

Significance
The enzyme transglutaminase 2 (TG2) is the target of autoantibodies characteristic of the gluten-sensitive enteropathy celiac disease, and intact enzyme activity seems to be required for the disease-causing immune response. TG2 activity is regulated through conformational changes. Ca2+ binding is required for enzyme activity, whereas oxidation inactivates the enzyme. Using hydrogen/deuterium exchange monitored by mass spectrometry, we have studied differences between active and inactive forms of TG2 in solution and found that oxidation prevents Ca2+-induced structural changes. Further, we have characterized the TG2 binding of a panel of monoclonal autoantibodies derived from disease lesion plasma cells. Autoantibody binding affected the structure of TG2, and mapping of the targeted epitopes suggests a possible mechanism for the induction of the autoimmune response.
Keywords: celiac disease, transglutaminase 2, autoantibodies, hydrogen/deuterium exchange
Abstract
The multifunctional enzyme transglutaminase 2 (TG2) is the target of autoantibodies in the gluten-sensitive enteropathy celiac disease. In addition, the enzyme is responsible for deamidation of gluten peptides, which are subsequently targeted by T cells. To understand the regulation of TG2 activity and the enzyme’s role as an autoantigen in celiac disease, we have addressed structural properties of TG2 in solution by using hydrogen/deuterium exchange monitored by mass spectrometry. We demonstrate that Ca2+ binding, which is necessary for TG2 activity, induces structural changes in the catalytic core domain of the enzyme. Cysteine oxidation was found to abolish these changes, suggesting a mechanism whereby disulfide bond formation inactivates the enzyme. Further, by using TG2-specific human monoclonal antibodies generated from intestinal plasma cells of celiac disease patients, we observed that binding of TG2 by autoantibodies can induce structural changes that could be relevant for the pathogenesis. Detailed mapping of two of the main epitopes targeted by celiac disease autoantibodies revealed that they are located adjacent to each other in the N-terminal part of the TG2 molecule.
Celiac disease is a gluten-sensitive enteropathy occurring in genetically predisposed individuals and is driven by CD4+ T cells reactive to peptides derived from dietary cereal gluten proteins (1). The disease is tightly associated with the production of autoantibodies against the enzyme transglutaminase 2 (TG2) (2), although it is unclear whether the autoantibodies play a pathogenic role. As with other members of the transglutaminase enzyme family, TG2 catalyzes the Ca2+-dependent cross-linking of proteins through the formation of Nε(γ-glutamyl)lysine isopeptide bonds in a reaction known as transamidation (3). Alternatively, a small-molecule primary amine or water can substitute for lysine in the reaction. When water is used as a substrate, glutamine is converted into glutamic acid, and the reaction is termed deamidation. Gluten peptides are rich in glutamine residues that can be targeted by TG2, and TG2-mediated gluten deamidation plays a central role in the activation of gluten-reactive T cells in celiac disease (4). Thus, knowing how TG2 activity is regulated is key to our understanding of the initiation of the immune response.
TG2 is ubiquitously expressed and is produced in the cytosol of human cells. A substantial amount of the enzyme, however, is exported to the extracellular environment through an unconventional mechanism (5). Once outside the cell, TG2 is in a Ca2+-rich environment supportive of transamidation/deamidation, and the extracellular enzyme is believed to be involved in cross-linking of matrix proteins (6, 7). In the cytosol, TG2 presumably works as GTPase (8). Binding of GTP/GDP inhibits transamidation/deamidation (9, 10). Thus, a high intracellular concentration of GTP in combination with a low Ca2+ concentration prevents transglutaminase activity in the cytosol under normal physiological conditions (10, 11).
Depending on the binding of effectors, TG2 can adopt two distinct conformations (10, 12, 13). Crystal structures of the enzyme in a complex with GDP or GTP reveal a “closed” conformation, in which the two C-terminal β-barrel domains are folded in on the catalytic core domain and cover the active site (14, 15). Conversely, when a peptide inhibitor was irreversibly attached to the active site cysteine, the enzyme adopted an “open” conformation, where the four structural domains were aligned to give an extended structure (16). The closed and open conformations are thought to reflect intracellular and extracellular TG2, respectively. Crystallization of TG2 demonstrated that the open conformation can form a vicinal disulfide bond (16), which has been shown to inactivate the enzyme and presumably serves as a redox switch regulating the activity in the extracellular environment (17, 18). In its active state, TG2 binds up to six Ca2+ ions (10, 19). The open conformation, however, was crystallized without bound Ca2+ and, hence, does not likely reflect the true structure of the catalytically active enzyme.
Hydrogen/deuterium exchange monitored by mass spectrometry (HDX-MS) has proven to be a valuable tool for studying both dynamic changes in protein conformation and protein–protein interactions (20). The method uses the exchange of backbone amide hydrogens with deuterium when proteins are placed in a D2O solution. Backbone amide hydrogens engaged in stable hydrogen bonds are protected against exchange with the solvent (21). According to the thermodynamic principle, all protecting hydrogen bonds will eventually break as the protein cycles through partially unfolded states. Therefore, over time, all backbone amides become transiently exposed to the solvent and exchange competent. In this way, the deuterium uptake kinetics directly reflects the backbone dynamics of the protein structure. For a protein–protein complex, the binding interface will usually exhibit a decreased deuterium uptake due to having reduced backbone dynamics and being shielded from the solvent (22).
