

Abstract
A novel cytochrome P450 enzyme TxtE was recently shown to catalyze the direct aromatic nitration of L-tryptophan. This unique chemistry induced us to ask whether TxtE could serve as a platform for engineering new nitration biocatalysts, as a replacement for current harsh synthetic methods. As a first step toward this goal and to better understand the wild-type enzyme, we have obtained high-resolution structures of TxtE in its substrate-free and substrate-bound forms. We have also screened a library of substrate analogs for spectroscopic indicators of binding and for production of nitrated products. From these results, we find that the wild-type enzyme accepts moderate decoration of the in dole ring, but the amino acid moiety is crucial for binding and correct positioning of the substrate and therefore less amenable to modification. A carbonyl must be present to recruit the αB′1 helix of the protein to seal the binding pocket, and a nitrogen atom is essential for catalysis.
Keywords: aromatic nitration, crystal-structure determination, cytochromes, enzyme catalysis
Introduction
Nature has evolved specialized protein machinery to carry out a vast array of synthetic transformations with a high degree of specificity and catalytic proficiency. In an effort to create new and ‘greener’ chemistries, researchers have borrowed enzymes from their native biological environments and engineered them to catalyze desired synthetic transformations over a large reaction space under mild, aqueous conditions.[1–3]
One transformation for which the available biocatalysts are limited is aromatic nitration. Conventional methods for the nitration of organic compounds employ harsh reaction conditions, most commonly direct aromatic nitration utilizing nitric and sulfuric acids.[4–6] Oxidation is a common side reaction under these conditions and formation and isolation of the desired isomer can be intractable depending on the complexity of the substrate. In contrast, biological nitration typically proceeds through the oxidation of an amine or, to a lesser extent, through unselective and/or inefficient direct aromatic nitration.[7–11]
The cytochrome P450 TxtE from Streptomyces scabies is the first enzyme reported to efficiently catalyze a direct and regioselective aromatic nitration.[12] This enzyme is partnered with a nitric oxide synthase and is believed to generate a reactive peroxynitrite intermediate that subsequently disproportionates to nitrate L-tryptophan, forming 4-nitro-L-tryptophan as an intermediate in the production of the S. scabies phytotoxin thaxtomin A. The in vivo activity can be recapitulated in vitro with recombinant TxtE, using molecular oxygen, a nitric oxide donor, and L-tryptophan as substrates and spinach ferredoxin (Fd) with ferredoxin NADP+ reductase (Fr) as artificial electron donors (Figure 1).[12]
Figure 1.
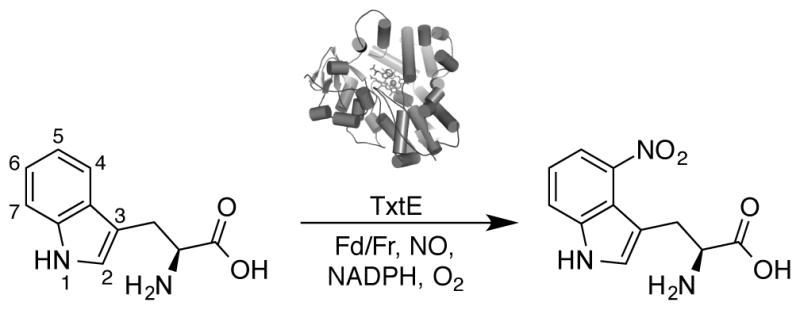
L-Tryptophan nitration catalyzed by TxtE as described by Barry et al.[12] Abbreviations: spinach ferredoxin (Fd) and spinach ferredoxin NADP+ reductase (Fr).
Given its unique chemistry, TxtE could be an ideal platform to engineer new aromatic nitration catalysts that could provide a more versatile and ‘greener’ alternative to traditional synthetic methods. In order to do this, a better understanding of the wild-type enzyme is required. To this end, we have assessed the scope of tryptophan-like substrates that TxtE can bind and nitrate. We have also obtained the first high-resolution structures of substrate-free and tryptophan-bound TxtE. In light of these results, we offer new insights into the substrate properties required for catalysis and provide information that can guide future protein engineering efforts.
Results and Discussion
Structural Determination of Substrate-Free and L-Tryptophan-Bound Forms of TxtE
TxtE has been crystallized previously with the inhibitor imidazole bound in the active site.[13] Here, we were able to crystallize the enzyme in a substrate-free state using a mixture of lithium sulfate and ammonium sulfate as a precipitant. Diffraction data were collected both with and without the addition of L-tryptophan. The crystals diffracted remarkably well, yielding resolutions of 1.18 Å and 1.22 Å, respectively. Full crystallographic statistics are shown in Table 1. As previously reported, the crystal structure of TxtE shows the canonical fold for a cytochrome P450, a mostly alpha-helical trigonal prism (Figure 2a). The structures described here are in agreement with the previously reported structure (backbone RMSD 0.237 Å and 0.219 Å, respectively, to chain A of 4L36), while providing a higher resolution picture of the protein active site in the absence and presence of the native substrate. A detailed discussion of the fold and active site structure can be found in the report of Yu et al.;[13] here we will focus on how the enzyme interacts with its substrate, which can only be seen in the current structures.
Table 1.
Crystallographic parameters of substrate free TxtE and L-tryptophan bound TxtE.
| Parameter | Substrate-free | +L-tryptophan |
|---|---|---|
| PDB ID: 4TPN | PDB ID: 4TPO | |
| Data Collection | ||
| Space Group | P212121 | P212121 |
| a (Å) | 65.7 | 65.8 |
| b (Å) | 79.4 | 79.7 |
| c (Å) | 112.4 | 112.6 |
| α, β, γ (°) | 90, 90, 90 | 90, 90, 90 |
| Resolution (Å) | 37.61 – 1.18 (1.24–1.18) | 37.69 – 1.22 (1.29–1.22) |
| Rmerge (%) | 4.8 (136.2) | 12.6 (99.5) |
| <I>/<σI> | 13.9 (1.1) | 5.6 (1.0) |
| Completeness (%) | 94.9 (81.1) | 98.0 (90.1) |
| Redundancy | 4.9 (4.4) | 3.6 (3.0) |
| Refinement | ||
| No. reflections | 173,667 | 161,654 |
| Rwork/Rfree (%) | 16.3/17.1 | 13.9/16.1 |
| No. atoms | ||
| Protein | 3014 | 3112 |
| Ligand | 93 | 96 |
| Water | 394 | 409 |
| RMSD | ||
| Bond lengths (Å) | 0.018 | 0.026 |
| Bond angles (°) | 1.661 | 2.299 |
| Ramachandran plot (%) | ||
| Favored | 337 | 372 |
| Allowed | 11 | 9 |
| Outliers | 1 | 1 |
Values in parentheses represent statistics from the highest resolution shell.
Figure 2.

