

Abstract
Bacterial communities colonizing the reproductive tracts of primates (including humans) impact the health, survival and fitness of the host, and thereby the evolution of the host species. Despite their importance, we currently have a poor understanding of primate microbiomes. The composition and structure of microbial communities vary considerably depending on the host and environmental factors. We conducted comparative analyses of the primate vaginal microbiome using pyrosequencing of the 16S rRNA genes of a phylogenetically broad range of primates to test for factors affecting the diversity of primate vaginal ecosystems. The nine primate species included: humans (Homo sapiens), yellow baboons (Papio cynocephalus), olive baboons (Papio anubis), lemurs (Propithecus diadema), howler monkeys (Alouatta pigra), red colobus (Piliocolobus rufomitratus), vervets (Chlorocebus aethiops), mangabeys (Cercocebus atys) and chimpanzees (Pan troglodytes). Our results indicated that all primates exhibited host-specific vaginal microbiota and that humans were distinct from other primates in both microbiome composition and diversity. In contrast to the gut microbiome, the vaginal microbiome showed limited congruence with host phylogeny, and neither captivity nor diet elicited substantial effects on the vaginal microbiomes of primates. Permutational multivariate analysis of variance and Wilcoxon tests revealed correlations among vaginal microbiota and host species-specific socioecological factors, particularly related to sexuality, including: female promiscuity, baculum length, gestation time, mating group size and neonatal birth weight. The proportion of unclassified taxa observed in nonhuman primate samples increased with phylogenetic distance from humans, indicative of the existence of previously unrecognized microbial taxa. These findings contribute to our understanding of host–microbe variation and coevolution, microbial biogeography, and disease risk, and have important implications for the use of animal models in studies of human sexual and reproductive diseases.
Introduction
The human vaginal microbiome has been relatively well characterized (Fredricks et al., 2005; Oakley et al., 2008; Kim et al., 2009; Hummelen et al., 2010; Srinivasan et al., 2010; Ravel et al., 2011; Gajer et al., 2012). Most human vaginal tracts are predominantly colonized by Lactobacillus species (Srinivasan and Fredricks, 2008; Ma et al., 2012). Production of lactic acid by vaginal lactobacilli lowers the pH of the vaginal, creating an acidic (pH⩽4.5) environment. This microbially mediated vaginal acidity provides an important barrier that restricts the colonization of potential pathogens (Boskey et al., 1999). Perturbations to the composition of the human vaginal microbiome are often associated with diseased states, including bacterial vaginosis (BV) (Fredricks et al., 2005; Oakley et al., 2008; Marrazzo et al., 2010; Srinivasan et al., 2010), susceptibility to sexually transmitted infectious diseases (Sewankambo et al., 1997; Wiesenfeld et al., 2003), tubal infertility (Wiesenfeld et al., 2012; van Oostrum et al., 2013) and adverse pregnancy outcomes (White et al., 2011; Ganu et al., 2013).
In contrast to humans, there is a paucity of information on the vaginal microbiomes of nonhuman primates (NHPs), and information about NHP vaginal microbiota is essential for understanding the factors that underlie microbial coevolution with their hosts, and more specifically, for testing adaptive hypotheses for the human microbiome. Given the known importance of the vaginal microbiome for female sexual and reproductive health and their role as microbial colonists for newborns (Tannock et al., 1990; Mandar and Mikelsaar, 1996; Leitich and Kiss, 2007; CDC, 2009; Biasucci et al., 2010; Dominguez-Bello et al., 2010; Marrazzo et al., 2010), host-vaginal microbiome interactions could comprise a strong selection force having a pivotal role in the evolution of humans and other primates. Primates are ideal hosts for studying variation in vaginal microbiota, because primate species differ markedly in their diet, anatomy, mating systems, gestational durations, birthing difficulties, and commensurate risks of maternal injury and subsequent infection (Rosenberg and Trevathan, 2002; Campbell et al., 2011).
Primate vaginal microbiomes may also vary with exogenous host factors such as phylogeny, geography, life history characteristics and sexuality. The degree to which these factors explain variation in vaginal microbiota remains to be tested. However, if predictions can be drawn from other host systems, such as primate gut microbiomes (Ley et al., 2008; Ochman et al., 2010; Yildirim et al., 2010), patterns of microbial abundance and diversity are expected to be most similar between closely related primate species and differences should increase with phylogenetic distance (that is, vaginal microbial patterns reflect primate phylogeny). An alternate hypothesis is that vaginal microbial abundance and diversity instead display consistent adaptive patterns related to differences in behaviors and ecologies. For example, given the diversity in mating systems among NHPs, which vary from monogamy to polygynandrous mating systems, microbial diversity is predicted to covary with the level of promiscuity.
Thus, the aim of our study was to examine microbial diversity and composition patterns characteristic of primate vaginal tracts and test for factors that account for the variation of patterns. Our specific goals were to: (1) identify microbial taxa from primate vaginal tracts, (2) conduct phylogenetic analyses of the respective microbial communities and (3) determine whether microbial phylogenies deduced from the samples correspond with differences in host taxonomy, body size, sexuality or substrate use (that is, the primary surface (arboreal vs terrestrial) on which primates locomote). The inclusion of several distinct primate species lineages enabled us to address a number of questions regarding the correlation of phylogenetic, socioecological, morphological and/or genetic factors with the composition similarity, richness and diversity of the vaginal microbiome among primates (See inset Box 1).
Box 1. Socioecological factors affect the diversity of vaginal microbiomes.
Origin.
The country of origin of the samples (listed in Table 1) was tested as a discrete variable.
Substrate.
Because most microbes reside in soils (Fierer and Jackson, 2006; Fierer et al., 2007) and primates vary substantially in the degree of terrestriality and arboreality, primate substrate use may affect vaginal microbial diversity.
Group size.
This measure refers to the collective number of individuals that are observed together (Strier, 2007). We hypothesize that group size will covary with microbial richness and diversity due to the opportunities for close social interaction, including: mating, grooming, aggression, spatial proximity and resource sharing.
Body mass.
We examined the relationship of vaginal microbiota with the average adult female body mass (data from Smith and Jungers (1997)).
Vaginal size.
Male and female genitalia have coevolved (Chapman et al. 2003), and mammalian vaginal cavity size correlates with baculum length (Patterson and Thaeler, 1982). Without direct estimates of vaginal cavity size available for most NHPs, we instead used baculum length as a parameter to estimate the impact of the size of the vaginal cavity on primate microbiomes.
Sexual swelling.
The fleshy skin of the anogenital region of some female primates tumesces (inflates) and detumesces (deflates) in response to hormonal signals. During tumescence, the swelling grows in size (Aykroyd and Zuckerman, 1938), thus increasing the surface area for microbes to colonize.
Promiscuity.
The use of broad mating system categories to measure levels of promiscuity is coarse. Thus, two additional measures were applied as proxies for promiscuity: (1) testes mass, a commonly used measure of male mating competition (Moller et al., 1998; Nunn et al., 2000; Moller et al., 2001), and (2) mating group size, a measure of sexual exposure to microbes. The vaginal microbial community may depend on the number of sexually active individuals (that is, mating partners) in a group. For example, microbial communities and strains are shared between mating partners (Jaspers and Overmann, 2004; Marrazzo et al., 2009; Danielsson et al., 2011; Eren et al., 2011) and sexual activity is a strong risk factor for dysbiosis (Fethers et al., 2008). In humans, microbial ecologies vary considerably between women depending on their mating group size, and BV is most common in women who have new or multiple sexual partners (Fethers et al., 2008, 2009; Eren et al., 2011). Extrapolating to mating systems, in polygynous groups, where males mate with multiple females, females may be exposed to vaginal microbes from other females. Thus, vaginal microbial communities may depend on the number of sexually active individuals of both sexes (mating group size). We are aware that measurement error in mating group size estimates exists (for example, extra pair or extra group mating may be difficult to measure), but estimates among diverse mating systems (differing levels of promiscuity) should vary sufficiently to diminish their effects.
Social structure.
