

Abstract
Dendritic cells (DCs) have several characteristics that make them an ideal vehicle for tumor vaccines, and with the first US FDA-approved DC-based vaccine in use for the treatment of prostate cancer, this technology has become a promising new therapeutic option. However, DC-based vaccines face several barriers that have limited their effectiveness in clinical trials. A major barrier includes the activation state of the DC. Both DC lineage and maturation signals must be selected to optimize the antitumor response and overcome immunosuppressive effects of the tumor microenvironment. Another barrier to successful vaccination is the selection of target antigens that will activate both CD8+ and CD4+ T cells in a potent, immune-specific manner. Finally, tumor progression and immune dysfunction limit vaccine efficacy in advanced stages, which may make DC-based vaccines more efficacious in treating early-stage disease. This review underscores the scientific basis and advances in the development of DC-based vaccines, focuses on current barriers to success and highlights new research opportunities to address these obstacles.
Keywords: cancer vaccines, dendritic cells, immunotherapy
Dendritic cell immunobiology
Why dendritic cells for tumor vaccines?
Dendritic cells (DCs) have unique characteristics that have made them an ideal choice for antitumor vaccines. They are considered the most effective antigen-presenting cell (APC) [1] responsible for primarily sensitizing naive T cells to specific antigens. DCs are ten- to 100-times more potent than their fellow APCs, B cells and monocytes, in inducing T-cell proliferation [2,3]. Additionally, DCs play an important role in the establishment of immunologic memory [4,5]. In contrast with monocytes and B cells, DCs are able to use soluble protein antigens to sensitize naive T cells in vitro [2,6,7]. Using these soluble proteins, DCs have successfully sensitized both CD4+ [7,8] and CD8+ T cells inducing antigen-specific cytotoxic T lymphocytes (CTLs) [6]. This capability gives developers of DC-based vaccines a wider range of potential antigen targets that can be effectively used to sensitize T cells. With respect to their use against cancer, the ability of DCs to prime T cells to attack tumor cells has been demonstrated in vitro [5,9] as well as in various animal models [10–12].
Another benefit of DCs is their role as the principal APC with the ability to cross-prime, meaning that in addition to their ability to present via the classical pathways of presenting exogenous antigens on MHC class II molecules and endogenous antigens on MHC class I molecules [13], they can also route exogenous antigens into the pathway for presentation on MHC class I molecules, which is necessary for the generation of CTLs [14–17]. The capability for presenting exogenous tumor antigens on MHC class I molecules has been demonstrated in vitro and in mouse models [18,19], and has been shown to occur with both infectious and tumor antigens [20,21]. While macrophages and B cells have exhibited some ability to cross-prime, they do so to a much lesser extent than DCs. Receptors involved in various steps in antigen internalization and processing have been associated with the ability of particular DCs to cross-prime in contrast with macrophages and B cells [22]. For example, DCs express fewer lysosomal proteases than macrophages [23] and also inhibit lysosomal acidification via NOX-2-mediated alkalization of phagosomes and endosomes [24,25], thereby preventing activation of lysosomal proteases and maintaining extracellular antigens available in the intracellular compartment for cross- presentation. This enhanced antigen stability in DCs increases their ability for cross-presentation [26]. In addition to higher antigen stability, cross-priming depends on endocytic receptors that target internalized antigen to storage compartments. For example, DEC-205 is an endocytic receptor associated with effective and prolonged cross-presentation in CD8+ murine DCs [27–29]. Mannose receptor-mediated endocytosis has also been associated with targeting internalized antigens for endosomes that can be processed for cross-priming [22,30] as have other endocytic receptors, including dectin-1 [31], and neonatal Fc receptor [32] among others [33–36]. More recently, CD40 on monocyte-matured DCs was shown to be highly efficient at cross-presentation when compared with mannose receptor and DEC-205 despite less efficient antigen internalization [33]. Another requirement for successful cross-presentation is functional transporter associated with antigen processing (TAP) complexes, in particular endosomal TAP complexes [37], to mediate peptide transport from the cytosol into phagosomes for loading onto MHC class I molecules [38–40]. While more than one pathway can mediate cross-presentation [41], having operational pathways for cross-presentation make DCs that have this capability important for activating CTLs using exogenous antigen in vaccine constructs. Such DCs have been shown to be capable of priming CTLs even in the absence of sensitized CD4+ cells [42,43].
The capabilities of the DC as an immune effector cell, in particular its role as a potent and versatile APC, make it well suited to be the vehicle of an antitumor vaccine. However, it is important to recognize that DCs not only prime naive T cells for antigen recognition but also regulate the nature of the subsequent immune response. DCs can induce a diversity of T-cell behaviors varying from potent antitumor or antimicrobial activity to regulation of immune tolerance, which can be a serious factor limiting vaccine success [44]. Some of this variability in behavior relates to their heterogeneous lineage and understanding this lineage helps us to choose appropriate DCs for use in vaccination.
DC lineages & the choice of cell lineage for vaccine construct
DCs develop from a variety of precursors. A choice of DC subset to use for vaccination should weigh both logistic concerns in terms of obtaining adequate numbers of cells for vaccination as well as biologic factors as to how these DCs differentiate, mature and function as part of the immune response. The ontogeny of DCs is fairly complicated and a complete mapping and understanding of DC development is yet to be fully realized. Labeling schemes for DC subsets vary in the literature and are evolving. This inconsistency further complicates understanding DC development. We will outline the current knowledge of DC subsets and how their characteristics impact choice of DC lineage for vaccine production.
Mouse models have examined different DC lineages to elucidate their tendencies to induce a certain immune response. Mouse cells have been separated into CD8+ and CD8− subsets, although this CD8 positivity has not been found in human DCs. CD8+ murine DCs have been shown to induce a T helper (Th)1 response while CD8− DCs have a tendency to promote a Th2 response, suggesting that DC lineages have certain biases [45,46]. Variability among these murine lineages has also been found in their ability to cross-prime. In particular, DCs of the CD8+ lineage are capable of cross-presentation [47, 48]. This functional divide in mice suggests the need to be aware of similar differences in the capabilities of human DCs of different lineages. For example, evidence suggests that not all human DC subsets are able to undertake cross-presentation [27], which is an important part of mediating CD8+ immunity to exogenous tumor antigen. Various tissue DCs have demonstrated the ability to cross-present [49,50] as have DCs derived from monocytes [15]. As outlined above, further study is being conducted into the receptors on DCs that are important for cross-presentation along with ways of manipulating these receptors to enhance cross-presentation [51]. In addition to monitoring cell subsets for receptor expression, antigens can be targeted towards receptors that are present to increase cross-presentation efficiency [28]. Investigation into which human DC subsets are able to cross-prime will help identify candidate lineages for creating effective vaccines while a more in-depth understanding of the receptors and cellular machinery involved in cross-presentation may not only help identify cells with the intrinsic capacity to cross-prime, but provide targets for engineering DCs to more effectively engage in cross-presentation.
Historically, attempts have been made to classify various DC subsets both in terms of their differentiation pathways as well as based on their tendency to elicit a particular immune response, with type 1 DCs (DC1s) and type 2 DCs (DC2s) or pre-DC1s and pre-DC2s designated based on their tendency to polarize T cells towards a Th1 or Th2 response, respectively [52,53]. However, the plasticity of DCs and their ability to elicit different outcomes despite these tendencies have led to ongoing evolution of classification systems used in the literature. We will attempt to elucidate and clarify classification schemes as we discuss various DC subsets and their applications in cancer vaccines. Currently, the distinction between conventional DCs (cDCs) that arise directly from DC progenitor cells and nonconventional DCs (non-cDCs) is one major separation in DC ontogeny noted in the literature [54]. Previously, attempts were made to distinguish DCs of myeloid origin from those of lymphoid origin. Myeloid DCs were originally thought of as cDCs and considered to have a tendency to elicit Th1-type immune responses. However, subsequently, it was discovered that DCs, whether of myeloid or lymphoid origin, can give rise to any DC subset with Flt3L identified as an essential component of DC development [55–58]. In another prior classification attempt, cDCs were distinguished from plasmacytoid DCs (pDCs), although more recently pDCs and monocyte-derived DCs have both been grouped together as non-cDCs, given that in steady state in vivo, they do not give rise to the DCs that reside in various lymphoid and non-lymphoid tissues [59] but rather differentiate into DCs under certain environmental conditions.
Whereas monocyte-derived DCs and pDCs are both migratory in nature, DCs that fall within the category of cDCs can be both migratory or reside in lymphoid tissues [60]. Migratory DCs have been of greater interest for potential use in vaccines given that they are characterized by their ability to take up antigen in the periphery and subsequently travel to draining lymph nodes to interact with T cells [60]. However, given the limited exploration into their use, examination of tissue DCs may reveal additional useful DC subpopulations for incorporation into vaccine strategies.
Separate from cDCs, stem cells also produce cells capable of following a different development path but that, under certain conditions, can differentiate into DCs. Some literature has termed these cells immediate DC precursors, or pre-DCs, in contrast with cell lines that differentiate earlier from hematopoietic progenitors into immature DCs, although this terminology is somewhat inconsistent across the literature with some using the term pre-DC to refer to committed DC progenitors [59]. Immediate DC precursors exist in lymphoid and solid organs and replenish or give rise to resident DCs. These resident DC precursors, or ‘resident pre-DCs’ [54], have not been commonly incorporated into vaccine constructs. Some literature has also designated circulating monocytes and pDCs as pre-DCs [52,53], although they have more recently also been classified as non-cDCs [54]. Initially, monocyte-derived DCs and pDCs were deemed pre-DC1 and pre-DC2 for suggested tendencies to mature into DCs that polarized T cells to a Th1 and Th2 response, respectively. The subsequent realization that there exists plasticity in their development and immunologic outcomes has abrogated use of this terminology, and the terms DC1 and DC2 are used more to describe mature phenotype and functional outcome rather than being closely associated with particular DC lineages. Found in the blood, and thus migratory in nature, the non-cDCs – pDCs and monocytes – are of potential interest for use in vaccines.
Human pDCs express CD4, the IL-3 receptor, are CD11c−, and have been most distinctly characterized by their production of type 1 interferon in response to viral infection [61]. As noted above, pDCs have at times been designated as pre-DC2s in the literature [62], in contrast with monocytes that have been designated as pre-DC1s. This pre-DC2 designation resulted because pDCs that were shown to develop in the presence of IL-3 and CD40L [62,63] did not produce high levels of IL-12, an important cytokine for causing Th1 polarization of the immune system. Rather, pDCs have demonstrated a tendency to polarize T cells towards a Th2 profile [64] or even induce suppressor T cells [64,65] via induction of Tregs [66]. Some clinical studies have found tumor-associated pDC infiltration in the primary tumor and in draining lymph nodes to be associated with poorer outcomes in various cancers including ovarian cancer, breast cancer, prostate cancer and melanoma, and these pDCs were shown to be less effective at immune activation, probably due to dysfunction caused by the tumor microenvironment [52,66–68]. Zou et al. showed tumor recruitment of pDCs to the tumor microenvironment in ovarian cancer that in turn induced poorly functioning T cells when compared with their counterpart pDCs found in blood [52]. While TGFβ and PD-1/PD-L1 have been implicated in mediating tolerogenic properties of tumor-associated pDCs, FOXO3 has also recently been implicated as a key component in inducing immunosuppressive activities in tumor-associated DCs, primarily pDCs. These mediators of the tolerogenic behaviors of pDCs provide targets to block that could potentially enhance the antitumor function of pDCs [68,69]. Furthermore, opportunities exist to take advantage of the functional plasticity of pDCs. Despite a historical association to produce a Th2 response, different signals can alter the pathway of their development. For example, viral infection can induce their production of type 1 interferons, indicating the capability of pDCs to play a role in the inflammatory immune response, and pDCs have been shown to differentiate to more closely resemble inflammatory cDCs in the setting of viral infection [70,71]. While the murine pDC counterparts of human pDCs have been shown to possess a more immature phenotype than mouse cDCs, particular signals can elicit a mature phenotype from these pDCs with subsequent successful priming of T cells. Brawand et al. compared murine cDCs and pDCs, revealing different patterns of Toll-like receptor (TLR) expression with cDCs expressing high amounts of TLR2, TLR3, and TLR4 while pDCs had higher expression of TLR7 and TLR9. Stimulation with CD40L and CPG, a TLR9 agonist, led to a more mature phenotype of the pDCs with increased expression of costimulatory molecules and secretion of IL-12, albeit at lower levels than their cDC counterparts. Furthermore, when activated with CPG and CD40L, the pDCs were able to sensitize naive T cells to specific antigens at levels comparable to cDCs [63]. These findings suggest that while pDCs may not have as strong a tendency as cDCs towards mediating a specific inflammatory response, particular signals administered to pDCs may make them just as viable a DC subset for use in a vaccine [72]. The ability of pDCs to induce Th1- or Th2-type immune responses despite a tendency towards Th2 polarization suggests that environmental signals have a greater impact on the mature DC phenotype. Taking advantage of the plasticity of pDCs may provide a population of malleable DCs that can be manipulated to optimize their use in cancer vaccines.
Another set of non-cDCs, those derived from circulating moncoytes, has been well-studied for use in DC-based vaccines. Several biologic factors favor the use of monoctye-derived DCs. They are present in circulation and are able to take up antigen and travel to draining lymph nodes for antigen presentation. They are easily converted into immature DCs (iDCs) with GM-CSF and IL-4 [73]. Monocyte-derived CD11c+ iDCs demonstrate a high phagocytic ability in contrast with CD11c– pDCs [73] thereby optimizing their ability to take up antigen for presentation. Furthermore, this subset has a tendency to be easily matured into a DC1 phenotype, earning them the designation of pre-DC1s in some of the literature describing DC ontogeny [64].
In addition to their favorable biology, widespread use of monocyte-derived DCs is largely logistical given the easy accessibility to peripheral blood monocytes in humans when compared with tissue sources of DCs. Leukapheresis is an efficient way to obtain sufficient numbers of peripheral blood monocytes for the generation of a vaccine and for additional in vitro study [74]. Logistic limitations change when using a mouse model where DCs from bone marrow, spleen or lymph nodes may in fact be more easily accessible.
While monocyte-derived DCs are used for vaccine development, utilization of other DC lineages is possible given evidence that environmental signals more strongly influence mature phenotype. Use of particular DC lineages for vaccine production must be shaped by knowledge of their receptor expression and the signals required to induce a desired phenotype [75]. While in vitro and mouse studies suggest efficacy of T-cell sensitization for a variety of DC subtypes including monocyte-derived DCs, pDCs, Langerhans cells [76,77] and other interstitial DCs [78], there is a paucity of clinical trials testing the efficacy of these varying subtypes (see Table 1) and few comparative studies [79]. In particular, in vitro studies of Langerhans cells have been very promising in demonstrating sensitization [77] as have pDCs [80], even when compared with monocyte-derived DCs [81]. However, human trials using pDCs have yet to be conducted. Comparative clinical study of DC lineages is an area where further study could reveal more effective combinations of functional DCs for incorporation into a DC-based vaccine.
Table 1.
Recent clinical trials using different sources of dendritic cells and varying maturation strategies.
| Maturation regimen | DC phenotype | Description: cancer type, stage, sample size and antigen used | Important findings | Ref. |
|---|---|---|---|---|
| Immature DCs | ||||
| GM-CSF and IL-4 | Immature |
|
|
[87] |
| GM-CSF and IL-4 | Immature |
|
|
[271] |
| Mature versus immature DCs | ||||
| GM-CSF and IL-4 IL-1β, TNF-α, IL-6, PGE2 | Immature Mature |
|
|
[272] |
| pDCs | ||||
| No human clinical trials | ||||
| MoDCs | ||||
| IL-1β, TNF-α, IL-6, PGE2 | Mature |
|
|
[273] |
| IL-1β, TNF-α, IL-6, PGE2 | Mature |
|
|
[274] |
| IL-1β, TNF-α, IL-6, PGE2 | Mature |
|
|
[275] |
| TNF-α,IL-6, IL-1β, PGE2 | Mature |
|
|
[276] |
| TNF-α, IL-1β, PGE2 | Mature |
|
|
[277] |
| TNF-α, IL-6, IL-1β, PGE2 | Mature |
|
|
[278] |
| IFN-α, TNF-α, polyinosinic:polycytidylic acid | DC1 |
|
|
[279] |
| IL-1, TNF, IFN and poly-l:C | DC1 |
|
|
[280] |
| LPS and IFN-γ | DC1 |
|
|
[157,213] |
| MoDCs and LCs | ||||
| CD40L or inflammatory cytokines: IL-1 b, IL-6, TNF-α, andPGE2 | DC1 |
|
|
[79] |
| CD34+ hematopoietic-derived DCs (includes LCs and interstitial DCs) | ||||
| GM-CSF, FLT3-L, TNF | Not specified |
|
|
[281] |
| Interstitial DCs | ||||
| No human clinical trials | ||||
These trials demonstrate a bias towards use of monocyte-derived DCs, matured with inflammatory cytokines in an attempt to elicit a DC1-driven immune response.
CTL: Cytotoxic T lymphocyte; DC: Dendritic cell; DCIS: Ductal carcinoma in situ; GBM: Glioblastoma multiforme; GM-CSF: Granulocyte-macrophage colony stimulating factor; KLH: Keyhole limpet hemocyanine;
LC: Langerhans cell; LPS: Lipopolysaccharide; MoDC: Monocyte-derived dendritic cell; pDC: Plasmacytoid DC; Peg-IFN: Pegylated interferon.
DC maturation & immune tolerance
DCs in circulation and in peripheral tissues are largely found in an immature form. Upon receiving appropriate maturation signals, DCs upregulate chemokine receptors to facilitate migration to nearby lymph nodes [82], increase surface expression of MHC molecules to enhance antigen presentation and upregulate costimulatory models necessary for amplification of the T-cell response [83,84]. In addition to antigen uptake and T-cell interactions, DCs require additional danger signals to become fully activated. Based on the type of maturation signals the DCs receive, they mature into various phenotypes, and these phenotypes affect their interactions with T cells and the cytokines they will secrete.
In addition to playing a role in activating the immune system, DCs can also induce immune tolerance, which is a potential barrier to a successful vaccine strategy. Evidence has suggested that DCs that are not fully matured will be prone to inducing tolerance [85]. Studies that support this role have linked immature DCs to the promotion of regulatory T-cell development conferring peripheral self-tolerance [86,87]. Jonuleit et al. used peripheral monocytes to generate immature DCs by culturing them with GM-CSF and IL-4. A mature population was also developed from these peripheral monocytes by further activating them with inflammatory cytokines, IL-1, TNFα, IL-6 and PGE2. When these populations were used to stimulate naive CD4+ T cells, the mature population induced a proliferative response on restimulation and a cytokine profile characteristic of a Th1-polarized immune response. In contrast, the immature DCs induced a profile more consistent with a Treg population characterized by nonproliferation upon restimulation and IL-10 secretion. This phenotype was not completely reversed with subsequent stimulation by mature DCs nor with IL-2 [86]. While this immune suppression described by Jonuleit et al. was not antigen specific, Dhodapkar et al. subsequently demonstrated that injection of antigen-pulsed iDCs leads to antigen-specific immune suppression, even inhibiting pre-existing antigen-specific T-cell function and leading to antigen-specific IL-10 secretion by CD8+ T cells in humans in vivo [87]. These findings suggest that using DCs that are not fully matured will be ineffective in vaccination against tumor antigens and may even promote immune tolerance, indicating that vaccines should incorporate signals to achieve full maturation and activation of DCs prior to vaccine administration. In fact, taking advantage of immature DCs to try to promote anergy has been applied in efforts to subdue to immune system in the settings of transplantation and automimmunity [88]. Other researchers aiming for immunosuppression have even engineered DCs to lack expression of certain mature features, such as CD80/86, important costimulatory molecules expressed on mature DCs whose absence has been shown to lead to anergic T-cell development [89].