A) The overall fold of TxtE, with simplified helices and loops, including the heme (pink) and bound tryptophan molecule (light blue). The I-helix runs from the top to the bottom in yellow, with an interruption for the kink caused by the distal threonine. The B–C loop with the pair of B′ helices is in light blue at the top of the structure. B) Structure of the TxtE active site with L-tryptophan bound. TxtE protein is shown in green, the heme cofactor is shown in pink, and L-tryptophan is highlighted in sky blue. The polar contacts between the amino acid and the protein scaffold are shown with yellow dotted lines. The water that distally ligates the heme (and its hydrogen bond network back to Thr 250) is also shown, despite being present at an occupancy of 20%. An Fo-Fc omit map of the active site is shown in Figure S1.
Yu et al. used AutoDock to propose three L-tryptophan binding poses.[13] The binding orientation observed in our study closely resembles their first two proposals. As shown in Figure 2b, the tryptophan (which is bound to approximately 80% occupancy) binds with its indole side chain facing into the distal face of the heme, displacing the water that ligates the heme (this water is still visible in the tryptophan-bound structure, refined at 20% occupancy). The amino acid moiety faces toward the B-C loop containing the pair of B′ helices. Figure 3 shows a schematic of the interactions involved in tryptophan binding. The alpha amine of the amino acid is directly coordinated by Asn293 and Thr296 of the pre-β3 loop, and also forms a water-mediated hydrogen bond with Glu394. Thr296 is also involved in coordination of the carboxylate, as is Glu394 through a different bridging water molecule. The carboxylate also interacts with Arg59, which forms the start of the αB′1 helix, and Tyr89 at the end of the αB′2-αC loop. Residues Met88 and Phe395 define the hydrophobic cleft for the substrate’s indole moiety. The C-4 position of the indole ring, the site of nitration, faces away from the heme and toward the hydroxyl of Tyr89 and the solvent channel that was identified previously[13] and confirmed in our structures. In the substrate-free structure, three ordered waters define this solvent channel as reported,[13] whereas in the tryptophan-bound structure a tube of indistinct density is visible that could not be modeled as waters. The binding of tryptophan also displaces two waters present in the substrate-free structure, one that fills the amine-binding pocket and one that takes the place of the carboxylate oxygen trans to it. The imidazole-bound structure of Yu et al. contains both these waters, as well as another that takes the place of the second carboxylate oxygen.[13]
Figure 3.

A schematic of interactions in the L-tryptophan binding pocket. Hydrogen bonds, direct and water-mediated, are shown (as determined by contact distances and angles) as dashed lines. Nonpolar contacts are shown with arches. The minor conformation of Asn293 is shown with dashed lines. For clarity, hydrogen atoms on heteroatoms are not shown, except on water molecules.
Interestingly, in the substrate-free structure, the density for the αB′1 helix was insufficiently clear to model residues 61–72, which suggests that this portion of the protein is disordered. The density can be resolved in the substrate-bound structure, though at 80% occupancy (mirroring that of the tryptophan itself) and with considerably elevated B-factors, suggesting that it is still fairly flexible. Rearrangements of the B-C loop are known to be crucial for substrate binding in other P450s.[14–15] Thus we suggest that substrate binding-induced organization of this loop also occurs in TxtE. This hypothesis is further supported by the change in the rotamer observed at Arg59, which anchors the αB′1 helix and provides a key interaction with the substrate carboxylate. Upon binding of L-tryptophan, Arg59 moves to coordinate to both the substrate carboxylate and the propionate of the heme, as shown in Figure 4, resulting in an almost 4-Å shift of Gly60, and presumably imparting enough order to the helix to make it crystallographically resolvable. In the imidazole-bound structure, the αB′1 helix is fully resolved, with Arg59 facing downward in a conformation similar to that observed in this tryptophan-bound structure. As shown in Figure 4c and discussed above, a more complete network of waters, anchored by the heme-bound imidazole, takes the place of the tryptophan heteroatoms and may be responsible for the recruitment of Arg59 and the helix.
Figure 4.