Primate species were grouped into male-dominant (adult males dominate females in social interactions), female-dominant (adult females dominate males in social interactions) or egalitarian relationships (dominance can be held by both males and females, depending on context (Strier, 2007)).
Obstetric traits.
In humans, the vaginal microbiome is thought to have a role in preterm birth (White et al., 2011). Therefore, we considered gestation length and neonatal mass in the context of the vaginal microbiota.
Materials and methods
Ethics statement
Approvals for collection and subsequent processing and analysis of the NHP and human samples were obtained from the University of Illinois Institutional Animal Care and Use Committee (Protocol Nos: 08044 and 11046), the University of Illinois Institutional Review Board (Protocol No. 05079), the University of Illinois Institutional Biosafety Committee (Protocol No. IBC-82) and Carle Foundation Hospital (Protocol No. 05-04).
Sample collection and processing
The nine primate species are listed in Table 1, along with their origin, classification and sample size. Experienced field collaborators obtained vaginal swab samples from tranquilized primates and completed detailed datasheets for each primate sampled. All primates were observed to be healthy, and none included here was observed to be menstruating at the time of sampling. Sterile swabs (Copan Diagnostics, Corona, CA, USA) were used to collect microbes from the vaginal cavities. Each swab was immediately placed in a sterile 8-ml screw cap tube prefilled with 2–3 ml RNAlater (Ambion, Grand Island, NY, USA; Cat. No. 7020). Tubes were flash frozen or placed on ice, then transferred to −80 °C freezers on arrival in the United States, and stored there until sample processing. To verify host origin, we amplified and sequenced mitochondrial 12S rRNA genes from all species and the COXII gene from genomic DNA (gDNA) extracted from these samples, using previously published primers (Kocher et al., 1989; Ruvolo et al., 1991). Comparison of the 12S rRNA and COXII gene sequences with Genbank using nucleotide–nucleotide basic local alignment search tool confirmed that the samples collected were from the designated primate species. Human samples were collected from healthy volunteers with no signs of BV, as described in Kim et al. (2009). The clinical data associated with the human subjects included in this study were recently published (Yeoman et al., 2013). It should be noted that two of the human samples, although apparently healthy and asymptomatic for BV, exhibited Nugent scores of 6 (hm403) and 9 (hm409) (Kim et al., 2009).
Table 1. Origin, classification and sample size of primate species included in this study.
| Common name | Species name | Origin | N | Classification |
|---|---|---|---|---|
| Humans | Homo sapiens | IL, USA | 9 | Humans |
| Chimpanzees | Pan troglodytes | Uganda | 12 | Apes |
| Vervets | Chlorocebus aethiops | St Kitts | 6 | OWM |
| Vervetsa | Chlorocebus aethiops | NC, USA | 6 | OWM |
| Mangabeysa | Cercocebus atys | GA, USA | 6 | OWM |
| Olive baboonsa | Papio anubis | TX, USA | 6 | OWM |
| Yellow baboons | Papio cynocephalus | Kenya | 6 | OWM |
| Red colobus | Piliocolobus rufomitratus | Uganda | 6 | OWM |
| Howlers | Alouatta pigra | Guatemala | 5 | NWM |
| Lemurs (Sifakas) | Propithecus diadema | Madagascar | 6 | Prosimian |
Abbreviations: NWM, New World Monkey; OWM, Old World Monkey.
Captive animals.
DNA extraction and PCR
gDNA from vaginal samples was extracted as previously described (Yildirim et al., 2010). Briefly, the frozen samples stored in 8-ml screw cap tubes were thawed on ice, topped up with sterile phosphate-buffered saline and vortexed at full speed for 1 min. The tube contents were then transferred to 1.5 ml Eppendorf tubes (Eppendorf Snap cap; 1.5 ml; Fisher Scientific Cat. No. 05-402-25, Waltham, MA, USA) and centrifuged for 5 min at maximum speed to remove any particulate matter. Subsequent steps included: incubation with lysozyme (10 mg ml−1 in 20 mM Tris-HCl, pH 7.4, 100 mM EDTA, 50 mM NaCl, 0.2% Tween), addition of 10% SDS, freeze–thaw cycling, incubation with proteinase-K (10 mg ml−1), addition of 5 M NaCl and incubation on ice to precipitate remaining protein, centrifugation, incubation with RNAse (30 U mg−1), followed by phenol–chloroform extraction and alcohol precipitation to precipitate gDNA. gDNA concentration and purity was estimated by spectrometry (NanoDrop, ND-2000, ThermoScientific, Wilmington, DE, USA) and gel electrophoresis.
The hypervariable regions V1–V3 of the 16S rRNA genes of each of the gDNA samples were amplified by PCR using primers 27F-YM (5′-AGAGTTTGATYMTGGCTCAG-3′) and 534R (5′-ATTACCGCGGCTGCTGG-3′) (Baker et al., 2003). Each 50 μl PCR reaction contained 45 μl of Platinum PCR SuperMix (Invitrogen, Grand Island, NY, USA; Cat. No. 11306-016), 1.5–3.0 mM MgCl2, 200 pmol of each primer and 20–50 ng of template gDNA. PCR was performed on a Bio-Rad thermocycler (DNA Engine, Hercules, CA, USA) using the following cycling steps: 5 min at 95 °C, followed by 20–28 cycles for 40 s at 94 °C, 30 s at 60 °C, 30 s at 72 °C, and a final 10-min extension at 72 °C. Amplicons from eight separate PCR reactions were pooled using standard protocols, run on 1.5% agarose gel, excised and purified using Qiagen gel extraction kit (Qiagen, Valencia, CA, USA). The purified PCR products were then visualized on 1.5% agarose gel and submitted to the core sequencing facility at the University of Illinois, Urbana, for 454 pyrosequencing using GS FLX Titanium series reagents (454 Life Sciences, Roche Diagnostics, Branford, CT, USA). Library quality checks using standard protocols were performed.
The 16S rRNA sequence data sets generated in this study were submitted to the short read archive of NCBI with the accession number SRP040592.
Data analysis
The sequence files in FASTA format were processed using mothur software (Schloss et al., 2009). For quality filtering, sequences that had average quality scores of <20 (Q<20) over a 50-bp sliding window, lacked an accurate primer sequence, contained ambiguous base call or possessed more than eight homopolymers were excluded from analyses. Sequences were then aligned against the SILVA alignment template (Pruesse et al., 2007). Potential chimeric sequences were detected by using chimera.slayer embedded in the mothur software (http://www.mothur.org/wiki/Chimera.slayer) and removed. A total of 907 454 high-quality sequences (1639–90 192 reads per sample, median=10493.5) were used for subsequent analyses. The high-quality sequences were pre-clustered as previously described (Huse et al., 2010) to further reduce any potential influence of sequencing errors. All community diversity parameters (number of sequences for each sample, Shannon, Chao, coverage and Simpson) were calculated using mothur software (see Supplementary Table S1). Operational taxonomic units (OTUs) were determined using average neighbor clustering of sequences with 97% sequence identity. To assess the taxonomic distributions across each primate sample, a weighted sequence from each OTU was selected (that is, the representative was found by selecting the sequence that has the smallest total distance to all other sequences in that OTU) and subsequently was classified by locally running an RDP classifier program (70% bootstrap threshold; (Wang et al., 2007)). OTUs with only one sequence read (singletons) were removed.
Phylogenetic analysis
A parsimony-based approach adapted from Ochman et al. (2010) was used to test phylogenetic congruence of the vaginal microbiome with their host phylogeny. Using this approach, bacterial OTU frequencies obtained at 99.0% similarity cutoffs were log normalized by coding into one of the eight ordered states, zero being OTU absent in a given sample. Parsimony uninformative sites were eliminated from the matrix. This data matrix (containing 7575 OTUs) was then subjected to a heuristic maximum parsimony tree search using PAUP version 4.0 b.10 (Sinauer Associates, Inc., Sunderland, MA, USA), with default settings and ordered character status.