Alternatively activated DCs can also lead to immune tolerance, such as those used by Arce et al. whereby lentivirus vectors were used to selectively activate ERK in DCs that subsequently led to antigen-specific Treg differentiation [90] showing that inappropriately activated DCs can also induce T-cell tolerance. Perona-Wright et al. demonstrated that DC maturation induced with lipopolysaccharide (LPS) in the presence of IL-10 still led to expression of mature cell-surface markers such as CD80 and CD86 but that this expression was less stable and became downregulated more quickly than those DCs matured without IL-10 present. Similarly, IL-12 secretion was more short-lived and ultimately these cells induced tolerance [91]. These and other signals have been shown to alternatively activate DCs to a tolerogenic state [92]. Awareness of such signals must direct the choice of maturation signals used, as well as facilitate targeting of any such factors that may be present in the tumor microenvironment mediating tolerance. Furthermore, maturation alone is likely not sufficient to assure immune activation. Banjeree et al. demonstrated that vaccination of myeloma patients with cytokine-matured DCs led to expansion of a Treg population despite the use of mature DCs. Being an in vivo study, this suggests a role of the tumor environment in affecting the behavior of these DCs, but also indicates that the signals used to lead to a mature phenotype impact the mature DCs’ ability to be immune-activating or -suppressing [93]. Thus, for cancer vaccines, assuring full maturation as well as appropriate activation of DCs is an important part of overcoming the barrier of immune tolerance. The tolerogenic outcomes in these studies point out the need to confirm DC maturity and phenotype through cell-surface markers and cytokine secretion prior to vaccine administration.
DCs as immune suppressors
To better understand how to use DCs in a vaccine construct, the role of DCs as potential mediators of immune suppression should be explored further. Immune suppressor cells exist in both the lymphoid population, such as the well-characterized Treg population, as well as among myeloid cells. DCs, when in the immature or resting state, have the capability of mediating immune tolerance via induction of Tregs, which in turn secrete immunosuppressive cytokines such as IL-10, suppressing both T-cell proliferation and DC activation [94]. However, myeloid-derived suppressor cells (MDSCs) also play an important role in immune tolerance, and are of particular interest as a potential barrier to successful vaccination with DC-based vaccines as they may arise from the same cell populations used to construct the vaccines and may be recruited by signals used as adjuvants with these vaccines.
MDSCs are defined by their myeloid origin, immature state [95,96] and, of course, their biologic role in immune suppression. MDSCs have been shown to exert their effect on T cells via multiple factors including reactive oxygen species [97], inducible nitric oxide synthase, nitric oxide [98], TGFβ [99,100], IL-10 [101] and prostaglandin E2 [102]. In a similar characterization scheme to that of mice, MDSCs have been divided into two main subsets: human granulocytic MDSC and monocytic MDSC, although in reality these MDSCs have been found to be quite heterogenous [103,104]. These different human subsets utilize distinct mechanisms with granulocytic monocytic MDSCs utilizing reactive oxygen species and M-MDSCs secreting TGF-β [105]. The monocytic subset is CD14+, like the peripheral monocytes used to develop many of the DC-based vaccines in humans, and has been identified in association with various cancers such as melanoma [102] and prostate cancer [106]. One recruiter of MDSCs, GM-CSF, is of note in that it has been used as an adjuvant in vaccine therapy, and the suppressive effect of excess GM-CSF has been demonstrated in humans. Melanoma patients that were injected with autologous melanoma cell-derived heat-shock protein peptide complex gp96 in the presence of low-dose GM-CSF in an attempt to promote DC development and accumulation were found to display a decreased CD8+-mediated T-cell response and decreased antitumor effect when compared with those treated without the GM-CSF injection. Under randomized controlled conditions, these differences were associated with an increased population of MDSCs, specifically CD14+CD11b+ cells that were secreting TGFβ, and this difference was in turn linked to the administration of GM-CSF [102]. This study suggests the importance of maturing sufficient numbers of DCs and injecting them in an already activated state rather than using GM-CSF in vivo given its potential to recruit a variety of cells including MDSCs.
Tolerogenic DCs that maintain antigen- specific Treg populations have also been described. Their formation is promoted by TGFβ, IL-10, IL-27, vitamin D3 and IDO and, similar to MDSCs, they promote immune tolerance via secretion of TGFβ, IL-10 or IDO [107–109]. Tregs, which are characterized by expression of Foxp3 [110], have the ability to induce immune tolerance among other T cells [111] as well as promote tolerogenic DCs. The importance of TGFβ in the interplay between Tregs and tolerogenic DCs has been well-characterized [108,112].
Aside from assuring appropriate activation of DCs administered in a vaccine so as to avoid an immunosuppressive phenotype, the vaccine must overcome immune suppression being carried out by regulatory cells present in the microenvironment such as MDSCs, Tregs and tumor-associated immunosuppressive macrophage (M2) cells [113]. Tumors themselves condition the microenvironment to promote immune tolerance, a concept addressed in part upon discussing the role of the tumor microenvironment in inducing tolerogenic behavior in pDCs. Immunosuppressive cytokines such as GM-CSF, VEGF, IL-13, IL-6 and IL-10 can negatively impact the function of T cells and promote formation of MDSCs [98,114]. In addition to factors secreted by the tumor, the bidirectional communication between Tregs and tolerogenic DCs contributes to persistent immune tolerance.
Using properly matured and activated DCs is one way to prevent these DCs from having a tolerogenic effect and overcome the suppressive tumor microenvironment. In fact, DCs have been shown to reverse peripheral T-cell tolerance against a variety of antigens including tumor antigens [115–117]. The importance of maturing DCs to a particular phenotype is highlighted by studies showing that the suppressive phenotype of MDSCs is enhanced by Th2 cytokines whereas Th1 cytokines can overcome this inhibition and increase antigen-specific T-cell cytotoxicity [98,118]. These findings suggest that a Th1 immune response will have the capability for overcoming immune suppression induced by the tumor. In addition to engineering DC-based vaccines to overcome these barriers, pharmacologic methods for combating MDSCs are under investigation by inhibiting compounds such as inducible nitric oxide synthase or arginase that mediate their suppressive function [119 –121]. Coupling such pharmacologic therapies to target a variety of regulatory cells found in the tumor microenvironment with immunotherapy in the form of a properly activated DC vaccine could increase vaccine success.
Maturation signals
As previously noted, DCs require signals in addition to the antigen, in order to achieve a fully mature and activated state. These can be inflammatory signals from the local microenvironment, pathogen-related molecules and signals from T cells [73]. Inflammatory signals include TNF, IL-1 and prostaglandins, used to generate ‘classical cytokine-generated DCs’ that have demonstrated successful in vitro sensitization [86,122] and been applied successfully in some human trials (see Table 1). Still, this method has its limitations. Generating these DCs requires at least 1 week of culture, which increases the risk of bacterial contamination in addition to being less physiologic [1], and some in vivo studies have demonstrated cytokine-matured DCs leading to expansion of the immunosuppressive Treg population [93]. There are now effective protocols for rapidly maturing DCs within 24–48 h [123] using both serum-containing [124] and serum-free media [123]. Deviating from the classical cytokine method has opened the possibility of eliciting more effective danger signals for activating the DCs. While inflammatory cytokines physiologically mimic conditions that may be found in infection, so do pathogen-related signals including LPS, bacterial DNA and dsRNA [125,126]. In particular, TLR agonists have been explored due to their role in activating innate immunity and their subsequent potential for activating DCs. When LPS, a TLR4 agonist, has been used as one of the activating agents, high levels of IL-12, whose role in effective T-cell sensitization will be discussed, are produced [126]. A subsequent comparative in vitro study has reinforced these findings, suggesting that use of TLR agonists, in contrast with pure stimulation by the classical inflammatory cytokine milieu, is a more effective strategy [127]. Clinical trials using TLR agonists as a maturation technique are still few (Table 1), with the bias towards cytokine-matured DCs, and further trials will provide an opportunity to compare outcomes between cytokine-matured and TLR-activated DCs in order to determine the most effective method to activate DCs in such a manner as to prevent and even combat immune tolerance. In addition to exogenous signals to activate TLRs, researchers have been exploring alternative methods for delivering maturation signals via the downstream cell signaling pathways by use of various different molecules as well as by engineering DCs to have constitutively active TLR signalling [128]. For example, DCs modified to express constitutively active TLR4 with and without CD40L have demonstrated both maturation and the ability to stimulate an immune-specific CTL response [129]. These highlight alternative methods for taking advantage of the TLR signaling pathway.
DC phenotype in vaccine design
Thus far, we have seen that there is a high degree of plasticity in DC lineage and that external signals greatly impact DC maturation. We have also discussed the importance of fully activating DCs, such as with the use of TLR agonists, in order to prevent or even abrogate immune tolerance. The ability of DCs to drive the immune system depends on both functional maturation, discussed above, as well as the mature phenotype [86]. Choice of maturation signals is considered, not only for achieving full maturation, but also to determine the subsequent polarization of the immune response.
It has long been established that the immune response can be polarized into different categories, initially described as the Th1- and Th2-type response [130]. These labels reflect active CD4+ Th cell subtypes and refer to the nature of the associated immune response. We have already discussed tolerogenic DCs and their ability to induce Tregs and thus an immunosuppressive response. More recently, a Th17 helper cell has also been described. DCs have been shown to affect the development of these different helper cells via the elaboration of cytokines [131]. The DCs that induce these types of response can thus be labeled as DC1s, DC2s and DC17s. In particular, the cytokines that most greatly impact this polarization are those that are present during antigen presentation when DCs are interacting with T cells via the T-cell receptor (TCR) [132,133]. We will discuss these categories of DCs, the signals that induce their development, the subsequent signals they provide and their impact on the immune system (Figure 1) as well as their role in antitumor immunity and cancer vaccines.
Figure 1. Dendritic cell phenotype and subsequent T-cell polarization.
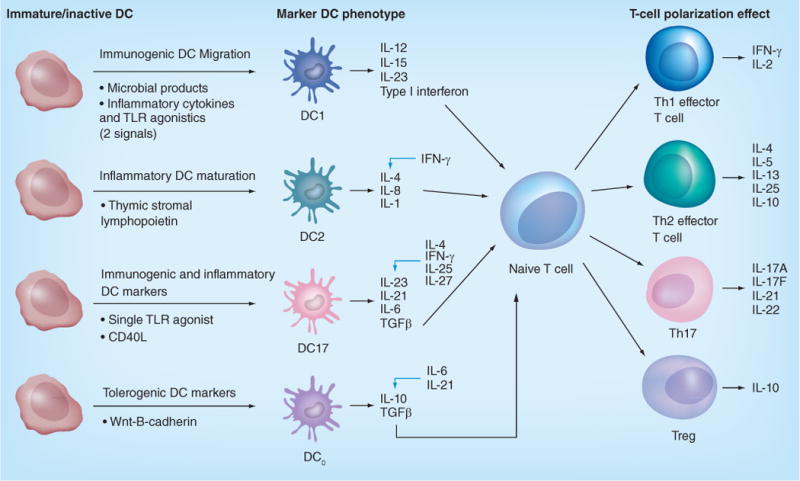
Various factors affect the mature DC phenotype that in turn lead to the production of different cytokines, which then polarize T cells, alter the cytokine milieu and affect the ultimate nature of the immune response. DCs of different phenotypes secrete cytokines that exert their effect on naive T cells to polarize them towards phenotypes including Th1, Th2, Th17 and the immunosuppressive Treg cells. These T cells in turn are characterized by their own corresponding cytokine secretion profile; black arrows indicate activating factors; blue arrows indicate inhibition. DC: Dendritic cell; TLR: Toll-like receptor.
DC1-induced Th1 polarization
The DC1 phenotype is so named because it induces Th1 helper subsets. Th1 cells are characterized by high IFN-γ secretion, and have been associated with immunity against intracellular pathogens [130,131], autoimmunity [134,135] and antitumor immunity, particularly when compared with Th2 cells [136–138]. IFN-γ enhances the activity of cytotoxic CD8+ lymphocytes, an action thought to be a large part of the antitumor effectiveness of Th1 cells. Other cytokines produced by Th1 cells include TNF-α, a mediator of inflammation; and IL-2, which leads to expansion of lymphocyte populations. Th1 cells play a role in humoral immunity by inducing antibody class-switching and the production of IgG by B cells, including complement-fixing IgG1. The association of Th1 cells with autoimmunity also suggests an ability to overcome the barrier of immune tolerance, given that many tumor antigens are self-differentiation antigens. We have already addressed the improved ability of Th1 cytokines to overcome immune suppression when compared with Th2 cytokines. Furthermore, we have found that Th1-driven T cells are more sensitive at detecting MHC class I–tumor antigen complexes than their Th2-driven counterparts [139]. This role of Th1 cells in anticancer immunity has made DC1s the primary DC phenotype for vaccination.
Cytokines that favor induction of a Th1 response both in vitro and in vivo include IL-12 and IFN-γ [140]. DCs with the capability of inducing a strong Th1 response are characterized by the secretion of high amounts of IL-12. Both inflammatory cytokines and TLR agonists can be combined to mature DCs to a Th1-polarizing phenotype [141,142], and high IL-12 production requires two such danger signals [141,143]. Although various combinations have been used with success [141], we have had particular success using a combination of IFN-γ and LPS [144], leading to the highest levels of IL-12 secretion by DCs [126]. We have found that IL-12 is a key component of the antitumor effects of DC1s in addition to its well-described role in initiating the Th1-polarized response [145,146].
IL12 is a member of the small family of cytokines. It can be secreted in a free or homodimeric subunit or as a p70 heterodimer [147], but it is the p70 heterodimer composed of a p40 and p35 subunit and secreted by DCs that has the ability to polarize CD4+ T cells to the Th1 phenotype [144,148,149]. IL-12 itself seems to have an inherent antitumor effect, in that it possesses antiangiogenic capabilities [150] and can activate natural killer cells [151,152], which play a key role in attacking tumor cells that have decreased or absent MHC expression. However, IL-12 also enhances adaptive immunity and improves sen-sitization to tumor antigens [144]. Our laboratory has demonstrated that DC1s, characterized by high IL-12 secretion, improve CD8+ recognition of tumor-derived peptide antigens. When compared with CD8+ T cells sensitized by DC2s, those sensitized by DC1s showed enhanced recognition of tumor cells expressing the target antigen and increased tumor lysis, which were linked to the impact of IL-12 during DC–T-cell interactions. Furthermore, activation by DC1s increased the functional avidity of CD8+ T cells, the mechanism of which was also linked to the presence of IL-12 [144]. By using DC1s that were characterized by high IL-12 secretion, antigens to which T cells previously could not be sensitized [153], or that could only be sensitized with multiple stimulations [154], were sensitized over the course of one stimulation with DC1s in a 6–7 day culture of DC1s with CD8+ T cells. This increased T-cell sensitization in turn resulted in tumor recognition and killing, in contrast with earlier studies that identified antigen recognition but without tumor killing when HER2/neu peptide was injected with Freund’s incomplete adjuvant [155]. Clinically, we have found that using DC1s that secrete high amounts of IL-12 and timing our injections to take advantage of this IL-12 secretion has yielded promising clinical results [156,157].
DC2-induced Th2 polarization
DC2s, in turn, refer to DCs that mature so as to have a cytokine profile favoring production of Th2 CD4+ cells and their associated immune response. Cytokines that favor production of the Th2 subset include IL-4 and anti-IFN-γ. These Th2 CD4+ cells in turn secrete IL-4, IL-5, IL-6 and IL-10. The Th2 arm of the immune system is characterized by its role in combating parasites while also promoting allergic reactions and asthma. This is because IL-4 and IL-5 activate mast cells and eosinophils that lead to elevated levels of IgE. Th2 cells also produce B-cell growth and differentiation factors, associating the Th2 arm of the immune system with humoral responses although, as discussed, Th1 polarization induces antibody class-switch and thus also affects humoral immunity. Some have suggested a role of the Th2 response in antitumor activity via the activation of eosinophils [158], and Mattes et al. found that Th2 cells were capable of clearing lung and visceral metastases of B16 melanoma transfected to express chicken protein, OVA, in C57Bl/6 mice. This tumor regression was associated with an influx of eosinophils into tumors and was dependent on eotaxin, an eosinophil chemokine, and STAT6 [159]. Despite findings that both Th1 and Th2 cells can have an antitumor effect [136], Th2 cells have generally been deemed less effective than their Th1 counterparts in combating cancer [160,161]. Furthermore, Th2 cytokines can inhibit Th1 activity and in that way may be detrimental. Aspord et al. linked Th2 polarization, particularly IL-13 secretion, with promoting early tumor development in breast cancer. This high IL-4- and IL-13-secreting Th phenotype depended on induction by DCs that had been influenced by the tumor microenvironment, illustrating the importance of DC activation in directing immune polarization [162]. DeNardo et al. used tissue analysis and mouse models to demonstrate that Th2 CD4+ cells characterized by IL-4 secretion promoted invasion and subsequent pulmonary metastases in breast cancer via their effects on macrophages, with one effect being increased expression of TGFβ and EGF [163]. The beneficial findings of Th1-polarized immunity and the mixed or potentially harmful influence of Th2 immunity has biased vaccine production towards the promotion of Th1 polarization via the production of DC1-based vaccines.
DC17-induced Th17 polarization
DC17s refer to DCs matured so as to induce a Th17 response. Th17 cells are a more recent discovery and are so named due to their production of IL-17. These Th17 cells appear to be generated by TGFβ and IL-6 via STAT-3-dependent signaling [164], and are also driven by IL-23 [165] as well as IL-1β. Interestingly, IFN-γ and IL-4, which promote Th1 formation, have been implicated in the inhibition of IL-23-dependent IL-17 production [137]. Among the literature addressing factors that shape development of Th17 cells, the role of IL-23 in inducing IL-17 production by T cells is by now fairly well-established [165–167]. We have demonstrated the ability to develop DCs that produce IL-23 by treating iDCs generated from monocytes with a single TLR agonist. Using LTA, LPS or R848, which are agonists against TLR2, TLR4 and TLR7/8, respectively, we successfully produced DCs that secreted IL-23. This IL-23 secretion by monocyte-derived DCs was enhanced by rapid culture of monocytes in the absence of IL-4, given that IL-4 inhibits IL-23-dependent IL-17 production [168]. These DC17s produced IL-23 and induced secretion of IL-17A when co-cultured with CD4+ T cells that were able to subsequently demonstrate an antigen-specific Th17 response and could also induce antigen-specific CD8+ T-cell secretion of IL-17A [167]. These findings show the successful production of functional DC17s in the laboratory that can be used to further study this branch of the immune response. These laboratory-produced DC17s could also feasibly be incorporated into a vaccine should further evidence support their role in antitumor immunity.
