

Significance
In the context of the nucleotide-binding domain and leucine rich repeat pyrin containing 1b (NLRP1b) inflammasome activation by anthrax lethal toxin, we reveal a new role for full-length caspase-1. We directly demonstrate that the caspase-1 45-kDa zymogen is able to process pro–IL-1β and to induce pyroptosis, and that apoptosis-associated speck-like protein containing a CARD is dispensable for the activity of the NLRP1b inflammasome. This is in contrast to the NLRP3 inflammasome activity, which is inhibited in the absence of caspase-1 autoproteolyis. Our data, which highlight differential requirements for caspase-1 autoproteolysis in NLRP1b and NLRP3 inflammasome function, may have implications for pathogen recognition and response.
Keywords: interleukin-1beta, pyroptosis, lethal toxin, macrophage, dendritic cell
Abstract
Inflammasomes are caspase-1–activating multiprotein complexes. The mouse nucleotide-binding domain and leucine rich repeat pyrin containing 1b (NLRP1b) inflammasome was identified as the sensor of Bacillus anthracis lethal toxin (LT) in mouse macrophages from sensitive strains such as BALB/c. Upon exposure to LT, the NLRP1b inflammasome activates caspase-1 to produce mature IL-1β and induce pyroptosis. Both processes are believed to depend on autoproteolysed caspase-1. In contrast to human NLRP1, mouse NLRP1b lacks an N-terminal pyrin domain (PYD), indicating that the assembly of the NLRP1b inflammasome does not require the adaptor apoptosis-associated speck-like protein containing a CARD (ASC). LT-induced NLRP1b inflammasome activation was shown to be impaired upon inhibition of potassium efflux, which is known to play a major role in NLRP3 inflammasome formation and ASC dimerization. We investigated whether NLRP3 and/or ASC were required for caspase-1 activation upon LT stimulation in the BALB/c background. The NLRP1b inflammasome activation was assessed in both macrophages and dendritic cells lacking either ASC or NLRP3. Upon LT treatment, the absence of NLRP3 did not alter the NLRP1b inflammasome activity. Surprisingly, the absence of ASC resulted in IL-1β cleavage and pyroptosis, despite the absence of caspase-1 autoprocessing activity. By reconstituting caspase-1/caspase-11−/− cells with a noncleavable or catalytically inactive mutant version of caspase-1, we directly demonstrated that noncleavable caspase-1 is fully active in response to the NLRP1b activator LT, whereas it is nonfunctional in response to the NLRP3 activator nigericin. Taken together, these results establish variable requirements for caspase-1 cleavage depending on the pathogen and the responding NLR.
Anthrax is a zoonotic disease caused by the Gram-positive bacterium Bacillus anthracis. B. anthracis provokes a shock-like syndrome that can prove fatal to the host (1) and has recently gained notoriety as a potential bioterrorism agent. Anthrax pathogenicity relies on its ability to secrete three virulence proteins, which combine with each other to form two toxins. The protective antigen (PA) combines with the edema factor (EF) to form the edema toxin (2, 3). EF is an adenylate cyclase that causes edema of the infected tissue. The binary combination of PA with lethal factor (LF) gives rise to the most virulent factor, called lethal toxin (LT), responsible for the systemic symptoms and death of the infected animal. To escape the host immune response, LT impairs the host innate immunity by killing macrophages (4–6). The PA protein interacts with LF and binds to cell surface receptors, enabling endocytosis of the LT complex. In the acidic compartment, PA forms pores allowing the delivery of LF to the cytosol. LF is a zinc metalloprotease that was shown to cleave the N-terminal region of many MAP kinase kinases and to induce apoptosis of macrophages. LT also triggers pyroptosis through the formation of a caspase-1–activating platform, named “inflammasome” (6–8).
Inflammasomes are multiprotein complexes of the innate immune response that control caspase-1 activity and pro–IL-1β and pro–IL-18 maturation. Most inflammasomes are composed of specific cytosolic pathogen recognition receptors (PRRs), as well as the apoptosis-associated speck-like protein containing a caspase activation and recruitment domain (CARD) (ASC) adaptor protein that enables the recruitment and activation of the caspase-1 protease. Once caspase-1 is oligomerized within an inflammasome platform, the enzyme undergoes autoproteolysis to form heterodimers of active caspase-1 (9–12). In the mouse, at least five distinct inflammasomes have been described, distinguished by the PRR that induces the complex formation. The PRRs capable of participating in inflammasome platform formation are either members of the nod-like receptor (NLR) family (e.g., NLRP1, NLRP3, or NLRC4) or of the PYrin and HIN (PYHIN) family (e.g., AIM2) (13, 14). ASC is composed of a pyrin domain (PYD) and a caspase activation and recruitment domain (CARD). ASC interacts with a PYD-containing PRR via its PYD domain and recruits the CARD domain of caspase-1 via its CARD domain. Thus, ASC is essential to the formation of the inflammasome by receptors such as NLRP3 or AIM2 (15–18). However, its presence is dispensable for NLRC4, which contains a CARD in place of a PYD, allowing direct interaction with the CARD domain of caspase-1 (19, 20).