To construct the host phylogeny, we used recently published sequence data (Perelman et al., 2011), representing 54 nuclear gene regions and 61 primate genera. Sequences were not available for four primate species used in this data set (Papio cynocephalus, Cercocebus atys, Piliocolobus rufomitratus and Alouatta pigra). To adjust for this, the most closely related species of the same genus was used instead (Papio hamadryas, C. torquatus, Piliocolobus badius and A. caraya, respectively). The alignment was subjected to heuristic maximum parsimony to facilitate comparison with the phylogeny of vaginal microbiome. Bootstrap analysis with identical settings for each method of phylogenetic reconstruction was used to support the placement of nodes within the phylogeny (1000 iterations) and values greater than 50% were retained.
To further assess phylogenetic associations between vaginal microbial communities and their hosts, UniFrac analysis of the mothur-selected representative OTU sequences was performed using QIIME software (EC2 image, v1.3; Caporaso et al., 2010. UniFrac provides a phylogeny-based analysis with a quantitative measure of evolutionary distance between OTUs in microbial communities (Lozupone and Knight, 2005). The OTU-representative sequences were aligned using PyNast (Caporaso et al., 2010) against the Greengenes template alignment (DeSantis et al., 2006) to build a phylogenetic tree for measuring UniFrac distance metrics. Jackknifing with unweighted Unifrac was performed with 100 replicates to measure robustness of individual clusters.
As an alternate interrogation of phylogenetic concordance, we examined correlations between the host phylogeny and measures of β-diversity. We reasoned that if host species and their vaginal microbiota shared an evolutionary history, then the distance matrix obtained from pairwise combinations of samples from each species should correlate with the host distance matrix. To calculate a correlation value between the phylogenetic relationship and the Bray–Curtis dissimilarity index of each primate sample, we randomly permuted the vaginal sample matrix (one sample point from each host species included in each permutation) and calculated the Pearson correlation coefficients with the host phylogeny distance matrix. This process was repeated for 450 000 permutations. Distribution of these correlations is shown as a density plot using a Gaussian kernel in R, which provides the extent of association between phylogenetic relationship and microbiome similarity across primate samples.
Multivariate analysis of community structures and diversity
To analyze multivariate ecological data, we used the following techniques: a Bray–Curtis dissimilarity matrix was calculated based on the standardized and square root-transformed read abundance data. An unconstrained ordination technique (nonmetric multidimensional analysis) was performed to display overall similarities in microbial community structures among samples. Multivariate null hypothesis of no difference among a priori-defined groups was tested using permutational multivariate analysis of variance (PERMANOVA) and analysis of similarities (ANOSIM). Permutational analysis of multivariate dispersions (PERMDISP) (Anderson, 2006) was used to test heterogeneity of community structure. Primer V6 (PRIMER-E Ltd, Ivybridge, UK) and PERMANOVA+ were used to perform the ordinations, PERMDISP function and PERMANOVA test.
Wilcoxon signed-rank test available in R package was used to study the individual effects of various categorized ecological factors (see below) on the Shannon diversity values. Wilcoxon test was also used to compare the diversity values across the primate vaginal microbiomes.
Socioecological effects on microbial community composition
The relative contributions of each of the following factors to vaginal microbial composition were tested as a discrete variable (listed in Table 2), using PERMANOVA (MANOVA using distance matrices) with the Adonis function of the vegan package in R (Anderson, 2001): country of origin of the samples, substrate (terrestriality versus arboreality), group size, average female body mass, reproductive traits (gestation length, neonatal mass, vaginal cavity size (derived from baculum length), sexual swelling), mating system (sexual promiscuity, testes mass, mating group size, social structure).
Table 2. Factors affecting variation in primate vaginal microbiota.
| Factor | R2 a | ANOSIM Rb |
|---|---|---|
| Species | 42.7 | 0.95 |
| Sample origin | 21.56±0.75 | 0.49 |
| Sexual swelling | 10.58±0.99 | 0.408 |
| Promiscuity | 10.56±0.41 | 0.33 |
| Social structure | 9.37±1.16 | 0.6 |
| Testes mass | 6.14±0.37 | 0.22 |
| Body weight | 6.1±0.28 | 0.22 |
| Neonatal weight | 6.04±0.29 | 0.22 |
| Gestation duration | 5.86±0.24 | 0.56 |
| Mating group size | 5.45±0.37 | 0.32 |
| Baculum length | 5.53±0.22 | 0.33 |
| Group size | 5.52±0.22 | 0.71 |
Abbreviation: ANOSIM, analysis of similarities.
Permutational multivariate analysis of variance using distance matrices. Values represent the average percent R2 variation explained by the factor after all potential factor-ordered combinations were tested to account for shared R2. Values are presented with their 95% confidence intervals and all were significant based on F-tests of sequential sums of squares from permutations of the raw data (P<0.001).
ANOSIM values based on groupings of high, medium and low thresholds for continuous variables (Supplementary Table S2) or otherwise their discrete variables. Analyses were based on 10 000 permutations and all values were significant (P<0.001).
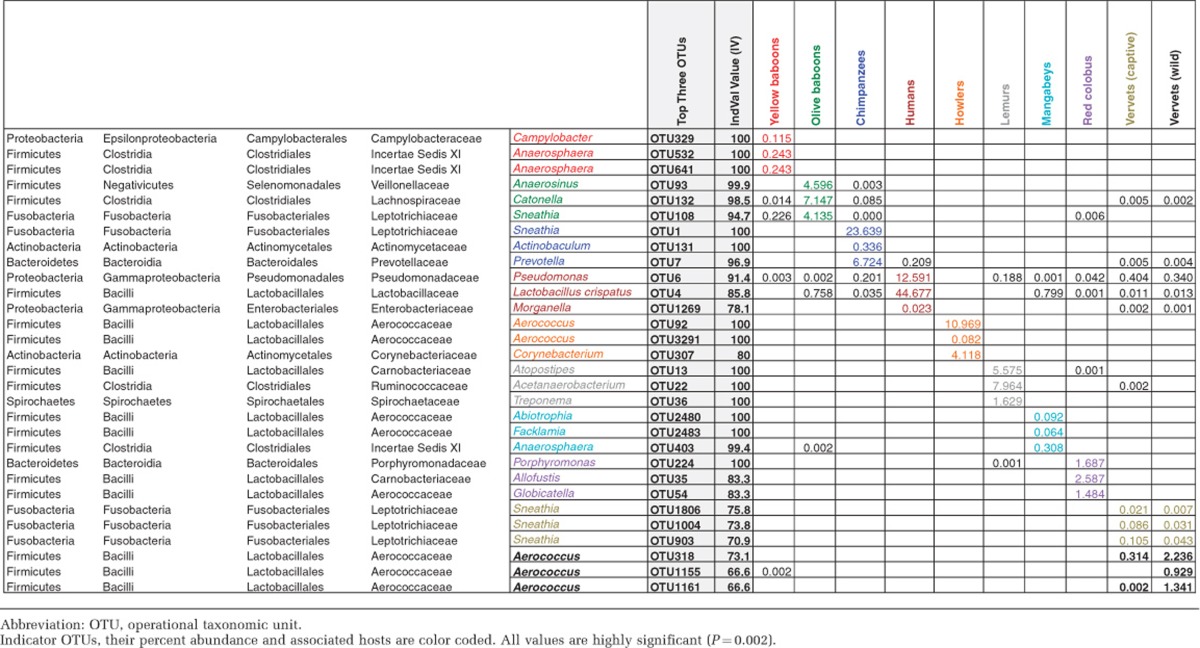
PERMANOVA assigns R2 to the first listed factor before determining R2 from the remaining explanatory space to the next factor. Any shared R2 is attributed to the first listed factor. To circumvent these order-based effects, all factors were tested in all possible combinations and are reported as averages with 95% confidence intervals. In PERMANOVA, ANOSIM based on Bray–Curtis dissimilarities was also used to examine the significance of each variable in driving the overall vaginal microbial composition. To identify the indicator species (Dufrene and Legendre, 1997) that characterize each of the phylogenetic host groups (species), we used indicator species analysis in PC-ORD software (v.6; MJM Software Design, Gleneden Beach, OR, USA). OTUs with IV>0.3 and P<0.05 were selected for the analysis and for each species the top three OTUs with the highest IV values were identified.
