

Abstract
Ulcerative colitis (UC) and Crohn’s disease (CD) are two forms of chronic inflammatory bowel disease. CD4 T cells play a central role in the pathogenesis of both diseases. Smoking affects both UC and CD but with opposite effects, ameliorating UC and worsening CD. We hypothesized that the severity of gut inflammation could be modulated through T-cell nicotinic acetylcholine receptors (nAChRs) and that the exact clinical outcome would depend on the repertoire of nAChRs on CD4 T cells mediating each form of colitis. We measured clinical and immunologic outcomes of treating BALB/c mice with oxazolone- and TNBS-induced colitides by nicotine. Nicotine attenuated oxazolone colitis, which was associated with increased percentage of colonic Tregs and a reduction of Th17 cells. TCR stimulation of naïve CD4+CD62L+ T cells in the presence of nicotine upregulated expression of Foxp3. In marked contrast, nicotine worsened TNBS colitis, and this was associated with increased Th17 cells among colonic CD4 T cells. Nicotine upregulated IL-10 and inhibited IL-17 production, which could be abolished by exogenous IL-12 that also abolished the nicotine-dependent upregulation of Tregs. The dichotomous action of nicotine resulted from the up- and downregulation of anti-inflammatory α7 nAChR on colonic CD4 T cells induced by cytokines characteristic of the inflammatory milieu in oxazolone (IL-4), and TNBS (IL-12) colitis, respectively. These findings help explain the dichotomous effect of smoking in patients with UC and CD, and underscore the potential for nicotinergic drugs in regulating colonic inflammation.
Keywords: oxazolone colitis, TNBS colitis, Tregs, Th17 cells, α7 nicotinic receptor, IL-4, IL-12
Introduction
Ulcerative colitis (UC) and Crohn’s disease (CD) are chronic inflammatory disorders of the gastrointestinal tract that result in significant morbidity. A search for novel and more efficient approaches to treat patients with these two forms of inflammatory bowel disease (IBD) is one of the most important tasks of contemporary clinical medicine. UC is characterized by contiguous mucosal inflammation in the rectum and colon causing epithelial cell destruction and sloughing leading to ulceration. CD, in contrast, is manifested by transmural, granulomatous inflammation occurring anywhere in the alimentary canal but is usually centered in the terminal ileum and ascending colon.
Both UC and CD are epidemiologically related to smoking (1–4). Most patients with UC are non-smokers, and patients with a history of smoking usually acquire their disease after they have stopped smoking (5–7). Upon cessation of smoking, patients with UC experience more severe disease progression that can be ameliorated by returning to smoking (8–10). In contrast, patients with CD experience severe disease when smoking, requiring an immediate and complete cessation of any tobacco usage (3, 11).
Nicotine appears to be the key mediator of the dichotomous effect of smoking on UC and CD, because its administration in transdermal patches inhibits inflammation associated with UC but not in patients with CD (8, 12–15). These observations suggested that the effects of smoking in patients with IBD are mediated, at least in part, by the nicotinic acetylcholine receptors (nAChRs) activated by nicotine. The effects of nicotine are complex and include its specific actions on the central nervous system, gastrointestinal tract and immune system. It is believed that the therapeutic effects of nicotine in UC are mediated by the nAChRs of gut immune cells (12). The nAChRs expressed by T cells play an important role in T cell development, differentiation and function (reviewed in (16)). We have recently reported that, on the one hand, nicotinergic stimulation alters cytokine production, and, on the other hand, that immune cytokines themselves can alter the structure and function of T cell nAChRs (17, 18).
The pathogenesis of IBD is unknown but is thought to reflect an interaction of genetic and environmental factors. Despite having a common basis in overresponsiveness to mucosal antigens, the two diseases have considerably different pathophysiologies. CD is associated with a Th1/Th17 T cell–mediated response induced by IL-12, and possibly IL-23, with concomitant increased production of IL-2, IL-17, IL-18 and IFN-γ, whereas UC is associated with an atypical Th2-mediated response characterized by NKT cell secretion of IL-13, and increased production of IL-4 and IL-5 (19–27).