Past studies have determined that certain mouse strains are more sensitive than others to LT cytotoxicity, and genetic studies identified NLRP1b as the factor conferring mouse strain susceptibility to anthrax LT (21). The mouse genome contains three different NLRP1 isoforms (a, b, and c) and a functional NLRP1b was found to be expressed by the mouse strains sensitive to LT (e.g., BALB/c or 129 background). Expression of NLRP1b was shown to mediate IL-1β release and caspase-1–mediated cell death in response to LT (7, 21, 22). Mouse NLRP1b differs structurally from human NLRP1 in that it lacks the N-terminal PYD (23). The absence of the PYD suggests that NLRP1b can directly engage caspase-1 without a requirement for ASC. However, studies dissecting the mechanism of NLRC4 inflammasome activation demonstrated that ASC is required for the amplification of caspase-1 autoprocessing and IL-1β secretion but not for pyroptosis (19, 20). Cell lysis mediated by LT was shown to be dependent on sodium and potassium fluxes (24), and high extracellular potassium inhibited IL-1β secretion upon LT treatment, suggesting a role for the NLRP3 inflammasome in LT sensing (22, 25). Therefore, we investigated whether NLRP3 and/or ASC were required for caspase-1 activation in response to LT. The NLRP3, ASC, and caspase-1 mouse knockout strains were backcrossed into the BALB/c background and the response of macrophages and dendritic cells (DCs) to LT intoxication was studied. Our data reveal that (i) in response to LT, ASC is dispensable for caspase-1 activation, but uncleavable caspase-1 is fully active; and (ii) upon activation of the NLRP3 inflammasome, uncleavable caspase-1 is inactive.
Results
Secretion of Mature IL-1β in Response to LT Is Independent of the NLRP3 Inflammasome in Murine Macrophages.
In response to LT, NLRP1b is activated to form an inflammasome, resulting in caspase-1 activation. To study whether ASC and/or NLRP3 were also required to activate caspase-1 in response to LT, we isolated peritoneal macrophages and differentiated macrophages from bone marrow progenitors (BMDMs) from BALB/c WT, caspase-1/caspase-11-, ASC-, or NLRP3-deficient mice. In mouse macrophages, lipopolysaccharide (LPS) is used as the priming signal, also called signal 1 that is necessary to induce the expression of the caspase-1 substrate, pro–IL-1β, and to increase the amount of NLRP3. The cells were then incubated with LT for 6 h and inflammasome activation was monitored by assessing the secretion of mature IL-1β and cleaved caspase-1. As expected, LT induced secretion of mature IL-1β and cleaved caspase-1 (p20) in the supernatant of primed WT cells (Fig. 1 A and B). Secretion of IL-18 was also significantly increased upon LT treatment (Fig. S1A). In nonprimed macrophages, the level of cleaved caspase-1 secreted into the supernatant was similar to that under priming conditions (Fig. 1 A and B). Upon LT treatment, NLRP3−/− macrophages secreted a similar amount of IL-1β, IL-18, and cleaved caspase-1 to WT cells, demonstrating that NLRP3 was dispensable for NLRP1b inflammasome activation (Fig. 1 and Fig. S1A). LT treatment triggered significant IL-1β maturation in the cell supernatants of ASC−/− peritoneal macrophages and BMDMs, suggesting that ASC was not required for NLRP1b inflammasome formation to induce IL-1β maturation (Fig. 1 A and B). Intriguingly, in the absence of ASC, cleaved caspase-1 was not detected in the cell supernatant of either peritoneal macrophages or BMDMs, whether the cells were primed or not. Similarly, cleaved caspase-1 was not detected in the ASC−/− cell lysates, suggesting that caspase-1 did not undergo autoproteolysis (Fig. S1B). As previously described, IL-1β and caspase-1 p20 secretions were abolished in NLRP3−/− and ASC−/− peritoneal macrophages and BMDMs in response to the NLRP3 activator nigericin (15). Finally, primed caspase-1/caspase-11−/− macrophages treated with LT did not produce mature IL-1β, demonstrating that caspase-1 is the effector of NLRP1b activation.
Fig. 1.

LT induces IL-1β secretion but not caspase-1 autoprocessing in ASC-deficient macrophages and BMDCs. Nonprimed or LPS-primed mouse peritoneal macrophages (A) or BMDMs (B) or BMDCs (C) of indicated genotypes were treated with B. anthracis lethal toxin (LT, 0.5 μg/mL, 6 h) or nigericin (Nig, 10 μM, 2 h) or left untreated (−); caspase-1 and IL-1β cleavage, and ASC and NLRP3 protein levels were assessed by Western blot. Actin was used as a loading control. These results were replicated three times independently.
LT Induces IL-1β Maturation in the Absence of Caspase-1 Cleavage in ASC−/− Bone Marrow Derived Dendritic Cells.