Finally, we hypothesized that certain factors (for example, promiscuity) may increase vaginal microbial diversity without impacting specific taxa, or even the overall microbiome. To test this hypothesis, we tiered each variable over three discrete ranges, representing low, medium and high (see Supplementary Table S2), and used a Wilcoxon test to determine whether changes in diversity were significantly different among the tiers. Diversity was evaluated using Shannon's diversity index.
Results
Comparative analysis reveals distinct vaginal microbiota among humans and NHPs
Humans exhibited significantly lower richness and diversity estimates (Figure 1 and Supplementary Table S1) (P<2.392 × 10−5, Kruskal–Wallis test). Rarefied Chao1 estimates indicated that a median of 62 OTUs (range 32–144) occupy the human vagina. Chao1 estimates among NHPs ranged from a median of 220 OTUs for vervets to 1309 for mangabeys.
Figure 1.

Shannon–Weaver diversity indices generated using the 16S rRNA gene pyrosequencing data. Ba, yellow baboons; Bc, olive baboons; Ch, chimpanzees; Lm, lemurs; Hw, black howler monkeys; Mb, mangabeys; RC, red colobus; Vc, vervets captive; and Vw, vervets wild.
Across all primates, most classifiable bacteria could be assigned to the phyla Firmicutes, Fusobacteria, Bacteroidetes, Proteobacteria or Actinobacteria (Supplementary Figure S1). Firmicutes were detected in all primate species, typically as the most abundant taxa present, and comprised 20–30% of the total microbiota among NHPs (γ-diversity). Exceptions were observed in samples from chimpanzees and captive vervets, where members of the Fusobacteria outnumbered Firmicutes. The Fusobacteria were highly underrepresented (<0.1%) in lemur and howler monkey samples compared with other NHPs in this study. These two NHP species are in the clades distantly related to Old World Monkeys and Apes. At the genus level, Sneathia and Aerococcus spp. were consistently enriched across all NHPs, although other genera, including Anaerococcus, Porphyromonas, Fusobacterium, Atopobium and Prevotella, were also commonly found (Figure 2). Lactobacillus species dominated human vaginal samples (range 65.9–98.1%), consistent with previous reports (Zhou et al., 2007; Kim et al., 2009; Hummelen et al., 2010; Srinivasan et al., 2010; Ravel et al., 2011).
Figure 2.

Heatmap of the relative abundance of 16S rRNA sequences displayed at taxonomic genus level. The column Z-score indicates differences between primate samples in terms of the relative abundances of bacterial phylotypes associated with the primate samples; white color indicates relative abundance of phylotypes having column average. Blue color tones represent relative abundances less than the average abundance; and red color tones representing relative abundances above the column average. Sample and bacterial phylotypes were clustered using average linkage hierarchical clustering of a distance matrix based on Bray–Curtis distance. Phylotypes with relative abundance >1% and observed at least in three animal subjects across all animals shown. Samples from each group were color coded on the column side bar as follows: yellow baboons (blue), olive baboons (gray), chimpanzees (yellow), humans (purple), howlers (orange), lemurs (green), mangabeys (cyan), red colobus (pink), vervets captive (magenta) and vervets wild (black) (see Supplementary Tables S4 for genus-level relative abundances).
Of particular note, the relative abundances of lactobacilli were significantly lower in NHPs, whereas the majority of the human female populations have Lactobacillus as the dominant vaginal bacteria (60–93%, depending on the ethnicity (Zhou et al., 2010; Ravel et al., 2011)). Chimpanzees, the closest human relatives, had <3.5% lactobacilli. Among NHPs, mangabeys displayed the greatest numbers of lactobacilli, yet this was still <5%. Species-level assignments using speciateIT tool (Ravel et al., 2011) showed that the majority of NHPs are associated with multiple species of Lactobacilli (L. acidophilus, L. animalis, L. crispatus, L. fornicalis, L. gasseri, L. iners, L. mucosae, L. reuteri, L. ruminis, and L. salivarius), which is consistent with a recent report (Gravett et al., 2012). Notably, few or no reads of Lactobacilli species were found in the samples from arboreal monkeys (howlers, lemurs and red colobus). Reads classified to the genus Gardnerella appeared to be under-sampled from human samples (and possibly from NHP samples too) in this study, compared with previously published reports (Kim et al., 2009; Ravel et al., 2013). This may be attributed to not including the primer cocktail (27F-YM+3), as described in Frank et al., 2008.
Recent studies (Zhou et al., 2010; Ravel et al., 2011; Ravel et al., 2013) have revealed Lactobacillus spp. in low abundance in the vaginal tract of a subpopulation of apparently healthy women with high Nugent scores, who have been categorized as ‘asymptomatic BV' (ABV) in contrast to ‘symptomatic BV' (SBV) subjects. Indeed, two human subjects (Hm403 and Hm409) in our study could be categorized as ABV, as they had reduced relative abundance of Lactobacilli, 65.9 and 3.0% with corresponding Nugent scores of 6 and 9, respectively (Kim et al., 2009; Yeoman et al., 2013). We considered the possibility that the ABV-like and SBV-like human microbiomes might be more similar to that of NHPs. To address this, we compared the vaginal microbial composition of the NHPs in our study with that of human subjects with low abundance of Lactobacillus spp. (ABV and SBV), by including a recently published data set (Ravel et al., 2013) of ABV and SBV samples. Clustering analysis of pairwise binary Jaccard (Supplementary Figure S2) and Bray–Curtis (Supplementary Figure S3) dissimilarities of genus-level abundance distributions between samples indicated that both ABV and SBV fall in a distinct cluster and are more similar to NHPs. Abundance-weighted clustering using the Jaccard index showed identical results, although Bray–Curtis index placed ABV–SBV group closer to healthy humans (data not shown), which may result from using different data analysis tools in the two studies.
Sequences that could not be classified (<70% Bayesian bootstrap cutoff) to any hierarchical taxonomic level using the RDP classifier were grouped as unclassified taxa. A significantly higher proportion of sequences from lemurs could not be classified (13±8%) at the phylum level compared with other primate microbiomes. Further, a substantial proportion of phylum-resolved taxa from lemurs (68±11%) lacked genus-level resolution. This suggests that the vaginal microbiome of lemurs is comprised of largely novel bacterial taxa not previously characterized. Other NHPs showed comparatively higher proportions of phylum-level classification, yet a significant portion of these sequences could not be further classified at the genus level (Figure 2), indicating that vaginal phylotypes of NHPs are underrepresented in databases. The degree of genus-level resolution appeared to be associated with phylogenetic distance from humans. For example, most sequences from chimpanzee samples were well resolved at the genus level (unclassified range 0.4–18.3%), making this group second to humans in terms of percent-classified taxa (unclassified range 0.01–5.6%).
The vaginal microbiome is host specific
Comparison of vaginal microbial communities using Bray–Curtis dissimilarity indices revealed species-specific distinctions (Figure 3). ANOSIM supported species-specific vaginal microbiota (R=0.95, P<0.0001). A nonparametric PERMANOVA (Anderson, 2001) was used to test compositional differences of the vaginal microbiota among species. Pairwise PERMANOVA comparisons revealed significant differences in the composition of vaginal microbiota among primate species (p(perm)<0.001). However, such analysis did not support differences between captive and wild vervets (pseudo-t=0.943, p(perm)=0.503).
Figure 3.

Ordination using nonmetric multidimensional scale analysis, calculated based on Bray–Curtis similarity distances, was used to visually assess variation in patterns of microbial diversity. Each data point represents 16S rRNA gene sequence data from a single individual color coded by host species.
We used three distinct methods to test for phylogenetic congruence of the host and compositional changes in the vaginal microbiome. The phylogenetic analysis indicated that, although the community tree separated clades of bacterial communities according to their host origin, the most parsimonious tree (P-score=73 685) showed weak congruence with host phylogeny (Figures 4a and b). Low bootstrap values (<70%) were observed at inner nodes, corresponding to intraspecific branch points, and starkly contrasted with high bootstrap values (90–100%) in the interspecific outer nodes, supporting the host-specific characteristic of vaginal microbiomes.