Here, we describe the use of HDX-MS to map conformational changes in TG2 induced by Ca2+ and GTP, and we address the structural implications of cysteine oxidation. By using a panel of TG2-specific monoclonal antibodies (mAbs) (23), we have also mapped the main epitopes targeted by celiac disease autoantibodies. Their locations might explain why one epitope seems to be favored in the response and indicate what drives the activation of autoreactive B cells in celiac disease.
Results
Ca2+ Ions Stabilize the Structure of TG2.
Because of the large difference in hydrodynamic radius between open and closed TG2, the two conformational states can be separated by nondenaturing PAGE (12, 24). In agreement with earlier reports, we observed that TG2, produced recombinantly in Sf9 insect cells, adopts a closed conformation in the presence of GTP, whereas Ca2+ in combination with a specific active-site inhibitor induces an open conformation (Fig. 1A). If no effectors were added to the solutions, the protein was found to populate both conformational states, with the majority of TG2 molecules existing in the open form. Although it is possible that purified TG2 can be associated with effectors derived from the expression system, we will refer to the protein with no added effectors as “effector-free” in the following. Distinct conformational states were also observed when analyzing the intact, deuterated proteins by using MS (Fig. 1B and Table S1). When labeling TG2 in the presence of GTP or inhibitor-bound TG2 (iTG2) in the presence of Ca2+, we observed a single isotopic distribution in each mass spectrum, indicating that the proteins only populated one conformational state in solution. In the absence of externally added effectors, TG2 gave rise to two isotopic distributions when analyzed by HDX-MS, indicating that the protein adopted two states with different dynamics. The state with the lower deuterium uptake, state A, was found to mimic the GTP-bound state. However, the state displaying a greater deuterium uptake, state B, did not match the deuterium uptake kinetics of Ca2+-bound iTG2. The Ca2+-bound protein incorporated a smaller amount of deuterium than state B in effector-free TG2, indicating a stabilization of the protein structure. This effect could either be due to the binding of Ca2+ or the presence of an inhibitor in the active site. However, state B was found to match the deuterium uptake of iTG2 labeled in the absence of Ca2+, indicating that the inhibitor by itself did not have a pronounced effect on the structure of TG2 in the open conformation (Fig. S1). Hence, the observed deuterium uptake patterns show that the binding of Ca2+ ions has a stabilizing effect on the open form of TG2.
Fig. 1.

Detection of open and closed TG2 conformations. (A) Separation of open and closed TG2 by nondenaturing PAGE. TG2 was either left untreated or incubated with GTP or Ca2+ before loading. To avoid extensive autocross-linking of active TG2, the peptide inhibitor Ac-P(DON)LPF-NH2 was included in the sample with Ca2+. This treatment yields TG2 with the substrate-mimicking inhibitor bound irreversibly to the active site cysteine (iTG2) (16). (B) Deconvoluted mass spectra showing the mass of full-length TG2 or iTG2 before (solid black line) and after 10-s (dashed red line) or 1,000-s (dashed blue line) incubation in 90% (vol/vol) D2O. Incubation was done in the absence of effectors or with GTP or Ca2+ present. Covalent attachment of inhibitor peptide to the active site cysteine results in a 638-Da mass increase. The spectra are aligned accordingly, so that deuterium uptake can be compared directly for the different states.
Binding of Ca2+ or GTP Stabilizes Distinct Regions in TG2.
To locate regions in TG2 that were impacted by effector binding, we compared the deuterium incorporation at the peptide level (local-exchange analysis) in the effector-free protein and in the Ca2+-bound and GTP-bound forms. For this purpose, the labeled protein was digested with pepsin before MS analysis, resulting in 70.3% sequence coverage. A complete list of identified peptides can be found in Table S2, and deuterium uptake plots for the N-terminal, core, and C-terminal domains are shown in Fig. S2.
Many of the peptides obtained from the catalytic core domain of TG2 incorporated more deuterium and, thus, appeared more dynamic in the effector-free state than in either of the effector-bound states. This difference indicates that both Ca2+ and GTP have a stabilizing effect on the structure of the core domain. When comparing the deuteration patterns for Ca2+-bound iTG2 and GTP-bound TG2, we observed that the GTP-bound state exhibited less dynamic behavior in the C-terminal domains and regions of the core domain which, according to the reported crystal structures, are involved in interdomain interactions when the enzyme adopts its closed conformation (Fig. 2A and Table S2). Other parts of the core domain, however, appeared to have a less dynamic structure in the Ca2+-bound state. This behavior was observed for peptide fragments covering residues 289–311 and 354–369. The first region comprises parts of the substrate binding pocket and a putative Ca2+-binding site. Both Ca2+ binding and the presence of inhibitor in the active site are therefore plausible explanations for the observed protection. The second region contains a loop adjacent to another, high-affinity, Ca2+ site (19). Based on the crystal structures of the Ca2+bound forms of the homologous proteins TG3 and factor XIIIa, this loop is predicted to undergo structural changes upon Ca2+ binding (25, 26). Thus, our observations with HDX-MS correspond well with previously reported models of transglutaminases.
Fig. 2.