To determine whether the phenotype observed in mouse macrophages was conserved in mouse DC, bone marrow dendritic cells (BMDCs) from WT, ASC-, NLRP3-, and caspase-1/caspase-11–deficient mice were generated and exposed to LT. WT BMDCs released a similar amount of cleaved caspase-1 into the cell supernatant whether or not the cells were exposed to a priming signal; however, only LPS-primed cells secreted a large amount of mature IL-1β (Fig. 1C). As expected, caspase-1/caspase-11−/− BMDCs primed with LPS did not produce active IL-1β (Fig. 1C). In NLRP3−/− BMDCs, LT induced similar levels of caspase-1 activation to WT cells, as illustrated by the amount of p20 subunit secreted into the cell supernatant. Furthermore, in the presence of LPS + LT, NLRP3−/− cells efficiently secreted IL-1β, demonstrating that NLRP3 does not participate in NLRP1b inflammasome formation in response to LT (Fig. 1C). ASC−/− BMDCs stimulated with LT did not secrete processed caspase-1 as illustrated by the absence of p20 in the cell supernatant, regardless of the presence of a priming signal. As observed in macrophages, when the cells were primed with LPS, mature IL-1β was secreted despite the lack of processed caspase-1 in the cell supernatant (Fig. 1C). Thus, the absence of ASC in primed BMDCs prevents caspase-1 autoprocessing but does not affect the pro–IL-1β maturation and secretion (Fig. 1C and Fig. S1C). As described earlier, the absence of NLRP3 and ASC abolished the IL-1β production in response to nigericin treatment.
LT-Induced Pyroptosis Is Independent of NLRP3 and ASC.
In addition to its role in IL-1β maturation, LT is a potent inducer of pyroptosis, a caspase-1–dependent cell death (21). To address the question of whether ASC and NLRP3 are required for LT-induced pyroptosis, peritoneal macrophages isolated from WT, caspase-1/caspase-11−/−, ASC−/−, and NLRP3−/− mice, were treated with LT and cell death was monitored over 6 h. As shown in Fig. 2A, when exposed to LT, WT macrophages died rapidly, beginning at 2 h posttreatment and reaching more than 90% cell death by 6 h. ASC−/− and NLRP3−/− cells were as sensitive as WT cells to LT-induced pyroptosis, indicating that neither ASC nor NLRP3 participated in LT-induced cell death. LT intoxication was easily visible 2 h posttreatment by phase contrast microscopy as illustrated by the presence of bright blebs on sensitive cells (Fig. S2A). As described previously in the literature, caspase-1/caspase-11−/− macrophages were fully protected from pyroptosis and showed no morphological signs of intoxication (Fig. 2A and Fig. S2A) (21). Similar experiments were carried out in BMDMs (Fig. 2B) and BMDCs (Fig. 2C), and, as described for peritoneal macrophages, ASC and NLRP3 were not required for the pyroptosis triggered by LT. The absence of caspase-1 protected both BMDMs and BMDCs from death. Next, an in vivo model was used to assess in situ the macrophage sensitivity to LT intoxication. Thioglycollate-treated animals were injected intraperitoneally on day 3 with LT and euthanized 3 h later to quantify the number of macrophages present in the peritoneal cavity. As observed in vitro, LT induced the death of peritoneal macrophages in vivo of WT and ASC−/− mice, whereas peritoneal macrophages of caspase-1/caspase-11−/− mice were mostly not affected, but did not induce the death of neutrophils (Fig. 2D and Fig. S2B).
Fig. 2.

ASC and NLRP3 are dispensable for LT-induced cell death. LPS-primed mouse peritoneal macrophages (A) or BMDMs (B) or BMDCs (C) of indicated genotypes were treated with LT (0.5 μg/mL) for 6 h. Cell death was measured by cytotoxic assay (lactate dehydrogenase, LDH release) upon LT treatment over a time course of 6 h. Quantification by flow cytometry of peritoneal macrophages and neutrophils in the peritoneum of WT and ASC−/− mice (Left) or WT and caspase-1/caspase-11−/− mice (Right) injected with PBS or LT for 3 h is shown (D). Data represent mean ± SEM *P < 0.01 (Mann–Whitney test) (N.S., nonsignificant).
Uncleaved Caspase-1 Is Able to Induce Pyroptosis and IL-1β Maturation upon LT Stimulation.
