

Significance
Atherosclerosis, the major cause of death and illness in industrialized nations, develops in regions of arteries in which fluid flow patterns are disturbed and endothelial cells fail to align in the direction of flow. In contrast, regions of laminar flow in which cells are aligned are protected. The current work shows that the transmembrane proteoglycan syndecan 4 is required for endothelial cell alignment in the direction of flow and for the protective effect of high laminar flow, yet other flow responses are intact. The data therefore identify a role for syndecan 4 in flow direction sensing, show that sensing flow direction is separable from sensing flow magnitude, and provide new support for the key role of cell alignment in atheroprotection.
Keywords: mechanotransduction, polarity, shear stress, atherosclerosis
Abstract
Atherosclerotic plaque localization correlates with regions of disturbed flow in which endothelial cells (ECs) align poorly, whereas sustained laminar flow correlates with cell alignment in the direction of flow and resistance to atherosclerosis. We now report that in hypercholesterolemic mice, deletion of syndecan 4 (S4−/−) drastically increased atherosclerotic plaque burden with the appearance of plaque in normally resistant locations. Strikingly, ECs from the thoracic aortas of S4−/− mice were poorly aligned in the direction of the flow. Depletion of S4 in human umbilical vein endothelial cells (HUVECs) using shRNA also inhibited flow-induced alignment in vitro, which was rescued by re-expression of S4. This effect was highly specific, as flow activation of VEGF receptor 2 and NF-κB was normal. S4-depleted ECs aligned in cyclic stretch and even elongated under flow, although nondirectionally. EC alignment was previously found to have a causal role in modulating activation of inflammatory versus antiinflammatory pathways by flow. Consistent with these results, S4-depleted HUVECs in long-term laminar flow showed increased activation of proinflammatory NF-κB and decreased induction of antiinflammatory kruppel-like factor (KLF) 2 and KLF4. Thus, S4 plays a critical role in sensing flow direction to promote cell alignment and inhibit atherosclerosis.
Syndecan 4 (S4) is a transmembrane heparan sulfate proteoglycan that serves as a coreceptor for extracellular matrix proteins and growth factors (1–3). S4−/− mice are viable and fertile (4, 5) but show defective wound healing consequent to impaired angiogenesis (6). They also have higher mortality after LPS injection (7) and exhibit defective muscle repair and myofiber organization as a result of inefficient differentiation and migration of muscle satellite cells (8). We and others have also demonstrated that S4 plays a critical role in the control of cell polarity, by controlling Rho GTPase activity (9–11), as well as in planar cell polarity (12). S4 has also been recently identified as a putative mechanosensor (13).
Atherosclerosis is an inflammatory disease of large to midsized arteries that is the major cause of illness and death in developed nations and is rapidly increasing in developing nations (14, 15). It is linked to a variety of risk factors including high LDL cholesterol level and triglycerides, diabetes, smoking, hypertension, sedentary lifestyle, and inflammatory mediators. However, atherosclerotic lesions occur selectively in regions of arteries that are subject to disturbances in fluid shear stress (FSS), the frictional force flowing blood exerts on the endothelium. Regions of arteries with lower flow magnitude, flow reversal, and other complex spatial/temporal flow patterns are predisposed to atherosclerosis. Systemic risk factors appear to synergize with local biomechanical factors in the initiation and progression of atherosclerotic lesions (16).
The importance of S4 in endothelial biology prompted us to test its role in atherogenesis. Surprisingly, S4 deletion not only drastically increased atherosclerotic plaque burden in hypercholesterolemic mice but also caused plaque to form in regions that are normally resistant to disease. These findings led us to investigate the role of S4 in flow signaling. Our results showed that S4 is specifically required in alignment of endothelial cells (ECs) in flow and suggest that loss of this atheroprotective mechanism leads to increased atherosclerosis in S4−/− mice.
Results
Widespread Atherosclerotic Lesions in Hypercholesterolemic S4−/− Mice.
S4 KO mice were crossed into the hypercholesterolemic low-density lipoprotein receptor (LDLR)−/−/apolipoprotein B (apoB)100/100 (DKO) background. DKO/S4+/+ and DKO/S4−/− male mice at 12 wk were put on the lipid-enriched Paigen diet without cholate (PD) for 16–20 additional weeks to induce atherosclerotic lesions (17–20). In DKO/S4+/+ mice, small lesions in the descending aorta were visible after 16 wk of PD (Fig. 1 A and B), specifically localized near the branch points for small intercostal arteries, a known site of flow disturbance (21). These lesions covered, on average, 9% of the total aortic surface. In DKO/S4−/− aortas, widespread lesions near the intercostal bifurcations were also visible after 16 wk of PD, covering up to 18% of the total area (P = 0.0028). Surprisingly, nascent lesions also appeared in other regions, away from intercostal branches, that are normally atheroresistant. After 20 wk of PD, DKO/S4+/+ mice showed larger lesions that were still highly localized to branch points, whereas DKO/S4−/− mice showed massive appearance of lesions over much of the aorta (Fig. 1A). Lesions were also visible inside the intercostal arteries, where the flow profile is laminar (Fig. 1C). No significant difference in HDL and LDL levels was observed in the two strains, excluding a contribution of impaired lipoprotein metabolism: 239 ± 30 mg/dL LDL for DKO/S4−/− (n = 6) and 236 ± 6 mg/dL for DKO/S4+/+ (n = 4), and 46 ± 4 mg/dL HDL for DKO/S4−/− (n = 6) and 44 ± 6 for DKO/S4+/+ (n = 4; results are expressed as mean ± SD).