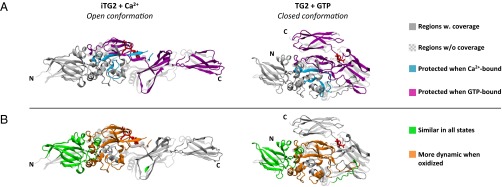
TG2 regions that undergo effector-induced structural changes. The relevant regions are highlighted on both the open conformation (PDB ID code 2Q3Z) and the closed conformation (PDB ID code 1KV3) crystal structure, containing bound peptide inhibitor and GDP, respectively (bound molecules are shown in red stick representation). Importantly, the open structure does not contain bound Ca2+ ions and is therefore not identical to the protein studied here. Coloring matches the one used in Table S2. (A) Regions with lower deuterium uptake in Ca2+-bound iTG2 compared with GTP-bound TG2 are shown in turquoise, whereas regions displaying the opposite pattern are shown in magenta. (B) Orange regions displayed increased deuterium uptake, when iTG2 had been oxidized before the addition of Ca2+. Regions where the deuterium uptake was similar in all investigated states are shown in green.
Disulfide Bond Formation in iTG2 Prevents the Stabilizing Effects of Ca2+.
Cysteine oxidation inactivates TG2 (17, 27, 28), and intramolecular disulfide bond formation presumably leads to TG2 inactivation in the extracellular environment (18, 29). To study the structural properties of oxidized TG2, we also performed local-exchange analysis on iTG2, which had been incubated with oxidized glutathione before the addition of Ca2+. The peptides 354–369, 355–369, and 370–378 could no longer be observed after oxidation (Table S2), indicating that Cys370 was engaged in disulfide bond formation, which prevented enzymatic cleavage of the peptide bond between residues 369 and 370. Similarly, the peptides 511–532, 517–532, and 548–555 were found to be sensitive to oxidation, strongly indicating that a bond between Cys524 and Cys554, which are in close proximity according to the crystal structures, had also formed. Local-exchange analysis revealed that oxidized iTG2 exhibited an increased deuterium uptake, compared with Ca2+-bound iTG2, in peptides covering the core domain (Fig. 2B and Table S2). Interestingly, within the region 167–354, comprising most of the core domain, oxidized iTG2 instead matched the behavior of the effector-free protein (Table S2). Thus, even in the presence of Ca2+, oxidized iTG2 had a core domain structure resembling that of the effector-free protein.
The N-Terminal Domain of TG2 Is Unaffected by Effector Binding and Oxidation State.
Although changes in the deuterium uptake could be observed in the C-terminal β-barrel domains and the catalytic core domain, when TG2 was exposed to different effectors, the peptides derived from the N-terminal β-sandwich domain exhibited deuteration patterns, which were almost identical under all of the investigated conditions, including the oxidized state (Fig. 2B and Table S2). Interestingly, epitopes recognized by a panel of TG2-specific mAbs were found to cluster in the N-terminal part of TG2 (30). Despite the constant dynamics of the N-terminal domain, some mAbs bind with greater affinity to the open conformation than to the closed conformation (ref. 30 and Fig. S3A). One explanation for this apparent discrepancy could be that the proximity of the C-terminal domains inhibits antibody binding to parts of the N-terminal domain when TG2 adopts the closed, GTP-bound conformation. Alternatively, core domain regions, which are close to the N-terminal domain, could take part in antibody binding. However, oxidation of TG2 before incubation with Ca2+ did not affect mAb binding, indicating that prevention of Ca2+-induced structural changes in the core domain does not compromise antibody binding (Fig. S3B).
Binding of Autoantibodies Can Induce Structural Changes in TG2.
The panel of TG2-specific mAbs described above was generated from small intestinal biopsies of celiac disease patients (23). Based on competitive ELISA experiments, we recently showed that 49 of 57 mAbs could be placed into one of four distinct epitope groups (epitope 1–4, targeted by 30, 6, 11, and 2 mAbs, respectively), which correlated with mAb VH-segment use. Some of the epitopes were partly overlapping, and they all appeared to be clustered closely together (30). To study antibody binding to TG2 in more detail, we used HDX-MS to identify TG2 regions with altered deuterium uptake after incubation with mAbs. Hence, mAbs targeting two nonoverlapping epitopes (epitope 1 and epitope 2) were incubated with iTG2 in the presence of Ca2+, and the resulting immune complexes were subjected to HDX-MS analysis. We selected two mAbs reacting with each epitope and chose the Ca2+-bound enzyme as antigen, because this state is assumed to represent the extracellular form of TG2, which the immune system encounters.
Digestion of labeled, mAb-bound iTG2 revealed that several peptides in all four structural domains displayed altered deuterium uptake (Fig. S4 and Table S2). Although the effect in most cases was small, the widespread changes indicate that we not only observe localized shielding of the epitope by the bound mAbs, but also an additional, allosteric effect on the TG2 structure. Because the presence of an inhibitor in the active site presumably locks TG2 in an open conformation (Fig. S1), we also tested the effect of mAb binding on the effector-free enzyme by monitoring the distribution of TG2 molecules between state A and state B (Fig. 3). Interestingly, in the presence of epitope-1 mAbs, the conformational equilibrium of TG2 was shifted in favor of the closed form, state A, whereas epitope-2 mAbs caused the enzyme to only populate the open conformation, state B. Although these observations were made in the absence of effectors, they demonstrate the potential of antibody binding to change the TG2 structure, suggesting that celiac disease autoantibodies could have an impact on the function of the enzyme.
Fig. 3.

Deconvoluted mass spectra of intact effector-free TG2 before (solid black line) and after (dashed colored lines) 1,000-s incubation in 90% (vol/vol) D2O. Deuterium incorporation was done in the presence or absence of TG2-reactive mAbs recognizing epitope 1 (mAbs 679-14-E06 and 693-10-B06) or epitope 2 (mAbs 693-1-F06 and 693-1-A03). Different mAbs recognizing the same epitope have the same effect on the distribution of TG2 molecules between closed (state A) and open (state B) conformations.