Altogether, these observations suggest that caspase-1 may be fully active in its 45-kDa form and in the absence of autoprocessing. To clarify whether caspase-1 activity is involved in IL-1β secretion and pyroptosis in ASC−/− cells, WT and ASC−/− macrophages were pretreated with carbobenzoxy-valyl-alanyl-aspartyl-[O-methyl]-fluoromethylketone (z-VAD-fmk), a pan-caspase inhibitor, or Z-Tyr-Val-Ala-Asp(OMe)-fluoromethylketone (z-YVAD-fmk), a caspase-1–specific inhibitor, before the addition of the toxin. In WT cells, z-VAD-fmk and z-YVAD-fmk completely inhibited both the NLRP3 inflammasome activity in response to nigericin, and the NLRP1b inflammasome activity upon LT treatment, as illustrated by the absence of mature IL-1β and cleaved caspase-1 in the cell supernatants (Fig. 3A). LT-induced IL-18 secretion was also reduced (Fig. S3A). Similarly, in ASC−/− cells incubated with LT, a complete inhibition of IL-1β and IL-18 secretion was observed in the presence of z-VAD-fmk or z-YVAD-fmk. Both caspase inhibitors protected WT and ASC−/− macrophages from LT intoxication (Fig. 3B). These results indirectly suggest that caspase-1 is enzymatically active in ASC−/− cells despite its inability to undergo autoproteolysis. Caspase-1/caspase-11−/− BMDMs were reconstituted with retroviruses expressing either WT or different mutant forms of caspase-1 to directly and definitively establish whether, in response to LT, noncleaved caspase-1 is capable of triggering IL-1β and IL-18 maturation and secretion and pyroptosis. Caspase-1 was expressed at similar levels using WT caspase-1, caspase-1 DEAD (mutated at the catalytic site), and uncleavable caspase-1 C71 (catalytically active but mutated in all processing sites) (Fig. 4A) (20). Transduction of caspase-1/caspase-11–deficient cells with WT caspase-1–expressing plasmid restored LT sensitivity, as assessed by cell death (Fig. 4B). Furthermore, the expression of WT caspase-1 resulted in the secretion of IL-1β and IL-18 and autoprocessed caspase-1 upon LT and nigericin treatments (Fig. 4A and Fig. S3B). As expected, cells reconstituted with the enzyme-dead version did not produce any active IL-1β in response to LT or to nigericin and were not sensitive to LT intoxication. Interestingly, the expression of the uncleavable C71 caspase-1 mutant was able to restore pyroptosis and the production of mature IL-1β and IL-18, demonstrating that the caspase-1 45 kDa is fully active in response to LT treatment (Fig. 4A and Fig. S3B). As described for BMDMs, the transduction of caspase-1/caspase-11–deficient BMDCs with the uncleavable C71 caspase-1 mutant restored both IL-1β secretion and pyroptosis in response to LT (Fig. 4 C and D). Notably, in both BMDMs and BMDCs, C71 caspase-1 did not restore nigericin-induced cleavage and secretion of IL-1β, suggesting different mechanisms of caspase-1 activation in the NLRP1b and NLRP3 inflammasomes (Fig. 4 A and C).
Fig. 3.

LT-induced cell death and IL-1β secretion rely on caspase-1 catalytic activity in ASC-deficient cells. LPS-primed mouse peritoneal macrophages were treated or not (−, NT) with LT (0.5 μg/mL) in the presence or absence of the pan-caspase inhibitor z-VAD-fmk (50 μM) or caspase-1 inhibitor z-YVAD-fmk (50 μM) for 6 h. Protein expression was assessed by Western blot (A) and cell death was determined by measuring the LDH released in cell supernatants (B). Peritoneal macrophages were isolated from WT or ASC−/− mice.
Fig. 4.
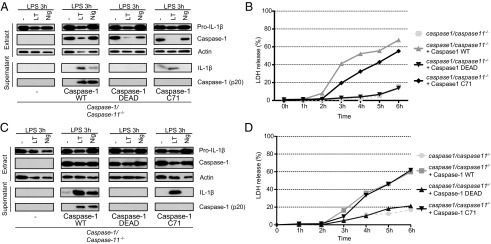
Caspase-1 autoproteolysis is not required to induce cell death nor to induce IL-1β cleavage and secretion upon LT treatment. LPS-primed BMDMs (A and B) or BMDCs (C and D) from caspase-1/caspase-11−/− mice, transduced or not (−) with the indicated caspase-1 constructs were treated with LT (0.5 μg/mL) for 6 h or with nigericin (Nig, 10 μM) for 2 h or left untreated (−). Protein expression was assessed by Western blot (A and C), LDH release was measured in cell supernatants of LT-treated cells (B and D).
LT Induces Speck Formation in WT and Caspase-1/Caspase-11−/− Cells.
Upon inflammasome activation, ASC assembles into a large multimeric structure called a “speck” detectable by immunofluorescence as a single perinuclear focus (26, 27). The polymerized ASC recruits and dimerizes caspase-1 within the specks, allowing caspase-1 activation by autoproteolysis of the enzyme (27). Recently, efficient cytokine production was associated with autoprocessing of caspase-1 within specks upon NLRC4 inflammasome activation (20). LT treatment of macrophages induced caspase-1 cleavage and secretion, suggesting active ASC speck formation. We therefore wondered whether the sensing of LT by the NLRP1b inflammasome could induce ASC speck formation. In WT macrophages treated with LT, ASC specks were detected 2 h posttreatment by immunofluorescence (Fig. 5A). At the same time point, caspase-1 was also found to form a perinuclear focus (Fig. 5B). Colabeling of ASC and caspase-1 clearly demonstrated that foci of caspase-1 were localized in very close proximity to ASC specks (Fig. 5C). In the absence of caspase-1, ASC was still able to form a speck (Fig. 5A). However, when ASC was absent from the cells, no caspase-1 focus was detected, suggesting that ASC specks recruit caspase-1 to the foci in response to LT (Fig. 5B).
Fig. 5.

LT induces ASC speck formation in a caspase-1–independent manner. LPS-primed peritoneal macrophages were treated with LT (0.5 μg/mL) or not (−) for 2 h. Cells were then fixed in 2% (wt/vol) paraformaldehyde and immunostained for ASC (green, A) or caspase-1 (red, B). Confocal microscopy sequential imaging of ASC immunostaining (green) and caspase-1 immunostaining (red) was performed on LPS-primed macrophages treated or not (−) with LT (0.5 μg/mL) (C). Hoechst (blue) was used to stain nuclei.