Fig. 1.

Effect of S4 deletion on atherosclerosis. (A) Thoracic aortas from male DKO hypercholesterolemic mice with or without S4 (S4+/+ or S4−/−) after 16 or 20 wk on PD. Aorta were stained with Oil red O to reveal the atherosclerotic lesions (in white/red). (B) Quantification of the lesions area, normalized to the total area of the thoracic aorta (n = 6 for DKO/S4+/+ and n = 7 for DKO/S4−/−; P < 0.01). (C) Detailed view of the intercostal arteries (white stars) after 20 wk of PD.
Deletion of S4 Inhibits Endothelial Alignment in Vivo.
The presence of lesions in areas that are normally resistant suggested impairment of flow-dependent atheroprotective mechanisms by loss of S4. We first examined EC alignment, which is highly correlated with atheroresistant regions of arteries (22, 23) and plays an important role in the activation of antiinflammatory versus proinflammatory pathways by flow (24). Staining the descending thoracic aorta for β-catenin to mark EC boundaries in wild-type C57BL/6 mice revealed uniformly elongated ECs that were well aligned in the direction of blood flow. In contrast, the same region in S4 KO mice showed poor alignment in the direction of flow, with markedly less elongation and many misaligned cells (Fig. 2A). The average cell shape index (4π area/perimeter2) was 0.33 ± 0.01 in wild-type mice and 0.58 ± 0.01 in S4 KO mice (n > 300 cells, four arteries). ECs in wild-type mice had well-organized actin stress fibers that were highly oriented in the direction of the flow, whereas in S4 KO mice, actin stress fibers were present but were poorly organized and misaligned (Fig. 2B; n > 300 cells, four arteries). Together, these data demonstrate a drastic loss of alignment in the direction of flow in S4 KO mice.
Fig. 2.

Endothelial morphology in the thoracic aorta. En face view of the thoracic aorta from wild-type or S4−/− mice. Tissue was stained for beta-catenin to mark cell borders (A) or with phalloidin and DAPI to mark stress fibers and nuclei (B). Stress fiber alignment: stress fibers were considered “aligned” if their direction was parallel to the direction of the cell major axis (within 30° of the major axis) and “misaligned” if not (n < 300 cells, four arteries per condition).
S4 Knock-Down Inhibits Flow Alignment in Vitro.
We then tested alignment under flow in vitro in ECs stably expressing S4 shRNA, which decreased S4 levels by 65–80% (Fig. 3C). These cells formed a confluent monolayer with normal actin cytoskeletal organization, similar to control cells with scrambled shRNA (Fig. 3A). Cells were subjected to steady laminar FSS (12 dynes⋅cm−2) for 16 h and then fixed, the nuclei stained, and alignment quantified by measuring the angle between the major axis of the nucleus and the flow direction. Nuclear orientation is highly correlated with stress fibers and cell orientation (25) (Fig. S1A) and is more accurately and easily measurable. S4-depleted cells showed a striking failure to align in the direction of flow (Figs. 3 A and B). A second shRNA sequence gave similar effects, rescued by expression of shRNA-resistant murine S4 (Fig. 3 C and D). In fact, rescued cells aligned somewhat better than control, untreated cells, probably because of the moderately higher S4 levels (Fig. 3C). We also noted that in preparations in which S4 expression was 5–10-fold above endogenous levels, flow-induced alignment was inhibited (Fig. S1C), indicating that S4 must be present at physiological levels. We also tested the response of cells over a wide range of shear stress magnitudes: scrambled shRNA cells aligned between 10 and 20 dynes⋅cm−2, whereas cells depleted for S4 never aligned in the flow direction (Fig. S1B).
Fig. 3.
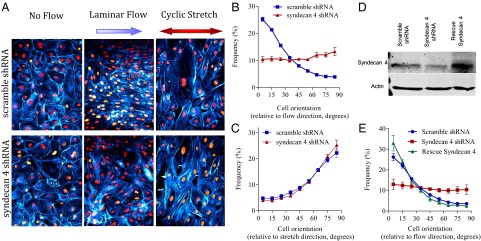
S4 is required for endothelial cell polarity in flow. (A) HUVECs stably expressing scrambled shRNA or S4 shRNA (#119) were untreated (no flow) or subjected to laminar FSS at 12 dynes/cm2 for 16 h [(B) quantification, n > 3,000 cells/experiment from 15 independent experiments] or 10% uniaxial cyclic stretch (1 Hz) for 4 h [(C) quantification, n > 2,000 cells/experiment from seven independent experiments]. Cells were fixed and labeled with DAPI (red) and phalloidin (cyan). (D) Rescue of S4 knockdown (shRNA #121) by adenoviral reexpression of rat S4. Western blot of S4 and actin as a loading control; all samples were sheared for 16 h. (E) Nuclear orientation was quantified to characterize cell orientation relative to flow direction (n > 3,000 cells, from at least four independent experiments). Values are means ± SEM.