Epitope Mapping of Anti-TG2 Autoantibodies.
Three peptides in the N-terminal domain of TG2 displayed a reduction in deuterium uptake, which was specific for mAbs targeting either epitope 1 or epitope 2 and was evident at all time points (Fig. 4A and Fig. S4). Thus, the peptides 5–12 and 27–40 were consistently protected more by epitope-1 than epitope-2 mAbs, whereas peptide 13–26 displayed the opposite pattern. These regions therefore represented the most likely candidates for taking part in epitopes 1 and 2, respectively. We previously reported that epitope 2 can be disrupted by introduction of the triple mutation R19S E153S M659S (30). This triple mutation was first described by Simon-Vecsei et al., who showed that it inhibits the binding of serum antibodies from celiac disease patients (31). Of the residues that were mutated in the previous study, Arg19 is located within the peptide we identified as an epitope-2 candidate by HDX-MS. We therefore tested the effect of the R19S mutation on mAb binding in ELISA by using recombinant TG2 produced in Escherichia coli. Importantly, this protein displayed the same behavior as TG2 produced in Sf9 insect cells when analyzed by HDX-MS, indicating that the expression system does not affect the folding of the enzyme (Fig. S5). The R19S mutant showed a dramatic loss of reactivity toward epitope-2 mAbs (Fig. 4B), comparable to what can be observed for the triple mutant (Fig. S6A). The binding of epitope-1 mAbs, however, was largely unaffected, indicating that the R19S mutation selectively targets epitope 2 and, to some extent, the partly overlapping epitope 3 (Fig. 4B).
Fig. 4.

Identification of epitopes targeted by TG2-specific autoantibodies. (A) Mapping of epitopes using HDX-MS. Regions protected from deuterium incorporation are shown on the open TG2 structure (PDB ID code 2Q3Z) with a peptide inhibitor (red stick representation) attached to the active-site cysteine residue. iTG2 was incubated in 90% (vol/vol) D2O containing 5 mM CaCl2 for 10, 100, or 1,000 s in the presence or absence of mAb and digested with pepsin before analysis. Regions that were protected by mAbs recognizing epitope 1 (mAbs 679-14-E06 and 693-10-B06) but not by mAbs recognizing epitope 2 (mAbs 693-1-F06 and 693-1-A03) are shown in blue, whereas regions with the opposite pattern are shown in green. Residues that were found to be important for mAb binding are shown in red stick representation. (B) Confirmation of epitopes by mutational analysis. The reactivity of 53 mAbs to wild-type (WT) and mutant TG2 was tested in ELISA. Signals obtained with the indicated mutants are given relative to the signals obtained with WT TG2. The majority of the mAbs could be placed into one of three main epitope groups (epitope 1–3) based on their ability to compete with each other for binding (30). Two mAbs have been assigned to a minor epitope (epitope 4, open circles) and five mAbs presumably target other epitopes. Red bars indicate medians.
To confirm the location of epitope 1, we constructed TG2 variants harboring triple mutations in either of the two candidate regions identified by HDX-MS (Fig. S6B). Both triple mutations affected binding of epitope-1 mAbs, suggesting that both regions contribute to the epitope. To further dissect epitope 1, we assessed the binding of mAbs to TG2 containing single amino acid mutations in the two identified regions. Glu8 in the first region and Lys30 in the second region were found to be the most important residues for binding. Mutation of each of these residues selectively abolished most of the binding for epitope-1 mAbs (Fig. 4B).
We have provided evidence that epitope 1 overlaps with a binding site for fibronectin (30). The fibronectin binding site is known to be in the N-terminal domain of TG2 and could consist of several separate stretches of amino acids (32–34). In one study, Asp94 and Asp97 were shown to be important for the interaction (33). However, we observed no compromising effect of the previously described D94A and D97A mutations on the binding of epitope-1 mAbs (Fig. 4B and Fig. S6C). Interestingly, most epitope-3 mAbs lost reactivity upon introduction of the D94A mutation (Fig. 4B), suggesting that epitope 3, which does not overlap with the fibronecting binding site (30), is located around Asp94.
Because the epitope groups that we have assigned reflect targeted surface areas rather than specific interactions, not all mAbs in each group showed the same loss of reactivity toward the TG2 mutants (Fig. 4B). Thus, for some antibodies, other residues than the ones identified are expected to be important for binding. We cannot rule out that neighboring residues in the core domain are involved, but our results indicate that the main epitopes targeted by celiac disease autoantibodies are clustered in the N-terminal domain of TG2 with the four residues reported in Fig. 4 collectively being important for binding most of the antibodies. The introduced mutations selectively disrupted individual epitopes and did not affect the overall conformation of TG2 as judged by nondenaturing PAGE analysis (Fig. S7A). Interestingly, for a number of mAbs, the effect of the mutations was decreased if binding was assessed in the presence of Ca2+ using inhibitor-bound TG2 as antigen (Fig. S7B). Thus, Ca2+ binding to TG2 can shield the effect of introduced mutations, suggesting that there could be one or more Ca2+ binding sites in the vicinity of the antibody binding sites.
Discussion
TG2 plays a central role in celiac disease, both through its enzymatic activity, which leads to gluten deamidation, and as the target of autoantibodies. Conformational changes in TG2 control the enzyme’s activity and are also important for autoantibody binding. Describing the different structural states of TG2 and knowing how the enzyme conformation is regulated are therefore relevant for our understanding of celiac disease pathogenesis.