Discussion
In mouse macrophages of sensitive strains, LT triggers the formation of the NLRP1b inflammasome, culminating in pyroptosis and in the secretion of mature IL-1β and active caspase-1 (7, 21). We and others could inhibit NLRP1b inflammasome activity by adding extracellular potassium to the culture medium (Fig. S4) (22, 25), suggesting a role for NLRP3 and/or ASC in the formation of the NLRP1b inflammasome. To study whether these proteins contribute to LT detection by the NLRP1b inflammasome, we examined the role of NLRP3, ASC, and caspase-1 in response to LT intoxication in both macrophages and DCs in the mouse BALB/c background.
The data presented here show that LT induces similar levels of autoprocessed caspase-1 in primed and nonprimed cells (Figs. 1 and 2), suggesting that the assembly of the NLRP1b inflammasome does not require any upstream Toll-like receptors (TLRs) signaling (signal 1) for optimal activation. This mode of activation differs from the NLRP3 inflammasome activation. Although the NLRP3 inflammasome can form and activate caspase-1 without an additional signal, studies showed that the TLR priming signal significantly increases the amount of NLRP3 protein and therefore the amount of caspase-1 activated within the inflammasome platform (28, 29).
However, our results show that NLRP3 is not required for IL-1β maturation and pyroptosis in response to LT in mouse macrophages and in BMDCs. As such, our results extend those of Kovarova et al. who described that NLRP3 is dispensable for NLRP1b inflammasome activation in macrophages of 129 background mice (30). Using ASC-deficient macrophages and BMDCs, we observed that upon LT stimulation, IL-1β secretion and pyroptosis proceeded normally despite the absence of caspase-1 autoprocessing (Figs. 1 and 2). As described in the literature, caspase-1/caspase-11−/− macrophages and DCs were fully protected from death and did not secrete any IL-1β. With regard to macrophage cell death, cells were as sensitive in vivo as in vitro; ASC−/− macrophages died in response to LT treatment, whereas caspase-1/caspase-11−/− macrophages were protected (Fig. 2D). Intriguingly, whereas ASC−/− cells secreted a similar level of IL-1β to WT cells, we were not able to detect any cleaved caspase-1 (p20) in the cell supernatants. However, IL-1β production was dependent on caspase-1 activity, as IL-1β secretion was inhibited by the pan-caspase inhibitor z-VAD-fmk and by the caspase-1 inhibitor z-YVAD-fmk (Fig. 3A). Similarly, pyroptosis was inhibited in ASC−/− cells with both caspase inhibitors (Fig. 3B). Our results suggest that in response to LT and in the absence of ASC, caspase-1 zymogen (p45) is active despite its inability to undergo autoproteolysis.
Caspase-1 belongs to the family of initiator caspases, which includes caspase-8, -9, and -2. Initiator caspases are found in the cell cytosol as monomeric zymogens with little activity (31). Their activation is thought to follow the proximity-induced dimerization model (32, 33). Local increase in caspase-1 zymogen through dimerization increases the catalytic activity required to initiate its own activation by autoproteolysis. Once proteolysed, the caspase-1 p20 and p10 subunits assemble into enzymatically active p20/p10 dimers that cleave caspase-1 substrates such as pro–IL-1β and pro–IL-18 (9, 10). Inflammasomes form the molecular platforms that allow caspase-1 dimerization, activation, and autoproteolysis (11). Our data indicate a possible role for the zymogen form of caspase-1 in IL-1β processing and pyroptosis in ASC-deficient cells. A study by Broz et al. suggested that specific inflammasome receptors had distinct molecular platforms with distinct modes of activation of caspase-1 depending on the presence of ASC (20). Inflammasomes with a PRR containing a CARD domain, such as NLRC4, form large specks in the presence of ASC, where caspase-1 is autoproteolysed and cleaves pro–IL-1β and pro–IL-18. A second type of NLRC4 inflammasome platform that functions independently of ASC, activates caspase-1 through CARD–CARD interactions without inducing its autoprocessing activity. This platform controls caspase-1–induced pyroptosis (20). A similar mode of action was proposed for the CARD-containing NLRP1b receptor. However, our study demonstrates another mechanism of activation. In the absence of ASC and caspase-1 autoproteolysis, the NLRP1b inflammasome was still capable of inducing both cytokine maturation and cell death. In addition, by reconstituting caspase-1/caspase-11–deficient BMDMs and BMDCs with a vector expressing the uncleavable C71 or the enzyme dead caspase-1 mutant, we showed that uncleavable caspase-1 promoted IL-1β secretion and pyroptosis in response to LT (Fig. 4). The efficient reconstitution of caspase-1/caspase-11–deficient cells with WT caspase-1 excluded a role for caspase-11 in response to LT.