Rearrangement of the TxtE aB’1 helix upon L-tryptophan binding through residue Arg59. A) The structure of substrate-free TxtE is shown in lime green; B) The structure of L-tryptophan-bound TxtE is shown in purple, including indications of the polar contacts made by Arg59 in its new conformation; C) The previously reported imidazole-bound TxtE structure (PDB ID: 4L36) is shown in cyan, including the imidazole-anchored network of waters taking the place of the tryptophan amino acid motif; and D) overlay of (A) and (B), showing the movement of Arg59 and Gly60.
An additional detail visible in our high-resolution structures is that Asn293 has two rotamers. The major rotamer (60% in the substrate-free structure, 80% in the tryptophan-bound structure) is involved in binding the amine, but the other rotamer (shown in dashed lines in Figure 3) forms a hydrogen bond with the indole nitrogen of tryptophan, and may be involved in the catalytic cycle.[12]
The observed binding mode of tryptophan provides a starting point for our survey of the enzyme’s substrate preferences (Figure 1). The indole side chain is held in place through nonspecific hydrophobic interactions. Thus, we hypothesized that the side-chain might be amenable to substitutions and moderate decorations, subject to the electronic requirements of the reaction itself. The amino acid moiety of the substrate is involved in a complex network of hydrogen bonds involving both the amine and the carboxylate with amino acids of four different loops of the protein and with the propionates of the heme cofactor. With this structural information in hand, we have explored to what extent the native substrate can be modified while maintaining productive nitration chemistry.
Overview of the Spectroscopic and Functional Characterization of TxtE Substrate Scope
Consistent with previous reports,[12] we observed that recombinant TxtE carries out the regioselective nitration of its native substrate L-tryptophan to generate 4-nitro-L-tryptophan, in purified enzyme form as well as in cell lysate (Figure S2). Binding and reactivity were assessed for each of 28 substrates tested (Tables 2–5). Because TxtE is a cytochrome P450, the binding mode of a small molecule can be inferred by determining the spectral shift of the Soret peak from a differential UV-visible spectrum (Figure S13–S15).[16] A type I shift from 420 nm to 390 nm is observed when the incoming substrate displaces the axial water ligand from the heme iron. This substrate binding results in conversion from low to high spin iron, which is essential for initiating the catalytic cycle.[16] L-tryptophan binds TxtE and produces a type I shift (Figure S13).[12] Alternately, a type II spectral shift, from 420 nm to 425–435 nm, is characteristic of a ligand that directly coordinates to the iron center and in most cases prevents oxygen-driven catalysis.[16,17] Nitration reactions were carried out with overexpressed TxtE in E. coli cell lysates at least in triplicate with a vector (no-enzyme) control. The spinach ferredoxin/ferredoxin NADP+ reductase system and the nitric oxide donor diethylamine NONOate were used as previously described.[12] Productive chemistry was assessed qualitatively by the formation of yellow color corresponding to a nitro product (in most cases) and was confirmed by LC-MS analysis. Some substrates were converted to tryptophan by other enzymes present in the cell lysate so purified protein was used instead.
Table 2.
Chemical structures, spectral shifts, and nitration chemistry of L-tryptophan derivatives with blocked heteroatom contacts.
| Substratea | Structure | Spectral Shift | Nitrationb |
|---|---|---|---|
| 1 |

|
Type I | ++ |
| 2 |

|
Type I | ND |
| 3 |

|
Type I | ND |
| 4c |

|
Type I | ++d |
| 5 |

|
Type II | ND |
Reaction conditions can be found in the experimental section. Differential UV-visible spectra can be found in Figure S13 and LC-MS chromatograms for nitrated substrates can be found in Figures S2–S3.
Abbreviations: not detected (ND).
Purified protein was used.
Product formation was observed above background.
Table 5.
Chemical structures, spectral shifts, and nitration chemistry of L-tryptophan derivatives with modified amino acid motifs.
| Substratea | Structure | Spectral Shift | Nitrationb |
|---|---|---|---|
| 19 |