Many murine models of mucosal inflammation mimicking human IBD forms have been described, leading to a profound increase in our understanding of the immunologic basis of colonic inflammation (28). Experimental animal models have convincingly demonstrated that CD4 T cells play a central role in IBD (28). A hapten-induced colitis in mice caused by intrarectal (i.r.) instillation of trinitrobenzene sulfonic acid (TNBS) is a Th1 T cell–mediated colitis that captures many of the features of CD (29–32). In contrast, a hapten-induced colitis caused by i.r. instillation of oxazolone (4-ethoxymethylene-2-phenyl-2-oxazoline-5-one) is driven by the production of Th2 cytokines and reproduces many of the features of UC (33, 34). The potential for nicotinergic drugs in regulating colonic inflammation has been recently documented in vagotomized mice with experimental IBD, wherein the nAChR agonist nicotine alleviated and the antagonist hexamethonium aggravated colitis (35, 36). However, recent studies on the clinical and immunologic outcomes of administration of nicotinergic agents to mice with TNBS-induces colitis gave conflicting results (37, 38), suggesting the need for an in depth analysis of the nicotinergic immunopharmacology in experimental IBDs.
We postulated that the severity of gut inflammation in mice with experimental IBD could be regulated through T cell nAChRs and that the exact clinical outcome would depend on the particular type of T cell-mediated experimental colitis. In the present study, we measured the clinical and the immunologic outcomes of treating BALB/c mice with oxazolone- and TNBS-induced colitides by nicotine. Nicotine attenuated oxazolone colitis and worsened TNBS colitis, which was associated with increased percentage of colonic Treg cells in mice with oxazolone-, but not TNBS-induced colitis. The dichotomous action of nicotine could result from up- and downregulation of the expression of anti-inflammatory α7 nAChR in colonic CD4 T cells under the influence of the cytokines characteristic of oxazolone and TNBS colitides, respectively.
Materials and Methods
Induction of colitis
Oxazolone and TNBS colitides in BALB/c mice were induced using standard techniques (34, 39). Specific pathogen-free 6–8 week-old female mice, weighting 20–25 g, were purchased from The Jackson Laboratory (Bar Harbor, ME) and maintained in the Animal Facility at the University of California, Irvine. All experiments were approved by Institutional Animal care and Use Committee. Animals were lightly anesthetized with Kethamine (100 mg/kg) and Xylazine (10 mg/kg), and a 3.5-F silicon catheter (Harvard Apparatus, Holliston, MA) was inserted 4 cm into the lumen of the colon, through which 100 μl of 3% oxazolone or 3% TNBS in 47.5% ethanol in saline were slowly infused. To ensure distribution of the instilled solutions within the entire colon and cecum, the mice were held vertically for 30 seconds after removing the catheter. The mice treated i.r. with a vehicle, i.e., 47.5% ethanol in saline, served as disease controls. The α7 nAChR knockout mice used in the oxazolone colitis experiments were from The Jackson Laboratory. The nicotine saline solution was freshly prepared from nicotine hydrogen tartrate salt (Sigma-Aldrich, Inc, St. Louis, MO) and subcutaneously (s.c.) injected into each mouse. For nicotine treatment, mice received nicotine twice a day at the total dose of 7.5 mg/kg for 5 days starting one day prior to the administration of haptenating agents. Mice were sacrificed on day 2 or day 5 after hapten infusion. Twenty mice per each treatment condition were used in experiments.
Evaluations of colitis severity
To grade the clinical severity of colitis, mice were assessed daily for body weight, stool consistency, rectal bleeding and presence of blood in the stool by a guaiac paper test. The clinical disease activity index (DAI) was computed based on scores for weigh loss, stool consistency, hematochezia and rectal bleeding using standard protocols (31, 40, 41), as detailed in Table 1. To grade the severity of intestinal histopathology, the entire colon was excised from each mouse, opened longitudinally, fixed in 10% neutral buffered formalin, embedded in paraffin, cut into 5 μm sections, and stained with hematoxylin and eosin (H&E). The degree of inflammation and epithelial injury was graded in a blinded fashion by the same pathologist (Dr. Edwards). The histology activity index (HAI) was assessed to quantify colitis severity based on standard criteria for assessment of inflammation and injury in mice with experimental IBD, such as appearance and extent of inflammatory cell infiltration, goblet cell mucin depletion, mucosal thickening and the extent of damage (31, 34, 42, 43), as described in Table 2.