While preparing this paper, a study by Van Opdenbosch et al. reported observations similar to ours in C57BL/6 mice expressing a NLRP1b transgene (34). They described IL-1β cleavage and pyroptosis in ASC−/− macrophages. However, their work, which also suggested that uncleaved caspase-1 is active in ASC−/− macrophages, was largely based on the use of a caspase-1 inhibitor, which, like most pharmacological inhibitors, may lack complete selectivity. In the work presented here, we sought to overcome this limitation and, by reconstituting caspase-1 knockout cells expressing endogenous NLRP1b with different caspase-1 mutant constructs, we directly and definitively demonstrated for the first time to our knowledge that the uncleavable version of caspase-1 accomplishes efficient IL-1β maturation and pyroptosis upon LT stimulation.
Our work suggests that the NLRP1b–caspase-1 interaction through their CARD domains confers a structural change allowing full-length caspase-1 to become activated. The NLRP1b inflammasome may function like the apoptosome (Fig. S5). A recent biochemical analysis reported that the electrostatic surface charges of hNLRP1 and caspase-1 CARDs share similarities with the surfaces of Apaf-1 and caspase-9 CARDs (35). Caspase-9 was shown to be active in its full-length form when oligomerized within the apoptosome in the absence of autoproteolysis (36, 37). Further biochemical experiments are required to understand how full-length caspase-1 is activated within the NLRP1b inflammasome.
In addition, we show that the C71 uncleavable mutant was not effective in response to the NLRP3 inflammasome activator nigericin despite the presence of ASC. This result implies that the NLRP3 inflammasome fully relies on caspase-1 autoproteolysis for activity. The NLRP3 inflammasome requires the presence of ASC to activate caspase-1. Indeed, it has been widely described that in response to NLRP3 activators, the IL-1β secretion is abolished and the pyroptosis is strongly impaired in ASC−/− macrophages (16, 19). NLRP3 bearing only a PYD and no CARD domain requires ASC, containing both a PYD and a CARD, to recruit caspase-1 and to form an inflammasome. Caspase-1 is then autoproteolysed and activated within specks. Thus, NLRP1b and NLRP3 inflammasomes differentially activate caspase-1.
Our study also shows that in WT cells treated with LT, ASC specks are formed and foci of caspase-1 colocalized with the specks (Fig. 5). In the absence of ASC, the foci of caspase-1 are not detected, whereas in the absence of caspase-1, ASC specks are still formed. In response to LT, ASC specks are most likely responsible for the autoproteolysis of caspase-1 as previously described for other inflammasomes (20, 27). Recent studies demonstrated that ASC PYD and MAVS CARD domains function like prion domains by inducing polymerization of the PYD and CARD domains (38). Our results suggest that the NLRP1b CARD domain is not able to induce polymerization of caspase-1 because no focus was detected in the absence of ASC. However, ASC speck formation is most likely dependent on the NLRP1b CARD domain. ASC polymerization was shown to be dependent on low intracellular potassium concentration (27). ASC assembly and the NLRP3 inflammasome activation are inhibited by increasing the extracellular potassium concentration above 90 mM (25, 27). Despite the inhibitory effect of extracellular potassium addition on the NLRP1b inflammasome activation by LT, caspase-1 activity was not dependent on its autoproteolysis within the ASC speck. Low intracellular potassium may be required for NLRP1b and caspase-1 interaction. Indeed, it has previously been demonstrated that the apoptosome assembly is sensitive to potassium concentration (39). The NLRP1b inflammasome can efficiently activate caspase-1 in the absence of ASC and in the absence of caspase-1 autoproteolysis. It is therefore tempting to speculate that ASC may have additional functions yet to be described. Similarly, caspase-1 autoprocessing appears not to be critical for its activity. Thus, it remains to be elucidated why caspase-1 autoproteolyses in WT cells in response to LT and whether the autoproteolysis is associated with unknown function.
In conclusion, our data, which highlight differential requirements for caspase-1 autoproteolysis in NLRP1b and NLRP3 inflammasome function, may have implications for pathogen recognition and response.
Materials and Methods
Reagents.
Nigericin (N7143) and KCl (P9541) from Sigma, ultra-pure LPS (Escherichia coli 0111:B4) from Invivogen, anthrax lethal factor (batch 1692A1B), and protective antigen (batch 17117A1B) from List Biological Laboratories. z-YVAD-fmk and z-VAD-fmk are from Bachem.
Mice.
NLRP3−/− mice were obtained from J. Tschopp (16), ASC−/− mice from V. M. Dixit (19), and caspase-1−/− mice from R. A. Flavell (40). The three transgenic strains were backcrossed in BALB/c/Ola background for at least nine generations. WT animals were littermates from the caspase-1/caspase-11−/−, ASC−/−, or NLRP3−/− colonies. Animals were housed in individually ventilated cages under specific pathogen-free conditions, and studies were conducted under protocols in accordance with the animal care guidelines of the European Union laws and were validated by the local Animal Ethic Evaluation Committee (CECCAPP).
Infection.
For transduction of primary bone marrow cells, retroviral particles were generated using Phoenix-Eco packaging cells and used to transduce bone marrow cells at day 2 and day 3 during cell differentiation as described by Broz et al. (20). Cells were stimulated after 7 d of differentiation as described above. pMSC2.2-expressing vectors for caspase-1 wild type, caspase-1 DEAD, and caspase-1 C71 were obtained from P. Broz.