|
Type I | ND |
| 20 |

|
Type II | ND |
| 21 |

|
Type I | ND |
| 22 |

|
Type I | ND |
| 23 |

|
Type II | ND |
| 24 |

|
Type I | ++ |
| 25 |

|
Type II | ND |
| 26c |

|
Type I | ND |
| 27c |

|
Type I | ++ |
| 28 |

|
Type I | ND |
| 29 |

|
Type I | ++ |
Reaction conditions can be found in the experimental section. Differential UV-visible spectra can be found in Figure S15 and LC-MS chromatograms for nitrated substrates can be found in Figures S10–S12.
Abbreviations: not detected (ND).
Purified protein was used.
Disrupting the Heteroatom Bond Contacts of L-Tryptophan
First, we disrupted heteroatom bond contacts in L-tryptophan (substrate 1) to identify the components of the molecule that are required for nitration. Methylation of the indole nitrogen of L-tryptophan in substrate 2, which eliminates a labile hydrogen atom and adds steric bulk, completely abolishes nitration chemistry while still maintaining type I binding (Table 2). The basicity and bulk of the amino acid nitrogen was altered through acetylation (substrate 3) and methylation (substrate 4). Titration of TxtE with substrates 3 and 4 results in characteristic type I spectra. Uniquely among the compounds tested in this study, nitration of substrate 4 is observed even in the absence of TxtE. However, higher rates of conversion are observed in the presence of TxtE (Figure S3). No nitrated product is detected for substrate 3, where acetylation causes a larger shift in both the steric bulk and the pKa of the nitrogen than methylation alone. Lastly, methyl esterification of the carboxylic acid in the amino acid moiety (substrate 5) results in a type II spectral shift and no productive nitration. These results indicate that alkylation or acetylation of the heteroatom bond contacts in L-tryptophan alters how these substrates are anchored in the active site of the enzyme. With the exception of substrate 4, no nitrated products are observed, suggesting that significant modification of the L-tryptophan heteroatoms prevents productive catalysis.
Modification of the Indole Moiety of L-Tryptophan
We next investigated substrates that retained the amino acid moiety but were altered in the indole side chain with ring decorations and heteroatom substitutions. TxtE tolerates methyl substitution at the 5- (substrate 6) and 7- (substrate 7) positions on the indole side chain, resulting in nitro product formation, but substrate 7 is nitrated to a lesser extent, perhaps due to a steric clash above the heme pocket (Table 3, Figure S5). Surprisingly, methyl substitution at the 4-position (substrate 8), the site of nitration on the native substrate, does not prevent the nitration reaction and nitrated product is still observed (Figure S6), which suggests that the enzyme can display alternative regioselectivity when this position is occluded. These results prompted us to test the effects of electron-withdrawing substituents with 5- and 6-fluoro-tryptophan analogs (substrates 9 and 10), which are both nitrated by TxtE (Figure S7 and S8).
Table 3.
Chemical structures, spectral shifts, and nitration chemistry of L-tryptophan derivatives with modified side chains.
| Substratea | Structure | Spectral Shift | Nitrationb |
|---|---|---|---|
| 6 |

|
Type I | ++ |
| 7 |

|
Type I | + |
| 8 |

|
Type I | ++ |
| 9 |

|
Type I | ++ |
| 10 |

|
Type I | ++ |
| 11 |

|
Type I | ++ |
| 12 |

|
Type I | ND |
| 13 |

|
Type I | ND |
Reaction conditions can be found in the experimental section. Differential UV-visible spectra can be found in Figure S14 and LC-MS chromatograms for nitrated substrates can be found in Figures S4–S9.
Abbreviations: not detected (ND).
From here, we explored substitution at the 5-position with hydrogen bonding groups. Substrate 11 has a methoxy substitution that increases the steric bulk beyond the methyl substitution alone, but the reaction still proceeds (Figure S9). In contrast, substrate 12, 5-hydroxy-L-tryptophan, is not nitrated, although it binds in a type I fashion similar to the other tryptophan derivatives. Likewise, amino substitution (substrate 13) prevents nitration. Thus, TxtE can nitrate substrates with different ring substituents, but the aromatic ring cannot be decorated with certain polar functional groups as these moieties prevent productive nitration chemistry, perhaps due to unfavorable interactions with the protein scaffold, the heme center, or the catalytic intermediates. The regioselectivity for the nitration of non-native substrates is currently under investigation.
The effects of heteroatom substitution in the indole ring were also tested (Table 4). The introduction of a nitrogen atom at the 7-position of indole (7-azatryptophan, substrate 14) results in no nitration activity. This atom is centered above the heme center, and we speculate that introduction of the nitrogen atom may interfere with the formation of the peroxynitrite intermediate or with compound II of the catalytic cycle. Replacement of the indole nitrogen with a less electronegative sulfur or carbon atom does not lead to nitration (substrates 15 and 16), similar to substrate 2. We also tested other aromatic amino acids, including L-phenylalanine (substrate 17) and L-histidine (substrate 18), but observed no nitration product. These results suggest that the enzyme requires an indole ring system with a labile hydrogen atom above the iron center that could stabilize or be abstracted to drive the catalytic cycle. This reasoning is consistent with a mechanism previously proposed by Barry et al.[12]
Table 4.
Continuation of Table 3. Chemical structures, spectral shifts, and nitration chemistry of L-tryptophan derivatives with modified side chains.
| Substratea | Structure | Spectral Shift | Nitrationb |
|---|---|---|---|
| 14 |