Table 1.
The Disease Activity Index (DAI) scoring system used in this study.
| Score | Weight loss (%) | Stool consistency | Blood in stool (hematochezia) | Rectal bleeding |
|---|---|---|---|---|
| 0 | none | normal | none | absent |
| 1 | 1–10 | loose stool | haemaoccult positive | present |
| 2 | 11–20 | diarrhea | gross blood | n/a |
| 3 | ≥20 | n/a | n/a | n/a |
Table 2.
The Histologic Activity Index (HAI) scoring system used in this study.
| Score | Infiltrate | Extent of inflammatory infiltrate | Goblet cell mucin depletion | Mucosal thickness | Length of bowel damage |
|---|---|---|---|---|---|
| 0 | normal/physiologic | none | none | normal | none |
| 1 | minimal elevation | single or rare, scattered foci | minimal | minimal, focal thickening | <20% |
| 2 | expanded within or beyond lamina propria | patchy, moderately abundant | moderate | moderate, multifocal thickening | 21–40% |
| 3 | crypt abscesses or submucosal involvement | extensive | extensive | extensive | 41–60% |
| 4 | inflammatory crypt destruction with erosion of the mucosa | n/a | n/a | n/a | >61% |
Immunofluorescence staining
The 8 μm thick cryostat sections of mouse colons were fixed with cold acetone, blocked with 5% fetal calf serum (FCS), and used in the indirect immunofluorescence assays following a standard procedure (44). The V450 rat anti-mouse CD4 (BD Bioscience, San Jose, CA) and Alexa Fluor 488 anti-mouse CD25 (BioLegend, San Diego, CA) were used for double staining, and PE rat anti-mouse Foxp3 (BD Bioscience) was added for triple-staining. For the CD4/IL-17 double staining, we used V450 rat anti-mouse CD4 (BD Bioscience) and a combination of rat anti-mouse IL-17 and FITC-labeled rabbit anti-rat antibody (both from Abcam, Cambridge, MA). To identify the CD4 T cells expressing α4 or α7 nAChRs, we used rabbit anti-α4 or anti-α7 antibodies (both from Abcam, Cambridge, MA) visualized with Alexa Fluor 488 goat anti-rabbit antibody (Invitrogen, Eugene, OR), and rat anti-mouse CD4 antibody (BioLegend) visualized with Alexa Fluor 555 goat anti-rat antibody (Invitrogen). The slides were thrice washed, mounted using the ProLong Gold anti-fade reagent (Invitrogen) and analyzed with a Nikon eclipse Ti fluorescence microscope. Samples without primary antibodies served as negative controls. In each slide, the positive cells were counted in ten high-power 60x microscopic fields.
Isolation of lamina propria (LP) and spleen mononuclear cells (MCs)
LPMCs were isolated from freshly obtained colonic specimens using modification of previously described protocols (45). Briefly, the colons were opened longitudinally and cut into pieces. After incubation in DMEM with 2 mM dithiothreitol (DTT; Bio-Rad, Hercules, CA) for 30 min, vortexing and passing through a cell strainer, the tissue was digested in DMEM containing collagenase D/Dispase (5 mg/mL) and DNase I (5 mg/ml) (both from Roche, Indianapolis, IN). The LP cells released from the colonic tissue were harvested and subjected to Percoll (Sigma-Aldrich Co) gradient separation. The LPMCs were collected at the 40/80 interphase. Spleen MCs were extracted by Ficoll (Sigma-Aldrich Co) gradient centrifugation. The obtained MCs were washed and resuspended in either the FACS buffer or culture medium.