To determine whether S4 is generally required for alignment in response to mechanical stimulation, cells were subjected to cyclic uniaxial stretch. S4 knock-down cells aligned perpendicularly to the direction of the force, similar to control cells (Fig. 3 A and B). Thus, the failure to align in flow does not reflect a general defect in mechanical or cytoskeletal responses. In addition, we did not detect any difference in the endothelial glycocalyx after S4 knock-down in human umbilical vein endothelial cells (HUVECs; Fig. S2A). Heparan sulfate, the major proteoglycan component of S4, mostly localized on the basal side of scrambled shRNA cells, and its organization and localization were not affected by S4 depletion (Fig. S2B). These observations argue against direct roles for the endothelial glycocalyx or heparan sulfate organization in sensing flow direction.
S4 Knock-Down Cells Still Respond to Flow.
We have reported that multiple EC responses to flow, including cell alignment, require a complex of proteins at cell–cell junctions consisting of platelet endothelial cell adhesion molecule 1 (PECAM-1), vascular endothelial (VE)-cadherin, and VEGF receptor 2 (VEGFR2) (26). Flow triggers activation of the VEGFR2 tyrosine kinase, which mediates downstream events, including activation of NF-κB. We therefore examined activation of these events by flow. Phosphorylation at VEGFR2 tyrosine 1054 at 45 s after flow was increased similarly in S4 shRNA and control shRNA cells (Fig. 4A). Activation of NF-κB by flow was modestly, but significantly, higher in S4 knock-down cells compared with controls (Fig. 4C; P < 0.05). Thus, signaling through the junctional complex does not require S4.
Fig. 4.
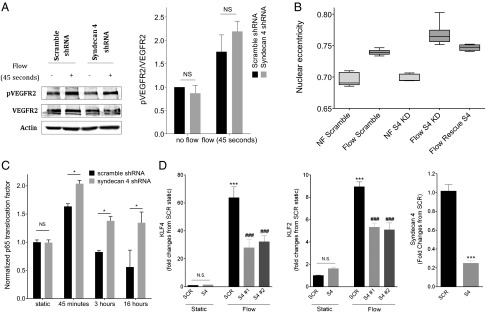
Effect of S4 knock-down on fluid shear-stress responses. (A) Western blot of phosphorylated and total VEGFR2, with actin as a loading control (n = 4, mean ± SEM). (B) Average nuclear eccentricity for all the nuclei in each experiment was quantified using the Matlab Eccentricity function (n ≥ 4 individual experiments, n > 3,000 nuclei per experiment; NF, no flow). (C) To assess NF-κB translocation, cells were stained for p65, imaged, and analyzed as described in Methods. The translocation factor is the total nuclear p65 fluorescence relative to the total cytosolic p65 fluorescence. Results for each experiment were then normalized to the static scramble cells. At least 2,000 cells were analyzed per condition, in at least three independent experiments (*P < 0.05, values are mean ± SEM). (D) S4, KLF2, and KLF4 message levels, relative to the static scramble mRNA for each individual experiment. mRNA levels were normalized to GAPDH (n = 4; ***P < 0.001 versus static scramble; ###P < 0.001 versus flow scrambled). SCR, scrambled shRNA; S4 #1, S4 shRNA #119; S4 #2, S4 shRNA #121. Values are mean ± SEM.
Fluid shear stress also induces elongation of the cell body and nucleus (27, 28). Remarkably, after flow for 16 h, nuclear eccentricity increased in S4 knock-down cells to a slightly greater extent than in control cells (Fig. 4B), despite their random orientation (Fig. 3). Together, these results underscore the highly specific defect in sensing flow direction after S4 depletion.
S4 Promotes Flow-Dependent Atheroprotective Pathways.
Alignment of ECs in the direction of flow is an important adaptive mechanism by which inflammatory pathways are down-regulated and antiinflammatory pathways are activated (24). This point is evident in Fig. 4C, where laminar flow at later times reduces p65 nuclear translocation below the levels seen in unstimulated conditions in wild-type cells. In contrast, in S4 knock-down cells, p65 translocation remained above the no-flow baseline and was strikingly higher than for control cells in flow for 16 h. To further assess proinflammatory versus antiinflammatory mechanisms, we measured levels of the antiinflammatory transcription factors kruppel-like factor (KLF) 2 and KLF4, which are induced by sustained laminar flow (29, 30). Induction by flow was substantially less in S4 shRNA cells compared with control cells (Fig. 4D; P < 0.05). Taken together, these results show that ECs lacking S4 fail to align in flow and have higher NF-κB activity and lower antiinflammatory KLF2 and KLF4 expression. These effects thereby may provide a mechanism for increased atherosclerosis in S4−/− mice, especially at normally atheroresistant regions of the vasculature.
Discussion
Although atherosclerosis is strongly associated with systemic risk factors such as high LDL cholesterol or diabetes, the localization of atherosclerotic lesions within arteries is highly correlated with areas of disturbed blood flow, characterized by low-magnitude FSS and directional changes during the cardiac cycle (31, 32). In contrast, high laminar shear inhibits the inflammatory, oxidative, and thrombotic pathways that promote atherosclerosis. The transcription factors Klf2 and Klf4 are major mediators of the atheroprotective phenotype in high laminar flow (29, 30), whereas NF-κB is a major proinflammatory transcription factor that promotes atherosclerosis (33). In vitro, onset of high-laminar FSS applied to ECs transiently activates the inflammatory transcription factor NF-κB; however, over several hours, cells align in the direction of flow and NF-κB declines to levels below baseline (34). Cell alignment in the direction of flow has therefore been proposed to be an adaptive mechanism that alters the way forces act on the cells (35). In contrast, cells in disturbed flow do not align, Klf2 and Klf4 remain low (36, 37), and NF-κB and other inflammatory pathways remain high (22, 23, 34). Poor endothelial alignment is also a marker for susceptibility to atherosclerosis in vivo (38).