|
Type I | ND |
| 15 |

|
Type I | ND |
| 16 |

|
Type I | ND |
| 17 |

|
Type I | ND |
| 18 |

|
Type I | ND |
Reaction conditions can be found in the experimental section. Differential UV-visible spectra can be found in Figure S14.
Abbreviations: not detected (ND).
Modification of the L-Tryptophan Amino Acid Moiety
Finally, we evaluated to what extent the amino acid motif can be removed or modified (Table 5). L-tryptophan was broken down into three substrates: indole, tryptamine, and indole-3-propionic acid (substrates 19, 20, and 21). No nitrated product was observed for any of these. A known type II binder to cytochrome P450s, [16] tryptamine’s lack of reactivity is not surprising. Nor is it surprising that indole does not react, as it is quite small and does not provide any of the hydrogen-bonding atoms. However, indole-3-propionic acid’s lack of reactivity indicates that the hydrogen bonds to the amine part of the amino acid may be essential. Similarly, indole-3-propanol (substrate 22) does not have an amine and is not nitrated.
Interestingly, reduction of the carboxylic acid of the amino acid to an alcohol (substrate 23) yields a type II binder as seen with substrates 3 and 5. This indicates that an interaction between Arg59 and the carboxylate of L-tryptophan is important for binding the substrate in a configuration that prevents the indole nitrogen or the amino acid amine from coming in close proximity to the iron center, which results in a type II shift and prevents catalysis. Further, increasing the steric bulk on the amino acid with substrates 24 and 25 does not prevent binding but nitrated product is only observed for substrate 24 (Figure S10).
From here, we substituted the heteroatoms in the amino acid moiety of L-tryptophan, resulting in indole-3-lactic acid (substrate 26) and L-tryptophan amide (substrate 27). Both bind in a type I fashion, but substrate 26 does not get nitrated while 27 does (Figure S11). In the lactic acid substrate, the hydroxyl group can donate only one hydrogen bond instead of two or three from the amine (depending on the protonation state), so binding to Asn293 and Thr296 is necessarily reduced. Substrates 25 and 28, which also lack an amino acid amine are also not nitrated. However, substrate 27 can be nitrated as it has the requisite amine and carbonyl motifs to facilitate binding to TxtE in the correct configuration and induce the observed conformational change. We next tested indole-3-acetamide (substrate 29), a smaller compound that has an amide. It binds in a type I mode and is nitrated, and we suggest it represents the minimal tryptophan derivative that can be nitrated by wild-type TxtE (Figure S12). Substrate 29 has a primary amide that we speculate can bind in the pocket formed by residues Asn293 and Thr296. Additionally, the Arg59 residue is likely able to hydrogen bond with the substrate carbonyl as it did with the carboxylate, either directly or in a water-mediated fashion, closing the active site and positioning the substrate for catalysis. A second heteroatom in the form of a carboxylate (substrate 1) or an amide (substrate 27) is not required. Collectively, these examples demonstrate that hydrogen bond contacts to residues Asn293 and Thr296 from the amino acid amine gate catalysis, and one carbonyl interaction with residue Arg59 is required to position the substrate for catalysis.
Determination of Binding Constants for L-Tryptophan and Derivatives
Lastly, we sought to determine to what extent differences in binding affinity were responsible for the observed pattern of substrate reactivity. Dissociation constants (Kd) were determined for purified enzyme with L-tryptophan (substrate 1), indole-3-propionic acid (substrate 21), indole-3-lactic acid (substrate 26), tryptophan amide (substrate 27), and indole-3-acetamide (substrate 29) by examination of the differential UV-visible spectra (Table 6, Figure S16). We measured for the recombinant TxtE a Kd for L-tryptophan of 39 μM, slightly lower than the previously reported Kd values of 44 to 60 μM.[12,13] If the L-tryptophan amine is removed (substrate 21) or replaced with a hydroxyl group (substrate 26), binding affinity is maintained or even increased (Kd ~ 11 μM and 45 μM, respectively), but no nitration is observed. This suggests that while the amine moiety is required for catalysis as described above, it does not provide a significant improvement in binding affinity.
Table 6.
Dissociation constants of selected substrates with TxtE.
| Substrate | Structure | Nitrationa | Kdb |
|---|---|---|---|
| 1 |