The role of Th17 cells in tumor immunology has been controversial and is still under investigation. While some literature has implied a role of Th17-related cytokines in tumor promotion due to the presence of IL-23 mRNA in various cancers [169] and Th17 cells in the tumor microenvironment and draining lymph nodes of several tumors [170,171], these findings are non-specific and do not truly indicate whether or not Th17 immune responses promote or combat cancer, especially given that some studies have found other tumors to be associated with decreased Th17 cell levels [172–174]. Such associations are difficult to interpret without a better understanding of the immunobiology of the Th17 response.
One finding supportive of a protumor effect is that Th17 cells play a role in angiogenesis [175]. In particular, IL-17 is implicated in neovascularization via STAT3 signaling [176]; by mechanisms such as enhancing the mitogenic effects of bFGF, HGF and VEGF on vascular endothelial cells [177]; and by inducing increased tumor secretion of IL-8 [178]. They have also been shown to elaborate matrix metalloproteases in murine models [169], although one may argue that this increases mobility for infiltrating inflammatory cells and not only for tumor cells. There is a suggestion that the inflammation induced by Th17 cells may in fact be procarcinogenic [169,179], with one study showing that IL-23 deficiency conferred a protective effect in mice [169]. Another recent study of PTEN-deficient mice in which a prostate tumor cell line was implanted compared tumor behavior in mice that expressed IL-17 and in those genetically engineered to lack IL-17 expression. Mice that did not express IL-17 developed smaller tumors, exhibited less cellular proliferation as measured by Ki67 staining and had slower progression to invasive disease, possibly due in part to decreased elaboration of MMP7 when compared with the IL-17-expressing mice. These findings suggest a role of IL-17 in promoting tumor growth and invasion [180]. Part of the controversy arises from mixed findings even within the same study, suggesting that the role of IL-17 may not be so clear cut. For example, Benchetrit F et al. found that when IL-17 was transfected into human tumor cell lines, these tumors had increased progression in nude mice due to neovascularization. However, the opposite effect was found in immunocompetent mice, suggesting that in the context of a functioning immune system, this pro-angiogenic effect does not tell the entire story, and that IL-17 may in fact contribute to an antitumor response [181].
Indeed, there also exists growing evidence that the Th17 arm of the immune system enhances immunity against cancer. In contrast with findings that IL-23 deficiency was protective in animal models [182], other murine studies indicated that systemic IL-23 enhanced the antitumor activity of T cells [183,184]. Furthermore, IL-23 has been shown to induce a CTL memory response [185] and DCs producing IL-23 have demonstrated the promotion of antitumor immunity [186]. Findings that Th17 cells, like their Th1 counterparts, are involved in autoimmunity [187] also suggest the potential ability to overcome the immune tolerance induced by tumors. One striking demonstration of Th17-polarized antitumor immunity was exhibited by Muranski et al., who compared the therapeutic effects of adoptive transfer of antigen-specific Th1, Th17, and non-polarized Th0 cells on the treatment of a B16 melanoma in a mouse model. The Th17-polarized T cells were the most effective at inducing tumor regression, even demonstrating the ability to completely eradicate tumor and promote long-term survival [188]. However, the underlying mechanisms reflect the complexity of the Th17 response. The antitumor effects of the Th17-polarized T cells were found to be dependent on IFN-γ, and neutralization of IFN-γ abrogated tumor rejection by the Th17 cell population. IFN-γ, while secreted by some Th17 cells, is more intimately associated with Th1 polarization and is secreted at higher concentrations by Th1 cells. One explanation for the ability of Th17 cells to mediate improved antitumor immunity in this particular model was attributed to potential improved survivability of the Th17 population. Another explanation is that there is a degree of plasticity in the Th response [158,189]. We have shown that DCs that produce IL-12 have a tendency to polarize the Th response to a primarily Th1 response even in the presence of IL-23 secretion, whereas DCs that produce IL-23 in the absence of IL-12 elicit a Th17-polarized response [167]. The presence of IL-12 may cause an alteration in the Th17 response from initial IL-17 secretion to more IFN-γ secretions; [188] a shift that has been observed in other studies [190].
The immunobiology of Th17 polarization and its role in antitumor immunity remains controversial. As this role becomes better-elucidated, the applications of DC17s in DC-based vaccines will become clearer. The possibility of interplay between the Th1 and Th17 arms seems possible given the plasticity of the Th response and evidence that IFN-γ remains important in antitumor immunity whether mediated by Th1 or Th17 cells. Further studies, in particular clinical trials in humans, are necessary to better clarify the antitumor effects of these responses, given there is a lack of human trials examining the use of DC17s and comparing outcomes among DC phenotypes (Table 1). Improving our understanding of the Th17 immune response, its role in antitumor immunity and its interplay with the Th1 immune response provides an opportunity to choose the most effective immune phenotype or combination of phenotypes for a vaccine to induce antitumor activity.
Importance of incorporating CD4+ Th cells for effective antitumor immunity
There has been a historical bias towards optimizing MHC class I-restricted CD8+ CTL responses in antitumor immunotherapy, compared with focusing on the class II-restricted CD4+ Th-cell responses [191]. This is because early murine studies pointed to a comparatively greater dependence on CTLs for tumor rejection, and the ease of CTL isolation from human tumors gave credence to the idea that CTLs were the primary effectors for antitumor immunity. However, growing evidence suggests that CD4+ Th cells have a more fundamental role in antitumor immunity beyond priming CTL responses. Early clinical trials of DC-based vaccines have focused on activating CD8+ T cells, and this failure to incorporate CD4+ T-cell activation could be a contributing factor to the poor results of these clinical trials [192–194]. Dranoff et al. found that CD4+ Th depletion immediately prior to tumor challenge in the presence of vaccine-primed CTLs resulted in loss of ability to reject tumor, indicating a more immediate role for these cells in antitumor immunity [195]. Meanwhile, Hung et al. demonstrated that CD8−/− mice could be immunized to reject tumors in a CD4+-dependent manner [196]. It is now clear that CD4+ Th cells mediate antitumor effects through a variety of mechanisms, including direct cytotoxic antitumor activity, production and modulation of the antitumor cytokine response, potentiation of long-term CTL survival and memory, and activation of other immune effector cells.
CD4+ Th cells can have direct tumoricidal effects via induction of various apoptotic mechanisms in tumor cells including Fas/Fas ligand interactions, which has been demonstrated in Burkitt’s lymphoma; [197] and, via TNF-related apoptosis-inducing ligand (TRAIL) [198], demonstrated in melanoma and T-cell lymphoma. They can also lyse tumors by utilization of the granzyme–perforin-dependent cytolytic pathway [199]. For tumors that attempt to escape immune surveillance via downregulation of MHC class I molecules, these direct tumorcidal effects of CD4+ T cells are important for mediating tumor lysis. Recently, using a model of murine hepatocellular carcinoma, it was established that CD4+-mediated immunity was essential for the elimination of premalignant senescent tumor cells, the suppression of which was important in preventing development of hepatocellular carcinoma [200].
As we discussed, CD4+ Th cells elaborate cytokines that polarize the immune response, some of which have direct antitumor activity. Th1 cells produce IFN-γ, which activates tumor macrophages to produce nitric oxide and superoxide, both of which play an important role in tumor killing [191]. Furthermore, IFN-γ inhibits tumorigenesis via STAT1-dependent suppression of cell cycle progression [1]. In a recent study, changes in the cytokine milieu elicited by CD4+ Th cells abrogated angiogenesis and triggered cellular senescence in a murine model of Myc inactivation, suggesting that such CD4+ Th-dependent mechanisms could be involved in eliminating residual tumor burden and prolonging tumor-free survival [201].
The function of CD4+ Th cells in priming and activating CD8+ CTLs has been extensively studied and thus will only be addressed briefly [202]. Although DCs are able to directly activate CD8+ T cells [42,43], CD4+ T cells help augment this activation [203] as well as maintain the CD8+ T-cell pool and enhance their cytolytic activity [192,203]. Furthermore, CD4+ T cells play a key role in eliciting antigen-specific CTL memory, which is perhaps one of the more crucial ways in which they impact CD8+ CTLs [204]. CD4+ Th cells probably impart an irreversible blueprint onto CTLs that allows sustained secondary expansion of CD8+ memory CTLs [205]. In one study, when CD8+ T cells were primed in the absence of CD4+ T cells, decreased expansion of secondary effector CTLs was observed as a result of TRAIL-induced apoptosis [202], reinforcing the notion that CD4+ T-cell help is critical in the generation of memory CTLs [206,207].
CD4+ Th cells also activate other immune cells. They enhance DC activation via a CD40–CD40L interaction that promotes survival of the DC and further augments the capability of the DCs to prime CTLs [208]. CD4+ cells primed by DCs are also able to sensitize B cells to produce antibodies [209], and thus their incorporation into a vaccine opens up the possibility for taking advantage of antibody-mediated immunity should an extracellular target antigen be used.
The beneficial role of CD4+ T cells in antitumor immunity supports their use as part of a vaccine construct. CD4+ T cells can be incorporated into a DC-based vaccine by choosing antigens that sensitize CD4+ T cells via presentation by DCs on MHC class II molecules. A vaccine will be most effective when using antigens to sensitize both CD4+ and CD8+ T cells. This synergistic effect has been supported by in vivo studies using mice [210,211]. Recently, methods to use a single full-length, tumor-associated antigen (TAA) loaded onto DCs have been shown to successfully sensitize both CD4+ and CD8+ T cells. Van Nuffel et al. used mRNA encoding a full-length TAA fused to sorting signals that direct the antigen for processing and subsequent presentation on both MHC class I and class II molecules [212]. Additional methods are being explored to assure sensitization of CD4+ cells as part of vaccine constructs, particularly since many tumors do not express HLA class II molecules. We will address some of these strategies when discussing antigen choice and additional methods for engineering DC-based vaccines.
Clinically, we have seen preliminary success in sensitizing only CD4+ T cells in ductal carcinoma in situ (DCIS) patients. Due to the restrictive nature of MHC class I alleles and our use of class I peptides that bind HLA-A2, HLA-A2− patients only receive DCs pulsed with promiscuous MHC class II peptides, in contrast with HLA-A2+ patients who receive both class I and class II peptides. Clinically, these patients, like their HLA-A2+ counterparts, demonstrate increased tumor infiltration by lymphocytes, durable immunity [213] and therapeutic benefit from this treatment [157], although close comparison of these two groups will more clearly elaborate whether and to what extent there is a therapeutic difference among those vaccinated with DCs pulsed with MHC class II molecules alone, as opposed to those vaccinated using both MHC class I and class II molecules.
Choosing a target antigen
General principles
Identifying effective immunogenic antigens is one of the biggest challenges in DC-based vaccine development. Many types of TAAs may be used, but they should fulfil four basic criteria. Ideally, they should be specific to cancer, preferably mutated in the cancer of interest to increase immunogenicity and minimally expressed on normal tissues to avoid autoimmunity; common in the cancer of interest; play a role in tumor progression or survival; and be capable of eliciting an antigen-specific immune response. Most tumor antigens are not specific to the cancer of interest but rather are overexpressed or dysregulated differentiation antigens and are thus less immunogenic. Choosing an epitope of the tumor antigen that will elicit sufficient antigen-specific T-cell sensitization remains one of the most difficult criteria to fulfill, and we will focus on overcoming this barrier. Immunogenicity of an antigen is dependent on several factors, including binding affinity for HLA or the TCR, as well as the ability of T cells to recognize the antigen as nonself. There are methods for altering antigens to improve immunogenicity by increasing binding affinities or coupling them with other immunogenic factors.
Technical considerations
In DC-based vaccines, peptides are a major antigen source. Given the large number of possible target peptides that can be derived from each protein, it is time- and cost-prohibitive to systematically test all possible peptides for the ability to induce antigen-specific sensitization [214]. When analyzing a protein sequence to choose peptide antigens, major considerations include the MHC class type and the HLA allele, which refers to genetic variation among MHC receptors.
MHC class I receptors display intracellular proteins to CD8+ T cells. Class I molecules have a ‘closed’ binding groove, meaning that both the N and C termini of a peptide are bound within the groove, limiting peptide size to eight to ten amino acids [215]. This rigid binding conformation allows for easier identification of amino acid sequences where the side chains are likely to fit into the known pockets of the binding groove. Consequently, there are more than 30 online-accessible algorithms for predicting peptide–MHC class I binding [216]. For MHC class I, positions 2 and 9 in a 9-mer peptide are called anchor residues, as the amino acids in those positions play key roles in peptide–MHC binding. Substitution of amino acids at anchor residues has been shown to improve peptide–MHC binding and, more importantly, enhance CD8+ T-cell sensitization [217], providing an option for overcoming lack of immunogenicity of a peptide due to limited binding.
MHC class II receptors, in contrast with the MHC class I receptors expressed by all cells, are only found on APCs and, unlike class I receptors, have an ‘open’ binding groove. Although the core of the binding groove still fits only nine amino acids, the N- and C-termini of a peptide are not bound in the groove itself, allowing the class II receptor to accommodate peptides up to 30 amino acids in length [218]. The open binding groove permits promiscuous binding, meaning that the peptide can slide through multiple registers of nonamer epitopes. This flexibility in binding has made the prediction of CD4+ T- cell epitopes more difficult [219]. Recent analyses of MHC class II algorithms showed both poor sensitivity and specificity in predicting T-cell epitopes, especially when compared with class I algorithms [220]. At least 25% of peptides known to elicit a strong CD4+ response were not predicted to be strong binders by the algorithms studied [215]. Further research is needed to better understand MHC class II receptors and binding patterns and refine predictive algorithms.
Limitations of peptide antigens
Besides limitations due to MHC binding, the efficacy of peptide antigens can be limited by self-anergy, HLA allele restriction and TCR binding. Developing methods to overcome these limitations will lead to the improved efficacy and broader applicability of peptide-based DC vaccines.
Often, the target protein for a DC vaccine is an overexpressed self-protein, such as HER2 in breast cancer. Being a self-protein, these epitopes are subject to thymic-negative self-selection. Given that high-affinity binding leads to thymic-negative selection, peptides that are predicted to bind with greatest affinity and stability by MHC prediction algorithms may be at greater risk for self-anergy. To address this limitation, peptides with both high- and medium-binding affinity should be considered as candidates when screening peptides for potential use in a vaccine based on binding affinity.
As mentioned, the difference in HLA alleles must be considered when choosing antigens. Peptide binding is specific to particular HLA alleles and will only benefit the population that expresses that allele. To overcome this barrier, some researchers suggest using longer peptides containing multiple epitopes that can bind different alleles. The promiscuous binding of MHC class II molecules makes this feasible, as the large binding groove can accommodate a longer sequence of amino acids and yet there is variation in the nine core amino acids. Identifying a peptide that harbors binding sites for multiple alleles will widen its applicability to more patients. In vitro assays looking at a multi-epitope approach that included tumor-associated peptide antigens found in multiple cancers and that also had reactivity in activating multiple subtypes of the HLA-A2 allele, indicated the possibility of identifying or synthesizing epitopes capable of treating a broader population base [221]. Both in vitro and in vivo testing of a synthetic peptide containing overlapping epitopes to allow the promiscuous binding of multiple MHC class II alleles demonstrated antigen-specific responses, suggesting this as an alternate approach for widening the target population treated with a single antigen [222].
Despite advances in understanding the MHC–peptide complex, it has been much more difficult to fully characterize the MHC–peptide–TCR complex that is ultimately responsible for T-cell activation. Recent studies have shown that specific amino acid residues within MHC-bound peptides provided key contact points with TCRs and influenced immunogenicity [223]. Mutations of these contact points abrogate T-cell activation more than 50% of the time. The spectrum of TCRs in an individual is a function of the predominant MHC class II receptors and self-peptides present in the thymus during development. Therefore, contact points have to be elicited on an individual basis. This is a growing area of inquiry, and deeper understanding of these interactions will improve prediction of immunogenic antigens.
Viral vectors, fusion peptides & DC–tumor fusion
To address concerns regarding immunogenicity, some early vaccine techniques, rather than using DCs, injected patients with recombinant virus expressing tumor antigens, taking advantage of the immune response to the viral pathogen to confer antitumor immunity. Mouse models demonstrated therapeutic benefit from vaccination with recombinant adenoviruses, and a subsequent human trial in which adenovirus vectors expressing melanoma differentiation antigens MART-1 and gp100 demonstrated a good safety profile, although with mixed therapeutic efficacy. These mixed therapeutic results could depend, in part, on the tumor antigens used, as they lack a role in disease progression, as well as the development of neutralizing antibodies against the viral vector [224].
In a similar attempt to augment tumor antigen immunogenicity, Shahabi et al. made a chimeric human HER2/neu gene expressed as a fusion protein to a non-hemolytic fragment of listeriolysin O (LLO) and tested the immunologic and therapeutic effects in mice. Taking advantage of the pathogenic response to the Listeria fragment seemed to enhance immunogenicity to the HER2/neu protein in their transgenic mouse model [225]. While it is ideal to choose a TAA that is mutated in the cancer of interest and is involved in tumor progression, such as mutated BRAF, or EGF receptor variant III (EGFRvIII), which is expressed in several tumor types but has not been detected in normal tissues [226], this mutated status does not always confer the desired immunogenicity. These otherwise ideal antigens can thus be selected for incorporation into a fusion peptide. Using recombinant DNA technology, Duan et al. created a fusion protein comprising an immunogenic portion of EGFRvIII that was inserted into an immunodominant loop of hepatitis B core antigen and used to vaccinate mice using Freund’s incomplete adjuvant. Vaccination with this fusion peptide yielded an antigen-specific T-cell response as measured by IFN-γ secretion, induced a cytotoxic response to tumor cells and mediated a protective effect on mice challenged with tumors [226]. These fusion proteins can also be used in DC-based vaccines. Sipuleucel-T, brand name Provenge® (Dendreon Corporation, WA, USA), is the first US FDA-approved DC-based vaccine therapy and uses a fusion protein consisting of a prostate differentiation antigen, prostate acid phosphatase, linked to GM-CSF. It has been shown to confer a survival benefit in patients with metastatic castration-resistant prostate c ancer [227,228].
This idea to link tumor antigens with peptides that enhance their immunogenicity is the basis of another platform for modifying antigens that can be incorporated into DC-based vaccines known as the Ligand Epitope Antigen Presentation System. Antigenic peptide epitopes, such as infectious antigens or tumor antigens, are linked to immune cell-binding ligands, which are peptides that can interact with receptors on leukocytes and thus promote immunogenicity and direct the subsequent nature of the immune response [229]. Much like the TLR agonists used to activate DCs, these peptides mimic pathogenic invasion. Studies utilizing the J peptide from β2-microglobulin linked with a herpes simplex viral epitope demonstrated the induction of DC1s that secreted high amounts of IL-12 and induced an IFN-γ-secreting Th1 phenotype in both mouse and human DCs. These DCs treated with J-linked peptides did not require further treatment with TLR agonists and demonstrated antigen-specific T-cell responses [229,230]. Furthermore, adoptive transfers of DCs treated with J peptide linked to herpes simplex viral antigen in mice conferred therapeutic protection against subsequent infectious challenge [231]. Additional work testing tumor antigens, which present more of an immunogenic challenge, is necessary to determine the utility of this platform in DC-based cancer vaccines. Consistent with limitations of peptide vaccines, the peptide antigen being targeted must have inherent immunogenicity and is limited by restriction of the HLA allele [229,232].