The results presented here identify S4 as a potent antiatherosclerotic molecule. In particular, the appearance of plaques in normally atheroresistant regions of arteries was striking. We cannot completely exclude that lesions may propagate into atheroresistant areas because of the effects of lesions on downstream flow. However, the observed phenotype is not observed in other mouse models of severe atherosclerosis, even after a prolonged high-fat diet (39–42). Moreover, mislocalized plaque was evident even at earlier times. The increase and broad distribution of plaque correlated with loss of EC alignment in the direction of flow. S4 suppression also inhibited flow-dependent alignment in vitro, accompanied by elevated NF-κB activity and decreased Klf2 and Klf4 expression at longer times in high laminar shear. This effect was highly specific, as loss of S4 inhibited neither signals through the PECAM-1/VE-cadherin/VEGFR2 complex involved in shear stress sensing (26) nor alignment of ECs in cyclic stretch. Remarkably, nuclei in S4-depleted ECs elongated in flow, although without any preferred direction. These results lead to the conclusion that S4 is specifically required for sensing flow direction, which is independent of other aspects of flow mechanotransduction. Although ECs are generally thought to have multiple flow sensors (16), to our knowledge, this work provides the first evidence that flow direction sensing is separate from sensing flow magnitude.
The alignment defect in vivo, together with recent results showing that alignment is critical for the switch from proinflammatory to antiinflammatory signaling (24), suggest the hypothesis that loss of alignment leads to an activated endothelium, which increases susceptibility to atherosclerotic risk factors. It is tempting to speculate further that these findings may relate to the minority of cases in which atherosclerotic plaque occurs in regions of coronary arteries where flow patterns are expected to be laminar (43). Examining EC alignment and S4 expression in human specimens could test whether loss of this mechanism is a factor in the diffuse atherosclerosis seen in some patients (44–47).
In summary, these data reveal a highly specific role for S4 in sensing flow direction. Its loss, in vitro, leads to misaligned cells that show proinflammatory “priming,” which we hypothesize is similar to what is normally seen in atheroprone regions of disturbed flow (33). These cells are then susceptible to further activation by global risk factors, leading to formation of atherosclerotic lesions. How S4 mediates shear stress direction sensing is presently unknown. Indeed, the highly specific role of S4 in direction sensing underscores our ignorance about the mechanisms of this process. S4 can cooperate with integrins in adhesion to extracellular matrix and subsequent signaling (48); however, in the context of flow signaling, our data show they clearly act on distinct pathways. S4 has been reported to interact with polarity proteins (12), to control polarized recycling of integrins (49, 50), and to control polarized activation of Rac1 (9). It is attractive to speculate that the interactions between S4 and integrins might therefore be involved in the integration of adhesion/signaling pathways and polarity pathways in responses to flow. These questions await further research. The identification of S4 effector pathways is therefore an important question for future work that may provide a means for understanding more generally how ECs sense flow direction.
Methods
Cell Culture.
HUVECs, in which each batch was pooled from three different donors, were obtained from the Yale Vascular Biology and Therapeutics program. Cells were cultured in M199 medium supplemented with 20% FBS, 50 µg⋅mL−1 endothelial cell growth supplement prepared from bovine hypothalamus, 100 μg⋅mL−1 heparin, 100 U⋅mL−1 penicillin, and 100 μg⋅mL−1 streptomycin. They were used between passages 3 and 5.
Lentivirus Generation.
Lentivirus for stable shRNA expression was generated as described (51). Briefly, packaging plasmids (Addgene) were mixed with shRNA plasmid (Mission shRNA, Sigma-Aldrich) in Optimem medium (Invitrogen) and Lipofectamine 2000 (Invitrogen) with the following ratios: 5 μg pMDL/pRRE, 2.5 μg pRSV-Rev, 2.5 μg pCMV-VSG, and 10 μg shRNA. The mixture was transferred to 90% confluent 293T cells in 10-cm dishes for 6 h. The medium was replaced with regular DMEM 10% FBS and collected after 48 h. Medium containing virus was filtered through a 0.45-μm filter and used immediately for HUVEC transduction. The target sequences for human S4 were: #119, targets 3UTR: CCGGGCCAGGTTCTTCTTGAGCTTTCTCGAGAAAGCTCAAGAAGAAGAACCTGGCTTTTTG; and #121, targets controlled dangerous substance: CCGGCCCGTTGAAGAGAGTGAGGATCTCGAGATCCTCACTCTCTTCAACGGGTTTTTG.
Stable Knock-Down.
To achieve stable knockdown, HUVECs at passage 2 were seeded on gelatin-coated 10-cm plates and transduced at 70% confluence with freshly produced lentivirus carrying scrambled or S4 shRNAs also containing a puromycin-resistance gene. After 6 h, the medium was replaced with complete M199 medium. Forty-eight hours after infection, cells were treated with puromycin (1 μg⋅mL−1) for 3 d. Selected cells were maintained in complete M199 medium with puromycin (0.4 μg⋅mL−1) and used for no more than two more passages.
Adenoviral Expression.