Figure 4.

(a) Unrooted tree displaying the phylogenetic relationship between humans and NHPs. The aligned sequences based on 54 concatenated primate genes were used to build the maximum parsimony tree (Perelman et al., 2011). (b) Unrooted maximum parsimony tree showing phylogenetic positions of vaginal microbiomes. Vaginal samples are color coded to match with their host species in Figure 4a. Black: yellow baboons, blue: olive baboons, green: chimpanzees, brown: lemurs, magenta: howler monkeys, purple: mangabeys, cyan: red colobus, orange: vervets (captive and wild) and red: humans.
Second, an unweighted pair group method with arithmetic clustering of UniFrac distances clearly distinguished primate vaginal microbial communities according to the primate host, with the exception of howlers. Howlers were placed among the human and chimpanzee clade in the consensus tree. Jackknifing did not robustly support the majority of nodes in the consensus tree (<50%), which is consistent with the parsimony test described above (data not shown).
Finally, we found little correlation between host phylogeny and the Bray–Curtis dissimilarity matrix of vaginal microbiota. The highest correlation coefficient observed was −0.6 and the majority centered on −0.4 with some poor combinations nearing zero (Supplementary Figure S4). In addition, when we repeated the analysis after pooling the sequences produced from each sample according to host species, the correlation coefficient between host phylogeny matrix and vaginal microbiome similarity matrix was −0.46, −0.49 or −0.54, depending on the transformation method of OTU abundances for the Bray–Curtis calculations (square root, double square root and log transformation, respectively; data not shown). However, the two-tailed probability (P=0.0876) for even the highest correlation coefficient (−0.6) was higher than expected by chance, suggesting that statistical relation is possibly caused by a third variable. These findings indicate that, although the primate vaginal microbiomes are host specific, our data do not conclusively support that the host and microbiome phylogenies are congruent.
Multivariate dispersion test (PERMDISP function in Primer-E) comparing all groups supported the null hypothesis that all groups show equal dispersions in their respective ‘locations' (F=1.7285, df1=9, df2=58, P(perm)=0.344, 9999 permutations); however, individual pairwise tests indicated that the dispersions of community assemblages for chimpanzees, howlers and mangabeys were significantly larger than that of other NHPs (Supplementary Table S3), suggesting that there are other, presumably ecological factors confounding species-specific variation.
Host species was, by far, the overriding factor determining vaginal microbial community composition. PERMANOVA analysis (Table 2) indicated that primate species identity was sufficient to explain 42.7% of the variation in microbial composition observed (the total explainable variation was 44.1% based on all factors tested). Host species-specific factors affecting variation in microbial composition included (in the order from most to least correlated): geographic origin, female swelling and promiscuity, social structure, testes, body, and neonate weight, gestation duration, baculum length, group size and mating group size (Table 2). These analyses indicated that these factors shared responsibility for driving much of the variation in microbial composition between host species. However, these factors did not provide additional explanatory power beyond that of host species identity, with the exception of neonate weight, which explained an additional 1.4% of the total variation in microbial composition.
Several socioecological factors were also found to correlate more broadly with microbial species diversity within the vaginal microbiome (Supplementary Figures S5A–J); female promiscuity, mating group size and male testes mass were associated with increasing microbiome diversity (P<4.3 × 10−5, P<2.9 × 10−6, P<4.4 × 10−4, respectively). In contrast, increased group size and gestation duration were found to associate with decreased diversity (P<2 × 10−2 and P<7.7 × 10−3, respectively).
To identify the specific phylotypes that are significantly correlated with each host species, we employed indicator species analysis and tested ‘IndVal IV' values. Table 3 shows the top three indicator OTUs with highest IndVal IV values. Sneathia, Anaerosphaera and Aerococcus are significantly associated with multiple NHPs, and L. crispatus, Morganella and Pseudomonas spp. are the top three phylotypes associated with humans. Aerococcus and Abiotrophia (the top indicator OTUs in mangabeys) are known to be among the lactic acid-producing bacteria (Pfeiler and Klaenhammer, 2007) within the order Lactobacillales.
Table 3. Top three indicator OTUs in primates and humans.
Discussion
Our analyses indicate that ca. 43% of the variation in vaginal microbiome composition among NHP species and humans can be explained by species-specific socioecological factors, such as geographic origin, promiscuity, mating group size and testes mass, of which the majority of factors concern sexuality, specifically the number of mating partners. These findings underline the importance of physical, specifically sexual, contact between the hosts in the composition, as well as possible transmission and adaptive function of microbes within a population. Indeed, mutualistic and pathogenic bacteria use very similar mechanisms in colonizing hosts (Hentschel et al., 2000). Pathogenic microbes spread effectively among socially structured populations (Nunn et al., 2011) and more readily cross host species boundaries when the host species are closely related and inhabit the same geographical region (Woolhouse et al., 2001; Davies and Pedersen, 2008). Geographical vicinity (that is, habitat sharing) gives microbes opportunity to colonize susceptible sympatric host populations when they come in contact. Strikingly, even though humans and NHPs share pathogenic gut microbes, domesticated animals pose greater risk of transmitting pathogenic microbes to humans than NHPs due to habitat sharing (Pedersen et al., 2007).
We observed significant overlap among the explanatory socioecological factors. For example, primate sexual swellings are associated with multi-male mating and promiscuity (Stumpf et al., 2011). Testes mass is also associated with mating group size and promiscuity (Harcourt et al., 1981; Preston et al., 2003). Promiscuity and larger mating group sizes facilitate mucosal contact and transmission of microbes among many mating partners, whereas perineal swellings increase the length and volume of the vagina, and consequently increase exposure to microbes. These factors may account for some of the distinctness of the vaginal microbiomes in primate host species that vary socioecologically.
We found that more promiscuous primate species showed greater microbial diversity, which is consistent with microbial analyses in mice (MacManes, 2011). Promiscuity is likely to be a driving factor leading to systemic differences in the primate immune system (Nunn et al., 2000), as well as the rate of molecular evolution of genes impacting immune function (Wlasiuk and Nachman, 2010). This raises expectations that mating system, and in particular promiscuity, may influence the composition of vaginal microbiomes both directly and indirectly, which in turn may have a role in shaping the evolution of mating behavior (Immerman, 1986; Loehle, 1995; Thrall et al., 1997, 2000; Kokko et al., 2002; Sharon et al., 2010). Interestingly, primate vaginal microbiomes appeared unaffected by factors such as diet or captivity (see Supplementary Information), unlike primate gut microbiota (Amato et al., 2013; Amato, 2013). This suggests that while host species identity characterizes variation in microbial composition in the gut, vagina, and presumably other locales, host-specific factors particularly relevant to that locale (e.g. diet or sexuality) are what drive the host interspecific differences in microbiome variation.
We found a number of unclassifiable taxa observed in NHP vaginal microbiomes, particularly among species more evolutionarily distant from humans, suggesting existence of novel taxa. Unclassifiable taxa were most prevalent within lemurs, where a significant portion of reads could not be classified into any known phylum. Lemurs are evolutionarily primitive primates and the sexual behavior of lemurs is unusual in that members of this species copulate only briefly over a 2–4 day period. During the rest of the year, the vagina is sealed and thus is largely isolated from the environment (Ankel-Simons, 2007). In this respect, the vaginal microbiome of lemurs is quite distinct from any other microbiome we examined.
Even though there was considerable intraspecific variation of vaginal microbiomes among NHPs in this study (alpha diversity, Supplementary Table S1) and others (Rivera et al., 2010; Hashway et al., 2014), interspecific differences exceed intraspecific diversity. Despite exhibiting the lowest richness, the human vaginal microbiome showed a significantly broader degree of dispersion in beta diversity (Supplementary Figure S6), suggesting the presence of individuals with highly distinct microbial compositions within this group. Indeed, the human subjects Hm409 and Hm403 in our data set, similar to the ABVs (Ravel et al., 2013), represent distinct subsets within humans with microbiomes characterized by low abundance of Lactobacilli spp., yet enriched in anaerobic bacteria commonly found in the vaginal tracts of NHPs. These findings are consistent with previous observations on vaginal microbiota of rhesus macaque and humans (Spear et al., 2010).