In order to circumvent limitations of the peptide antigens based on HLA specificity, one method is to use irradiated tumor cell lysates fused with DCs. The use of irradiated tumor cells eliminates the tumorigenic capacity, while the application of electrofusion to create DC–tumor fusion hybrids offers the advantage of utilizing multiple tumor antigens that can be processed and presented on both MHC class I and class II molecules and on different HLA alleles. Using DC fusion hybrids allows us to take advantage of as yet undefined TAAs, while use of tumor lysates that contain known mutations will permit targeting of specific known antigens. The ability to take advantage of both defined and undefined TAAs will help in cases where there are limited TAAs identified and may help to identify new antigen targets. Meanwhile, many studies assessing the therapeutic effects of DC–tumor fusion hybrids as well as factors for enhancing their efficacy have been conducted in mouse models [233–237], and in vitro models have demonstrated the ability of using these DC fusion hybrids to induce antigen-specific T cells using human DCs [238,239]. Human trials utilizing DC fusion are limited. One small trial in patients with renal cell carcinoma demonstrated safety and immune sensitization, although clinical results were equivocal. Further clinical trials will be required to determine the effectiveness of the DC-tumor fusion technique as an effective methodology in DC-based vaccines [240].
Genetic engineering of DCs
Genetically engineering DCs has been another approach to overcome HLA restriction by genetically altering DCs to express the desired TAA. DCs expressing any HLA allele can present these TAAs, as they are processed within the DC and packaged endogenously onto the HLA types inherent to those DCs [241]. Additional genetic manipulations of the pathways for packaging these TAAs onto MHC molecules can further enhance their immunogenicity for stimulation of CD8+ CTLs [242] and CD4+ T-cell responses [192,243], thereby overcoming concerns regarding the HLA restriction inherent in choosing peptide antigens. These techniques can also be used to assure dual stimulation of both CD8+ and CD4+ T cells. Electroporating DCs with TAA epitopes linked to signals that target the proteins to lysosomes within the cells simultaneously with HLA class I and class II molecules leads to the expression of TAAs on both HLA class I and class II molecules [244]. Genetically altering DCs to express tumor antigens has been carried out using both viral vectors and the nonviral electroporation method. Both result in good expression of tumor antigens by genetically modified DCs, although electroporated cells may have less immune potency, which has been attributed to decreased IL-12 production [245]. Still, clinical trials utilizing DCs electroporated with mRNA encoding the tumor antigen have shown good immunologic response, although mixed clinical results, suggesting the need for larger clinical trials with earlier stage patients [246,247].
Other genetic alterations made experimentally to DCs in order to enhance immunogenicity of the antigen use conferred during DC–T cell interactions include increasing their expression of important costimulatory molecules such as CD40L, CD70, TNF family ligands and OX40L, and causing the constitutive activation of TLR4 [241]. This expression has been carried out by transduction using various viruses [248–250] and by using mRNA electroporation [251]. Increased in vitro T-cell sensitization to TAAs was found when DCs were engineered to upregulate expression of CD40L [252,253], RANK/RANKL [254] and OX40L [255], among others. Upregulation of these molecules augments an activation state that is favorable both for conferring immunity and for overcoming tolerogenic influences in the tumor microenvironment.
In a similar fashion, researchers have worked on genetically manipulating DCs to increase secretion of activating cytokines, such as IL-12 [256,257] or, conversely, to downregulate molecules that function in immunosuppression. Murine and in vitro human studies have demonstrated that silencing A20, ubiquitin-editing enzyme that can adversely affect TLR and TNF receptor signaling [241,258], increases expression of costimulatory molecules, augments secretion of inflammatory cytokines, enhances the Th1 immune response and improves CD8+ T- cell antigen recognition [259,260]. Other molecules whose activities have been genetically silenced in DCs in mouse models include SOCS1 [261] and DIgR2 [262]. Silencing these and other regulatory molecules on DCs is another method being explored to overcome immune inhibition in combating tumors. With new advances in genetic engineering, opportunities to alter DCs provide a potential for overcoming the limitations of peptide binding and HLA allele restrictions as well as for combating tumor-related immune suppression.
Rationale for vaccination in early stages of carcinogenesis
A majority of the initial cancer immunotherapy trials have been performed in end-stage cancer patients, and the results of such trials have been disappointing (Table 1) [263]. Advanced tumors are heterogeneous and have diverse, often redundant pathways of immune escape. Consequently, efforts at targeted elimination of individual escape mechanisms result in limited success. This is evident in the resistance that develops to a wide range of pharmacologic therapies that target specific cellular proliferation pathways, such as to imatinib in chronic myelogenous leukemia or small molecule BRAF inhibitors in melanoma [264,265]. While utilizing antigens that will target multiple complementary intracellular signaling pathways or by combining vaccines with pharmacologic therapy targeting these alternate pathways may be one way to address multiple mutations, the more advanced the cancer, the more alternate pathways will be in effect.
The systemic immunosuppressive milieu of the tumor microenvironment in advanced cancer contributes to immune failure, posing another way by which advanced stage disease is more difficult to treat with immunotherapy. Standard treatments utilized in advanced-stage cancer, including systemic chemotherapy regimens and radiation, are known to be immunosuppressive, and may preclude optimal responses to cancer immunotherapy [263,266]. In one study, dose-intensive chemotherapy resulted in rapid CD4+ T-cell depletion in adult populations, followed by protracted and suboptimal CD4+ recovery. When stimulated by mitogens, these post-chemotherapy CD4+ cells were more prone to apoptosis compared with cells from normal donors, suggesting that conventional antitumor treatment modalities compromise antitumor immune function [266]. There are, of course, exceptions regarding the immunologic effects of chemotherapy with various conventional and targeted chemotherapeutics that induce immunogenic cell death [267], as well as therapies that take advantage of the immune response, for example, antibodies designed to block immunosuppressive pathways such as anti-CTLA-4 and anti-PD1 [268]. While some of these therapies can be taken advantage of and combined with immunotherapy, the immunosuppressive effects of many anticancer treatments and the declining health in patients with advancing cancer compromises their ability to mount an effective antitumor immune response [263].
In recent years, there has been a paradigm shift away from administering cancer vaccines in advanced-stage patients and a move towards using cancer vaccines to treat earlier stages of carcinogenesis, before tumor- and treatment-mediated immunosuppressive environments can be established, and before the accumulation of mutations that activate redundant pathways for tumor proliferation. Preliminary application of this strategy has yielded promising results. In a transgenic murine model of prostate adenocarcinoma, therapeutic vaccination directed against two different prostate cancer-associated antigens at the earliest stage of carcinogenesis elicited long-term protection against spontaneous prostate cancer development [269]. Vaccination of premalignant cervical intraepithelial neoplasia lesions can cause their complete eradication or partial regression to a lower-grade lesion [270]. Our group has applied this concept in administering a HER2/neu pulsed DC vaccine to HER2/neu-overexpressing DCIS patients. Recently published results suggested that anti-HER2/neu vaccination induces decline or eradication of HER2/neu expression, particularly in estrogen-independent DCIS [157]. Owing to these promising results, this DC vaccine for treating HER2-positive DCIS is entering a large Phase III trial. These findings suggest that DC-based vaccines will have improved success when used as a treatment in early disease and as an adjuvant treatment to currently available modalities.
Conclusion
DC vaccines must be engineered to overcome immune tolerance and necessitate maturation of DCs to an activation state best poised to overcome this barrier. Antigens are currently limited by restricted peptide binding that decreases the population that can benefit from a particular vaccine construct, as well as by their ability to elicit sufficient immune response. Many strategies are under investigation to address these limitations, including methods to genetically engineer DCs used in DC-based vaccines. Furthermore, the paradigm shift to administer vaccines in earlier-stage cancer takes into consideration tumor biology, and is proving to be a more advantageous strategy. The barriers to effective DC-based vaccines provide opportunities for further research that continues to improve their efficacy and applicability.
Future perspective
As knowledge grows about DC and tumor biology, we have been better able to understand the limitations of DC-based vaccines and also determine where further investigation needs to be focused in order to improve this modality. We may expect to see a wider range of DC lineages incorporated into vaccine strategies as well as more complex manipulations of DC phenotype in order to take advantage of Th1- and/or Th17- polarized immune responses. Technology that allows us to alter DCs and antigens will help overcome the barrier of HLA restriction such that these vaccines will be used to treat a broader population of cancer patients. Despite the barriers that we have identified, success of clinical trials treating early disease suggest that DC-based vaccines are on their way to becoming an early adjuvant treatment modality for several cancers.
Executive summary.
Dendritic cell immunobiology: why dendritic cells for tumor vaccines?
-
■
Dendritic cell (DC) biology makes them an ideal vehicle for an antitumor vaccine.
-
■
Despite their promise, results of many clinical trials of DC-based vaccines have been disappointing.
DC lineages & the choice of cell lineage for vaccine construct
-
■
DC lineages have tendencies to develop particular mature phenotypes, but plasticity in their development is such that environmental signals may have a stronger impact on their ultimate behavior.
-
■
Though monocyte-derived DCs have been favored for vaccine production, further study of DC lineages, in particular identifying those capable of cross-priming, could identify new subsets that could be incorporated into vaccine strategies.
DC maturation & immune tolerance: DCs as immune suppressors
-
■
Incomplete or inappropriate maturation of DCs can lead to immune tolerance, and DC vaccine strategies must take into account the need to combat the suppressive influence of myeloid-derived suppressor cells.
Maturation signals
-
■
Toll-like receptor agonists or alternate methods that take advantage of Toll-like receptor signaling pathways are under investigation as promising methods for maturing and activating DCs.
DC Phenotype in vaccine design
-
■
DCs can elicit Th1, Th2, Th17 or Treg phenotypes. The role of Th17s is still under investigation, and comparative clinical trials may be required to better understand its role in tumor immunology and its potential for incorporation into DC-based vaccine strategies.
Importance of incorporating CD4+ Th cells for effective antitumor immunity
-
■
Increasing evidence supports the importance of CD4+ T cells in antitumor immunity, advocating for their incorporation into DC-based vaccines via the identification and use of MHC class II antigens.
-
■
In addition to choosing a maturation strategy to overcome immune tolerance, DCs can be engineered to overcome tolerance.
Choosing a target antigen: general principles
-
■
Antigen targets are identified based on being mutated or upregulated in the cancer of interest, their role in tumor progression and survival and their ability to elicit an antigen-specific immune response.
Technical considerations & limitations of peptide antigens
-
■
Identifying peptides that bind MHC class I and class II molecules is one barrier to choosing peptide antigens, with other limitations including self-anergy, restriction of peptide antigens to specific HLA alleles and limited immunogenicity for other reasons, such as T-cell receptor binding.
Viral vectors, fusion peptides & DC–tumor fusion
-
■
Research is investigating ways to overcome the barriers of HLA restriction, such as via DC–tumor fusion or genetic engineering of DCs, as well as ways to improve immunogenicity such as by linking tumor antigens to other immunogenic molecules.
Genetic engineering of DCs
-
■
Genetic engineering provides opportunities for improving the efficacy and broadening the applicability of DC-based vaccines by providing methods for circumventing limitations posed by antigen choice and by addressing immune tolerance.
The rationale for vaccination in early stages of carcinogenesis
-
■
DC-based vaccines are more effective in early-stage disease as a result of inherent tumor biology, which is supported by clinical trials yielding better success with treatment of early-stage disease.
Acknowledgments
The authors would like to thank Robin Noel for graphic design assistance and Pennies in Action for their continued support of cancer vaccine research.
The authors recieved an NIH grant R01-CA096997-04A.
Footnotes
Financial & competing interests disclosure
The authors have no other relevant affliations or financial involvement with any organization or entity with a financial interest in or financial confict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
References
Papers of special note have been highlighted as:
■ of interest
■■ of considerable interest
- 1.Koski GK, Cohen PA, Roses RE, Xu S, Czerniecki BJ. Reengineering dendritic cell-based anti-cancer vaccines. Immunol Rev. 2008;222:256–276. doi: 10.1111/j.1600-065X.2008.00617.x. [DOI] [PubMed] [Google Scholar]
- 2.Fong L, Engleman EG. Dendritic cells in cancer immunotherapy. Annu Rev Immunol. 2000;18:245–273. doi: 10.1146/annurev.immunol.18.1.245. [DOI] [PubMed] [Google Scholar]
- 3.Inaba K, Romani N, Steinman RM. An antigen-independent contact mechanism as an early step in T cell-proliferative responses to dendritic cells. J Exp Med. 1989;170(2):527–542. doi: 10.1084/jem.170.2.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ludewig B, Oehen S, Barchiesi F, Schwendener RA, Hengartner H, Zinkernagel RM. Protective antiviral cytotoxic T cell memory is most efficiently maintained by restimulation via dendritic cells. J Immunol. 1999;163(4):1839–1844. [PubMed] [Google Scholar]
- 5.Mayordomo JI, Zorina T, Storkus WJ, et al. Bone marrow-derived dendritic cells pulsed with synthetic tumour peptides elicit protective and therapeutic antitumour immunity. Nat Med. 1995;1(12):1297–1302. doi: 10.1038/nm1295-1297. [DOI] [PubMed] [Google Scholar]
- 6.Mehta-Damani A, Markowicz S, Engleman EG. Generation of antigen-specific CD8+ CTLs from naive precursors. J Immunol. 1994;153(3):996–1003. [PubMed] [Google Scholar]
- 7.Mehta-Damani A, Markowicz S, Engleman EG. Generation of antigen-specific CD4+ T cell lines from naive precursors. Eur J Immunol. 1995;25(5):1206–1211. doi: 10.1002/eji.1830250511. [DOI] [PubMed] [Google Scholar]
- 8.Guery JC, Ria F, Adorini L. Dendritic cells but not B cells present antigenic complexes to class II-restricted T cells after administration of protein in adjuvant. J Exp Med. 1996;183(3):751–757. doi: 10.1084/jem.183.3.751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Celluzzi CM, Mayordomo JI, Storkus WJ, Lotze MT, Falo LD., Jr Peptide-pulsed dendritic cells induce antigen-specific CTL-mediated protective tumor immunity. J Exp Med. 1996;183(1):283–287. doi: 10.1084/jem.183.1.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Paglia P, Chiodoni C, Rodolfo M, Colombo MP. Murine dendritic cells loaded in vitro with soluble protein prime cytotoxic T lymphocytes against tumor antigen in vivo. J Exp Med. 1996;183(1):317–322. doi: 10.1084/jem.183.1.317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Flamand V, Sornasse T, Thielemans K, et al. Murine dendritic cells pulsed in vitro with tumor antigen induce tumor resistance in vivo. Eur J Immunol. 1994;24(3):605–610. doi: 10.1002/eji.1830240317. [DOI] [PubMed] [Google Scholar]
- 12.Sancho D, Mourao-Sa D, Joffre OP, et al. Tumor therapy in mice via antigen targeting to a novel, DC-restricted C-type lectin. J Clin Invest. 2008;118(6):2098–2110. doi: 10.1172/JCI34584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lanzavecchia A. Mechanisms of antigen uptake for presentation. Curr Opin Immunol. 1996;8(3):348–354. doi: 10.1016/s0952-7915(96)80124-5. [DOI] [PubMed] [Google Scholar]
- 14.Norbury CC, Chambers BJ, Prescott AR, Ljunggren HG, Watts C. Constitutive macropinocytosis allows TAP-dependent major histocompatibility complex class I presentation of exogenous soluble antigen by bone marrow-derived dendritic cells. Eur J Immunol. 1997;27(1):280–288. doi: 10.1002/eji.1830270141. [DOI] [PubMed] [Google Scholar]