Rat S4 [previously known as ryudocan (52)] with a hemagglutinin tag after the signal peptide was cloned into the Ad-Cla (E1/E3 deleted) adenoviral vector provided by the Harvard Gene Therapy Initiative (hgti.med.harvard.edu) virus core laboratory, which subsequently used the construct for the production of replication-deficient adenovirus.
Shear Stress.
Cells were seeded on fibronectin-coated (20 μg⋅mL−1) slides. After reaching confluency, cells were starved with M199 medium containing 5% FBS with 100 U⋅mL−1 penicillin and 100 μg⋅mL−1 streptomycin for a minimum of 4 h. Shear stress with a calculated intensity of 12 dynes⋅cm−2 was applied in a parallel flow chamber (53). Alignment was examined after 16 h of steady laminar flow.
Uniaxial Cyclic Stretch.
Cells were seeded on fibronectin-coated Uniflex 6-well culture plates (Flexcell International Corporation) for 48 h. Plates were transferred to a Flexcell 5000 station and submitted to uniaxial stretch for 4 h (10% tension, 1Hz, sinusoidal waveform). Cells were immediately fixed with 3.7% paraformaldehyde (PFA) for 10 min. We then removed the central rectangular area of the well with a scalpel, processed for staining, and mounted on a glass slide.
Western Blot.
Cells were washed with cold PBS and proteins were extracted with Laemmli’s buffer. Samples were run on 8% SDS/PAGE and transferred onto nitrocellulose membranes. Membrane was blocked with StartingBlock buffer (Thermo Scientific) and probed with primary antibodies overnight at 4 °C: pVEGFR2 (Invitrogen), VEGFR2 (Cell Signaling), S4 (Abcam), and actin (Santa Cruz). DyLight conjugated fluorescent secondary antibodies (680 and 800 nm; Thermo Scientific) were used to detect primary antibodies. Bands were detected and quantified with an Odyssey infrared imaging system (Li-Cor).
Immunofluorescence.
Cells were fixed for 10 min with 3.7% PFA, permeabilized 10 min with 1% Triton X-100 in PBS, blocked for 30 min with StartingBlock buffer (Thermo Scientific), and then probed with p65 antibody (Cell Signaling) or phalloidin conjugated to 647 Alexa Fluor (Molecular Probes) and DAPI. Cells were mounted in Fluoromount G. Aortas were perfusion-fixed with 3.7% PFA, excised, and adventitial tissue was removed. The vessels were opened longitudinally and fixed again for 4 h at 4 °C. They were permeabilized with 0.1% Triton X-100 in PBS for 5 min, blocked for 1 h with 5% normal goat serum in PBS (Cell Signaling), and probed with beta-catenin antibody (Cell Signaling) or phalloidin-conjugated to 647 Alexa Fluor (Molecular Probes) and DAPI. Specimens were mounted, en face, using fluoromount G. Images were captured with a 20× objective (HUVEC cells) or 63× oil immersion objective (en face aorta) mounted on a Perkin-Elmer spinning disk confocal microscope.
Image Analysis.
Masks of the images were made using a combination of an adaptive histogram equalization algorithm with intensity and size thresholding. Cell orientation was calculated by taking the masks of the cell nuclei (determined from DAPI images), fitting them to an ellipse, and determining the angle between the flow direction and the major axis of the ellipse. Nuclei eccentricity was measured on the basis of the eccentricity of the fitted ellipse. Nuclear translocation was computed by taking the mask of the transcription factor stain (p65) and calculating the product of both the area and the intensity of the stain present in the nucleus and dividing it by the product of the total stain area and the total intensity of the stain. This yields a unitless metric we termed the translocation factor, which equals 1 in cells with complete nuclear translocation and 0 in cells with no translocation.
RNA Isolation and Real-time PCR.
Cells were washed with PBS and homogenized with a QIAshredder kit (Qiagen). Total RNA was extracted with an RNeasy Plus mini kit (Qiagen), which eliminates gDNA. cDNA synthesis was performed with an iScript cDNA synthesis kit (Bio-Rad). Quantitative real-time PCR was performed in triplicate, using an iQSYBR Green Supermix kit and CFX96TM real-time system (Bio-Rad). Thermocycling conditions were 95 °C for 3 min, followed by 45 cycles that used 95 °C for 10 s and 60 °C for 30 s. Gene expression was normalized with the housekeeping gene (GAPDH), and relative expression was calculated using the ΔΔCt method. Primers sequences: GAPDH: 5′-GAGTCAACGGATTTGGTCGT-3′ (sense) and 5′-GACAAGCTTCCCGTTCTCAG-3′ (antisense); S4: 5′- TGTTCTTCGTAGGCGGAGTC-3′ (sense) and 5′- CCCCACTACATCCTCATCGT-3′ (antisense); KLF2: 5′-GCCACTCACCGGTGTCC-3′ (sense) and 5′-CGGCCAGCGCTCCTG-3′ (antisense); and KLF4: 5′-ATCTCGGCCAATTTGGGGTT-3′ (sense) and 5′-TTGACGCAGTGTCTTCTCCC-3′ (antisense).
Mouse Strains and Diet.