The finding that Lactobacillus dominance, a trait believed critical to the health of human vaginal system (White et al., 2011), is not universal among primates illustrates the uniqueness of the human vaginal microbial ecosystem and provides added insight into what makes us human. The lactic acid-producing activity of the Lactobacillus spp. is thought to be an important factor in preventing the establishment of vaginal pathogens in humans (Boskey et al., 1999). Humans are unique in that women are characterized by continuous sexual receptivity and face considerable constraints on birth relative to NHPs. Unlike other primates, human gestations are longer, and the dimensions of the human pelvic outlet are smaller than the neonatal head (Rosenberg and Trevathan, 2002). Pregnant women are particularly vulnerable to microbial community disruptions (McGregor and French, 2000) and birth constraints greatly elevate risks of infection to both the fetus and mother. Although our correlational study cannot determine causes of the relationship between sexuality, birth and a distinctive microbial ecosystem, we hypothesize that humans effectively select microbial communities that minimize risks of sexual, gestational and post-parturition infection (see Stumpf et al. (2013) for additional hypotheses concerning human uniqueness).
This study is the first large, comprehensive and comparative analysis of the vaginal microbiomes from humans and NHPs. Our results provide insight into the selective factors affecting the vaginal microbiome across species, and highlight the diversity among primate vaginal microbiomes. These findings have implications for understanding host–microbe coevolution, disease risk and immunology. The uniqueness of the human vaginal microbiome has implications for understanding host–microbe relationships in humans, with potential applications to microbial dysbiosis and preterm birth, as well as for questioning the applicability of current NHP models to study sexually transmitted diseases in humans. These analyses also establish a baseline of the vaginal microbiome in primates, with conservation and captive management implications. Finally, these findings emphasize the need for greater understanding of the forces that shape our microbiomes and their impact on health, reproduction and disease.
Acknowledgments
This project was funded by UIUC RB and UIUC-Carle TR grants (to BA Wilson), NSF grant no. BCS-08-20709 (to RMS), and NSF HOMINID grant no. BCS-09-35347 (to SRL). We gratefully acknowledge Jeanne Altmann, Susan C Alberts, Jenny Tung, John Polk, and the late Abigail Salyers, as well as collaborator support from NCRR P40 grant no. RR019963 and VA contract no. VA247-P-0447 (to JDC), NIH grant no. 5R01RR016300 (to TT) grants R01RR016300 and R010D010980 to NBF, NSF grants BCS-0323553 and BCS-0323596 to SCA and JA, NSF grant BCS-0962807 to LCO, the Morris Animal Foundation support to TG, as well as the Office of the President of the Republic of Kenya, the Kenya Wildlife Service, and the Institute of Primate Research, Kenya, The Uganda Wildlife Authority, The Uganda National Council for Science and Technology, the Chimpanzee Sanctuary and Wildlife Conservation Trust, The Wildlife Conservation Society, The University of Veracruz, and Consejo Nacional de Areas Protegidas Guatemala. We also thank Michael Worobey and Anthony Yannarell for their guidance with phylogenetic and statistical analyses, respectively.
The authors declare no conflict of interest.
Footnotes
Supplementary Information accompanies this paper on The ISME Journal website (http://www.nature.com/ismej)
Supplementary Material
References
- Amato KR, Yeoman CJ, Kent A, Righini N, Carbonero F, Estrada A, et al. Habitat degradation impacts black howler monkey (Alouatta pigra) gastrointestinal microbiomes. ISME J. 2013;7:1344–1353. doi: 10.1038/ismej.2013.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amato KR.2013Co-evolution in context: the importance of studying gut microbiomes in wild animals Microb Sc Med 110–29.ISSN (Online) 2084-7653. [Google Scholar]
- Anderson MJ. A new method for non-parametric multivariate analysis of variance. Austral Ecology. 2001;26:32–46. [Google Scholar]
- Anderson MJ. Distance-based tests for homogeneity of multivariate dispersions. Biometrics. 2006;62:245–253. doi: 10.1111/j.1541-0420.2005.00440.x. [DOI] [PubMed] [Google Scholar]
- Ankel-Simons F.2007Primate Anatomy: an Introduction3rd ednElsevier Academic Press: San Diego, CA, USA [Google Scholar]
- Aykroyd OE, Zuckerman S. Factors in sexual skin oedema. J Physiol. 1938;94:13–25. doi: 10.1113/jphysiol.1938.sp003659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker GC, Smith JJ, Cowan DA. Review and re-analysis of domain-specific 16S primers. J Microbiol Methods. 2003;55:541–555. doi: 10.1016/j.mimet.2003.08.009. [DOI] [PubMed] [Google Scholar]
- Biasucci G, Rubini M, Riboni S, Morelli L, Bessi E, Retetangos C. Mode of delivery affects the bacterial community in the newborn gut. Early Hum Dev. 2010;86 (Suppl 1:13–15. doi: 10.1016/j.earlhumdev.2010.01.004. [DOI] [PubMed] [Google Scholar]
- Boskey ER, Telsch KM, Whaley KJ, Moench TR, Cone RA. Acid production by vaginal flora in vitro is consistent with the rate and extent of vaginal acidification. Infect Immun. 1999;67:5170–5175. doi: 10.1128/iai.67.10.5170-5175.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell C, Fuentes A, MacKinnon K, Bearder S, Stumpf R.2011Primates in Perspective2nd edn.Oxford University Press: Oxford [Google Scholar]
- Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CDC Trends in perinatal group B streptococcal disease - United States, 2000–2006. MMWR Morb Mortal Wkly Rep. 2009;58:109–112. [PubMed] [Google Scholar]
- Chapman T, Arnqvist G, Bangham J, Rowe L. Sexual conflict. TREE. 2003;18:41–48. [Google Scholar]
- Danielsson D, Teigen PK, Moi H. The genital econiche: focus on microbiota and bacterial vaginosis. Ann NY Acad Sci. 2011;1230:48–48. doi: 10.1111/j.1749-6632.2011.06041.x. [DOI] [PubMed] [Google Scholar]
- Davies TJ, Pedersen AB. Phylogeny and geography predict pathogen community similarity in wild primates and humans. Proc Biol Sci. 2008;275:1695–1701. doi: 10.1098/rspb.2008.0284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, et al. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol. 2006;72:5069–5072. doi: 10.1128/AEM.03006-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez-Bello MG, Costello EK, Contreras M, Magris M, Hidalgo G, Fierer N, et al. Proc Natl Acad Sci USA. 2010. pp. 11971–11975. [DOI] [PMC free article] [PubMed]
- Dufrene M, Legendre P. Species assemblages and indicator species: the need for a flexible asymmetrical approach. Ecol Monogr. 1997;67:345–366. [Google Scholar]
- Eren AM, Zozaya M, Taylor CM, Dowd SE, Martin DH, Ferris MJ. Exploring the diversity of Gardnerella vaginalis in the genitourinary tract of monogamous couples through subtle nucleotide variation. PLoS One. 2011;6:e26732. doi: 10.1371/journal.pone.0026732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fethers KA, Fairley CK, Hocking JS, Gurrin LC, Bradshaw CS. Sexual risk factors and bacterial vaginosis: a systematic review and meta-analysis. Clin Infect Dis. 2008;47:1426–1435. doi: 10.1086/592974. [DOI] [PubMed] [Google Scholar]
- Fethers KA, Fairley CK, Morton A, Hocking JS, Hopkins C, Kennedy LJ, et al. Early sexual experiences and risk factors for bacterial vaginosis. J Infect Dis. 2009;200:1662–1670. doi: 10.1086/648092. [DOI] [PubMed] [Google Scholar]