- 15.Albert ML, Pearce SF, Francisco LM, et al. Immature dendritic cells phagocytose apoptotic cells via αvβ5 and CD36, and cross-present antigens to cytotoxic T lymphocytes. J Exp Med. 1998;188(7):1359–1368. doi: 10.1084/jem.188.7.1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shen Z, Reznikoff G, Dranoff G, Rock KL. Cloned dendritic cells can present exogenous antigens on both MHC class I and class II molecules. J Immunol. 1997;158(6):2723–2730. [PubMed] [Google Scholar]
- 17.Pooley JL, Heath WR, Shortman K. Cutting edge: intravenous soluble antigen is presented to CD4 T cells by CD8− dendritic cells, but cross-presented to CD8 T cells by CD8+ dendritic cells. J Immunol. 2001;166(9):5327–5330. doi: 10.4049/jimmunol.166.9.5327. [DOI] [PubMed] [Google Scholar]
- 18.Zitvogel L, Regnault A, Lozier A, et al. Eradication of established murine tumors using a novel cell-free vaccine: dendritic cell-derived exosomes. Nat Med. 1998;4(5):594–600. doi: 10.1038/nm0598-594. [DOI] [PubMed] [Google Scholar]
- 19.Chandawarkar RY, Wagh MS, Srivastava PK. The dual nature of specific immunological activity of tumor-derived gp96 preparations. J Exp Med. 1999;189(9):1437–1442. doi: 10.1084/jem.189.9.1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang AY, Golumbek P, Ahmadzadeh M, Jaffee E, Pardoll D, Levitsky H. Role of bone marrow-derived cells in presenting MHC class I-restricted tumor antigens. Science. 1994;264(5161):961–965. doi: 10.1126/science.7513904. [DOI] [PubMed] [Google Scholar]
- 21.Heath WR, Carbone FR. Cross-presentation in viral immunity and self-tolerance. Nat Rev Immunol. 2001;1(2):126–134. doi: 10.1038/35100512. [DOI] [PubMed] [Google Scholar]
- 22■.Kreer C, Rauen J, Zehner M, Burgdorf S. Cross-presentation: how to get there – or how to get the ER. Front Immunol. 2011;2:87. doi: 10.3389/fimmu.2011.00087. Reviews various mechanisms underlying cross-presentation in dendritic cells (DCs) highlighting both the complexity and diversity of these cellular pathways and pointing out new research directions for understanding this important capability. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Delamarre L, Pack M, Chang H, Mellman I, Trombetta ES. Differential lysosomal proteolysis in antigen-presenting cells determines antigen fate. Science. 2005;307(5715):1630–1634. doi: 10.1126/science.1108003. [DOI] [PubMed] [Google Scholar]
- 24.Savina A, Jancic C, Hugues S, et al. NOX2 controls phagosomal pH to regulate antigen processing during crosspresentation by dendritic cells. Cell. 2006;126(1):205–218. doi: 10.1016/j.cell.2006.05.035. [DOI] [PubMed] [Google Scholar]
- 25.Mantegazza AR, Savina A, Vermeulen M, et al. NADPH oxidase controls phagosomal pH and antigen cross-presentation in human dendritic cells. Blood. 2008;112(12):4712–4722. doi: 10.1182/blood-2008-01-134791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hotta C, Fujimaki H, Yoshinari M, Nakazawa M, Minami M. The delivery of an antigen from the endocytic compartment into the cytosol for cross-presentation is restricted to early immature dendritic cells. Immunology. 2006;117(1):97–107. doi: 10.1111/j.1365-2567.2005.02270.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dudziak D, Kamphorst AO, Heidkamp GF, et al. Differential antigen processing by dendritic cell subsets in vivo. Science. 2007;315(5808):107–111. doi: 10.1126/science.1136080. [DOI] [PubMed] [Google Scholar]
- 28.Bonifaz LC, Bonnyay DP, Charalambous A, et al. In vivo targeting of antigens to maturing dendritic cells via the DEC-205 receptor improves T cell vaccination. J Exp Med. 2004;199(6):815–824. doi: 10.1084/jem.20032220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tsuji T, Matsuzaki J, Kelly MP, et al. Antibody-targeted NY-ESO-1 to mannose receptor or DEC-205 in vitro elicits dual human CD8+ and CD4+ T cell responses with broad antigen specificity. J Immunol. 2011;186(2):1218–1227. doi: 10.4049/jimmunol.1000808. [DOI] [PubMed] [Google Scholar]
- 30.Burgdorf S, Kautz A, Bohnert V, Knolle PA, Kurts C. Distinct pathways of antigen uptake and intracellular routing in CD4 and CD8 T cell activation. Science. 2007;316(5824):612–616. doi: 10.1126/science.1137971. [DOI] [PubMed] [Google Scholar]
- 31.Weck MM, Appel S, Werth D, et al. hDectin-1 is involved in uptake and cross-presentation of cellular antigens. Blood. 2008;111(8):4264–4272. doi: 10.1182/blood-2006-10-051375. [DOI] [PubMed] [Google Scholar]
- 32.Baker K, Qiao SW, Kuo TT, et al. Neonatal Fc receptor for IgG (FcRn) regulates cross-presentation of IgG immune complexes by CD8−C D11b+ dendritic cells. Proc Natl Acad Sci USA. 2011;108(24):9927–9932. doi: 10.1073/pnas.1019037108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chatterjee B, Smed-Sorensen A, Cohn L, et al. Internalization and endosomal degradation of receptor-bound antigens regulate the efficiency of cross presentation by human dendritic cells. Blood. 2012;120(10):2011–2020. doi: 10.1182/blood-2012-01-402370. [DOI] [PubMed] [Google Scholar]
- 34.Idoyaga J, Lubkin A, Fiorese C, et al. Comparable T helper 1 (Th1) and CD8 T-cell immunity by targeting HIV gag p24 to CD8 dendritic cells within antibodies to langerin, DEC205, and Clec9A. Proc Natl Acad Sci USA. 2011;108(6):2384–2389. doi: 10.1073/pnas.1019547108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Klechevsky E, Flamar AL, Cao Y, et al. Cross-priming CD8+ T cells by targeting antigens to human dendritic cells through DCIR. Blood. 2010;116(10):1685–1697. doi: 10.1182/blood-2010-01-264960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tacken PJ, Ginter W, Berod L, et al. Targeting DC-SIGN via its neck region leads to prolonged antigen residence in early endosomes, delayed lysosomal degradation, and cross-presentation. Blood. 2011;118(15):4111–4119. doi: 10.1182/blood-2011-04-346957. [DOI] [PubMed] [Google Scholar]
- 37.Burgdorf S, Scholz C, Kautz A, Tampe R, Kurts C. Spatial and mechanistic separation of cross-presentation and endogenous antigen presentation. Nat Immunol. 2008;9(5):558–566. doi: 10.1038/ni.1601. [DOI] [PubMed] [Google Scholar]
- 38.Ackerman AL, Giodini A, Cresswell P. A role for the endoplasmic reticulum protein retrotranslocation machinery during crosspresentation by dendritic cells. Immunity. 2006;25(4):607–617. doi: 10.1016/j.immuni.2006.08.017. [DOI] [PubMed] [Google Scholar]
- 39.Ackerman AL, Kyritsis C, Tampe R, Cresswell P. Early phagosomes in dendritic cells form a cellular compartment sufficient for cross presentation of exogenous antigens. Proc Natl Acad Sci USA. 2003;100(22):12889–12894. doi: 10.1073/pnas.1735556100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kovacsovics-Bankowski M, Rock KL. A phagosome-to-cytosol pathway for exogenous antigens presented on MHC class I molecules. Science. 1995;267(5195):243–246. doi: 10.1126/science.7809629. [DOI] [PubMed] [Google Scholar]
- 41.Rock KL, Shen L. Cross-presentation: underlying mechanisms and role in immune surveillance. Immunol Rev. 2005;207:166–183. doi: 10.1111/j.0105-2896.2005.00301.x. [DOI] [PubMed] [Google Scholar]
- 42.Young JW, Steinman RM. Dendritic cells stimulate primary human cytolytic lymphocyte responses in the absence of CD4+ helper T cells. J Exp Med. 1990;171(4):1315–1332. doi: 10.1084/jem.171.4.1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bhardwaj N, Bender A, Gonzalez N, Bui LK, Garrett MC, Steinman RM. Infuenza virus-inflected dendritic cells stimulate strong proliferative and cytolytic responses from human CD8+ T cells. J Clin Invest. 1994;94(2):797–807. doi: 10.1172/JCI117399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu YJ, Kanzler H, Soumelis V, Gilliet M. Dendritic cell lineage, plasticity and cross-regulation. Nat Immunol. 2001;2(7):585–589. doi: 10.1038/89726. [DOI] [PubMed] [Google Scholar]
- 45.Pulendran B, Smith JL, Caspary G, et al. Distinct dendritic cell subsets differentially regulate the class of immune response in vivo. Proc Natl Acad Sci USA. 1999;96(3):1036–1041. doi: 10.1073/pnas.96.3.1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Maldonado-Lopez R, De Smedt T, Michel P, et al. CD8α+ and CD8α− subclasses of dendritic cells direct the development of distinct T helper cells in vivo. J Exp Med. 1999;189(3):587–592. doi: 10.1084/jem.189.3.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.den Haan JM, Lehar SM, Bevan MJ. CD8(+) but not CD8(−) dendritic cells cross-prime cytotoxic T cells in vivo. J Exp Med. 2000;192(12):1685–1696. doi: 10.1084/jem.192.12.1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schnorrer P, Behrens GM, Wilson NS, et al. The dominant role of CD8+ dendritic cells in cross-presentation is not dictated by antigen capture. Proc Natl Acad Sci USA. 2006;103(28):10729–10734. doi: 10.1073/pnas.0601956103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Henri S, Poulin LF, Tamoutounour S, et al. CD207+ CD103+ dermal dendritic cells cross-present keratinocyte-derived antigens irrespective of the presence of Langerhans cells. J Exp Med. 2010;207(1):189–206. doi: 10.1084/jem.20091964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.del Rio ML, Rodriguez-Barbosa JI, Kremmer E, Forster R. CD103− and CD103+ bronchial lymph node dendritic cells are specialized in presenting and cross-presenting innocuous antigen to CD4+ and CD8+ T cells. J Immunol. 2007;178(11):6861–6866. doi: 10.4049/jimmunol.178.11.6861. [DOI] [PubMed] [Google Scholar]
- 51.Li Y, Wang LX, Pang P, et al. Tumor-derived autophagosome vaccine: mechanism of cross-presentation and therapeutic efficacy. Clin Cancer Res. 2011;17(22):7047–7057. doi: 10.1158/1078-0432.CCR-11-0951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zou W, Machelon V, Coulomb-L’Hermin A, et al. Stromal-derived factor-1 in human tumors recruits and alters the function of plasmacytoid precursor dendritic cells. Nat Med. 2001;7(12):1339–1346. doi: 10.1038/nm1201-1339. [DOI] [PubMed] [Google Scholar]
- 53.Shortman K, Liu YJ. Mouse and human dendritic cell subtypes. Nat Rev Immunol. 2002;2(3):151–161. doi: 10.1038/nri746. [DOI] [PubMed] [Google Scholar]
- 54.Kushwah R, Hu J. Role of dendritic cells in the induction of regulatory T cells. Cell Biosci. 2011;1(1):20. doi: 10.1186/2045-3701-1-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Karsunky H, Merad M, Cozzio A, Weissman IL, Manz MG. Flt3 ligand regulates dendritic cell development from Flt3+ lymphoid and myeloid-committed progenitors to Flt3+ dendritic cells in vivo. J Exp Med. 2003;198(2):305–313. doi: 10.1084/jem.20030323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schmid MA, Kingston D, Boddupalli S, Manz MG. Instructive cytokine signals in dendritic cell lineage commitment. Immunol Rev. 2010;234(1):32–44. doi: 10.1111/j.0105-2896.2009.00877.x. [DOI] [PubMed] [Google Scholar]
- 57.Waskow C, Liu K, Darrasse-Jeze G, et al. The receptor tyrosine kinase Flt3 is required for dendritic cell development in peripheral lymphoid tissues. Nat Immunol. 2008;9(6):676–683. doi: 10.1038/ni.1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.D’Amico A, Wu L. The early progenitors of mouse dendritic cells and plasmacytoid predendritic cells are within the bone marrow hemopoietic precursors expressing Flt3. J Exp Med. 2003;198(2):293–303. doi: 10.1084/jem.20030107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu K, Nussenzweig MC. Origin and development of dendritic cells. Immunol Rev. 2010;234(1):45–54. doi: 10.1111/j.0105-2896.2009.00879.x. [DOI] [PubMed] [Google Scholar]
- 60.Kushwah R, Hu J. Complexity of dendritic cell subsets and their function in the host immune system. Immunology. 2011;133(4):409–419. doi: 10.1111/j.1365-2567.2011.03457.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cella M, Jarrossay D, Facchetti F, et al. Plasmacytoid monocytes migrate to inflamed lymph nodes and produce large amounts of type I interferon. Nat Med. 1999;5(8):919–923. doi: 10.1038/11360. [DOI] [PubMed] [Google Scholar]
- 62.Grouard G, Rissoan MC, Filgueira L, Durand I, Banchereau J, Liu YJ. The enigmatic plasmacytoid T cells develop into dendritic cells with interleukin (IL)-3 and CD40-ligand. J Exp Med. 1997;185(6):1101–1111. doi: 10.1084/jem.185.6.1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Brawand P, Fitzpatrick DR, Greenfield BW, Brasel K, Maliszewski CR, De Smedt T. Murine plasmacytoid pre-dendritic cells generated from Flt3 ligand-supplemented bone marrow cultures are immature APCs. J Immunol. 2002;169(12):6711–6719. doi: 10.4049/jimmunol.169.12.6711. [DOI] [PubMed] [Google Scholar]
- 64.Rissoan MC, Soumelis V, Kadowaki N, et al. Reciprocal control of T helper cell and dendritic cell differentiation. Science. 1999;283(5405):1183–1186. doi: 10.1126/science.283.5405.1183. [DOI] [PubMed] [Google Scholar]
- 65.Ito T, Amakawa R, Inaba M, Ikehara S, Inaba K, Fukuhara S. Differential regulation of human blood dendritic cell subsets by IFNs. J Immunol. 2001;166(5):2961–2969. doi: 10.4049/jimmunol.166.5.2961. [DOI] [PubMed] [Google Scholar]
- 66.Matta BM, Castellaneta A, Thomson AW. Tolerogenic plasmacytoid DC. Eur J Immunol. 2010;40(10):2667–2676. doi: 10.1002/eji.201040839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gerlini G, Urso C, Mariotti G, et al. Plasmacytoid dendritic cells represent a major dendritic cell subset in sentinel lymph nodes of melanoma patients and accumulate in metastatic nodes. Clin Immunol. 2007;125(2):184–193. doi: 10.1016/j.clim.2007.07.018. [DOI] [PubMed] [Google Scholar]
- 68.Watkins SK, Zhu Z, Riboldi E, et al. FOXO3 programs tumor-associated DCs to become tolerogenic in human and murine prostate cancer. J Clin Invest. 2011;121(4):1361–1372. doi: 10.1172/JCI44325. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 69.Bronte V. Tolerogenic pDCs: spotlight on Foxo3. J Clin Invest. 2011;121(4):1247–1250. doi: 10.1172/JCI57190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zuniga EI, McGavern DB, Pruneda-Paz JL, Teng C, Oldstone MB. Bone marrow plasmacytoid dendritic cells can differentiate into myeloid dendritic cells upon virus infection. Nat Immunol. 2004;5(12):1227–1234. doi: 10.1038/ni1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Soumelis V, Liu YJ. From plasmacytoid to dendritic cell: morphological and functional switches during plasmacytoid pre-dendritic cell differentiation. Eur J Immunol. 2006;36(9):2286–2292. doi: 10.1002/eji.200636026. [DOI] [PubMed] [Google Scholar]
- 72.Krug A, Towarowski A, Britsch S, et al. Toll-like receptor expression reveals CpG DNA as a unique microbial stimulus for plasmacytoid dendritic cells which synergizes with CD40 ligand to induce high amounts of IL -12. Eur J Immunol. 2001;31(10):3026–3037. doi: 10.1002/1521-4141(2001010)31:10<3026::aid-immu3026>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 73.Banchereau J, Briere F, Caux C, et al. Immunobiology of dendritic cells. Annu Rev Immunol. 2000;18:767–811. doi: 10.1146/annurev.immunol.18.1.767. [DOI] [PubMed] [Google Scholar]
- 74.Wolf CE, Meyer M, Riggert J. Leukapheresis for the extraction of monocytes and various lymphocyte subpopulations from peripheral blood: product quality and prediction of the yield using different harvest procedures. Vox Sang. 2005;88(4):249–255. doi: 10.1111/j.1423-0410.2005.00562.x. [DOI] [PubMed] [Google Scholar]
- 75.Hornung V, Rothenfusser S, Britsch S, et al. Quantitative expression of toll-like receptor 1–10 mRNA in cellular subsets of human peripheral blood mononuclear cells and sensitivity to CpG oligodeoxynucleotides. J Immunol. 2002;168(9):4531–4537. doi: 10.4049/jimmunol.168.9.4531. [DOI] [PubMed] [Google Scholar]
- 76.Flacher V, Sparber F, Tripp CH, Romani N, Stoitzner P. Targeting of epidermal Langerhans cells with antigenic proteins: attempts to harness their properties for immunotherapy. Cancer Immunol Immunother. 2009;58(7):1137–1147. doi: 10.1007/s00262-008-0563-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ratzinger G, Baggers J, de Cos MA, et al. Mature human Langerhans cells derived from CD34+ hematopoietic progenitors stimulate greater cytolytic T lymphocyte activity in the absence of bioactive IL-12p70, by either single peptide presentation or cross-priming, than do dermal-interstitial or monocyte-derived dendritic cells. J Immunol. 2004;173(4):2780–2791. doi: 10.4049/jimmunol.173.4.2780. [DOI] [PubMed] [Google Scholar]
- 78.Santegoets SJ, Bontkes HJ, Stam AG, et al. Inducing antitumor T cell immunity: comparative functional analysis of interstitial versus Langerhans dendritic cells in a human cell line model. J Immunol. 2008;180(7):4540–4549. doi: 10.4049/jimmunol.180.7.4540. [DOI] [PubMed] [Google Scholar]
- 79.Romano E, Rossi M, Ratzinger G, et al. Peptide-loaded Langerhans cells, despite increased IL15 secretion and T-cell activation in vitro, elicit antitumor T-cell responses comparable to peptide-loaded monocyte-derived dendritic cells in vivo. Clin Cancer Res. 2011;17(7):1984–1997. doi: 10.1158/1078-0432.CCR-10-3421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80■.Aspord C, Charles J, Leccia MT, et al. A novel cancer vaccine strategy based on HLA-A*0201 matched allogeneic plasmacytoid dendritic cells. PLoS ONE. 2010;5(5):E10458. doi: 10.1371/journal.pone.0010458. Uniquely examined the possibility of using plasmacytoid DCs (pDCs) as part of a vaccine construct, and both in vitro assays as well as in vivo testing in a mouse model demonstrated the ability of pDCs to sensitize T cells to elicit an antigen-specific response. The findings of this study indicate the potential for pDCs to be applied successfully as part of DC-based anticancer vaccines. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chen W, Zhang Z, Shi M, et al. Activated plasmacytoid dendritic cells act synergistically with hepatitis B core antigen-pulsed monocyte-derived dendritic cells in the induction of hepatitis B virus-specific CD8 T-cell response. Clin Immunol. 2008;129(2):295–303. doi: 10.1016/j.clim.2008.07.026. [DOI] [PubMed] [Google Scholar]