S4−/− and LDLR−/− KO/ApoB 100/100 S4−/− mice were generated by heterozygous crossing and then nine backcrosses, including daughter–father crosses on a C57BL/6 background. B6;129S-Apobtm2Sgy Ldlrtm1Her/J (LDLR−/− ApoB100/100; DKO) mice (Jackson Laboratories) were crossed with S4−/− mice. LDLR−/− ApoB100/100 genotype was confirmed by PCR amplification of DNA extracted from tail snips, using primer sets and PCR conditions described by Jackson Laboratories. The S4−/− genotype was verified using forward primer 5′ GGAGAGTCGATTCGAGAG 3′ and reverse primer 5′ AAGCCATGCGTAGAACTC 3′. Thermocycler conditions were as follows: one cycle at 95 °C for 1 min, followed by 40 cycles of 95 °C for 45 s, 56 °C for 45 s, 72 °C for 60 s, and one cycle at 72 °C for 5 min. LDLR−/− ApoB100/100/ S4−/− mice were backcrossed seven times. Atherogenic LDLR−/− ApoB100/100/ S4−/− were fed normal chow diet for 12 wk, followed by 14–20 wk PD (Research Diets), as previously described (17–19). PD consists of 20% protein, 45% carbohydrate, 35% fat, and no cholate (54).
Statistical Analysis.
At least three independent experiments were performed for each condition. Statistical differences were tested by using either analysis of variance tests or nonpaired Student t tests, as indicated.
Supplementary Material
Acknowledgments
Lipid analysis was performed by the Yale Mouse Phenotypic Center, supported by a U24 DK059635 grant support. This work was supported by United States Public Health Services Grants PO1 HL107205 (to M.A.S. and M.S.) and HL069948 (to M.J.M.-K.). N.B. was supported by a Belgian American Educational Foundation postdoctoral fellowship, a Wallonie-Bruxelles International.World excellence scholarship, and an American Heart Association postdoctoral fellowship (14POST19020010). F.C. was supported by the American-Italian Cancer Foundation postdoctoral fellowship.
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1413725111/-/DCSupplemental.
References
- 1.Elfenbein A, Simons M. Syndecan-4 signaling at a glance. J Cell Sci. 2013;126(Pt 17):3799–3804. doi: 10.1242/jcs.124636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tkachenko E, Rhodes JM, Simons M. Syndecans: New kids on the signaling block. Circ Res. 2005;96(5):488–500. doi: 10.1161/01.RES.0000159708.71142.c8. [DOI] [PubMed] [Google Scholar]
- 3.Saoncella S, et al. Syndecan-4 signals cooperatively with integrins in a Rho-dependent manner in the assembly of focal adhesions and actin stress fibers. Proc Natl Acad Sci USA. 1999;96(6):2805–2810. doi: 10.1073/pnas.96.6.2805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ishiguro K, et al. Syndecan-4 deficiency impairs the fetal vessels in the placental labyrinth. Dev Dyn. 2000;219(4):539–544. doi: 10.1002/1097-0177(2000)9999:9999<::AID-DVDY1081>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 5.Ishiguro K, et al. Syndecan-4 deficiency impairs focal adhesion formation only under restricted conditions. J Biol Chem. 2000;275(8):5249–5252. doi: 10.1074/jbc.275.8.5249. [DOI] [PubMed] [Google Scholar]
- 6.Echtermeyer F, et al. Delayed wound repair and impaired angiogenesis in mice lacking syndecan-4. J Clin Invest. 2001;107(2):R9–R14. doi: 10.1172/JCI10559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ishiguro K, et al. Syndecan-4 deficiency leads to high mortality of lipopolysaccharide-injected mice. J Biol Chem. 2001;276(50):47483–47488. doi: 10.1074/jbc.M106268200. [DOI] [PubMed] [Google Scholar]
- 8.Cornelison DD, et al. Essential and separable roles for Syndecan-3 and Syndecan-4 in skeletal muscle development and regeneration. Genes Dev. 2004;18(18):2231–2236. doi: 10.1101/gad.1214204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Elfenbein A, et al. Suppression of RhoG activity is mediated by a syndecan 4-synectin-RhoGDI1 complex and is reversed by PKCalpha in a Rac1 activation pathway. J Cell Biol. 2009;186(1):75–83. doi: 10.1083/jcb.200810179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brooks R, Williamson R, Bass M. Syndecan-4 independently regulates multiple small GTPases to promote fibroblast migration during wound healing. Small GTPases. 2012;3(2):73–79. doi: 10.4161/sgtp.19301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tkachenko E, Elfenbein A, Tirziu D, Simons M. Syndecan-4 clustering induces cell migration in a PDZ-dependent manner. Circ Res. 2006;98(11):1398–1404. doi: 10.1161/01.RES.0000225283.71490.5a. [DOI] [PubMed] [Google Scholar]
- 12.Escobedo N, et al. Syndecan 4 interacts genetically with Vangl2 to regulate neural tube closure and planar cell polarity. Development. 2013;140(14):3008–3017. doi: 10.1242/dev.091173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bellin RM, et al. Defining the role of syndecan-4 in mechanotransduction using surface-modification approaches. Proc Natl Acad Sci USA. 2009;106(52):22102–22107. doi: 10.1073/pnas.0902639106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Perret F, Bovet P, Shamlaye C, Paccaud F, Kappenberger L. High prevalence of peripheral atherosclerosis in a rapidly developing country. Atherosclerosis. 2000;153(1):9–21. doi: 10.1016/s0021-9150(00)00376-2. [DOI] [PubMed] [Google Scholar]