|
++ | 39 ± 2 μM |
| 21 |

|
ND | 11 ± 4 μM |
| 26 |

|
ND | 45 ± 18 μM |
| 27 |

|
++ | 0.063 ± 0.025 μM |
| 29 |

|
++ | 1800 ± 544 μM |
Abbreviations: not detected (ND).
Differential UV-visible spectra and Kd plots can be found in Figure S16.
As previously discussed, removal of the carboxylate moiety altogether from the amino acid motif (substrates 20 and 23) results in non-productive type II binding. However, the carboxylate of L-tryptophan can be replaced by a terminal amide to retain productive chemistry, with substrate 27 binding to TxtE with high affinity of < 1 μM. Substrate 29 is a combination of these two observations. The nitrogen atom that we hypothesize is required for productive chemistry is present, and catalysis occurs, however, the carbonyl interaction with Arg59 is reduced resulting in much lower affinity (Kd ~ 1,800 μM). Taken together, a type I binder can have variable affinity as determined by interaction with Arg59, but can still be nitrated as long as it has an indole ring system with a labile hydrogen and a nitrogen atom that can hydrogen bond to Asn293 and Thr296.
Concluding Remarks
In this report, we have described the first substrate-free and L-tryptophan-bound structures of the cytochrome P450 enzyme TxtE. The tryptophan ring system sits in a hydrophobic pocket of the protein, with the indole nitrogen facing toward the distal face of the heme. Residues Asn293 and Thr296 form hydrogen bonds to the amine of the substrate amino acid, and the αB′1 helix reorganizes upon formation of a hydrogen bond between residue Arg59 and the tryptophan carboxylate, anchoring the substrate in a position for catalysis. Through a spectroscopic and functional substrate analog survey, we have determined that wild-type TxtE can nitrate non-natural substrates that are structurally similar to L-tryptophan with decorated indole side chains and modified amino acid moieties. We conclude that the substrate must meet the following criteria in order to be nitrated: it must bind in a type I mode, and it must also possess an indole ring system with a labile hydrogen atom and a properly-positioned hydrogen bond donor (nitrogen atom) and hydrogen bond acceptor (carbonyl). We find that substrates that bind weakly but meet these criteria can still be nitrated, while substrates that bind with high affinity but do not meet these criteria cannot be nitrated. An improved understanding of the molecular basis for TxtE’s substrate selectivity provides key information to guide future mechanistic investigations and engineering of TxtE for the nitration of diverse substrates.
Experimental Section
General
Substrates 7, 8, and 10 were purchased from MP Biomedicals. Substrate 16 was purchased from AK Scientific. Substrates 13, 15, 24, and 27 were purchased from Chem-Impex International. All other reagents and chemicals were purchased from Sigma-Aldrich and were used as received unless otherwise stated. If the substrate was purchased in the clean L-enantiomeric form, it is noted in the chemical structure (Tables 2–5). For tryptophan both the L-enantiomer and the racemic mixture were tested. Low-resolution mass spectral analyses were carried out using an Agilent 1290 UHPLC system with a 6140 quadrupole detector and an Eclipse Plus 2.1 × 50 mm C18 column (1.8 micron particle size).
TxtE Cloning, Expression, and Purification
The gene encoding Streptomyces scabies 87.22 TxtE was codon optimized for Escherichia coli (E. coli) using Gene Designer 2.0 (DNA 2.0), and then obtained as gBlocks from Integrated DNA Technologies. The gBlocks were amplified using Phusion High Fidelity DNA polymerase (Thermo Scientific) according to manufacturer’s instructions and Gibson cloned into pET28b+.[18] The His6-tagged construct was used to transform XL1-Blue chemically competent E. coli (Stratagene). After DNA sequencing (Laragen, Inc.), the correct construct was used to transform BL21 (DE3) E. coli for protein expression.
For expression, Hyper Broth™ (700 mL, 50 μg/mL kanamycin sulfate, 1-L flask) supplemented with glucose nutrient mixture (35 mL) (Athena Enzyme Systems) was inoculated with an overnight culture (35 mL, Terrific Broth, 50 mL in a 125-mL flask, 50 μg/mL kanamycin sulfate). After 3.5 to 4 hours of incubation at 37 °C and 225 rpm, the culture was cooled to 22–25 °C over approximately one hour. Then expression was induced with the addition of isopropyl beta-D-thiogalactopyranoside (IPTG, 0.5 mM final concentration, Research Products International Corp.) and 5-aminolevulinic acid hydrochloride (0.25 mM final concentration, Acros Organics) was added for the biosynthesis of heme. After 20–24 hours, the cells were harvested by centrifugation at 4 °C, 4,000 g for 10 min and the pellets were stored at −20 °C.
During protein purification, all samples were kept at 4 °C. The cell pellet was resuspended in Ni-NTA buffer A (25 mM Tris-HCl, 100 mM NaCl, 30 mM imidazole, pH 7, 4 mL/g of cell wet weight) and lysed by sonication (Sonicator 3000, Misonix, Inc.). The lysate was clarified by centrifugation at 15,000 g for 30 min, and the supernatant was loaded on a pre-equilibrated Ni-NTA column (GE Healthcare) using an AKTA purifier FPLC system (GE Healthcare). After washing with two column volumes (cv) of Ni-NTA buffer A, TxtE was eluted with a linear gradient (10-cv gradient) of buffer B (25 mM Tris-HCl, 100 mM NaCl, 300 mM imidazole, pH 7). The fractions that showed an absorbance at 280 nm and 417 nm were combined and buffer exchanged with 20 mM Tris-HCl pH 7.5 on a 10- or 30-kDA MWCO filter (EMD Millipore). The protein was aliquoted into PCR tubes, flash frozen over powdered dry ice, and stored at −80 °C until further use.
For qualitative binding assays and screening nitration reactions, E cloni® EXPRESS BL21 (DE3) Competent Cells (Lucigen) were transformed with pET28b+ and the pET28b+_TxtE construct and the proteins were expressed as described above. Cells were aliquoted into portions (40 mL) and harvested by centrifugation at 4 °C, 4,000 g for 10 min 20 to 24 hours after induction and stored at −20 °C until further use.
Protein concentration