Nicotine stimulation experiments
CD4+CD62L+ naïve T cells from intact BALB/c mice were enriched by using the CD4+CD62L+ T cell isolation kit II and MACS sorting (Miltenyi Biotec, Auburn, CA) following the protocol provided by the manufacturer. To stimulate T cells via the TCR/CD3 complex, 250 μl of 2×106 cells/ml were seeded in each well of the 24-well tissue culture plates (BD Falcon™, BD Biosciences) coated for 2 hrs at 37°C with 10 μg/ml of anti-mouse CD3 (clone 145-2C11) and 4.0 μg/ml of anti-mouse CD28 (clone 37.51) antibodies (both from BD Biosciences), and cultured for 5 days in RPMI 1640 supplemented with 10% FCS, 0.05 mM 2-mercaptoethanol, 10 mM HEPES buffer, 100 U/ml penicillin, 100 μg/ml streptomycin, 2 mM L-glutamine and 1 mM sodium pyruvate (all from Invitrogen) in a humid, 5% CO2 incubator at 37°C in the absence or presence of 100 μM of nicotine added on a daily basis. Some T cells were also exposed to 10 ng/ml of IL-4 or IL-12 (both from PeproTech, Rocky Hill, NJ). Changes in Foxp3 expression were analyzed by flow cytometry (see below) and immunoblotting, as follows. Briefly, after incubation, 1×106 cells were lysed in 1% NP-40 lysis buffer (Sigma-Aldrich Co), and the proteins were separated on 4–12% SDS-PAGE and transferred to nitrocellulose (Bio-Rad, Hercules, CA). After blocking with the Odyssey blocking solution, the membranes were incubated overnight with rabbit anti-mouse Foxp3 antibody (Abcam) followed by a goat anti-rabbit secondary antibody conjugated with LI-COR IRDye 800CW (LI-COR Biosciences, Lincoln, NE). For protein loading control, same membrane was re-probed with a β-actin antibody (Genway, San Diego, CA). The Odyssey Infrared Imaging System (LI-COR Biosciences) was used to scan the membranes and visualize protein bands. The ImageQuant5.1 software (Molecular Dynamics Inc., Sunnyvale, CA) was used for the semi-qualitative analysis of protein bands.
Cytokine stimulation experiments
After stimulation of CD4+CD62L+ naïve T cells by CD3/CD28 without any additions (control) or in the presence of 10 ng/ml of IL-4 or IL-12, as above described, the expression of α7 gene was quantitated at the mRNA and protein levels by real-time polymerase chain reaction (qPCR) and in-cell western (ICW) assays, respectively. Total RNA was extracted with the RNeasy Mini Kit (Qiagen, Valencia, CA) and the α7 mRNA expression was measured using the TaqMan® Gene Expression Assay (Mm01317884_m1) at the Applied Biosystems 7500 system in accordance to the manufacturer’s protocol, as described by us in detail elsewhere (46). Ubiquitin C (UBC, Mm01198158_m1) was included as endogenous reference gene, and the data were analyzed with a Sequence Detection Software version 1.2.3 (Applied Biosystems, Carlsbad, CA). The α7 protein level was determined in situ by ICW, as described by us in detail elsewhere (17), using anti-α7 antibody (Abcam) and secondary IRDye 800CW goat anti-rabbit antibody (LI-COR Biosciences, Lincoln, NE). Sapphire700 (LI-COR Biosciences) and DRAQ (Cell Signaling, Danvers, MA) was used to normalize for cell number/well. The receptor protein expression was quantitated using the LI-COR Odyssey Imaging System. The results of both qPCR and ICW assays were expressed as mean ± SD of α7 mRNA or protein relative to that of control, i.e., T cells stimulated by CD3/CD28 without IL-4 and IL-12, taken as 1.
Flow cytometry
Single, double or triple color flow cytometric analyses were performed using a BD FACSCalibur™ bench top analyzer and WinMDI software (http://facs.scripps.edu/software.html). The CD3PE/CY5, CD4FITC and CD8PE (all from eBioscience, Inc., San Diego, CA) antibodies were used as cell-surface markers of T cell subpopulations. The FITC IL-10, FITC IL-4, PE IL-17 and PE interferon-γ (IFN-γ), Alexa Fluor® 647 Foxp3 antibodies (all from BD Bioscience) were used for intracellular staining.