Studies performed in mice have shown that extracellular TG2 exists predominantly in a catalytically inactive form under normal conditions but can become activated through inflammation or in the presence of a reducing agent (18, 29). The latter observation suggests that the enzyme is inactivated by oxidation in the extracellular environment. In this regard, Cys370 in TG2 has been implicated in disulfide bond formation with its neighbor Cys371 or, alternatively, with Cys230 (16, 17, 35). Based on the disappearance of signals from the mass spectrum of TG2 following oxidation, we could confirm the formation of these disulfide bonds. In addition, we detected a previously undescribed disulfide bond between Cys524 and Cys554 (Table S2).
The mechanism by which TG2 is inactivated by cysteine oxidation is not clearly understood, but based on the negative effect of Ca2+ on the rate of oxidation (17, 27), it has been proposed that structural changes induced by disulfide bond formation interferes with Ca2+ binding and, thus, activation of the enzyme (17). Our data support a model in which oxidation negatively affects Ca2+ binding, because the core domain of oxidized TG2 remains dynamic in the presence of Ca2+, exhibiting a HDX pattern similar to effector-free TG2. This observation suggests that disulfide bond formation prevents the Ca2+-induced structural changes that are necessary for TG2 activation. Interestingly, Cys370 and Cys371 are situated in a loop, which displayed decreased dynamics in the Ca2+-bound protein. Thus, it is likely that structural changes in this loop play a key role in regulating the enzymatic activity.
In the absence of effectors, we observed that the binding of TG2 by mAbs induced large-scale conformational changes in the enzyme (Fig. 3). In the presence of Ca2+, mAb binding also appeared to cause structural changes in TG2, as several regions outside of the epitope displayed altered deuterium uptake upon mAb binding. TG2-reactive autoantibodies produced in celiac disease could therefore influence TG2 function. Indications of such an effect have been observed by the addition of celiac disease antibodies to cell culture systems (36–41). The mechanisms underlying the various biological effects reported are not well understood. Direct interference with enzymatic activity is not expected to play a role, because it has been shown that the TG2-reactive mAbs used in this study do not inhibit TG2-mediated transamidation/deamidation (23). However, TG2 has been implicated in a number of processes that do not depend on its catalytic activity, but rather on interactions with components of the extracellular matrix and the plasma membrane (42–45). It is therefore possible that TG2-reactive autoantibodies can play a pathogenic role in celiac disease by inducing conformational changes that interfere with the binding between TG2 and its interaction partners. Another intriguing possibility is that antibody binding renders the enzyme more or less susceptible to the formation of intramolecular disulfide bonds.
It was suggested that the transamidation activity of TG2 is directly involved in the activation of autoreactive B cells in celiac disease through the formation of isopeptide-linked B-cell receptors (BCRs) of the IgD isotype on the surface of TG2-targeting naïve cells (23). B cells that bind TG2 in a way that allows the enzyme to be catalytically active would then become selectively activated, and this property should be reflected in the location of the epitopes. Recently, we showed that epitope 1 is recognized by more than half of the mAbs cloned from TG2-specific plasma cells in the celiac disease lesion (30). The majority of these mAbs used the VH5-51 gene segment, which is dominating both in the mAb panel used here (23) and in an earlier reported panel of TG2-reactive antibody fragments obtained from phage display libraries (46). The overrepresentation of VH5-51 antibodies is most likely related to the location of the epitope they recognize. Thus, a possible explanation for the VH5-51 preference is that TG2 bound by an epitope 1-targeting BCR is oriented in a way that optimizes catalytic formation of isopeptide cross-links between BCRs on the cell surface. In this regard, it is noteworthy that the residues we have identified to be part of epitope 1 lie on an axis, which includes the active site (Fig. 4A). The location of epitope 1 thereby predicts that the active site will face toward the BCR upon binding and implies that BCR cross-linking can happen efficiently. The location of epitope 2, however, suggests that the active site will point in a different direction with respect to the BCR upon binding. This characteristic could be a reason why epitope 2 is only recognized by 10% of the mAbs in the TG2-specific panel (30) and, thus, appears to be targeted less frequently than epitope 1 by plasma cells in the celiac lesion.
In conclusion, our data demonstrate the use of HDX-MS for studying conformational changes and mapping of epitopes in TG2. Binding of Ca2+ and GTP was associated with stabilization of distinct regions in the catalytic core and C-terminal domains of the enzyme. Notably, oxidation prevented the Ca2+-induced changes. Binding of mAbs derived from celiac disease patients resulted in pronounced allosteric effects on TG2 structure, suggesting a process whereby celiac disease autoantibodies could alter the function of TG2. Finally, mapping of two of the main epitopes revealed that they are located close to each other in the N-terminal domain of TG2. We propose that the location of the preferentially targeted epitope 1 indirectly supports the enzymatic activity of BCR-bound TG2 by positioning the enzyme in an orientation that is optimal for cross-linking of BCRs on the cell surface. This effect could be instrumental for the activation of TG2-reactive B cells in celiac disease.
Materials and Methods
Proteins.
Recombinant human TG2 was either obtained from Phadia as purified protein produced in Sf9 insect cells or expressed in E. coli and purified as described (17). TG2 mutants were produced in E. coli. Mutations were introduced in the TG2 sequence by using the QuikChange Site-Directed Mutagenesis Kit (Stratagene) or by PCR amplification followed by subcloning into the pET-28a vector (Novagen) between the NdeI and HindIII sites. All mutations were verified by DNA sequencing (GATC Biotech). TG2-specific human mAbs were produced essentially as described (23, 47). Briefly, IgG1 and Igκ expression vectors containing the cloned heavy and light chain variable regions were cotransfected into HEK293 cells. Six days after transfection, antibodies were purified from the cell supernatants on Protein G Sepharose (GE Healthcare). The TG2-specific mouse mAb CUB7402 was obtained from NeoMarkers.