The concentration of TxtE was determined from ferrous carbon monoxide binding differential spectra using the reported extinction coefficient forcysteine-ligated P450 (ε = 91,000 M−1cm−1).[19] For crystallography, total protein concentration was determined with the Quick Start™ Bradford Protein Assay (Biorad).
Crystallization and Data Collection
High-throughput screening of crystallization conditions for TxtE was conducted at the Beckman Molecular Observatory at the California Institute of Technology, and the optimal conditions were further refined using focused screens. Crystals for diffraction were grown in a sitting-drop format in 24-well plate (Hampton Research) by mixing 1 μL of 15 mg/mL protein in 20 mM Tris buffer (pH 7.5) with 1 μL of the well solution containing 0.1 M sodium citrate buffer (pH 5.6), 1.25 M Li2SO4, and 0.5 M (NH4)2SO4. Moderately sized red-brown orthorhombic crystals appeared within a few days. To obtain substrate-bound structures, single crystals were transferred into 5-μL drops of mother liquor containing 5–10 mM of L-tryptophan and allowed to equilibrate for 1.5 – 4 hours at room temperature. All crystals were cryo-protected in mother liquor containing 25% (v/v) glycerol prior to flash-freezing in liquid nitrogen. Diffraction data were collected using a Dectris Pilatus 6M detector on beamline 12-2 at the Stanford Synchrotron Radiation Laboratory at 100 K. Diffraction datasets were indexed and integrated with XDS[20] and scaled using SCALA.[21]
Structure Solution
An initial substrate-free TxtE structure (not published) was solved to 1.62 Å by multiwavelength anomalous dispersion (MAD) using the anomalous signal from the heme iron K-edge. Three data sets were collected for this crystal, corresponding to the peak, the inflection point, and a high-energy remote. The heavy-atom location and phasing were performed with the SHELXC/D/E package.[22] After Parrot[23] density modification, Buccaneer[24] was used for initial chain tracing and model building, followed by automated refinement with Refmac5 (CCP4 suite) and manual refinement using Coot.[25] This model was then used as a starting model to solve a 1.18 Å substrate-free data set more thoroughly, using automated refinement with Refmac5 as well as with PHENIX refine[26] and manual refinement using Coot.[21] The higher resolution model was used as a starting model for the refinement of the L-tryptophan-bound structure. To minimize phase bias, the same R-Free set was used for all structures solved. Anisotropic B-factors were calculated for both high-resolution structures.
Qualitative Binding Assays
Cell pellets expressing the pET28b+ vector or the TxtE construct were resuspended in 25 mM Tris buffer (pH 8, 10 mL) and harvested by centrifugation at 4 °C, 4,000 g for 15 min. The supernatant was decanted and the pellet was resuspended in 25 mM Tris buffer (pH 8.0, 5 mL) and lysed by sonication (Sonicator 3000, Misonix, Inc.). The lysate was clarified by centrifugation at 4 °C, 20,817 g for 25 min, and the supernatant was transferred to pre-chilled Eppendorf tubes. The concentration of TxtE was determined as described above. TxtE protein (100 μL, 1.9 – 2.8 μM) was plated in a half area 96-well plate (Greiner Bio One). All substrates were prepared in dimethyl sulfoxide (DMSO) or 0.5 M sodium hydroxide at 25–50 mM and added to a final concentration of 250–500 μM (1 – 2 μL). The substrate was allowed to bind to the protein for 15 min and then spectra were recorded from 320 to 600 nm on a plate reader (Infinite M200, Tecan). For differential UV-visible spectra, the TxtE control spectrum was subtracted from the spectrum of TxtE plus substrate.
TxtE-Catalyzed Nitration Reactions
Clarified cell lysate expressing TxtE (70 μL, 1.9 – 2.8 μM) was plated in triplicate and cell lysate from pET28b+ expressing cells (70 μL) was plated in single replicate into a 96-well plate (Evergreen Scientific). For substrates 4, 26, and 27, purified TxtE (2 μM) in 25 mM Tris buffer, (pH 8) was used. All substrates were prepared in DMSO or 0.5 M sodium hydroxide at 250 mM and used at a final concentration of 5.4 mM (2 μL). The substrate was incubated with the protein for 15 min. Lyophilized spinach ferredoxin NADP+ reductase (Sigma) was reconstituted in 1 M Trizma (Sigma) at 17 U/mL. Spinach ferredoxin (Sigma) was used as received at concentrations of 1–3 mg/mL. A solution of spinach ferredoxin NADP+ reductase (1 μL/well, ~ 0.17U/mL final concentration) and spinach ferredoxin (1 μL/well, ~ 0.01 – 0.03 mg/mL final concentration) was prepared in 25 mM Tris buffer (pH 8, 8 μL) and added to each well. DiethylamineNONOate sodium salt hydrate (DEANO, Sigma) was prepared at 100 mM in 10 mM sodium hydroxide and stored at −80 °C and was further diluted to 10 mM in 25 mM Tris buffer (pH 8). Nicotinamide adenine dinucleotide phosphate (NADPH, Codexis, Inc.) was prepared fresh at 20 mM in 25 mM Tris buffer (pH 8). A solution of DEANO (10 mM, 515 μM final concentration) and NADPH (20 mM, 1 mM final concentration) were mixed in equal volumes and 10 μL were added to each well. The plate was covered, wrapped in foil, and the reactions proceeded with shaking. After 90 min, NADPH (5 μL/well, 1 mM final concentration) was added to each well and the reactions were allowed to continue overnight during which time some solutions became yellow. Each reaction was applied to a 0.5-mL 3-kDA MWCO filter (Millipore) and centrifuged at 20,817 g for 45 min. The filtrate (35 μL) was diluted with acetonitrile (70 μL) and transferred to a vial insert (Agilent Technologies) and analyzed by liquid chromatography mass spectrometry (LC-MS). For substrate 29, three reactions were combined in the workup for LC-MS analysis, because upon dilution with acetonitrile the yellow color was diluted and the product did ionize well. The extent of reactivity is denoted in Tables 2–6 as follows: (+) for the products that required an extract ion chromatogram of the mass chromatogram and (++) for products where the mass was readily detected in the mass chromatogram.
Determination of Dissociation Constants
