lazar I, Gamfi SL, et al. FAT10/diubiquitin-like protein-deficient mice exhibit minimal phenotypic differences. Mol Cell Biol. 2006;26(13):5180–5189. doi: 10.1128/MCB.00966-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gong P, Canaan A, Wang B, Leventhal J, Snyder A, Nair V, et al. The ubiquitin-like protein FAT10 mediates NF-kappaB activation. J Am Soc Nephrol. 2010;21(2):316–326. doi: 10.1681/ASN.2009050479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ebstein F, Lange N, Urban S, Seifert U, Kruger E, Kloetzel PM. Maturation of human dendritic cells is accompanied by functional remodelling of the ubiquitin-proteasome system. Int J Biochem Cell Biol. 2009;41(5):1205–1215. doi: 10.1016/j.biocel.2008.10.023. [DOI] [PubMed] [Google Scholar]
- 49.Ebstein F, Lehmann A, Kloetzel PM. The FAT10- and ubiquitin-dependent degradation machineries exhibit common and distinct requirements for MHC class I antigen presentation. Cell Mol Life Sci. 2012;69(14):2443–2454. doi: 10.1007/s00018-012-0933-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schmidtke G, Kalveram B, Groettrup M. Degradation of FAT10 by the 26S proteasome is independent of ubiquitylation but relies on NUB1L. FEBS Lett. 2009;583(3):591–594. doi: 10.1016/j.febslet.2009.01.006. [DOI] [PubMed] [Google Scholar]
- 51.Yokoi N, Komeda K, Wang HY, Yano H, Kitada K, Saitoh Y, et al. Cblb is a major susceptibility gene for rat type 1 diabetes mellitus. Nat Genet. 2002;31(4):391–394. doi: 10.1038/ng927. [DOI] [PubMed] [Google Scholar]
- 52.Yokoi N, Fujiwara Y, Wang HY, Kitao M, Hayashi C, Someya T, et al. Identification and functional analysis of CBLB mutations in type 1 diabetes. Biochem Biophys Res Commun. 2008;368(1):37–42. doi: 10.1016/j.bbrc.2008.01.032. [DOI] [PubMed] [Google Scholar]
- 53.Baschal EE, Sarkar SA, Boyle TA, Siebert JC, Jasinski JM, Grabek KR, et al. Replication and further characterization of a Type 1 diabetes-associated locus at the telomeric end of the major histocompatibility complex. Journal of diabetes. 2011;3(3):238–247. doi: 10.1111/j.1753-0407.2011.00131.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kohoutavá M, Günther E, Stark O. Genetic definition of a further gene region and identification of at least three different histocompatibility genes in the rat major histocompatibility system. Immunogenetics. 1980;11(5):483–490. doi: 10.1007/BF01567816. [DOI] [PubMed] [Google Scholar]
- 55.Mordes JP, Leif J, Novak S, DeScipio C, Greiner DL, Blankenhorn EP. The iddm4 locus segregates with diabetes susceptibility in congenic WF.iddm4 rats. Diabetes. 2002;51(11):3254–3262. doi: 10.2337/diabetes.51.11.3254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Institute of Laboratory Animal Research CoLS, National Research Council. Guide for the Care and Use of Laboratory Animals. The National Academies Press; 1996. [Google Scholar]
- 57.Mordes JP, Cort L, Norowski E, Leif J, Fuller JM, Lernmark A, et al. Analysis of the rat Iddm14 diabetes susceptibility locus in multiple rat strains: identification of a susceptibility haplotype in the Tcrb-V locus. Mamm Genome. 2009;20(3):162–169. doi: 10.1007/s00335-009-9172-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hong F, Breitling R, McEntee CW, Wittner BS, Nemhauser JL, Chory J. RankProd: a bioconductor package for detecting differentially expressed genes in meta-analysis. Bioinformatics. 2006;22(22):2825–2827. doi: 10.1093/bioinformatics/btl476. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.