- 82.Sallusto F, Schaerli P, Loetscher P, et al. Rapid and coordinated switch in chemokine receptor expression during dendritic cell maturation. Eur J Immunol. 1998;28(9):2760–2769. doi: 10.1002/(SICI)1521-4141(199809)28:09<2760::AID-IMMU2760>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 83.Caux C, Vanbervliet B, Massacrier C, et al. B70/B7-2 is identical to CD86 and is the major functional ligand for CD28 expressed on human dendritic cells. J Exp Med. 1994;180(5):1841–1847. doi: 10.1084/jem.180.5.1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.McLellan AD, Starling GC, Williams LA, Hock BD, Hart DN. Activation of human peripheral blood dendritic cells induces the CD86 co-stimulatory molecule. Eur J Immunol. 1995;25(7):2064–2068. doi: 10.1002/eji.1830250739. [DOI] [PubMed] [Google Scholar]
- 85.Albert ML, Jegathesan M, Darnell RB. Dendritic cell maturation is required for the cross-tolerization of CD8+ T cells. Nat Immunol. 2001;2(11):1010–1017. doi: 10.1038/ni722. [DOI] [PubMed] [Google Scholar]
- 86.Jonuleit H, Schmitt E, Schuler G, Knop J, Enk AH. Induction of interleukin 10-producing, nonproliferating CD4(+) T cells with regulatory properties by repetitive stimulation with allogeneic immature human dendritic cells. J Exp Med. 2000;192(9):1213–1222. doi: 10.1084/jem.192.9.1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Dhodapkar MV, Steinman RM, Krasovsky J, Munz C, Bhardwaj N. Antigen-specific inhibition of effector T cell function in humans after injection of immature dendritic cells. J Exp Med. 2001;193(2):233–238. doi: 10.1084/jem.193.2.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Jonuleit H, Schmitt E, Steinbrink K, Enk AH. Dendritic cells as a tool to induce anergic and regulatory T cells. Trends Immunol. 2001;22(7):394–400. doi: 10.1016/s1471-4906(01)01952-4. [DOI] [PubMed] [Google Scholar]
- 89.Tan PH, Yates JB, Xue SA, et al. Creation of tolerogenic human dendritic cells via intracellular CTLA4: a novel strategy with potential in clinical immunosuppression. Blood. 2005;106(9):2936–2943. doi: 10.1182/blood-2005-05-1826. [DOI] [PubMed] [Google Scholar]
- 90.Arce F, Breckpot K, Stephenson H, et al. Selective ERK activation differentiates mouse and human tolerogenic dendritic cells, expands antigen-specific regulatory T cells, and suppresses experimental inflammatory arthritis. Arthritis Rheum. 2011;63(1):84–95. doi: 10.1002/art.30099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Perona-Wright G, Anderton SM, Howie SE, Gray D. IL-10 permits transient activation of dendritic cells to tolerize T cells and protect from central nervous system autoimmune disease. Int Immunol. 2007;19(9):1123–1134. doi: 10.1093/intimm/dxm084. [DOI] [PubMed] [Google Scholar]
- 92.Loscher CE, Draper E, Leavy O, Kelleher D, Mills KH, Roche HM. Conjugated linoleic acid suppresses NF-κ B activation and IL-12 production in dendritic cells through ERK-mediated IL-10 induction. J Immunol. 2005;175(8):4990–4998. doi: 10.4049/jimmunol.175.8.4990. [DOI] [PubMed] [Google Scholar]
- 93.Banerjee DK, Dhodapkar MV, Matayeva E, Steinman RM, Dhodapkar KM. Expansion of FOXP3high regulatory T cells by human dendritic cells (DCs) in vitro and after injection of cytokine-matured DCs in myeloma patients. Blood. 2006;108(8):2655–2661. doi: 10.1182/blood-2006-03-011353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Mahnke K, Johnson TS, Ring S, Enk AH. Tolerogenic dendritic cells and regulatory T cells: a two-way relationship. J Dermatol Sci. 2007;46(3):159–167. doi: 10.1016/j.jdermsci.2007.03.002. [DOI] [PubMed] [Google Scholar]
- 95.Almand B, Clark JI, Nikitina E, et al. Increased production of immature myeloid cells in cancer patients: a mechanism of immunosuppression in cancer. J Immunol. 2001;166(1):678–689. doi: 10.4049/jimmunol.166.1.678. [DOI] [PubMed] [Google Scholar]
- 96.Gabrilovich D, Ishida T, Oyama T, et al. Vascular endothelial growth factor inhibits the development of dendritic cells and dramatically affects the differentiation of multiple hematopoietic lineages in vivo. Blood. 1998;92(11):4150–4166. [PubMed] [Google Scholar]
- 97.Kusmartsev S, Nefedova Y, Yoder D, Gabrilovich DI. Antigen-specific inhibition of CD8+ T cell response by immature myeloid cells in cancer is mediated by reactive oxygen species. J Immunol. 2004;172(2):989–999. doi: 10.4049/jimmunol.172.2.989. [DOI] [PubMed] [Google Scholar]
- 98.Serafini P, Borrello I, Bronte V. Myeloid suppressor cells in cancer: recruitment, phenotype, properties, and mechanisms of immune suppression. Semin Cancer Biol. 2006;16(1):53–65. doi: 10.1016/j.semcancer.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 99.Thomas DA, Massague J. TGF-β directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell. 2005;8(5):369–380. doi: 10.1016/j.ccr.2005.10.012. [DOI] [PubMed] [Google Scholar]
- 100.Valenti R, Huber V, Filipazzi P, et al. Human tumor-released microvesicles promote the differentiation of myeloid cells with transforming growth factor-β-mediated suppressive activity on T lymphocytes. Cancer Res. 2006;66(18):9290–9298. doi: 10.1158/0008-5472.CAN-06-1819. [DOI] [PubMed] [Google Scholar]
- 101.Loercher AE, Nash MA, Kavanagh JJ, Platsoucas CD, Freedman RS. Identification of an IL-10-producing HLA-DR-negative monocyte subset in the malignant ascites of patients with ovarian carcinoma that inhibits cytokine protein expression and proliferation of autologous T cells. J Immunol. 1999;163(11):6251–6260. [PubMed] [Google Scholar]
- 102.Filipazzi P, Valenti R, Huber V, et al. Identification of a new subset of myeloid suppressor cells in peripheral blood of melanoma patients with modulation by a granulocyte-macrophage colony-stimulation factor-based antitumor vaccine. J Clin Oncol. 2007;25(18):2546–2553. doi: 10.1200/JCO.2006.08.5829. [DOI] [PubMed] [Google Scholar]
- 103.Peranzoni E, Zilio S, Marigo I, et al. Myeloid-derived suppressor cell heterogeneity and subset definition. Curr Opin Immunol. 2010;22(2):238–244. doi: 10.1016/j.coi.2010.01.021. [DOI] [PubMed] [Google Scholar]
- 104.Ribechini E, Greifenberg V, Sandwick S, Lutz MB. Subsets, expansion and activation of myeloid-derived suppressor cells. Med Microbiol Immunol. 2010;199(3):273–281. doi: 10.1007/s00430-010-0151-4. [DOI] [PubMed] [Google Scholar]
- 105.Nagaraj S, Gabrilovich DI. Myeloid-derived suppressor cells in human cancer. Cancer J. 2010;16(4):348–353. doi: 10.1097/PPO.0b013e3181eb3358. [DOI] [PubMed] [Google Scholar]
- 106.Vuk-Pavlovic S, Bulur PA, Lin Y, et al. Immunosuppressive CD14+HLA-DRlow/-monocytes in prostate cancer. Prostate. 2010;70(4):443–455. doi: 10.1002/pros.21078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Awasthi A, Carrier Y, Peron JP, et al. A dominant function for interleukin 27 in generating interleukin 10-producing anti-inflammatory T cells. Nat Immunol. 2007;8(12):1380–1389. doi: 10.1038/ni1541. [DOI] [PubMed] [Google Scholar]
- 108.Lan Q, Zhou X, Fan H, et al. Polyclonal CD4+Foxp3+Treg cells induce TGFβ-dependent tolerogenic dendritic cells that suppress murine lupus-like syndrome. J Mol Cell Biol. 2012 doi: 10.1093/jmcb/mjs040. (Epub ahead of print) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Pallotta MT, Orabona C, Volpi C, et al. Indoleamine 2,3-dioxygenase is a signaling protein in long-term tolerance by dendritic cells. Nat Immunol. 2011;12(9):870–878. doi: 10.1038/ni.2077. [DOI] [PubMed] [Google Scholar]
- 110.Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. 2003;4(4):330–336. doi: 10.1038/ni904. [DOI] [PubMed] [Google Scholar]
- 111.Qin S, Cobbold SP, Pope H, et al. “Infectious” transplantation tolerance. Science. 1993;259(5097):974–977. doi: 10.1126/science.8094901. [DOI] [PubMed] [Google Scholar]
- 112.Cobbold SP. Future therapeutics for the induction of peripheral immune tolerance in autoimmune disease and organ transplantation. Immunotherapy. 2009;1(3):447–460. doi: 10.2217/imt.09.9. [DOI] [PubMed] [Google Scholar]
- 113.Schmieder A, Michel J, Schonhaar K, Goerdt S, Schledzewski K. Differentiation and gene expression profile of tumor-associated macrophages. Semin Cancer Biol. 2012;22(4):289–297. doi: 10.1016/j.semcancer.2012.02.002. [DOI] [PubMed] [Google Scholar]
- 114.Rabinovich GA, Gabrilovich D, Sotomayor EM. Immunosuppressive strategies that are mediated by tumor cells. Annu Rev Immunol. 2007;25:267–296. doi: 10.1146/annurev.immunol.25.022106.141609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Pulendran B, Smith JL, Jenkins M, Schoenborn M, Maraskovsky E, Maliszewski CR. Prevention of peripheral tolerance by a dendritic cell growth factor: flt3 ligand as an adjuvant. J Exp Med. 1998;188(11):2075–2082. doi: 10.1084/jem.188.11.2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Gong J, Chen D, Kashiwaba M, et al. Reversal of tolerance to human MUC1 antigen in MUC1 transgenic mice immunized with fusions of dendritic and carcinoma cells. Proc Natl Acad Sci USA. 1998;95(11):6279–6283. doi: 10.1073/pnas.95.11.6279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Steptoe RJ, Fu F, Li W, et al. Augmentation of dendritic cells in murine organ donors by Flt3 ligand alters the balance between transplant tolerance and immunity. J Immunol. 1997;159(11):5483–5491. [PubMed] [Google Scholar]
- 118.Bronte V, Apolloni E, Cabrelle A, et al. Identification of a CD11b(+)/Gr-1(+)/CD31(+) myeloid progenitor capable of activating or suppressing CD8(+) T cells. Blood. 2000;96(12):3838–3846. [PMC free article] [PubMed] [Google Scholar]
- 119.Terabe M, Shimizu M, Mabuchi A, et al. Unresponsiveness of intrahepatic lymphocytes to bacterial superantigen: rapid development of suppressive Mac-1(high) cells in the mouse liver. Hepatology. 2000;32(3):507–513. doi: 10.1053/JHEP.2000.9875. [DOI] [PubMed] [Google Scholar]
- 120.Fiorucci S, Santucci L, Gresele P, Faccino RM, Del Soldato P, Morelli A. Gastrointestinal safety of NO-aspirin (NCX-4016) in healthy human volunteers: a proof of concept endoscopic study. Gastroenterology. 2003;124(3):600–607. doi: 10.1053/gast.2003.50096. [DOI] [PubMed] [Google Scholar]
- 121.De Santo C, Serafini P, Marigo I, et al. Nitroaspirin corrects immune dysfunction in tumor-bearing hosts and promotes tumor eradication by cancer vaccination. Proc Natl Acad Sci USA. 2005;102(11):4185–4190. doi: 10.1073/pnas.0409783102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Sallusto F, Lanzavecchia A. Efficient presentation of soluble antigen by cultured human dendritic cells is maintained by granulocyte/macrophage colony-stimulating factor plus interleukin 4 and downregulated by tumor necrosis factor α. J Exp Med. 1994;179(4):1109–1118. doi: 10.1084/jem.179.4.1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Czerniecki BJ, Carter C, Rivoltini L, et al. Calcium ionophore-treated peripheral blood monocytes and dendritic cells rapidly display characteristics of activated dendritic cells. J Immunol. 1997;159(8):3823–3837. [PubMed] [Google Scholar]
- 124.Dauer M, Obermaier B, Herten J, et al. Mature dendritic cells derived from human monocytes within 48 hours: a novel strategy for dendritic cell differentiation from blood precursors. J Immunol. 2003;170(8):4069–4076. doi: 10.4049/jimmunol.170.8.4069. [DOI] [PubMed] [Google Scholar]
- 125.Lyakh LA, Koski GK, Telford W, Gress RE, Cohen PA, Rice NR. Bacterial lipopolysaccharide, TNF-α, and calcium ionophore under serum-free conditions promote rapid dendritic cell-like differentiation in CD14+ monocytes through distinct pathways that activate NK-κB. J Immunol. 2000;165(7):3647–3655. doi: 10.4049/jimmunol.165.7.3647. [DOI] [PubMed] [Google Scholar]
- 126.Koski GK, Lyakh LA, Rice NR. Rapid lipopolysaccharide-induced differentiation of CD14(+) monocytes into CD83(+) dendritic cells is modulated under serum-free conditions by exogenously added IFN-γ and endogenously produced IL-10. Eur J Immunol. 2001;31(12):3773–3781. doi: 10.1002/1521-4141(200112)31:12<3773::aid-immu3773>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 127.Vanderlocht J, Van Elssen CH, Senden-Gijsbers BL, et al. Increased tumor-specific CD8+ T cell induction by dendritic cells matured with a clinical grade TLR-agonist in combination with IFN-γ. Int J Immunopathol Pharmacol. 2010;23(1):35–50. doi: 10.1177/039463201002300104. [DOI] [PubMed] [Google Scholar]
- 128■.Breckpot K, Escors D. Dendritic cells for active anti-cancer immunotherapy: targeting activation pathways through genetic modification. Endocr Metab Immune Disord Drug Targets. 2009;9(4):328–343. doi: 10.2174/187153009789839156. Emphasizes the importance of DC activation state when developing DC-based vaccines and reviews the importance of Toll-like receptor (TLR) pathways in activating DCs. Furthermore, genetic modification to take advantage of activation pathways and the potential therapeutic role of this technology is addressed, with various strategies being reviewed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129■■.Bonehill A, Tuyaerts S, Van Nuffel AM, et al. Enhancing the T-cell stimulatory capacity of human dendritic cells by co-electroporation with CD40L, CD70 and constitutively active TLR4 encoding mRNA. Mol Ther J Am Soc Gene Ther. 2008;16(6):1170–1180. doi: 10.1038/mt.2008.77. Demonstrates successful enhancement of the ability of DCs to stimulate antigen-specific T-cell responses, a necessary component of DC-based vaccines, by genetically modifying DCs to take advantage of known stimulatory pathways. These results support genetically engineering DCs as a potential method for overcoming immune tolerance and improving the success of DC-based vaccines. [DOI] [PubMed] [Google Scholar]
- 130.Mosmann TR, Cherwinski H, Bond MW, Giedlin MA, Coffman RL. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J Immunol. 1986;136(7):2348–2357. [PubMed] [Google Scholar]
- 131.O’Garra A. Cytokines induce the development of functionally heterogeneous T helper cell subsets. Immunity. 1998;8(3):275–283. doi: 10.1016/s1074-7613(00)80533-6. [DOI] [PubMed] [Google Scholar]
- 132.Seder RA, Paul WE. Acquisition of lymphokine-producing phenotype by CD4+ T cells. Annu Rev Immunol. 1994;12:635–673. doi: 10.1146/annurev.iy.12.040194.003223. [DOI] [PubMed] [Google Scholar]
- 133.Abbas AK, Murphy KM, Sher A. Functional diversity of helper T lymphocytes. Nature. 1996;383(6603):787–793. doi: 10.1038/383787a0. [DOI] [PubMed] [Google Scholar]
- 134.O’Garra A, Murphy K. T-cell subsets in autoimmunity. Curr Opin Immunol. 1993;5(6):880–886. doi: 10.1016/0952-7915(93)90100-7. [DOI] [PubMed] [Google Scholar]
- 135.Liblau RS, Singer SM, McDevitt HO. Th1 and Th2 CD4+ T cells in the pathogenesis of organ-specific autoimmune diseases. Immunol Today. 1995;16(1):34–38. doi: 10.1016/0167-5699(95)80068-9. [DOI] [PubMed] [Google Scholar]
- 136.Nishimura T, Iwakabe K, Sekimoto M, et al. Distinct role of antigen-specific T helper type 1 (Th1) and Th2 cells in tumor eradication in vivo. J Exp Med. 1999;190(5):617–627. doi: 10.1084/jem.190.5.617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Ji Y, Zhang W. Th17 cells: positive or negative role in tumor? Cancer Immunol Immunother. 2010;59(7):979–987. doi: 10.1007/s00262-010-0849-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Nishimura T, Nakui M, Sato M, et al. The critical role of Th1-dominant immunity in tumor immunology. Cancer Chemother Pharmacol. 2000;46(Suppl):S52–S61. doi: 10.1007/pl00014051. [DOI] [PubMed] [Google Scholar]
- 139.Czerniecki BJ, Roses RE, Koski GK. Development of vaccines for high-risk ductal carcinoma in situ of the breast. Cancer Res. 2007;67(14):6531–6534. doi: 10.1158/0008-5472.CAN-07-0878. [DOI] [PubMed] [Google Scholar]
- 140.Feili-Hariri M, Falkner DH, Morel PA. Polarization of naive T cells into Th1 or Th2 by distinct cytokine-driven murine dendritic cell populations: implications for immunotherapy. J Leukoc Biol. 2005;78(3):656–664. doi: 10.1189/jlb.1104631. [DOI] [PubMed] [Google Scholar]
- 141.Napolitani G, Rinaldi A, Bertoni F, Sallusto F, Lanzavecchia A. Selected toll-like receptor agonist combinations synergistically trigger a T helper type 1-polarizing program in dendritic cells. Nat Immunol. 2005;6(8):769–776. doi: 10.1038/ni1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.de Jong EC, Vieira PL, Kalinski P, et al. Microbial compounds selectively induce Th1 cell-promoting or Th2 cell-promoting dendritic cells in vitro with diverse TH cell-polarizing signals. J Immunol. 2002;168(4):1704–1709. doi: 10.4049/jimmunol.168.4.1704. [DOI] [PubMed] [Google Scholar]
- 143.Snijders A, Kalinski P, Hilkens CM, Kapsenberg ML. High-level IL-12 production by human dendritic cells requires two signals. Int Immunol. 1998;10(11):1593–1598. doi: 10.1093/intimm/10.11.1593. [DOI] [PubMed] [Google Scholar]
- 144.Xu S, Koski GK, Faries M, et al. Rapid high efficiency sensitization of CD8+ T cells to tumor antigens by dendritic cells leads to enhanced functional avidity and direct tumor recognition through an IL-12-dependent mechanism. J Immunol. 2003;171(5):2251–2261. doi: 10.4049/jimmunol.171.5.2251. [DOI] [PubMed] [Google Scholar]
- 145.Trinchieri G. Interleukin-12: a proinflammatory cytokine with immunoregulatory functions that bridge innate resistance and antigen-specific adaptive immunity. Annu Rev Immunol. 1995;13:251–276. doi: 10.1146/annurev.iy.13.040195.001343. [DOI] [PubMed] [Google Scholar]
- 146.Hsieh CS, Macatonia SE, Tripp CS, Wolf SF, O’Garra A, Murphy KM. Development of TH1 CD4+ T cells through IL-12 produced by Listeria-induced macrophages. Science. 1993;260(5107):547–549. doi: 10.1126/science.8097338. [DOI] [PubMed] [Google Scholar]