- Fierer N, Jackson RB. The diversity and biogeography of soil bacterial communities. Proc Natl Acad Sci USA. 2006;103:626–631. doi: 10.1073/pnas.0507535103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fierer N, Breitbart M, Nulton J, Salamon P, Lozupone C, Jones R, et al. Metagenomic and small-subunit rRNA analyses reveal the genetic diversity of bacteria, archaea, fungi, and viruses in soil. Appl Environ Microbiol. 2007;73:7059–7066. doi: 10.1128/AEM.00358-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank JA, Reich CI, Sharma S, Weisbaum JS, Wilson BA, Olsen GJ. Critical evaluation of two primers commonly used for amplification of bacterial 16S rRNA genes. Appl Environ Microbiol. 2008;74:2461–2470. doi: 10.1128/AEM.02272-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredricks DN, Fiedler TL, Marrazzo JM. Molecular identification of bacteria associated with bacterial vaginosis. N Engl J Med. 2005;353:1899–1911. doi: 10.1056/NEJMoa043802. [DOI] [PubMed] [Google Scholar]
- Gajer P, Brotman RM, Bai G, Sakamoto J, Schutte UM, Zhong X, et al. Temporal dynamics of the human vaginal microbiota. Sci Transl Med. 2012;4:132ra152. doi: 10.1126/scitranslmed.3003605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganu RS, Ma J, Aagaard KM. The role of microbial communities in parturition: is there evidence of association with preterm birth and perinatal morbidity and mortality. Am J Perinatol. 2013;30:613–624. doi: 10.1055/s-0032-1329693. [DOI] [PubMed] [Google Scholar]
- Gravett MG, Jin L, Pavlova SI, Tao L. Lactobacillus and Pediococcus species richness and relative abundance in the vagina of rhesus monkeys (Macaca mulatta) J Med Primatol. 2012;41:183–190. doi: 10.1111/j.1600-0684.2012.00537.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harcourt AH, Harvey PH, Larson SG, Short RV. Testis weight, body weight and breeding system in primates. Nature. 1981;293:55–57. doi: 10.1038/293055a0. [DOI] [PubMed] [Google Scholar]
- Hashway SA, Bergin IL, Bassis CM, Uchihashi M, Schmidt KC, Young VB, et al. Impact of a hormone-releasing intrauterine system on the vaginal microbiome: a prospective baboon model. J Med Primatol. 2014;43:89–99. doi: 10.1111/jmp.12090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hentschel U, Steinert M, Hacker J. Common molecular mechanisms of symbiosis and pathogenesis. Trends Microbiol. 2000;8:226–231. doi: 10.1016/s0966-842x(00)01758-3. [DOI] [PubMed] [Google Scholar]
- Hummelen R, Fernandes AD, Macklaim JM, Dickson RJ, Changalucha J, Gloor GB, et al. Deep sequencing of the vaginal microbiota of women with HIV. PLoS One. 2010;5:e12078. doi: 10.1371/journal.pone.0012078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huse SM, Welch DM, Morrison HG, Sogin ML. Ironing out the wrinkles in the rare biosphere through improved OUT clustering. Environ Microbiol. 2010;12:1889–1898. doi: 10.1111/j.1462-2920.2010.02193.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Immerman R. Sexually transmitted disease and human evolution: survival of the ugliest. Hum Ethol Newsletter. 1986;4:6–7. [Google Scholar]
- Jaspers E, Overmann J. Ecological significance of microdiversity: identical 16S rRNA gene sequences can be found in bacteria with highly divergent genomes and ecophysiologies. Appl Environ Microbiol. 2004;70:4831–4839. doi: 10.1128/AEM.70.8.4831-4839.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TK, Thomas SM, Ho M, Sharma S, Reich CI, Frank JA, et al. Heterogeneity of vaginal microbial communities within individuals. J Clin Microbiol. 2009;47:1181–1189. doi: 10.1128/JCM.00854-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kocher TD, Thomas WK, Meyer A, Edwards SV, Paabo S, Villablanca FX, et al. Dynamics of mitochondrial DNA evolution in animals: amplification and sequencing with conserved primers. Proc Natl Acad Sci USA. 1989;86:6196–6200. doi: 10.1073/pnas.86.16.6196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokko H, Ranta E, Ruxton G, Lundberg P. Sexually transmitted disease and the evolution of mating systems. Evolution. 2002;56:1091–1100. doi: 10.1111/j.0014-3820.2002.tb01423.x. [DOI] [PubMed] [Google Scholar]
- Leitich H, Kiss H. Asymptomatic bacterial vaginosis and intermediate flora as risk factors for adverse pregnancy outcome. Best Pract Res Clin Obstet Gynaecol. 2007;21:375–390. doi: 10.1016/j.bpobgyn.2006.12.005. [DOI] [PubMed] [Google Scholar]
- Ley RE, Lozupone CA, Hamady M, Knight R, Gordon JI. Worlds within worlds: evolution of the vertebrate gut microbiota. Nat Rev Microbiol. 2008;6:776–788. doi: 10.1038/nrmicro1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loehle C. Social barriers to pathogen transmission in wild animal populations. Ecology. 1995;76:326–335. [Google Scholar]
- Lozupone C, Knight R. UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol. 2005;71:8228–8235. doi: 10.1128/AEM.71.12.8228-8235.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma B, Forney LJ, Ravel J. Vaginal microbiome: rethinking health and disease. Annu Rev Microbiol. 2012;66:371–389. doi: 10.1146/annurev-micro-092611-150157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacManes MD. Promiscuity in mice is associated with increased vaginal bacterial diversity. Naturwissenschaften. 2011;98:951–960. doi: 10.1007/s00114-011-0848-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandar R, Mikelsaar M. Transmission of mother's microflora to the newborn at birth. Biol Neonate. 1996;69:30–35. doi: 10.1159/000244275. [DOI] [PubMed] [Google Scholar]
- Marrazzo JM, Antonio M, Agnew K, Hillier SL. Distribution of genital Lactobacillus strains shared by female sex partners. J Infect Dis. 2009;199:680–683. doi: 10.1086/596632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrazzo JM, Martin DH, Watts DH, Schulte J, Sobel JD, Hillier SL, et al. Bacterial vaginosis: identifying research gaps proceedings of a workshop sponsored by DHHS/NIH/NIAID. Sex Transm Dis. 2010;37:732–744. doi: 10.1097/OLQ.0b013e3181fbbc95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGregor JA, French JI. Bacterial vaginosis in pregnancy. Obstet Gynecol Surv. 2000;55:1–19. doi: 10.1097/00006254-200005001-00001. [DOI] [PubMed] [Google Scholar]
- Moller AP, Merino S, Brown CR, Robertson RJ. Immune defense and host sociality: a comparative study of swallows and martins. Am Nat. 2001;158:136–145. doi: 10.1086/321308. [DOI] [PubMed] [Google Scholar]
- Moller H, Cortes D, Engholm G, Thorup J. Risk of testicular cancer with cryptorchidism and with testicular biopsy: cohort study. BMJ. 1998;317:729. doi: 10.1136/bmj.317.7160.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunn CL, Gittleman JL, Antonovics J. Promiscuity and the primate immune system. Science. 2000;290:1168–1170. doi: 10.1126/science.290.5494.1168. [DOI] [PubMed] [Google Scholar]
- Nunn CL, Thrall PH, Leendertz FH, Boesch C. The spread of fecally transmitted parasites in socially-structured populations. PLoS One. 2011;6:e21677. doi: 10.1371/journal.pone.0021677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakley BB, Fiedler TL, Marrazzo JM, Fredricks DN. Diversity of human vaginal bacterial communities and associations with clinically defined bacterial vaginosis. Appl Environ Microbiol. 2008;74:4898–4909. doi: 10.1128/AEM.02884-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochman H, Worobey M, Kuo CH, Ndjango JB, Peeters M, Hahn BH, et al. Evolutionary relationships of wild hominids recapitulated by gut microbial communities. PLoS Biol. 2010;8:e1000546. doi: 10.1371/journal.pbio.1000546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson BD, Thaeler CS., Jr The mammalian baculum: hypotheses on the nature of bacular variability. J Mammal. 1982;63:1–15. [Google Scholar]