ELISA assays
The IL-10, IL-17 and IFN-α were assayed by antibody sandwich ELISA in cell culture supernatants using the mouse ELISA Ready-SET-Go kit (eBioscience). ELISA assays of IL-10, IL-4, IL-12, IL-23 and IFN-γ in colon lysates were performed using mouse ELISA Immunoassay kits (R&D Systems, MN, USA). Total protein was extracted using the Total Protein Extraction Kit (Millipore, MA, USA) in accordance to the manufacturer’s protocol.
Statistical analysis
All experiments were performed in duplicates or triplicates. Statistical significance was determined using the Student’s t-test. The differences were deemed as significant when the calculated p value was <0.05.
Results
Oxazolone and TNBS induce acute forms of UC- and CD-like colitides, respectively, in BALB/c mice
Mice treated with either oxazolone or TNBS in both cases reproducibly developed a rapid onset colitis marked by weight loss and diarrhea. Mice were evaluated daily to assess the DAI. Their colons were removed post-mortem and examined macro- and microscopically. Macroscopic examination of colons from mice treated with either oxazolone or TNBS in both cases revealed striking hyperemia and focal mucosal necrosis (Fig. 1A). Colons from the TNBS-treated mice were hyperemic and contained practically no feces due to profound diarrhea. The peak of clinical disease in both forms of colitis occurred on the 5th day after i.r. instillation of the haptenating agent (Fig. 1B). Histological examination of the H&E sections of colons from mice with oxazolone colitis showed extensive goblet cell depletion as well as dense chronic inflammatory infiltrates in the lamina propria, with focal granuloma formation (Fig. 1C). The inflammation observed in colons from mice with TNBS-induced colitis also featured goblet cell depletion and lymphocytic infiltrates (Fig. 1C). Thus, the macro- and microscopic evaluations of colons from BALB/c mice treated with oxazolone and TNBS revealed clinical and pathologic correlations with UC and CD, respectively, consistent with previous reports (29, 33, 47, 48).
Figure 1. Clinical and histological correlations in mice with experimental colitides treated with nicotine.

A. Representative images of colons dissected from control BALB/c mice treated i.r. with 47.5% ethanol in saline (Control), mice with oxazolone-induced colitis either untreated (Ox) or treated with 7.5 mg/kg/day of nicotine (Ox+N), and untreated (T) vs. nicotine-treated (T+N) mice with TNBS colitis. The treatment with nicotine started from the day before instillation of a haptenating agent (i.e., at the day “zero”) and ended on the 4th day of experiment, i.e., the day before mice were sacrificed. Note: the colon from untreated mouse with oxazolone colitis has several necrotic areas, whereas in the nicotine-treated animal the inflammation is visible only in the distal segment. While the colon of mouse with TNBS colitis is diffusely hyperemic, it lacks overt necrosis. In contrast, the mouse with TNBS colitis treated by nicotine has developed necrotic areas, as seen in the proximal segment of colon.
B. Clinical evaluation of the effects of nicotine treatment on the severity of colitis in mice with oxazolone- and TNBS-induced colitides. The DAI values were computed as described in detail in Materials and Methods (Table 1). In oxazolone colitis, the differences are statistically significant (p<0.05) on days 4 and 5. In TNBS colitis, the statistical differences between the DAI values of nicotine-treated vs. untreated mice are significant (p<0.05) starting from the 3rd day after instillation of the haptenating agent. Each point represents the mean ± SD of data obtained in 20 mice. The DAI values in control mice instilled with the ethanol solution without haptenating agents were equal or close to zero (data not shown).
C. The representative images of the histologic findings in nicotine-treated vs. untreated mice with oxazolone- and TNBS-indiceds colities. The photomicrographs of H&E stained 5 μm sections of matching segments of colons were made at 10x magnification. Abbreviations are the same as as in panel “A”. Note: the extensive goblet cell depletion with submucosal involvement and lymphocytic infiltrations characteristic of oxazolone colitis were not present in mice treated with nicotine. In contrast, nicotine treatment aggravated colonic inflammation in TNBS colitis, leading to crypt destruction, goblet cells depletion and extensive lymphocytic infiltration.