- 147.Kobayashi M, Fitz L, Ryan M, et al. Identification and purification of natural killer cell stimulatory factor (NKSF), a cytokine with multiple biologic effects on human lymphocytes. J Exp Med. 1989;170(3):827–845. doi: 10.1084/jem.170.3.827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Moser M, Murphy KM. Dendritic cell regulation of TH1-TH2 development. Nat Immunol. 2000;1(3):199–205. doi: 10.1038/79734. [DOI] [PubMed] [Google Scholar]
- 149.Heufler C, Koch F, Stanzl U, et al. Interleukin-12 is produced by dendritic cells and mediates T helper 1 development as well as interferon-γ production by T helper 1 cells. Eur J Immunol. 1996;26(3):659–668. doi: 10.1002/eji.1830260323. [DOI] [PubMed] [Google Scholar]
- 150.Voest EE, Kenyon BM, O’Reilly MS, Truitt G, D’Amato RJ, Folkman J. Inhibition of angiogenesis in vivo by interleukin 12. J Natl Cancer Inst. 1995;87(8):581–586. doi: 10.1093/jnci/87.8.581. [DOI] [PubMed] [Google Scholar]
- 151.Chan SH, Perussia B, Gupta JW, et al. Induction of interferon γ production by natural killer cell stimulatory factor: characterization of the responder cells and synergy with other inducers. J Exp Med. 1991;173(4):869–879. doi: 10.1084/jem.173.4.869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Colombo MP, Trinchieri G. Interleukin-12 in anti-tumor immunity and immunotherapy. Cytokine Growth Factor Rev. 2002;13(2):155–168. doi: 10.1016/s1359-6101(01)00032-6. [DOI] [PubMed] [Google Scholar]
- 153.Dhodapkar MV, Young JW, Chapman PB, et al. Paucity of functional T-cell memory to melanoma antigens in healthy donors and melanoma patients. Clin Cancer Res. 2000;6(12):4831–4838. [PubMed] [Google Scholar]
- 154.Yang S, Linette GP, Longerich S, Haluska FG. Antimelanoma activity of CTL generated from peripheral blood mononuclear cells after stimulation with autologous dendritic cells pulsed with melanoma gp100 peptide G209-2M is correlated to TCR avidity. J Immunol. 2002;169(1):531–539. doi: 10.4049/jimmunol.169.1.531. [DOI] [PubMed] [Google Scholar]
- 155.Zaks TZ, Rosenberg SA. Immunization with a peptide epitope (p369–377) from HER-2/neu leads to peptide-specific cytotoxic T lymphocytes that fail to recognize HER-2/neu+ tumors. Cancer Res. 1998;58(21):4902–4908. [PubMed] [Google Scholar]
- 156.Czerniecki BJ, Koski GK, Koldovsky U, et al. Targeting HER-2/neu in early breast cancer development using dendritic cells with staged interleukin-12 burst secretion. Cancer Res. 2007;67(4):1842–1852. doi: 10.1158/0008-5472.CAN-06-4038. [DOI] [PubMed] [Google Scholar]
- 157■■.Sharma A, Koldovsky U, Xu S, et al. HER-2 pulsed dendritic cell vaccine can eliminate HER-2 expression and impact ductal carcinoma in situ. Cancer. 2012;118(17):4354–4362. doi: 10.1002/cncr.26734. A clinical trial of a DC-based vaccine applied in early-stage cancer with promising preliminary clinical results. This trial reflects and supports a shift towards administering cancer vaccines in earlier stage disease. Another unique aspect of this trial was the use of DC1-phenotype matured with TLR agonists in contrast to the more classically employed cytokine-matured DCs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Muranski P, Restifo NP. Does IL-17 promote tumor growth? Blood. 2009;114(2):231–232. doi: 10.1182/blood-2009-04-215541. [DOI] [PubMed] [Google Scholar]
- 159.Mattes J, Hulett M, Xie W, et al. Immunotherapy of cytotoxic T cell-resistant tumors by T helper 2 cells: an eotaxin and STAT6-dependent process. J Exp Med. 2003;197(3):387–393. doi: 10.1084/jem.20021683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Dunn GP, Bruce AT, Sheehan KC, et al. A critical function for type I interferons in cancer immunoediting. Nat Immunol. 2005;6(7):722–729. doi: 10.1038/ni1213. [DOI] [PubMed] [Google Scholar]
- 161.Dunn GP, Koebel CM, Schreiber RD. Interferons, immunity and cancer immunoediting. Nat Rev Immunol. 2006;6(11):836–848. doi: 10.1038/nri1961. [DOI] [PubMed] [Google Scholar]
- 162.Aspord C, Pedroza-Gonzalez A, Gallegos M, et al. Breast cancer instructs dendritic cells to prime interleukin 13-secreting CD4+ T cells that facilitate tumor development. J Exp Med. 2007;204(5):1037–1047. doi: 10.1084/jem.20061120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.DeNardo DG, Barreto JB, Andreu P, et al. CD4(+) T cells regulate pulmonary metastasis of mammary carcinomas by enhancing protumor properties of macrophages. Cancer Cell. 2009;16(2):91–102. doi: 10.1016/j.ccr.2009.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Harrington LE, Hatton RD, Mangan PR, et al. Interleukin 17-producing CD4+effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol. 2005;6(11):1123–1132. doi: 10.1038/ni1254. [DOI] [PubMed] [Google Scholar]
- 165.Langrish CL, Chen Y, Blumenschein WM, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201(2):233–240. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.McKenzie BS, Kastelein RA, Cua DJ. Understanding the IL-23-IL-17 immune pathway. Trends Immunol. 2006;27(1):17–23. doi: 10.1016/j.it.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 167■.Roses RE, Xu S, Xu M, Koldovsky U, Koski G, Czerniecki BJ. Differential production of IL-23 and IL-12 by myeloid-derived dendritic cells in response to TLR agonists. J Immunol. 2008;181(7):5120–5127. doi: 10.4049/jimmunol.181.7.5120. Elucidates signals that induce a DC17 phenotype and also demonstrates the ability to prime T cells using DC1s and DC17s to elicit Th1- and Th17-polarized antigen-specific responses, respectively. These findings lay groundwork for conducting further testing of the role of the Th17 response in cancer immunity. [DOI] [PubMed] [Google Scholar]
- 168.Acosta-Rodriguez EV, Napolitani G, Lanzavecchia A, Sallusto F. Interleukins 1β and 6 but not transforming growth factor-β are essential for the differentiation of interleukin 17-producing human T helper cells. Nat Immunol. 2007;8(9):942–949. doi: 10.1038/ni1496. [DOI] [PubMed] [Google Scholar]
- 169.Langowski JL, Zhang X, Wu L, et al. IL-23 promotes tumour incidence and growth. Nature. 2006;442(7101):461–465. doi: 10.1038/nature04808. [DOI] [PubMed] [Google Scholar]
- 170.Kryczek I, Wei S, Zou L, et al. Cutting edge: Th17 and regulatory T cell dynamics and the regulation by IL-2 in the tumor microenvironment. J Immunol. 2007;178(11):6730–6733. doi: 10.4049/jimmunol.178.11.6730. [DOI] [PubMed] [Google Scholar]
- 171.Sfanos KS, Bruno TC, Maris CH, et al. Phenotypic analysis of prostate-infiltrating lymphocytes reveals TH17 and Treg skewing. Clin Cancer Res. 2008;14(11):3254–3261. doi: 10.1158/1078-0432.CCR-07-5164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Kryczek I, Banerjee M, Cheng P, et al. Phenotype, distribution, generation, and functional and clinical relevance of Th17 cells in the human tumor environments. Blood. 2009;114(6):1141–1149. doi: 10.1182/blood-2009-03-208249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Yang ZZ, Novak AJ, Ziesmer SC, Witzig TE, Ansell SM. Malignant B cells skew the balance of regulatory T cells and TH17 cells in B-cell non-Hodgkin’s lymphoma. Cancer Res. 2009;69(13):5522–5530. doi: 10.1158/0008-5472.CAN-09-0266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Horlock C, Stott B, Dyson PJ, et al. The effects of trastuzumab on the CD4+CD25+FoxP3+ and CD4+I L17A+ T- cell axis in patients with breast cancer. Br J Cancer. 2009;100(7):1061–1067. doi: 10.1038/sj.bjc.6604963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Numasaki M, Fukushi J, Ono M, et al. Interleukin-17 promotes angiogenesis and tumor growth. Blood. 2003;101(7):2620–2627. doi: 10.1182/blood-2002-05-1461. [DOI] [PubMed] [Google Scholar]
- 176.Yu H, Kortylewski M, Pardoll D. Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nat Rev Immunol. 2007;7(1):41–51. doi: 10.1038/nri1995. [DOI] [PubMed] [Google Scholar]
- 177.Takahashi H, Numasaki M, Lotze MT, Sasaki H. Interleukin-17 enhances bFGF-, HGF- and VEGF-induced growth of vascular endothelial cells. Immunol Lett. 2005;98(2):189–193. doi: 10.1016/j.imlet.2004.11.012. [DOI] [PubMed] [Google Scholar]
- 178.Inozume T, Hanada K, Wang QJ, Yang JC. IL-17 secreted by tumor reactive T cells induces IL-8 release by human renal cancer cells. J Immunother. 2009;32(2):109–117. doi: 10.1097/CJI.0b013e31819302da. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Lin WW, Karin M. A cytokine-mediated link between innate immunity, inflammation, and cancer. J Clin Invest. 2007;117(5):1175–1183. doi: 10.1172/JCI31537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Zhang Q, Liu S, Ge D, et al. Interleukin-17 promotes formation and growth of prostate adenocarcinoma in mouse models. Cancer Res. 2012;72(10):2589–2599. doi: 10.1158/0008-5472.CAN-11-3795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Benchetrit F, Ciree A, Vives V, et al. Interleukin-17 inhibits tumor cell growth by means of a T-cell-dependent mechanism. Blood. 2002;99(6):2114–2121. doi: 10.1182/blood.v99.6.2114. [DOI] [PubMed] [Google Scholar]
- 182.Kortylewski M, Kujawski M, Wang T, et al. Inhibiting Stat3 signaling in the hematopoietic system elicits multicomponent antitumor immunity. Nat Med. 2005;11(12):1314–1321. doi: 10.1038/nm1325. [DOI] [PubMed] [Google Scholar]
- 183.Overwijk WW, de Visser KE, Tirion FH, et al. Immunological and antitumor effects of IL-23 as a cancer vaccine adjuvant. J Immunol. 2006;176(9):5213–5222. doi: 10.4049/jimmunol.176.9.5213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Kaiga T, Sato M, Kaneda H, Iwakura Y, Takayama T, Tahara H. Systemic administration of IL-23 induces potent antitumor immunity primarily mediated through Th1-type response in association with the endogenously expressed IL-12. J Immunol. 2007;178(12):7571–7580. doi: 10.4049/jimmunol.178.12.7571. [DOI] [PubMed] [Google Scholar]
- 185.Lo CH, Lee SC, Wu PY, et al. Antitumor and antimetastatic activity of IL-23. J Immunol. 2003;171(2):600–607. doi: 10.4049/jimmunol.171.2.600. [DOI] [PubMed] [Google Scholar]
- 186.Hu J, Yuan X, Belladonna ML, et al. Induction of potent antitumor immunity by intratumoral injection of interleukin 23-transduced dendritic cells. Cancer Res. 2006;66(17):8887–8896. doi: 10.1158/0008-5472.CAN-05-3448. [DOI] [PubMed] [Google Scholar]
- 187.Bettelli E, Oukka M, Kuchroo VK. T(H)-17 cells in the circle of immunity and autoimmunity. Nat Immunol. 2007;8(4):345–350. doi: 10.1038/ni0407-345. [DOI] [PubMed] [Google Scholar]
- 188.Muranski P, Boni A, Antony PA, et al. Tumor-specific Th17-polarized cells eradicate large established melanoma. Blood. 2008;112(2):362–373. doi: 10.1182/blood-2007-11-120998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Lee YK, Turner H, Maynard CL, et al. Late developmental plasticity in the T helper 17 lineage. Immunity. 2009;30(1):92–107. doi: 10.1016/j.immuni.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Suryani S, Sutton I. An interferon-γ-producing Th1 subset is the major source of IL-17 in experimental autoimmune encephalitis. J Neuroimmunol. 2007;183(1–2):96–103. doi: 10.1016/j.jneuroim.2006.11.023. [DOI] [PubMed] [Google Scholar]
- 191.Knutson KL, Disis ML. Tumor antigen-specific T helper cells in cancer immunity and immunotherapy. Cancer Immunol Immunother. 2005;54(8):721–728. doi: 10.1007/s00262-004-0653-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Bonehill A, Heirman C, Thielemans K. Genetic approaches for the induction of a CD4+ T cell response in cancer immunotherapy. J Gene Med. 2005;7(6):686–695. doi: 10.1002/jgm.713. [DOI] [PubMed] [Google Scholar]
- 193.Nestle FO, Alijagic S, Gilliet M, et al. Vaccination of melanoma patients with peptide- or tumor lysate-pulsed dendritic cells. Nat Med. 1998;4(3):328–332. doi: 10.1038/nm0398-328. [DOI] [PubMed] [Google Scholar]
- 194.Rosenberg SA, Yang JC, Schwartzentruber DJ, et al. Immunologic and therapeutic evaluation of a synthetic peptide vaccine for the treatment of patients with metastatic melanoma. Nat Med. 1998;4(3):321–327. doi: 10.1038/nm0398-321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Dranoff G, Jaffee E, Lazenby A, et al. Vaccination with irradiated tumor cells engineered to secrete murine granulocyte-macrophage colony-stimulating factor stimulates potent, specific, and long-lasting anti-tumor immunity. Proc Natl Acad Sci USA. 1993;90(8):3539–3543. doi: 10.1073/pnas.90.8.3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Hung K, Hayashi R, Lafond-Walker A, Lowenstein C, Pardoll D, Levitsky H. The central role of CD4(+) T cells in the antitumor immune response. J Exp Med. 1998;188(12):2357–2368. doi: 10.1084/jem.188.12.2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Schattner EJ, Mascarenhas J, Bishop J, et al. CD4+ T-cell induction of Fas-mediated apoptosis in Burkitt’s lymphoma B cells. Blood. 1996;88(4):1375–1382. [PubMed] [Google Scholar]
- 198.Thomas WD, Hersey P. TNF-related apoptosis-inducing ligand (TRAIL) induces apoptosis in Fas ligand-resistant melanoma cells and mediates CD4 T cell killing of target cells. J Immunol. 1998;161(5):2195–2200. [PubMed] [Google Scholar]
- 199.Echchakir H, Bagot M, Dorothee G, et al. Cutaneous T cell lymphoma reactive CD4+ cytotoxic T lymphocyte clones display a Th1 cytokine profile and use a fas-independent pathway for specific tumor cell lysis. J Invest Dermatol. 2000;115(1):74–80. doi: 10.1046/j.1523-1747.2000.00995.x. [DOI] [PubMed] [Google Scholar]
- 200.Kang TW, Yevsa T, Woller N, et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature. 2011;479(7374):547–551. doi: 10.1038/nature10599. [DOI] [PubMed] [Google Scholar]
- 201.Rakhra K, Bachireddy P, Zabuawala T, et al. CD4(+) T cells contribute to the remodeling of the microenvironment required for sustained tumor regression upon oncogene inactivation. Cancer Cell. 2010;18(5):485–498. doi: 10.1016/j.ccr.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Janssen EM, Droin NM, Lemmens EE, et al. CD4+ T-cell help controls CD8+ T-cell memory via TRAIL-mediated activation-induced cell death. Nat. 2005;434(7029):88–93. doi: 10.1038/nature03337. [DOI] [PubMed] [Google Scholar]
- 203.Marzo AL, Kinnear BF, Lake RA, et al. Tumor-specific CD4+ T cells have a major “post-licensing” role in CTL mediated anti-tumor immunity. J Immunol. 2000;165(11):6047–6055. doi: 10.4049/jimmunol.165.11.6047. [DOI] [PubMed] [Google Scholar]
- 204.Janssen EM, Lemmens EE, Wolfe T, Christen U, von Herrath MG, Schoenberger SP. CD4+ T cells are required for secondary expansion and memory in CD8+ T lymphocytes. Nature. 2003;421(6925):852–856. doi: 10.1038/nature01441. [DOI] [PubMed] [Google Scholar]
- 205.Cui W, Kaech SM. Generation of effector CD8+ T cells and their conversion to memory T cells. Immunol Rev. 2010;236:151–166. doi: 10.1111/j.1600-065X.2010.00926.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Bourgeois C, Rocha B, Tanchot C. A role for CD40 expression on CD8+ T cells in the generation of CD8+ T cell memory. Science. 2002;297(5589):2060–2063. doi: 10.1126/science.1072615. [DOI] [PubMed] [Google Scholar]
- 207.Shedlock DJ, Shen H. Requirement for CD4 T cell help in generating functional CD8 T cell memory. Science. 2003;300(5617):337–339. doi: 10.1126/science.1082305. [DOI] [PubMed] [Google Scholar]
- 208.Ridge JP, Di Rosa F, Matzinger P. A conditioned dendritic cell can be a temporal bridge between a CD4+ T-helper and a T-killer cell. Nature. 1998;393(6684):474–478. doi: 10.1038/30989. [DOI] [PubMed] [Google Scholar]
- 209.Sornasse T, Flamand V, De Becker G, et al. Antigen-pulsed dendritic cells can efficiently induce an antibody response in vivo. J Exp Med. 1992;175(1):15–21. doi: 10.1084/jem.175.1.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Gonzalez-Martin A, Gomez L, Lustgarten J, Mira E, Manes S. Maximal T cell-mediated antitumor responses rely upon CCR5 expression in both CD4(+) and CD8(+) T cells. Cancer Res. 2011;71(16):5455–5466. doi: 10.1158/0008-5472.CAN-11-1687. [DOI] [PubMed] [Google Scholar]
- 211.Takemoto S, Nishikawa M, Guan X, Ohno Y, Yata T, Takakura Y. Enhanced generation of cytotoxic T lymphocytes by heat shock protein 70 fusion proteins harboring both CD8(+) T cell and CD4(+) T cell epitopes. Mol Pharm. 2010;7(5):1715–1723. doi: 10.1021/mp1001069. [DOI] [PubMed] [Google Scholar]
- 212.Van Nuffel AM, Benteyn D, Wilgenhof S, et al. Dendritic cells loaded with mRNA encoding full-length tumor antigens prime CD4+ and CD8+ T cells in melanoma patients. Mol Ther. 2012;20(5):1063–1074. doi: 10.1038/mt.2012.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Koski GK, Koldovsky U, Xu S, et al. A novel dendritic cell-based immunization approach for the induction of durable Th1-polarized anti-HER-2/neu responses in women with early breast cancer. J Immunother. 2012;35(1):54–65. doi: 10.1097/CJI.0b013e318235f512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Gok M, Ozcerit AT. Prediction of MHC class I binding peptides with a new feature encoding technique. Cell Immunol. 2012;275:1–2. 1–4. doi: 10.1016/j.cellimm.2012.04.005. [DOI] [PubMed] [Google Scholar]
- 215.Chaves FA, Lee AH, Nayak JL, Richards KA, Sant AJ. The utility and limitations of current web-available algorithms to predict peptides recognized by CD4 T cells in response to pathogen infection. J Immunol. 2012;188(9):4235–4248. doi: 10.4049/jimmunol.1103640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Lin HH, Ray S, Tongchusak S, Reinherz EL, Brusic V. Evaluation of MHC class I peptide binding prediction servers: applications for vaccine research. BMC Immunol. 2008;9:8. doi: 10.1186/1471-2172-9-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Chen JL, Stewart-Jones G, Bossi G, et al. Structural and kinetic basis for heightened immunogenicity of T cell vaccines. J Exp Med. 2005;201(8):1243–1255. doi: 10.1084/jem.20042323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Liao WW, Arthur JW. Predicting peptide binding to major histocompatibility complex molecules. Autoimmun Rev. 2011;10(8):469–473. doi: 10.1016/j.autrev.2011.02.003. [DOI] [PubMed] [Google Scholar]