Supplementary Material
Acknowledgments
We thank V. M. Dixit (Genentech) for the ASC−/− mice; the late J. Tschopp (University of Lausanne) for the NLRP3−/− mice; R. A. Flavell (Yale School of Medicine) for the caspase-1/caspase-11−/− mice; Peter Broz (Basel University) for the gift of caspase-1–expressing vectors; T. Renno, S. Kabani, F. Martinon, and N. Rahim for critical reading of the manuscript; and C. Vanbelle and the Plateforme d'Expérimentation Animale en Cancérologie (AniCan) facility for technical help. This work was supported by the Institut Thématique Multi-Organismes (ITMO) Cancer and Ligue contre le Cancer comité de l’Ain. M.B. is supported by the Association Nationale de la Recherche et de la Technologie (ANRT). A.T. is supported by the Association “Institute for Arthritis Research” (aIAR).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1415756111/-/DCSupplemental.
References
- 1.Banks DJ, Ward SC, Bradley KA. New insights into the functions of anthrax toxin. Expert Rev Mol Med. 2006;8(7):1–18. doi: 10.1017/S1462399406010714. [DOI] [PubMed] [Google Scholar]
- 2.Bradley KA, Mogridge J, Mourez M, Collier RJ, Young JA. Identification of the cellular receptor for anthrax toxin. Nature. 2001;414(6860):225–229. doi: 10.1038/n35101999. [DOI] [PubMed] [Google Scholar]
- 3.Moayeri M, Leppla SH. The roles of anthrax toxin in pathogenesis. Curr Opin Microbiol. 2004;7(1):19–24. doi: 10.1016/j.mib.2003.12.001. [DOI] [PubMed] [Google Scholar]
- 4.Alileche A, Serfass ER, Muehlbauer SM, Porcelli SA, Brojatsch J. Anthrax lethal toxin-mediated killing of human and murine dendritic cells impairs the adaptive immune response. PLoS Pathog. 2005;1(2):e19. doi: 10.1371/journal.ppat.0010019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Agrawal A, et al. Impairment of dendritic cells and adaptive immunity by anthrax lethal toxin. Nature. 2003;424(6946):329–334. doi: 10.1038/nature01794. [DOI] [PubMed] [Google Scholar]
- 6.Park JM, Greten FR, Li Z-W, Karin M. Macrophage apoptosis by anthrax lethal factor through p38 MAP kinase inhibition. Science. 2002;297(5589):2048–2051. doi: 10.1126/science.1073163. [DOI] [PubMed] [Google Scholar]
- 7.Cordoba-Rodriguez R, Fang H, Lankford CSR, Frucht DM. Anthrax lethal toxin rapidly activates caspase-1/ICE and induces extracellular release of interleukin (IL)-1beta and IL-18. J Biol Chem. 2004;279(20):20563–20566. doi: 10.1074/jbc.C300539200. [DOI] [PubMed] [Google Scholar]
- 8.Duesbery NS, et al. Proteolytic inactivation of MAP-kinase-kinase by anthrax lethal factor. Science. 1998;280(5364):734–737. doi: 10.1126/science.280.5364.734. [DOI] [PubMed] [Google Scholar]
- 9.Elliott JM, Rouge L, Wiesmann C, Scheer JM. Crystal structure of procaspase-1 zymogen domain reveals insight into inflammatory caspase autoactivation. J Biol Chem. 2009;284(10):6546–6553. doi: 10.1074/jbc.M806121200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Walker NP, et al. Crystal structure of the cysteine protease interleukin-1 beta-converting enzyme: A (p20/p10)2 homodimer. Cell. 1994;78(2):343–352. doi: 10.1016/0092-8674(94)90303-4. [DOI] [PubMed] [Google Scholar]
- 11.Martinon F, Burns K, Tschopp J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. 2002;10(2):417–426. doi: 10.1016/s1097-2765(02)00599-3. [DOI] [PubMed] [Google Scholar]
- 12.Thornberry NA, et al. A novel heterodimeric cysteine protease is required for interleukin-1 beta processing in monocytes. Nature. 1992;356(6372):768–774. doi: 10.1038/356768a0. [DOI] [PubMed] [Google Scholar]
- 13.Strowig T, Henao-Mejia J, Elinav E, Flavell R. Inflammasomes in health and disease. Nature. 2012;481(7381):278–286. doi: 10.1038/nature10759. [DOI] [PubMed] [Google Scholar]
- 14.Schroder K, Tschopp J. The inflammasomes. Cell. 2010;140(6):821–832. doi: 10.1016/j.cell.2010.01.040. [DOI] [PubMed] [Google Scholar]
- 15.Mariathasan S, et al. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature. 2006;440(7081):228–232. doi: 10.1038/nature04515. [DOI] [PubMed] [Google Scholar]
- 16.Martinon F, Pétrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440(7081):237–241. doi: 10.1038/nature04516. [DOI] [PubMed] [Google Scholar]
- 17.Muruve DA, et al. The inflammasome recognizes cytosolic microbial and host DNA and triggers an innate immune response. Nature. 2008;452:103–7. doi: 10.1038/nature06664. [DOI] [PubMed] [Google Scholar]
- 18.Hornung V, et al. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature. 2009;458(7237):514–518. doi: 10.1038/nature07725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mariathasan S, et al. Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature. 2004;430(6996):213–218. doi: 10.1038/nature02664. [DOI] [PubMed] [Google Scholar]