D. Histopathologic analysis of the effects of treatment with nicotine on the colonic inflammation in mice with oxazolone- and TNBS-induced cotises. The HAI values were computed as described in detail in Materials and Methods (Table 2). Nicotine treatment protected the colons from inflammation induced by oxazolone, as can be judged from an approximately 2-fold decrease of HAI. Vice versa, the HAI value computed in mice with TNBS colitis treated by nicotine significantly increased, indicating that nicotine worsened TNBS-induced colonic inflammation. Asterisk = p<0.05 compared to the HAI value computed in colons of the nicotne-untreated mice with the respective form of colitis.
Nicotine attenuates the oxazolone colitis and exacerbates the TNBS colitis
To evaluate the effects of nicotine on the development and severity of experimental colitis, the mice were daily treated with nicotine solution administered s.c. starting on the day before instillation of each haptenating agent.
Nicotine alleviated oxazolone-induced colitis, as nicotine-treated mice showed significantly (p<0.05) decreased DAI values on days 4 and 5 and colons from nicotine-treated mice had only mild hyperemia restricted to the distal part (Figs. 1A,B). Histologically, nicotine treatment drastically diminished goblet cell mucin depletion and the intensity of lymphocytic infiltrates (Fig. 1C). Consequently, the calculated HAI value in colons from nicotine-treated mice was 2-fold less compared to that in colons from untreated mice with oxazolone colitis (Fig. 1D).
In marked contrast, the nicotine-treated mice with TNBS-induced colitis showed significantly (p<0.05) elevated DAI throughout the treatment period, and their colons had more severe macroscopic and microscopic inflammation than colons of the untreated mice with TNBS colitis (Figs. 1A,B). Nicotine treatment was associated with the appearance of extensive mucosal necrosis in the proximal colon, extensive crypt damage, complete goblet cell depletion, lamina propria destruction, and massive lymphocytic infiltration (Fig. 1C). HAI was significantly (p<0.05) elevated (Fig. 1D).
Thus, nicotine attenuated oxazolone-induced colitis but exacerbated TNBS-induced colitis, consistent with the notion that smoking/nicotine ameliorates UC and worsens CD (3, 12).
Nicotine alters both peripheral and colonic T lymphocyte populations in mice with experimental colitis
Because T cells play an important role in the pathogenesis of IBD (28), we analyzed major populations of T cells in the spleens and colons of experimental mice. The expression of CD3, CD4 and CD8 was measured by flow cytometry.
Untreated mice with oxazolone colitis showed an increased percentage of CD3+T cells among splenic MCs, and of CD4 T cells among splenic CD3+T cells (Fig. 2A). In the nicotine-treated mice, the numbers of these cells returned to the normal levels characteristic of control BALB/c mice. Mice with oxazolone colitis also had increased CD4 T cells and decreased CD8+T cells among lamina propria CD3+T cells. Treatment with nicotine led to a further increase of percentage of CD4 T cells among lamina propria CD3+T cells (Fig. 2B,C).
Figure 2. The effects of nicotine treatment of mice with oxazolone- and TNBS-induced colitides on the peripheral and colonic populations of T lymphocytes.

The FCM assays with T cell marker antibodies were performed using splenocytes and LPMCs freshly isolated from intact (control) and nicotine-treated and untreated mice with experimental colitides at the end of nicotine treatments, as detailed in the Materials and Methods section. The cells were triple stained for CD3, CD4 and CD8 and examined by flow cytometry. The contour plots were generated after gating on lymphocytes (by forward and side scatter) for CD3 staining or gating on CD3 T cells during the analyses of CD4/CD8 staining. Shown on the graphs are representative dot blots for the CD3 T cell population of spleen MCs (A) and the CD4 and CD8 T cell subpopulations of CD3 T cells from spleen (B) and colon (C). Note: treatment with nicotine decreased the percentage of spleen CD3 and CD4 T cells to the normal levels in mice with oxazolone colitis, and further elevated the percentage of CD3 T cells without altering the already elevated numbers of spleen CD4 T cells in mice with TNBS colitis.