TG2 Modifications.
To avoid autocross-linking of TG2 in the presence of Ca2+, the active site cysteine was irreversibly bound to the substrate-mimicking peptide Ac-P(DON)LPF-NH2 (where DON is the electrophilic amino acid 6-diazo-5-oxo-l-norleucine; ref. 16) (Zedira). TG2 was preincubated with 0.5 mM of the inhibitor for 30 min at room temperature before the reaction was started by the addition of CaCl2 to a final concentration of 5 mM. After 20 min of incubation, excess free inhibitor was removed by size-exclusion chromatography. To obtain the oxidized enzyme, iTG2 was incubated with 2 mM oxidized glutathione in the absence of Ca2+ for 3 h at 30 °C.
Deuterium Labeling.
All exchange reactions were performed at 25 °C in Tris-buffered saline (TBS) with or without 5 mM CaCl2. Buffer made with D2O was added to 90% (vol/vol) to initiate labeling at a final TG2 concentration of 0.06 mg/mL. After 10, 100, and 1,000 s, samples were collected and pH lowered to 2.5 by the addition of formic acid. The samples were then immediately snap-frozen in liquid nitrogen and stored at −80 °C until analyzed (SI Materials and Methods). Undeuterated control samples were treated in the same way. To study effector-bound TG2, the enzyme was pretreated at 2.6 mg/mL with 1 mM GTP or 5 mM CaCl2. The effect of antibody binding was assessed by incubating TG2 or iTG2 with a twofold molar excess (by antigen binding sites) of TG2-specific mAbs for at least 30 min at room temperature before labeling. Triplicate labelings were performed and analyzed for samples that did not contain antibodies. For antibody-interaction studies, a single labeling was performed, and each sample was analyzed twice.
Nondenaturing PAGE.
The preparations of TG2 and iTG2 that were used for deuterium labeling and antibody binding studies were also analyzed by nondenaturing PAGE as reported (12, 24). The enzyme was either left untreated or incubated with 1 mM GTP or 5 mM CaCl2 for 30 min at room temperature before loading of the gel. Electrophoresis was performed at 125 V for 75 min by using ice-cold running buffer and with the gel chamber submerged in an ice bath.
ELISAs.
Wild-type or mutant TG2 produced in E. coli was coated in microtiter plates at 3 µg/mL in TBS. Incubations with mAbs were done at 37 °C in TBS with 0.1% (vol/vol) Tween 20, followed by detection of bound antibody with alkaline phosphatase-conjugated rabbit anti-human IgG or goat anti-mouse IgG (Abcam). After addition of phosphatase substrate, absorbance was measured at 405 nm in a microplate reader (Thermo Scientific), and saturation binding curves were produced by nonlinear regression analysis of the OD values. In experiments where only a single mAb concentration was analyzed, the employed concentration was picked to fall within the linear range of the assay based on initial titration of each mAb. In cases where Ca2+ was present, 5 mM CaCl2 was included during coating of iTG2 and incubation with antibodies. TG2 produced in Sf9 insect cells was used to compare the recognition of open and closed or oxidized and reduced TG2 by mAbs. Open, Ca2+-bound iTG2 was generated as described above. Closed, GTP-bound TG2 was generated by incubating the enzyme with 1 mM GTP before coating and keeping the GTP concentration at 50 µM during coating and antibody incubations. Oxidized iTG2 was generated before coating and compared with nonoxidized iTG2 in the presence of 5 mM CaCl2.
Supplementary Material
Acknowledgments
We thank Patrick C. Wilson for providing TG2-specific human monoclonal antibodies. This work was supported by grants from the Research Council of Norway and the South-Eastern Norway Regional Health Authority, European Commission Grants MRTN-CT-2006-036032 and ERC-2010-Ad-268541 (to L.M.S.), and Carlsberg Foundation Grant 2012_01_0332 (to T.J.D.J.). Lundbeck Foundation Grant 95-310-13595 is greatly acknowledged for financial support (to S.M.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission. C.K. is a guest editor invited by the Editorial Board.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1407457111/-/DCSupplemental.