- 15.Go AS, et al. American Heart Association Statistics Committee and Stroke Statistics Subcommittee Heart disease and stroke statistics—2014 update: A report from the American Heart Association. Circulation. 2014;129(3):e28–e292. doi: 10.1161/01.cir.0000441139.02102.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Conway DE, Schwartz MA. Flow-dependent cellular mechanotransduction in atherosclerosis. J Cell Sci. 2013;126(Pt 22):5101–5109. doi: 10.1242/jcs.138313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Drinane M, et al. The antiangiogenic activity of rPAI-1(23) inhibits vasa vasorum and growth of atherosclerotic plaque. Circ Res. 2009;104(3):337–345. doi: 10.1161/CIRCRESAHA.108.184622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mollmark J, et al. Antiangiogenic activity of rPAI-1(23) promotes vasa vasorum regression in hypercholesterolemic mice through a plasmin-dependent mechanism. Circ Res. 2011;108(12):1419–1428. doi: 10.1161/CIRCRESAHA.111.246249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mollmark JI, et al. Fibroblast growth factor-2 is required for vasa vasorum plexus stability in hypercholesterolemic mice. Arterioscler Thromb Vasc Biol. 2012;32(11):2644–2651. doi: 10.1161/ATVBAHA.112.252544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Getz GS, Reardon CA. Diet and murine atherosclerosis. Arterioscler Thromb Vasc Biol. 2006;26(2):242–249. doi: 10.1161/01.ATV.0000201071.49029.17. [DOI] [PubMed] [Google Scholar]
- 21.Kazakidi A, Plata AM, Sherwin SJ, Weinberg PD. Effect of reverse flow on the pattern of wall shear stress near arterial branches. J R Soc Interface. 2011;8(64):1594–1603. doi: 10.1098/rsif.2011.0108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chien S. Mechanotransduction and endothelial cell homeostasis: The wisdom of the cell. Am J Physiol Heart Circ Physiol. 2007;292(3):H1209–H1224. doi: 10.1152/ajpheart.01047.2006. [DOI] [PubMed] [Google Scholar]
- 23.Hahn C, Schwartz MA. Mechanotransduction in vascular physiology and atherogenesis. Nat Rev Mol Cell Biol. 2009;10(1):53–62. doi: 10.1038/nrm2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang C, Baker BM, Chen CS, Schwartz MA. Endothelial cell sensing of flow direction. Arterioscler Thromb Vasc Biol. 2013;33(9):2130–2136. doi: 10.1161/ATVBAHA.113.301826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Versaevel M, Grevesse T, Gabriele S. Spatial coordination between cell and nuclear shape within micropatterned endothelial cells. Nat Commun. 2012;3:671. doi: 10.1038/ncomms1668. [DOI] [PubMed] [Google Scholar]
- 26.Tzima E, et al. A mechanosensory complex that mediates the endothelial cell response to fluid shear stress. Nature. 2005;437(7057):426–431. doi: 10.1038/nature03952. [DOI] [PubMed] [Google Scholar]
- 27.Dahl KN, Ribeiro AJ, Lammerding J. Nuclear shape, mechanics, and mechanotransduction. Circ Res. 2008;102(11):1307–1318. doi: 10.1161/CIRCRESAHA.108.173989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Deguchi S, Maeda K, Ohashi T, Sato M. Flow-induced hardening of endothelial nucleus as an intracellular stress-bearing organelle. J Biomech. 2005;38(9):1751–1759. doi: 10.1016/j.jbiomech.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 29.Hamik A, et al. Kruppel-like factor 4 regulates endothelial inflammation. J Biol Chem. 2007;282(18):13769–13779. doi: 10.1074/jbc.M700078200. [DOI] [PubMed] [Google Scholar]
- 30.SenBanerjee S, et al. KLF2 Is a novel transcriptional regulator of endothelial proinflammatory activation. J Exp Med. 2004;199(10):1305–1315. doi: 10.1084/jem.20031132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Malek AM, Alper SL, Izumo S. Hemodynamic shear stress and its role in atherosclerosis. JAMA. 1999;282(21):2035–2042. doi: 10.1001/jama.282.21.2035. [DOI] [PubMed] [Google Scholar]
- 32.Ku DN, Giddens DP, Zarins CK, Glagov S. Pulsatile flow and atherosclerosis in the human carotid bifurcation. Positive correlation between plaque location and low oscillating shear stress. Arteriosclerosis. 1985;5(3):293–302. doi: 10.1161/01.atv.5.3.293. [DOI] [PubMed] [Google Scholar]
- 33.Hajra L, et al. The NF-kappa B signal transduction pathway in aortic endothelial cells is primed for activation in regions predisposed to atherosclerotic lesion formation. Proc Natl Acad Sci USA. 2000;97(16):9052–9057. doi: 10.1073/pnas.97.16.9052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mohan S, Mohan N, Sprague EA. Differential activation of NF-kappa B in human aortic endothelial cells conditioned to specific flow environments. Am J Physiol. 1997;273(2 Pt 1):C572–C578. doi: 10.1152/ajpcell.1997.273.2.C572. [DOI] [PubMed] [Google Scholar]