Similar to oxazolone colitis, mice with TNBS colitis had an increased percentage of splenic CD3+ and CD4 T cells (Fig. 2A). However, the percent increase above the baseline in splenic CD3+ and CD4 T cells was greater than in oxazolone colitis. Alterations in lamina propria T-cell populations in TNBS colitis matched those in oxazolone colitis, i.e., increased CD4 T cells, decreased CD8+ T cells, and further elevation of CD4 T-cells following nicotine treatment (Fig. 2B,C).
Thus, the differences between the effects of nicotine on oxazolone- vs. TNBS-induced colitis were the following: 1) normailzation of the percentage of splenic CD3+ and CD4 T cells in the former, and 2) additional elevation of the percentage of CD3+T cells without changing the already elevated numbers of spleen CD4 T cells in TNBS colitis.
Distinct effects of nicotine on the numbers of Th17 T cells and Foxp3+ Tregs in the colons affected by oxazolone or TNBS colitis
The regulatory T cells (Tregs)/Th17 axis plays an important role in the control and development of IBD (49, 50). Therefore, to elucidate the role of CD4 T cells in mediating dichotomous effects of nicotine on the severity of oxazolone- and TNBS-induced colitides, we determined relative proportions of Tregs and Th17 T cells among LPMCs in nicotine-treated vs. untreated mice with each form of colitis. While nicotine treatment was associated with a significant (p<0.05) increase in colonic CD4+CD25+ T cells in both oxazolone- and TNBS-induced colitides (Fig. 3A), the percentage of CD25+Foxp3+ Tregs among colonic CD4 increased >13-fold in oxazolone colitis but remained practically unchanged in TNBS colitis (Fig. 3B). Nicotine induced the opposite response regarding Th17-positive colonic CD4 T cells, with a significant decrease in oxazolone colitis and an increase in TNBS colitis (p<0.05) (Fig. 3C).
Figure 3. The effects of nicotine treatment of mice with experimental IBD on the percentage of Tregs and Th17 cells among colonic CD4 T cells.

The cryosections of colons from mice with oxazolone- and TNBS-induced colitides treated with nicotine were subjected to a double or triple immunofluorescence staining for CD4/CD25, CD4/CD25/Foxp3 and CD4/Th17 on day 5 after hapten treatment, as described in Materials and Methods. The results are expressed as means ± SD of the percentage of CD25 (A), CD25/Foxp3 (B), and IL-17 (C) positive cells among colonic CD4 T cells, taken as 100%. The data was computed in at least 10 different microscopic fields at magnification 60x in the triplicates of tissue sections from three mice in each group. Asterisk = p<0.05 compared to nicotine-untreated mice with the respective form of colitis.
We also found that on day 2 both TNBS and TNBS+nicotine treatment groups featured similar T cell repertoire: ~80% of colonic T cells were CD4 positive, and ~90% of CD4+ T cells were also CD17 positive (not shown). These findings are consistent with the lack of significant differences between DAI values in the TNBS and TNBS+nicotine treatment groups on day 2 (Fig. 1B).
Thus, the ability of nicotine to ameliorate oxazolone-induced colitis was associated with an expansion of Tregs and reduction of CD4 Th17+ T cells among LPMCs. In contrast, the nicotine-dependent worsening of TNBS-induced colitis was associated with an elevation in lamina propria CD4 Th17+ T cells. Therefore, we next investigated the mechanism underlying the differential effects of nicotine in these two forms of experimental IBD.
Nicotinergic stimulation of naïve CD4+CD62L+ T cells facilitates Foxp3 expression
Since T cells express the nAChRs that meditate immunopharmacologic action of nicotine on these cells (17), we hypothesized that the immunomodulatory effects of nicotine in mice with experimental colitis are mediated, in part, by nAChR-dependent alterations of T cell development and function. To test this hypothesis, we exposed naïve CD4+CD62L+ T cells from intact BALB/c mice to 100 μM nicotine for 5 days, to match the in vivo nicotine treatment dose used in this study, and then evaluated changes in the expression of the transcription factor Foxp3.