A solution of purified TxtE (100 μL, 5 μM) in 25 mM Tris buffer (pH 8) was plated in a half area 96 well plate (Greiner Bio One) and each substrate was titrated at varying concentrations. Substrate 1 prepared in 25 mM Tris (pH 8) was added at the following final concentrations: 0, 12.5, 25, 50, 100, 200, 400, 600, 800, 1,000, and 1,200 μM. The volume change (6 μL) was the same for each well. Substrate 21 prepared in DMSO was added at the following concentrations: 0, 10, 50 200, 500, and 750 μM. The volume change did not exceed 1 μL of DMSO. Substrate 26 prepared in DMSO was added at the following final concentrations: 0, 10, 50, 100, 200, and 400 μM. the volume change did not exceed 1 μL of DMSO. Substrate 27 prepared in DMSO was added at the following final concentrations: 0, 0.1, 1, 10, 100, 250, and 500 μM. The volume change did not exceed 1 μL of DMSO except for 500 μM where 2 μL of DMSO was added. Substrate 29 prepared in DMSO was added at the following final concentrations: 0, 10 100, 500, 1,500, 2,500, and 5,000 μM. The volume change did not exceed 1 μL of DMSO. After at least 15 min of shaking, spectra were recorded from 325 to 525 nm with 5 or 10-nm step size on a plate reader (Infinite M200, Tecan) in triplicate. For each substrate concentration, differential UV-visible spectra were determined by subtracting the TxtE control spectrum from the Txte+substrate. The difference in the absorbance of each spectrum at the λmax and λmin was calculated in triplicate and the average with standard deviation was plotted versus the substrate concentration. Each data set was fitted to a binding isotherm model using KaleidaGraph.
Supplementary Material
Acknowledgments
We thank Dr. Jens Kaiser and Dr. PavleNikolovski of the Beckman Molecular Observatory (Caltech) for assistance with crystallography and Dr. Scott Virgil and the 3CS Center for Catalysis and Chemical Synthesis (Caltech) for assistance with LC-MS analyses. The authors thank the Resnick Sustainability Institute (Caltech) and Dow Chemical Company for support through the Dow-Resnick innovation program. S.C.D. and J.A.M. are supported by Ruth L. Kirschstein NRSA postdoctoral fellowships from the National Institutes of Health (5F32GM106618 and 5F32GM101792, respectively). J.K.B.C. acknowledges the support of the Resnick Sustainability Institute (Caltech). T.H. is supported by a postdoctoral fellowship from the Swiss National Science Foundation (Fellowship PBBSP2_146809). The Beckman Molecular Observatory is supported by the Gordon and Betty Moore Foundation, the Beckman Institute, and the Sanofi-Aventis Bioengineering Research Program (Caltech). The authors thank Dr. Fei Sun for sharing of the XL1-Blue E. coli strain and helpful discussions.
Footnotes
The content of this paper is solely the responsibility of the authors and does not represent the official views of any of the funding agencies.
References
- 1.Ran N, Zhao L, Chen Z, Tao J. Green Chem. 2008;10:361–372. [Google Scholar]
- 2.Bornscheuer UT, Huisman GW, Kazlauskas RJ, Lutz S, Moore JC, Robins K. Nature. 2012;485:185–194. doi: 10.1038/nature11117. [DOI] [PubMed] [Google Scholar]
- 3.Reetz MT. J Am Chem Soc. 2013;135:12480–12496. doi: 10.1021/ja405051f. [DOI] [PubMed] [Google Scholar]
- 4.Booth G. Nitro compounds, aromatic in Ullmann’s Encyclopedia of Industrial Chemistry. John Wiley & Sons; New York: 2000. [Google Scholar]
- 5.Ono N. The Nitro Group in Organic Synthesis. Wiley-VCH Publishers; New York: 2001. [Google Scholar]
- 6.Yan G, Yang M. Org Biomol Chem. 2013;11:2554–2556. doi: 10.1039/c3ob27354g. [DOI] [PubMed] [Google Scholar]
- 7.Buddha MR, Tao T, Parry RJ, Carne BR. J Biol Chem. 2004;279:49567–49570. doi: 10.1074/jbc.C400418200. [DOI] [PubMed] [Google Scholar]
- 8.Winkler R, Hertweck C. Chem Bio Chem. 2007;8:973–977. doi: 10.1002/cbic.200700042. [DOI] [PubMed] [Google Scholar]
- 9.Roncone R, Barbieri M, Monzani E, Casella L. Coord Chem Rev. 2010;250:1286–1293. [Google Scholar]
- 10.Ju K-S, Parales RE. Microbiol Mol Biol Rev. 2010;74:250–272. doi: 10.1128/MMBR.00006-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Parry R, Nishino S, Spain J. Nat Prod Rep. 2011;28:152–167. doi: 10.1039/c0np00024h. [DOI] [PubMed] [Google Scholar]
- 12.Barry SM, Kers JA, Johnson EG, Song L, Aston PR, Patel B, Krasnoff SB, Crane BR, Gibson DM, Loria R, Challis GL. Nat Chem Biol. 2012;8:814–816. doi: 10.1038/nchembio.1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yu F, Li M, Xu C, Wang Z, Zhou H, Yang M, Chen Y, Tang L, He J. PLoS One. 2013;8:e81526. doi: 10.1371/journal.pone.0081526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Poulos TL. Proc Natl Acad Sci USA. 2003;100:13121–13122. doi: 10.1073/pnas.2336095100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pochapsky TC, Kazanis S, Dang M. Antioxid Redox Signal. 2010;13:1273–1296. doi: 10.1089/ars.2010.3109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barry SM, Challis GL. Methods Enzymol. 2012;516:171–194. doi: 10.1016/B978-0-12-394291-3.00001-0. [DOI] [PubMed] [Google Scholar]
- 17.Peng CC, Pearson JT, Rock DA, Joswig-Jones CA, Jones JP. Arch Biochem Biophys. 2010;497:68–81. doi: 10.1016/j.abb.2010.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gibson DG. Methods Enzymol. 2011;498:349–361. doi: 10.1016/B978-0-12-385120-8.00015-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Omura T, Sato R. J Biol Chem. 1964;91:542–553. [Google Scholar]
- 20.Kabsch W. Acta Crystallogr D Biol Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Evans P. Acta Crystallogr D Biol Crystallogr. 2006;62:72–82. doi: 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- 22.Sheldrick GM. Acta Crystallogr D Biol Crystallogr. 2010;D66:479–485. doi: 10.1107/S0907444909038360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang KYJ, Cowtan K, Main P. Method Enzymol. 1997;277:53–64. doi: 10.1016/s0076-6879(97)77006-x. [DOI] [PubMed] [Google Scholar]
- 24.Cowtan K. Acta Crystallogr D Biol Crystallogr. 2006;D62:1002–1011. doi: 10.1107/S0907444906022116. [DOI] [PubMed] [Google Scholar]
- 25.Emsley P, Cowtan K. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 26.Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.