- Pedersen AB, Jones KE, Nunn CL, Altizer S. Infectious diseases and extinction risk in wild mammals. Conserv Biol. 2007;21:1269–1279. doi: 10.1111/j.1523-1739.2007.00776.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perelman P, Johnson WE, Roos C, Seuanez HN, Horvath JE, Moreira MA, et al. A molecular phylogeny of living primates. PLoS Genet. 2011;7:e1001342. doi: 10.1371/journal.pgen.1001342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeiler EA, Klaenhammer T. The genomics of lactic acid bacteria. Trends Microbiol. 2007;15:546. doi: 10.1016/j.tim.2007.09.010. [DOI] [PubMed] [Google Scholar]
- Preston BT, Stevenson IR, Pemberton JM, Coltman DW, Wilson K. Overt and covert competition in a promiscuous mammal: the importance of weaponry and testes size to male reproductive success. Proc Biol Sci. 2003;270:633–640. doi: 10.1098/rspb.2002.2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruesse E, Quast C, Knittel K, Fuchs B, Ludwig W, Peplies J, et al. SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 2007;35:7188–7196. doi: 10.1093/nar/gkm864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravel J, Gajer P, Abdo Z, Schneider GM, Koenig SS, McCulle SL, et al. Vaginal microbiome of reproductive-age women. Proc Natl Acad Sci USA. 2011;1:4680–4687. doi: 10.1073/pnas.1002611107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravel J, Brotman RM, Gajer P, Ma B, Nandy M, Fadrosh DW, et al. Daily temporal dynamics of vaginal microbiota before, during and after episodes of bacterial vaginosis. Microbiome. 2013;1:29. doi: 10.1186/2049-2618-1-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera AJ, Stumpf RM, Wilson B, Leigh S, Salyers AA. Baboon vaginal microbiota: an overlooked aspect of primate physiology. Am J Primatol. 2010;72:467–474. doi: 10.1002/ajp.20795. [DOI] [PubMed] [Google Scholar]
- Rosenberg K, Trevathan W. Birth, obstetrics and human evolution. BJOG. 2002;109:1199–1206. doi: 10.1046/j.1471-0528.2002.00010.x. [DOI] [PubMed] [Google Scholar]
- Ruvolo M, Disotell TR, Allard MW, Brown WM, Honeycutt RL. Resolution of the African hominoid trichotomy by use of a mitochondrial gene sequence. Proc Natl Acad Sci USA. 1991;88:1570–1574. doi: 10.1073/pnas.88.4.1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol. 2009;75:7537–7541. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sewankambo N, Gray RH, Wawer MJ, Paxton L, McNaim D, Wabwire-Mangen F, et al. HIV-1 infection associated with abnormal vaginal flora morphology and bacterial vaginosis. Lancet. 1997;350:546–550. doi: 10.1016/s0140-6736(97)01063-5. [DOI] [PubMed] [Google Scholar]
- Sharon G, Segal D, Ringo JM, Hefetz A, Zilber-Rosenberg I, Rosenberg E. Commensal bacteria play a role in mating preference of Drosophila melanogaster. Proc Natl Acad Sci USA. 2010;107:20051–20056. doi: 10.1073/pnas.1009906107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith RJ, Jungers WL. Body mass in comparative primatology. J Hum Evol. 1997;32:523–559. doi: 10.1006/jhev.1996.0122. [DOI] [PubMed] [Google Scholar]
- Spear GT, Gilbert D, Sikaroodi M, Doyle L, Green L, Gillevet PM, et al. Identification of rhesus macaque genital microbiota by 16S pyrosequencing shows similarities to human bacterial vaginosis: implications for use as an animal model for HIV vaginal infection. AIDS Res Hum Retroviruses. 2010;26:193–200. doi: 10.1089/aid.2009.0166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan S, Fredricks DN. The human vaginal bacterial biota and bacterial vaginosis. Interdiscip Perspect Infect Dis. 2008;2008:750479. doi: 10.1155/2008/750479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan S, Liu C, Mitchell CM, Fiedler TL, Thomas KK, Agnew KJ, et al. Temporal variability of human vaginal bacteria and relationship with bacterial vaginosis. PLoS One. 2010;5:e10197. doi: 10.1371/journal.pone.0010197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strier KB.2007Primate Behavioral Ecology3rd ednAllyn and Bacon: Needham, MA, USA [Google Scholar]
- Stumpf RM, Rivera A, Yildirim S, Yeoman C, Wilson BA, Polk JD, et al. The vaginal microbiome: Comparative perspectives and insights into human health and disease. Yearbook Phys Anthropol. 2013;57:119–134. doi: 10.1002/ajpa.22395. [DOI] [PubMed] [Google Scholar]
- Stumpf RM, Martinez-Mota R, Milich KM, Righini N, Shattuck MR. Sexual conflict in primates. Evol Anthropol. 2011;20:62–75. doi: 10.1002/evan.20297. [DOI] [PubMed] [Google Scholar]
- Tannock GW, Fuller R, Smith SL, Hall MA. Plasmid profiling of members of the family Enterobacteriaceae, lactobacilli, and bifidobacteria to study the transmission of bacteria from mother to infant. J Clin Microbiol. 1990;28:1225–1228. doi: 10.1128/jcm.28.6.1225-1228.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thrall PH, Antonovics J, Bever J. Sexual transmission of disease and host mating systems: within-season reproductive success. Am Nat. 1997;149:485–506. [Google Scholar]
- Thrall PH, Antonovics J, Dobson AP. Sexually transmitted diseases in polygynous mating systems: prevalence and impact on reproductive success. Proc Biol Sci. 2000;267:1555–1563. doi: 10.1098/rspb.2000.1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Oostrum N, De Sutter P, Meys J, Verstraelen H. Risks associated with bacterial vaginosis in infertility patients: a systematic review and meta-analysis. Hum Reprod. 2013;28:1809–1815. doi: 10.1093/humrep/det096. [DOI] [PubMed] [Google Scholar]
- Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol. 2007;73:5261–5267. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White BA, Creedon DJ, Nelson KE, Wilson BA. The vaginal microbiome in health and disease. Trends Endocrinol Metab. 2011;22:389–393. doi: 10.1016/j.tem.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiesenfeld HC, Hillier SL, Krohn MA, Landers DV, Sweet RL. Bacterial vaginosis is a strong predictor of Neisseria gonorrhoeae and Chlamydia trachomatis infection. Clin Infect Dis. 2003;36:663–668. doi: 10.1086/367658. [DOI] [PubMed] [Google Scholar]
- Wiesenfeld HC, Hillier SL, Meyn LA, Amortegui AJ, Sweet RL. Subclinical pelvic inflammatory disease and infertility. Obstet Gynecol. 2012;120:37–43. doi: 10.1097/AOG.0b013e31825a6bc9. [DOI] [PubMed] [Google Scholar]
- Wlasiuk G, Nachman MW. Promiscuity and the rate of molecular evolution at primate immunity genes. Evolution. 2010;64:2204–2220. doi: 10.1111/j.1558-5646.2010.00989.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolhouse ME, Taylor LH, Haydon DT. Population biology of multihost pathogens. Science. 2001;292:1109–1112. doi: 10.1126/science.1059026. [DOI] [PubMed] [Google Scholar]
- Yeoman CJ, Thomas SM, Miller ME, Ulanov AV, Torralba M, Lucas S, et al. A multi-omic systems-based approach reveals metabolic markers of bacterial vaginosis and insight into the disease. PLoS One. 2013;8:e56111. doi: 10.1371/journal.pone.0056111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yildirim S, Yeoman CJ, Sipos M, Torralba M, Wilson BA, Goldberg TL, et al. Characterization of the fecal microbiome from nonhuman wild primates reveals species specific microbial communities. PLoS One. 2010;5:e13963. doi: 10.1371/journal.pone.0013963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Brotman RM, Gajer P, Abdo Z, Schuette U, Ma S, et al. Recent advances in understanding the microbiology of the female reproductive tract and the causes of premature birth. Infect Dis Obstet Gynecol. 2010;2010:737425. doi: 10.1155/2010/737425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Brown CJ, Abdo Z, Davis CC, Hansmann MA, Joyce P, et al. Differences in the composition of vaginal microbial communities found in healthy Caucasian and black women. ISME J. 2007;1:121–133. doi: 10.1038/ismej.2007.12. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.