- 219.Lin HH, Zhang GL, Tongchusak S, Reinherz EL, Brusic V. Evaluation of MHC-II peptide binding prediction servers: applications for vaccine research. BMC Bioinformatics. 2008;9(Suppl. 12):S22. doi: 10.1186/1471-2105-9-S12-S22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Wang P, Sidney J, Dow C, Mothe B, Sette A, Peters B. A systematic assessment of MHC class II peptide binding predictions and evaluation of a consensus approach. PLoS Comput Biol. 2008;4(4):E1000048. doi: 10.1371/journal.pcbi.1000048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Kawashima I, Hudson SJ, Tsai V, et al. The multi-epitope approach for immunotherapy for cancer: identification of several CTL epitopes from various tumor-associated antigens expressed on solid epithelial tumors. Hum Immunol. 1998;59(1):1–14. doi: 10.1016/s0198-8859(97)00255-3. [DOI] [PubMed] [Google Scholar]
- 222.Kovjazin R, Volovitz I, Kundel Y, et al. ImMucin: a novel therapeutic vaccine with promiscuous MHC binding for the treatment of MUC1-expressing tumors. Vaccine. 2011;29(29–30):4676–4686. doi: 10.1016/j.vaccine.2011.04.103. [DOI] [PubMed] [Google Scholar]
- 223.Kosmrlj A, Read EL, Qi Y, et al. Effects of thymic selection of the T-cell repertoire on HLA class I-associated control of HIV infection. Nature. 2010;465(7296):350–354. doi: 10.1038/nature08997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Rosenberg SA, Zhai Y, Yang JC, et al. Immunizing patients with metastatic melanoma using recombinant adenoviruses encoding MART-1 or gp100 melanoma antigens. J Natl Cancer Inst. 1998;90(24):1894–1900. doi: 10.1093/jnci/90.24.1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Shahabi V, Seavey MM, Maciag PC, Rivera S, Wallecha A. Development of a live and highly attenuated Listeria monocytogenes-based vaccine for the treatment of Her2/neu-overexpressing cancers in human. Cancer Gene Ther. 2011;18(1):53–62. doi: 10.1038/cgt.2010.48. [DOI] [PubMed] [Google Scholar]
- 226.Duan XY, Han DG, Zhang MX, Wang JS. Generation of fusion protein EGFRvIII-HBcAg and its anti-tumor effect in vivo. J Exp Clin Cancer Res. 2009;28:133. doi: 10.1186/1756-9966-28-133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Cheever MA, Higano CS. PROVENGE (Sipuleucel-T) in prostate cancer: the first FDA-approved therapeutic cancer vaccine. Clin Cancer Res. 2011;17(11):3520–3526. doi: 10.1158/1078-0432.CCR-10-3126. [DOI] [PubMed] [Google Scholar]
- 228.Higano CS, Schellhammer PF, Small EJ, et al. Integrated data from 2 randomized, double-blind, placebo-controlled, Phase 3 trials of active cellular immunotherapy with sipuleucel-T in advanced prostate cancer. Cancer. 2009;115(16):3670–3679. doi: 10.1002/cncr.24429. [DOI] [PubMed] [Google Scholar]
- 229.Rosenthal KS, Taylor P, Zimmerman DH. J-LEAPS peptide and LEAPS dendritic cell vaccines. Microb Biotechnol. 2012;5(2):203–213. doi: 10.1111/j.1751-7915.2011.00278.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Taylor PR, Paustian CC, Koski GK, Zimmerman DH, Rosenthal KS. Maturation of dendritic cell precursors into IL12-producing DCs by J-LEAPS immunogens. Cell Immunol. 2010;262(1):1–5. doi: 10.1016/j.cellimm.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Taylor PR, Koski GK, Paustian CC, et al. J-LEAPS vaccines initiate murine Th1 responses by activating dendritic cells. Vaccine. 2010;28(34):5533–5542. doi: 10.1016/j.vaccine.2010.06.043. [DOI] [PubMed] [Google Scholar]
- 232.Buteau C, Markovic SN, Celis E. Challenges in the development of effective peptide vaccines for cancer. Mayo Clinic Proc Mayo Clin. 2002;77(4):339–349. doi: 10.4065/77.4.339. [DOI] [PubMed] [Google Scholar]
- 233.Lee WT, Tan C, Koski G, Shu S, Cohen P. Immunotherapy using allogeneic squamous cell tumor–dendritic cell fusion hybrids. Head Neck. 2010;32(9):1209–1216. doi: 10.1002/hed.21323. [DOI] [PubMed] [Google Scholar]
- 234.Shimizu K, Kuriyama H, Kjaergaard J, Lee W, Tanaka H, Shu S. Comparative analysis of antigen loading strategies of dendritic cells for tumor immunotherapy. J Immunother. 2004;27(4):265–272. doi: 10.1097/00002371-200407000-00002. [DOI] [PubMed] [Google Scholar]
- 235.Kuriyama H, Watanabe S, Kjaergaard J, et al. Mechanism of third signals provided by IL-12 and OX-40R ligation in eliciting therapeutic immunity following dendritic-tumor fusion vaccination. Cell Immunol. 2006;243(1):30–40. doi: 10.1016/j.cellimm.2006.11.002. [DOI] [PubMed] [Google Scholar]
- 236.Hayashi T, Tanaka H, Tanaka J, et al. Immunogenicity and therapeutic efficacy of dendritic-tumor hybrid cells generated by electrofusion. Clin Immunol. 2002;104(1):14–20. doi: 10.1006/clim.2002.5224. [DOI] [PubMed] [Google Scholar]
- 237.Kjaergaard J, Shimizu K, Shu S. Electrofusion of syngeneic dendritic cells and tumor generates potent therapeutic vaccine. Cell Immunol. 2003;225(2):65–74. doi: 10.1016/j.cellimm.2003.09.005. [DOI] [PubMed] [Google Scholar]
- 238.Parkhurst MR, DePan C, Riley JP, Rosenberg SA, Shu S. Hybrids of dendritic cells and tumor cells generated by electrofusion simultaneously present immunodominant epitopes from multiple human tumor-associated antigens in the context of MHC class I and class II molecules. J Immunol. 2003;170(10):5317–5325. doi: 10.4049/jimmunol.170.10.5317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Zhang Y, Ma B, Zhou Y, et al. Dendritic cells fused with allogeneic breast cancer cell line induce tumor antigen-specific CTL responses against autologous breast cancer cells. Breast Cancer Res Treat. 2007;105(3):277–286. doi: 10.1007/s10549-006-9457-8. [DOI] [PubMed] [Google Scholar]
- 240.Zhou J, Weng D, Zhou F, et al. Patient-derived renal cell carcinoma cells fused with allogeneic dendritic cells elicit anti-tumor activity: in vitro results and clinical responses. Cancer Immunol Immunother. 2009;58(10):1587–1597. doi: 10.1007/s00262-009-0668-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Boudreau JE, Bonehill A, Thielemans K, Wan Y. Engineering dendritic cells to enhance cancer immunotherapy. Mol Ther J Am Soc Gene Ther. 2011;19(5):841–853. doi: 10.1038/mt.2011.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Park MJ, Kim EK, Han JY, et al. Fusion of the human cytomegalovirus pp65 antigen with both ubiquitin and ornithine decarboxylase additively enhances antigen presentation to CD8(+) T cells in human dendritic cells. Hum Gene Ther. 2010;21(8):957–967. doi: 10.1089/hum.2009.216. [DOI] [PubMed] [Google Scholar]
- 243.Wu TC, Guarnieri FG, Staveley-O’Carroll KF, et al. Engineering an intracellular pathway for major histocompatibility complex class II presentation of antigens. Proc Natl Acad Sci USA. 1995;92(25):11671–11675. doi: 10.1073/pnas.92.25.11671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244■.Bonehill A, Heirman C, Tuyaerts S, et al. Messenger RNA-electroporated dendritic cells presenting MAGE-A3 simultaneously in HLA class I and class II molecules. J Immunol. 2004;172(11):6649–6657. doi: 10.4049/jimmunol.172.11.6649. Illustrates another application of genetically modifying DCs by engineering them to present antigen on both class I and II molecules. Simultaneous presentation addresses the limit of current peptide vaccines that may be unable to activate both CD4+and CD8+T cells. This study demonstrates both the feasibility of genetically modifying DCs to simultaneously present antigen on both class I and II molecules and also addresses how to optimize the efficacy of such simultaneous presentation. [DOI] [PubMed] [Google Scholar]
- 245.Dullaers M, Breckpot K, Van Meirvenne S, et al. Side-by-side comparison of lentivirally transduced and mRNA-electroporated dendritic cells: implications for cancer immunotherapy protocols. Mol Ther J Am Soc Gene Ther. 2004;10(4):768–779. doi: 10.1016/j.ymthe.2004.07.017. [DOI] [PubMed] [Google Scholar]
- 246.Coosemans A, Wolfl M, Berneman ZN, et al. Immunological response after therapeutic vaccination with WT1 mRNA-loaded dendritic cells in end-stage endometrial carcinoma. Anticancer Res. 2010;30(9):3709–3714. [PubMed] [Google Scholar]
- 247.Van Tendeloo VF, Van de Velde A, Van Driessche A, et al. Induction of complete and molecular remissions in acute myeloid leukemia by Wilms’ tumor 1 antigen-targeted dendritic cell vaccination. Proc Natl Acad Sci USA. 2010;107(31):13824–13829. doi: 10.1073/pnas.1008051107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Kikuchi T, Moore MA, Crystal RG. Dendritic cells modified to express CD40 ligand elicit therapeutic immunity against preexisting murine tumors. Blood. 2000;96(1):91–99. [PubMed] [Google Scholar]
- 249.Koya RC, Kasahara N, Favaro PM, et al. Potent maturation of monocyte-derived dendritic cells after CD40L lentiviral gene delivery. J Immunother. 2003;26(5):451–460. doi: 10.1097/00002371-200309000-00008. [DOI] [PubMed] [Google Scholar]
- 250.Feder-Mengus C, Schultz-Thater E, Oertli D, et al. Nonreplicating recombinant vaccinia virus expressing CD40 ligand enhances APC capacity to stimulate specific CD4+ and CD8+ T cell responses. Hum Gene Ther. 2005;16(3):348–360. doi: 10.1089/hum.2005.16.348. [DOI] [PubMed] [Google Scholar]
- 251.Tcherepanova IY, Adams MD, Feng X, et al. Ectopic expression of a truncated CD40L protein from synthetic post-transcriptionally capped RNA in dendritic cells induces high levels of IL-12 secretion. BMC Mol Biol. 2008;9:90. doi: 10.1186/1471-2199-9-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Knippertz I, Hesse A, Schunder T, et al. Generation of human dendritic cells that simultaneously secrete IL-12 and have migratory capacity by adenoviral gene transfer of hCD40L in combination with IFN-γ. J Immunother. 2009;32(5):524–538. doi: 10.1097/CJI.0b013e3181a28422. [DOI] [PubMed] [Google Scholar]
- 253.Bonehill A, Van Nuffel AM, Corthals J, et al. Single-step antigen loading and activation of dendritic cells by mRNA electroporation for the purpose of therapeutic vaccination in melanoma patients. Clin Cancer Res. 2009;15(10):3366–3375. doi: 10.1158/1078-0432.CCR-08-2982. [DOI] [PubMed] [Google Scholar]
- 254.Wiethe C, Debus A, Mohrs M, Steinkasserer A, Lutz M, Gessner A. Dendritic cell differentiation state and their interaction with NKT cells determine Th1/Th2 differentiation in the murine model of Leishmania major infection. J Immunol. 2008;180(7):4371–4381. doi: 10.4049/jimmunol.180.7.4371. [DOI] [PubMed] [Google Scholar]
- 255.Dannull J, Nair S, Su Z, et al. Enhancing the immunostimulatory function of dendritic cells by transfection with mRNA encoding OX40 ligand. Blood. 2005;105(8):3206–3213. doi: 10.1182/blood-2004-10-3944. [DOI] [PubMed] [Google Scholar]
- 256.Ojima T, Iwahashi M, Nakamura M, et al. The boosting effect of co-transduction with cytokine genes on cancer vaccine therapy using genetically modified dendritic cells expressing tumor-associated antigen. Int J Oncol. 2006;28(4):947–953. [PubMed] [Google Scholar]
- 257.Minkis K, Kavanagh DG, Alter G, et al. Type 2 Bias of T cells expanded from the blood of melanoma patients switched to type 1 by IL-12p70 mRNA-transfected dendritic cells. Cancer Res. 2008;68(22):9441–9450. doi: 10.1158/0008-5472.CAN-08-0900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Wang J, Ouyang Y, Guner Y, Ford HR, Grishin AV. Ubiquitin-editing enzyme A20 promotes tolerance to lipopolysaccharide in enterocytes. J Immunol. 2009;183(2):1384–1392. doi: 10.4049/jimmunol.0803987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Breckpot K, Aerts-Toegaert C, Heirman C, et al. Attenuated expression of A20 markedly increases the efficacy of double-stranded RNA-activated dendritic cells as an anti-cancer vaccine. J Immunol. 2009;182(2):860–870. doi: 10.4049/jimmunol.182.2.860. [DOI] [PubMed] [Google Scholar]
- 260.Song XT, Evel-Kabler K, Shen L, Rollins L, Huang XF, Chen SY. A20 is an antigen presentation attenuator, and its inhibition overcomes regulatory T cell-mediated suppression. Nat Med. 2008;14(3):258–265. doi: 10.1038/nm1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Shen L, Evel-Kabler K, Strube R, Chen SY. Silencing of SOCS1 enhances antigen presentation by dendritic cells and antigen-specific anti-tumor immunity. Nat Biotechnol. 2004;22(12):1546–1553. doi: 10.1038/nbt1035. [DOI] [PubMed] [Google Scholar]
- 262.Shi L, Luo K, Xia D, et al. DIgR2, dendritic cell-derived immunoglobulin receptor 2, is one representative of a family of IgSF inhibitory receptors and mediates negative regulation of dendritic cell-initiated antigen-specific T-cell responses. Blood. 2006;108(8):2678–2686. doi: 10.1182/blood-2006-04-015404. [DOI] [PubMed] [Google Scholar]
- 263.Gray A, Raff AB, Chiriva-Internati M, Chen SY, Kast WM. A paradigm shift in therapeutic vaccination of cancer patients: the need to apply therapeutic vaccination strategies in the preventive setting. Immunol Rev. 2008;222:316–327. doi: 10.1111/j.1600-065X.2008.00605.x. [DOI] [PubMed] [Google Scholar]
- 264.Roychowdhury S, Talpaz M. Managing resistance in chronic myeloid leukemia. Blood Rev. 2011;25(6):279–290. doi: 10.1016/j.blre.2011.09.001. [DOI] [PubMed] [Google Scholar]
- 265.Flaherty KT, Puzanov I, Kim KB, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363(9):809–819. doi: 10.1056/NEJMoa1002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Hakim FT, Cepeda R, Kaimei S, et al. Constraints on CD4 recovery postchemotherapy in adults: thymic insufficiency and apoptotic decline of expanded peripheral CD4 cells. Blood. 1997;90(9):3789–3798. [PubMed] [Google Scholar]
- 267.Galluzzi L, Senovilla L, Zitvogel L, Kroemer G. The secret ally: immunostimulation by anticancer drugs. Nat Rev Drug Discov. 2012;11(3):215–233. doi: 10.1038/nrd3626. [DOI] [PubMed] [Google Scholar]
- 268.Simeone E, Ascierto PA. Immunomodulating antibodies in the treatment of metastatic melanoma: the experience with anti-CTLA-4, anti-CD137, and anti-PD1. J Immunotoxicol. 2012;9(3):241–247. doi: 10.3109/1547691X.2012.678021. [DOI] [PubMed] [Google Scholar]
- 269.Garcia-Hernandez Mde L, Gray A, Hubby B, Klinger OJ, Kast WM. Prostate stem cell antigen vaccination induces a long-term protective immune response against prostate cancer in the absence of autoimmunity. Cancer Res. 2008;68(3):861–869. doi: 10.1158/0008-5472.CAN-07-0445. [DOI] [PubMed] [Google Scholar]
- 270.Einstein MH, Kadish AS, Burk RD, et al. Heat shock fusion protein-based immunotherapy for treatment of cervical intraepithelial neoplasia III. Gynecol Oncol. 2007;106(3):453–460. doi: 10.1016/j.ygyno.2007.04.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Geiger JD, Hutchinson RJ, Hohenkirk LF, et al. Vaccination of pediatric solid tumor patients with tumor lysate-pulsed dendritic cells can expand specific T cells and mediate tumor regression. Cancer Res. 2001;61(23):8513–8519. [PubMed] [Google Scholar]
- 272.Jonuleit H, Giesecke-Tuettenberg A, Tuting T, et al. A comparison of two types of dendritic cell as adjuvants for the induction of melanoma-specific T-cell responses in humans following intranodal injection. Int J Cancer. 2001;93(2):243–251. doi: 10.1002/ijc.1323. [DOI] [PubMed] [Google Scholar]
- 273.Schuler-Thurner B, Schultz ES, Berger TG, et al. Rapid induction of tumor-specific type 1 T helper cells in metastatic melanoma patients by vaccination with mature, cryopreserved, peptide-loaded monocyte-derived dendritic cells. J Exp Med. 2002;195(10):1279–1288. doi: 10.1084/jem.20012100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Lepisto AJ, Moser AJ, Zeh H, et al. A Phase I/ II study of a MUC1 peptide pulsed autologous dendritic cell vaccine as adjuvant therapy in patients with resected pancreatic and biliary tumors. Cancer Ther. 2008;6(B):955–964. [PMC free article] [PubMed] [Google Scholar]
- 275.Soleimani A, Berntsen A, Svane IM, Pedersen AE. Immune responses in patients with metastatic renal cell carcinoma treated with dendritic cells pulsed with tumor lysate. Scand J Immunol. 2009;70(5):481–489. doi: 10.1111/j.1365-3083.2009.02322.x. [DOI] [PubMed] [Google Scholar]
- 276.Rollig C, Schmidt C, Bornhauser M, Ehninger G, Schmitz M, Auffermann-Gretzinger S. Induction of cellular immune responses in patients with stage-I multiple myeloma after vaccination with autologous idiotype-pulsed dendritic cells. J Immunother. 2011;34(1):100–106. doi: 10.1097/CJI.0b013e3181facf48. [DOI] [PubMed] [Google Scholar]
- 277.Ardon H, Van Gool S, Lopes IS, et al. Integration of autologous dendritic cell-based immunotherapy in the primary treatment for patients with newly diagnosed glioblastoma multiforme: a pilot study. J Neurooncol. 2010;99(2):261–272. doi: 10.1007/s11060-010-0131-y. [DOI] [PubMed] [Google Scholar]
- 278.Ellebaek E, Engell-Noerregaard L, Iversen TZ, et al. Metastatic melanoma patients treated with dendritic cell vaccination, interleukin-2 and metronomic cyclophosphamide: results from a Phase II trial. Cancer Immunol Immunother. 2012;61(10):1791–1804. doi: 10.1007/s00262-012-1242-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Alfaro C, Perez-Gracia JL, Suarez N, et al. Pilot clinical trial of type 1 dendritic cells loaded with autologous tumor lysates combined with GM-CSF, pegylated IFN, and cyclophosphamide for metastatic cancer patients. J Immunol. 2011;187(11):6130–6142. doi: 10.4049/jimmunol.1102209. [DOI] [PubMed] [Google Scholar]
- 280.Okada H, Kalinski P, Ueda R, et al. Induction of CD8+ T-cell responses against novel glioma-associated antigen peptides and clinical activity by vaccinations with {α}-type 1 polarized dendritic cells and polyinosinic-polycytidylic acid stabilized by lysine and carboxymethylcellulose in patients with recurrent malignant glioma. J Clin Oncol. 2011;29(3):330–336. doi: 10.1200/JCO.2010.30.7744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Banchereau J, Palucka AK, Dhodapkar M, et al. Immune and clinical responses in patients with metastatic melanoma to CD34(+) progenitor-derived dendritic cell vaccine. Cancer Res. 2001;61(17):6451–6458. [PubMed] [Google Scholar]