- 35.Davies PF. Flow-mediated endothelial mechanotransduction. Physiol Rev. 1995;75(3):519–560. doi: 10.1152/physrev.1995.75.3.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ni CW, et al. Discovery of novel mechanosensitive genes in vivo using mouse carotid artery endothelium exposed to disturbed flow. Blood. 2010;116(15):e66–e73. doi: 10.1182/blood-2010-04-278192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang N, et al. Shear stress regulation of Krüppel-like factor 2 expression is flow pattern-specific. Biochem Biophys Res Commun. 2006;341(4):1244–1251. doi: 10.1016/j.bbrc.2006.01.089. [DOI] [PubMed] [Google Scholar]
- 38.Nerem RM, Levesque MJ, Cornhill JF. Vascular endothelial morphology as an indicator of the pattern of blood flow. J Biomech Eng. 1981;103(3):172–176. doi: 10.1115/1.3138275. [DOI] [PubMed] [Google Scholar]
- 39.Palinski W, et al. ApoE-deficient mice are a model of lipoprotein oxidation in atherogenesis. Demonstration of oxidation-specific epitopes in lesions and high titers of autoantibodies to malondialdehyde-lysine in serum. Arterioscler Thromb. 1994;14(4):605–616. doi: 10.1161/01.atv.14.4.605. [DOI] [PubMed] [Google Scholar]
- 40.Ma Y, et al. Hyperlipidemia and atherosclerotic lesion development in Ldlr-deficient mice on a long-term high-fat diet. PLoS ONE. 2012;7(4):e35835. doi: 10.1371/journal.pone.0035835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lichtman AH, et al. Hyperlipidemia and atherosclerotic lesion development in LDL receptor-deficient mice fed defined semipurified diets with and without cholate. Arterioscler Thromb Vasc Biol. 1999;19(8):1938–1944. doi: 10.1161/01.atv.19.8.1938. [DOI] [PubMed] [Google Scholar]
- 42.Sussan TE, et al. Disruption of Nrf2, a key inducer of antioxidant defenses, attenuates ApoE-mediated atherosclerosis in mice. PLoS ONE. 2008;3(11):e3791. doi: 10.1371/journal.pone.0003791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gould KL, et al. Frequency and clinical implications of fluid dynamically significant diffuse coronary artery disease manifest as graded, longitudinal, base-to-apex myocardial perfusion abnormalities by noninvasive positron emission tomography. Circulation. 2000;101(16):1931–1939. doi: 10.1161/01.cir.101.16.1931. [DOI] [PubMed] [Google Scholar]
- 44.Kate M, Sylaja PN, Kesavadas C, Thomas B. Imaging and clinical predictors of unfavorable outcome in medically treated symptomatic intracranial atherosclerotic disease. J Stroke Cerebrovasc Dis. 2014;23(5):973–978. doi: 10.1016/j.jstrokecerebrovasdis.2013.08.002. [DOI] [PubMed] [Google Scholar]
- 45.Gargiulo G, et al. Unexpected preserved brain perfusion imaging despite severe and diffuse atherosclerosis of supra-aortic trunks. Cardiovasc J Afr. 2013;24(3):e12–e14. doi: 10.5830/CVJA-2013-009. [DOI] [PubMed] [Google Scholar]
- 46.Botta L, Sonker U, Heijmen RH. Diffuse atherosclerosis of thoracic aorta involving supraaortic and coronary arteries: Single-stage surgical revascularization. Thorac Cardiovasc Surg. 2008;56(8):490–492. doi: 10.1055/s-2007-965712. [DOI] [PubMed] [Google Scholar]
- 47.Settepani F, Cappai A, Citterio E, Melis LC, Tarelli G. Unusable Radial Artery for Severe Atherosclerosis in a Young Patient. J Card Surg. 2014 doi: 10.1111/jocs.12356. [DOI] [PubMed] [Google Scholar]
- 48.Bass MD, Humphries MJ. Cytoplasmic interactions of syndecan-4 orchestrate adhesion receptor and growth factor receptor signalling. Biochem J. 2002;368(Pt 1):1–15. doi: 10.1042/BJ20021228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bass MD, et al. A syndecan-4 hair trigger initiates wound healing through caveolin- and RhoG-regulated integrin endocytosis. Dev Cell. 2011;21(4):681–693. doi: 10.1016/j.devcel.2011.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Morgan MR, et al. Syndecan-4 phosphorylation is a control point for integrin recycling. Dev Cell. 2013;24(5):472–485. doi: 10.1016/j.devcel.2013.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Corti F, Finetti F, Ziche M, Simons M. The syndecan-4/protein kinase Cα pathway mediates prostaglandin E2-induced extracellular regulated kinase (ERK) activation in endothelial cells and angiogenesis in vivo. J Biol Chem. 2013;288(18):12712–12721. doi: 10.1074/jbc.M113.452383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shworak NW, Kojima T, Rosenberg RD. Isolation and characterization of ryudocan and syndecan heparan sulfate proteoglycans, core proteins, and cDNAs from a rat endothelial cell line. Haemostasis. 1993;23(Suppl 1):161–176. doi: 10.1159/000216925. [DOI] [PubMed] [Google Scholar]
- 53.Tzima E, del Pozo MA, Shattil SJ, Chien S, Schwartz MA. Activation of integrins in endothelial cells by fluid shear stress mediates Rho-dependent cytoskeletal alignment. EMBO J. 2001;20(17):4639–4647. doi: 10.1093/emboj/20.17.4639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Paigen B, Morrow A, Brandon C, Mitchell D, Holmes P. Variation in susceptibility to atherosclerosis among inbred strains of mice. Atherosclerosis. 1985;57(1):65–73. doi: 10.1016/0021-9150(85)90138-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.