References
- 1.Sollid LM. Coeliac disease: Dissecting a complex inflammatory disorder. Nat Rev Immunol. 2002;2(9):647–655. doi: 10.1038/nri885. [DOI] [PubMed] [Google Scholar]
- 2.Dieterich W, et al. Identification of tissue transglutaminase as the autoantigen of celiac disease. Nat Med. 1997;3(7):797–801. doi: 10.1038/nm0797-797. [DOI] [PubMed] [Google Scholar]
- 3.Lorand L, Graham RM. Transglutaminases: Crosslinking enzymes with pleiotropic functions. Nat Rev Mol Cell Biol. 2003;4(2):140–156. doi: 10.1038/nrm1014. [DOI] [PubMed] [Google Scholar]
- 4.Molberg O, et al. Tissue transglutaminase selectively modifies gliadin peptides that are recognized by gut-derived T cells in celiac disease. Nat Med. 1998;4(6):713–717. doi: 10.1038/nm0698-713. [DOI] [PubMed] [Google Scholar]
- 5.Zemskov EA, Mikhailenko I, Hsia RC, Zaritskaya L, Belkin AM. Unconventional secretion of tissue transglutaminase involves phospholipid-dependent delivery into recycling endosomes. PLoS ONE. 2011;6(4):e19414. doi: 10.1371/journal.pone.0019414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aeschlimann D, Thomazy V. Protein crosslinking in assembly and remodelling of extracellular matrices: The role of transglutaminases. Connect Tissue Res. 2000;41(1):1–27. doi: 10.3109/03008200009005638. [DOI] [PubMed] [Google Scholar]
- 7.Hoffmann BR, Annis DS, Mosher DF. Reactivity of the N-terminal region of fibronectin protein to transglutaminase 2 and factor XIIIA. J Biol Chem. 2011;286(37):32220–32230. doi: 10.1074/jbc.M111.255562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nakaoka H, et al. Gh: A GTP-binding protein with transglutaminase activity and receptor signaling function. Science. 1994;264(5165):1593–1596. doi: 10.1126/science.7911253. [DOI] [PubMed] [Google Scholar]
- 9.Achyuthan KE, Greenberg CS. Identification of a guanosine triphosphate-binding site on guinea pig liver transglutaminase. Role of GTP and calcium ions in modulating activity. J Biol Chem. 1987;262(4):1901–1906. [PubMed] [Google Scholar]
- 10.Bergamini CM. GTP modulates calcium binding and cation-induced conformational changes in erythrocyte transglutaminase. FEBS Lett. 1988;239(2):255–258. doi: 10.1016/0014-5793(88)80928-1. [DOI] [PubMed] [Google Scholar]
- 11.Smethurst PA, Griffin M. Measurement of tissue transglutaminase activity in a permeabilized cell system: Its regulation by Ca2+ and nucleotides. Biochem J. 1996;313(Pt 3):803–808. doi: 10.1042/bj3130803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Murthy SN, Lomasney JW, Mak EC, Lorand L. Interactions of G(h)/transglutaminase with phospholipase Cdelta1 and with GTP. Proc Natl Acad Sci USA. 1999;96(21):11815–11819. doi: 10.1073/pnas.96.21.11815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Di Venere A, et al. Opposite effects of Ca(2+) and GTP binding on tissue transglutaminase tertiary structure. J Biol Chem. 2000;275(6):3915–3921. doi: 10.1074/jbc.275.6.3915. [DOI] [PubMed] [Google Scholar]
- 14.Liu S, Cerione RA, Clardy J. Structural basis for the guanine nucleotide-binding activity of tissue transglutaminase and its regulation of transamidation activity. Proc Natl Acad Sci USA. 2002;99(5):2743–2747. doi: 10.1073/pnas.042454899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jang TH, et al. Crystal structure of transglutaminase 2 with GTP complex and amino acid sequence evidence of evolution of GTP binding site. PLoS ONE. 2014;9(9):e107005. doi: 10.1371/journal.pone.0107005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pinkas DM, Strop P, Brunger AT, Khosla C. Transglutaminase 2 undergoes a large conformational change upon activation. PLoS Biol. 2007;5(12):e327. doi: 10.1371/journal.pbio.0050327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stamnaes J, Pinkas DM, Fleckenstein B, Khosla C, Sollid LM. Redox regulation of transglutaminase 2 activity. J Biol Chem. 2010;285(33):25402–25409. doi: 10.1074/jbc.M109.097162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jin X, et al. Activation of extracellular transglutaminase 2 by thioredoxin. J Biol Chem. 2011;286(43):37866–37873. doi: 10.1074/jbc.M111.287490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Király R, et al. Functional significance of five noncanonical Ca2+-binding sites of human transglutaminase 2 characterized by site-directed mutagenesis. FEBS J. 2009;276(23):7083–7096. doi: 10.1111/j.1742-4658.2009.07420.x. [DOI] [PubMed] [Google Scholar]
- 20.Wales TE, Engen JR. Hydrogen exchange mass spectrometry for the analysis of protein dynamics. Mass Spectrom Rev. 2006;25(1):158–170. doi: 10.1002/mas.20064. [DOI] [PubMed] [Google Scholar]
- 21.Skinner JJ, Lim WK, Bédard S, Black BE, Englander SW. Protein dynamics viewed by hydrogen exchange. Protein Sci. 2012;21(7):996–1005. doi: 10.1002/pro.2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Malito E, et al. Defining a protective epitope on factor H binding protein, a key meningococcal virulence factor and vaccine antigen. Proc Natl Acad Sci USA. 2013;110(9):3304–3309. doi: 10.1073/pnas.1222845110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Di Niro R, et al. High abundance of plasma cells secreting transglutaminase 2-specific IgA autoantibodies with limited somatic hypermutation in celiac disease intestinal lesions. Nat Med. 2012;18(3):441–445. doi: 10.1038/nm.2656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Murthy SN, et al. Conserved tryptophan in the core domain of transglutaminase is essential for catalytic activity. Proc Natl Acad Sci USA. 2002;99(5):2738–2742. doi: 10.1073/pnas.052715799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ahvazi B, Kim HC, Kee SH, Nemes Z, Steinert PM. Three-dimensional structure of the human transglutaminase 3 enzyme: Binding of calcium ions changes structure for activation. EMBO J. 2002;21(9):2055–2067. doi: 10.1093/emboj/21.9.2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stieler M, et al. Structure of active coagulation factor XIII triggered by calcium binding: Basis for the design of next-generation anticoagulants. Angew Chem Int Ed Engl. 2013;52(45):11930–11934. doi: 10.1002/anie.201305133. [DOI] [PubMed] [Google Scholar]