- 20.Broz P, von Moltke J, Jones JW, Vance RE, Monack DM. Differential requirement for Caspase-1 autoproteolysis in pathogen-induced cell death and cytokine processing. Cell Host Microbe. 2010;8(6):471–483. doi: 10.1016/j.chom.2010.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boyden ED, Dietrich WF. Nalp1b controls mouse macrophage susceptibility to anthrax lethal toxin. Nat Genet. 2006;38(2):240–244. doi: 10.1038/ng1724. [DOI] [PubMed] [Google Scholar]
- 22.Wickliffe KE, Leppla SH, Moayeri M. Anthrax lethal toxin-induced inflammasome formation and caspase-1 activation are late events dependent on ion fluxes and the proteasome. Cell Microbiol. 2008;10(2):332–343. doi: 10.1111/j.1462-5822.2007.01044.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martinon F, Gaide O, Pétrilli V, Mayor A, Tschopp J. NALP inflammasomes: A central role in innate immunity. Semin Immunopathol. 2007;29(3):213–229. doi: 10.1007/s00281-007-0079-y. [DOI] [PubMed] [Google Scholar]
- 24.Hanna PC, Kochi S, Collier RJ. Biochemical and physiological changes induced by anthrax lethal toxin in J774 macrophage-like cells. Mol Biol Cell. 1992;3(11):1269–1277. doi: 10.1091/mbc.3.11.1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pétrilli V, et al. Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ. 2007;14(9):1583–1589. doi: 10.1038/sj.cdd.4402195. [DOI] [PubMed] [Google Scholar]
- 26.Richards N, et al. Interaction between pyrin and the apoptotic speck protein (ASC) modulates ASC-induced apoptosis. J Biol Chem. 2001;276(42):39320–39329. doi: 10.1074/jbc.M104730200. [DOI] [PubMed] [Google Scholar]
- 27.Fernandes-Alnemri T, et al. The pyroptosome: A supramolecular assembly of ASC dimers mediating inflammatory cell death via caspase-1 activation. Cell Death Differ. 2007;14(9):1590–1604. doi: 10.1038/sj.cdd.4402194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guarda G, et al. Differential expression of NLRP3 among hematopoietic cells. J Immunol. 2011;186(4):2529–2534. doi: 10.4049/jimmunol.1002720. [DOI] [PubMed] [Google Scholar]
- 29.Bauernfeind FG, et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol. 2009;183(2):787–791. doi: 10.4049/jimmunol.0901363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kovarova M, et al. NLRP1-dependent pyroptosis leads to acute lung injury and morbidity in mice. J Immunol. 2012;189(4):2006–2016. doi: 10.4049/jimmunol.1201065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Renatus M, Stennicke HR, Scott FL, Liddington RC, Salvesen GS. Dimer formation drives the activation of the cell death protease caspase 9. Proc Natl Acad Sci USA. 2001;98(25):14250–14255. doi: 10.1073/pnas.231465798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boatright KM, et al. A unified model for apical caspase activation. Mol Cell. 2003;11(2):529–541. doi: 10.1016/s1097-2765(03)00051-0. [DOI] [PubMed] [Google Scholar]
- 33.Shi Y. Caspase activation: Revisiting the induced proximity model. Cell. 2004;117(7):855–858. doi: 10.1016/j.cell.2004.06.007. [DOI] [PubMed] [Google Scholar]
- 34.Van Opdenbosch N, et al. Activation of the NLRP1b inflammasome independently of ASC-mediated caspase-1 autoproteolysis and speck formation. Nat Commun. 2014;5:3209. doi: 10.1038/ncomms4209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jin T, Curry J, Smith P, Jiang J, Xiao TS. Structure of the NLRP1 caspase recruitment domain suggests potential mechanisms for its association with procaspase-1. Proteins. 2013;81(7):1266–1270. doi: 10.1002/prot.24287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chao Y, et al. Engineering a dimeric caspase-9: A re-evaluation of the induced proximity model for caspase activation. PLoS Biol. 2005;3(6):e183. doi: 10.1371/journal.pbio.0030183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rodriguez J, Lazebnik Y. Caspase-9 and APAF-1 form an active holoenzyme. Genes Dev. 1999;13(24):3179–3184. doi: 10.1101/gad.13.24.3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cai X, et al. Prion-like polymerization underlies signal transduction in antiviral immune defense and inflammasome activation. Cell. 2014;156(6):1207–1222. doi: 10.1016/j.cell.2014.01.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cain K, Langlais C, Sun XM, Brown DG, Cohen GM. Physiological concentrations of K+ inhibit cytochrome c-dependent formation of the apoptosome. J Biol Chem. 2001;276(45):41985–41990. doi: 10.1074/jbc.M107419200. [DOI] [PubMed] [Google Scholar]
- 40.Kuida K, et al. Altered cytokine export and apoptosis in mice deficient in interleukin-1 beta converting enzyme. Science. 1995;267(5206):2000–2003. doi: 10.1126/science.7535475. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.