- 27.Connellan JM, Folk JE. Mechanism of the inactivation of guinea pig liver transglutaminase by 5,5′-dithiobis-(2-nitrobenzoic acid) J Biol Chem. 1969;244(12):3173–3181. [PubMed] [Google Scholar]
- 28.Lorand L, Conrad SM. Transglutaminases. Mol Cell Biochem. 1984;58(1-2):9–35. doi: 10.1007/BF00240602. [DOI] [PubMed] [Google Scholar]
- 29.Siegel M, et al. Extracellular transglutaminase 2 is catalytically inactive, but is transiently activated upon tissue injury. PLoS One. 2008;3(3):e1861. doi: 10.1371/journal.pone.0001861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Iversen R, et al. Transglutaminase 2-specific autoantibodies in celiac disease target clustered, N-terminal epitopes not displayed on the surface of cells. J Immunol. 2013;190(12):5981–5991. doi: 10.4049/jimmunol.1300183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Simon-Vecsei Z, et al. A single conformational transglutaminase 2 epitope contributed by three domains is critical for celiac antibody binding and effects. Proc Natl Acad Sci USA. 2012;109(2):431–436. doi: 10.1073/pnas.1107811108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jeong JM, Murthy SN, Radek JT, Lorand L. The fibronectin-binding domain of transglutaminase. J Biol Chem. 1995;270(10):5654–5658. doi: 10.1074/jbc.270.10.5654. [DOI] [PubMed] [Google Scholar]
- 33.Hang J, Zemskov EA, Lorand L, Belkin AM. Identification of a novel recognition sequence for fibronectin within the NH2-terminal beta-sandwich domain of tissue transglutaminase. J Biol Chem. 2005;280(25):23675–23683. doi: 10.1074/jbc.M503323200. [DOI] [PubMed] [Google Scholar]
- 34.Wang Z, et al. Characterization of heparin-binding site of tissue transglutaminase: Its importance in cell surface targeting, matrix deposition, and cell signaling. J Biol Chem. 2012;287(16):13063–13083. doi: 10.1074/jbc.M111.294819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Han BG, et al. Crystal structure of human transglutaminase 2 in complex with adenosine triphosphate. Int J Biol Macromol. 2010;47(2):190–195. doi: 10.1016/j.ijbiomac.2010.04.023. [DOI] [PubMed] [Google Scholar]
- 36.Halttunen T, Mäki M. Serum immunoglobulin A from patients with celiac disease inhibits human T84 intestinal crypt epithelial cell differentiation. Gastroenterology. 1999;116(3):566–572. doi: 10.1016/s0016-5085(99)70178-2. [DOI] [PubMed] [Google Scholar]
- 37.Barone MV, et al. Humoral immune response to tissue transglutaminase is related to epithelial cell proliferation in celiac disease. Gastroenterology. 2007;132(4):1245–1253. doi: 10.1053/j.gastro.2007.01.030. [DOI] [PubMed] [Google Scholar]
- 38.Cervio E, et al. Sera of patients with celiac disease and neurologic disorders evoke a mitochondrial-dependent apoptosis in vitro. Gastroenterology. 2007;133(1):195–206. doi: 10.1053/j.gastro.2007.04.070. [DOI] [PubMed] [Google Scholar]
- 39.Myrsky E, et al. Coeliac disease-specific autoantibodies targeted against transglutaminase 2 disturb angiogenesis. Clin Exp Immunol. 2008;152(1):111–119. doi: 10.1111/j.1365-2249.2008.03600.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Myrsky E, et al. Celiac disease IgA modulates vascular permeability in vitro through the activity of transglutaminase 2 and RhoA. Cell Mol Life Sci. 2009;66(20):3375–3385. doi: 10.1007/s00018-009-0116-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Di Simone N, et al. Anti-tissue transglutaminase antibodies from celiac patients are responsible for trophoblast damage via apoptosis in vitro. Am J Gastroenterol. 2010;105(10):2254–2261. doi: 10.1038/ajg.2010.233. [DOI] [PubMed] [Google Scholar]
- 42.Turner PM, Lorand L. Complexation of fibronectin with tissue transglutaminase. Biochemistry. 1989;28(2):628–635. doi: 10.1021/bi00428a032. [DOI] [PubMed] [Google Scholar]
- 43.Akimov SS, Krylov D, Fleischman LF, Belkin AM. Tissue transglutaminase is an integrin-binding adhesion coreceptor for fibronectin. J Cell Biol. 2000;148(4):825–838. doi: 10.1083/jcb.148.4.825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xu L, Begum S, Hearn JD, Hynes RO. GPR56, an atypical G protein-coupled receptor, binds tissue transglutaminase, TG2, and inhibits melanoma tumor growth and metastasis. Proc Natl Acad Sci USA. 2006;103(24):9023–9028. doi: 10.1073/pnas.0602681103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Scarpellini A, et al. Heparan sulfate proteoglycans are receptors for the cell-surface trafficking and biological activity of transglutaminase-2. J Biol Chem. 2009;284(27):18411–18423. doi: 10.1074/jbc.M109.012948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Marzari R, et al. Molecular dissection of the tissue transglutaminase autoantibody response in celiac disease. J Immunol. 2001;166(6):4170–4176. doi: 10.4049/jimmunol.166.6.4170. [DOI] [PubMed] [Google Scholar]
- 47.Smith K, et al. Rapid generation of fully human monoclonal antibodies specific to a vaccinating antigen. Nat Protoc. 2009;4(3):372–384. doi: 10.1038/nprot.2009.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.