By immunoblotting, prior to stimulation with anti-CD3/CD28 antibodies, Foxp3 was undetectable in naïve CD4+CD62L+ T cells (Fig. 4A). CD3/CD28 stimulation upregulated Foxp3 expression, in keeping with the previously reported acquisition of T regulatory activity by stimulated naïve T cells (51, 52). In the presence of nicotine, anti-CD3/CD28 stimulation resulted in a further increase of Foxp3 (Fig. 4A). FCM analysis gave similar results. CD3/CD28 stimulation increased the number of Foxp3 positive T cells and nicotine intensified this effect (Fig. 4B).
Figure 4. The nicotinergic effects on the development of Tregs from the naïve CD4+CD62L+ T cells stimulated with anti-CD3/CD28 antibodies.
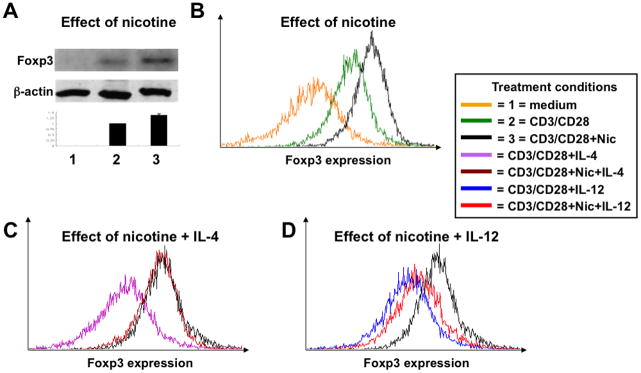
The enriched naïve CD4+CD62L+ T cells isolated from the spleens of intact BALB/c mice were cultured in growth medium alone (condition “1”) or stimulated with anti-CD3 and anti-CD28 antibodies in the absence (condition “2”) or presence of nicotine (Nic; condition “3”), 10 ng/ml of IL-4, 100 μM of nicotine plus 10 ng/ml of IL-4 or IL-12 or 100 μM of nicotine plus 10 ng/ml of IL-12 as detailed in Materials and Methods. After 5 days in culture, the expression of Foxp3 protein was analyzed by immunoblotting (A) and FCM (B–D). The graph in panel “A” demonstrates changes of Foxp3 concentration after its normalization for the level of the house keeping protein β-actin. In panels “B–D”, the changes in Foxp3 expression are shown as an overlay histogram representing different treatment conditions.
Thus, upregulation of Foxp3+ T cells is a normal outcome of TCR stimulation of naïve CD4+CD62L+ T cells in the presence of nicotine. This effect of nicotine could provide a mechanism for elevation of colonic CD25+Foxp3+ Tregs in mice with oxazolone colitis, but it remained unclear why such elevation did not occur in mice with TNBS colitis.
Contribution of the cytokine milieu to the dichotomous effect of nicotine on the development of Foxp3 CD4 T cells in oxazolone- vs. TNBS-induced colitides
It is well recognized that the cytokine milieu of oxazolone colitis differs from that of TNBS colitis, with IL-4 being the predominant cytokine of the former and IL-12—the principal cytokine of the latter form of experimental IBD (53). It is also known that nicotinergic signaling in non-neuronal cells can be altered by the environmental stimuli such as cytokines, hormones and growth factors (reviewed in (54, 55)). Therefore, we hypothesized that the difference in the cytokine milieu could lead to the dichotomous effects of nicotine treatment on oxazolone- vs. TNBS-induced colitis. To test this hypothesis, we reproduced in vitro the most distinctive features of oxazolone- and TNBS-specific gut inflammation using IL-4 and IL-12, respectively. IL-4 did not alter the stimulatory effect of nicotine on Foxp3 expression in naïve CD4+CD62L+ T cells stimulated with anti-CD3/CD28 antibodies (Fig. 4C). In marked contrast, the presence of IL-12 eliminated the ability of nicotine to upregulate Foxp3 (Fig. 4D). These results were consistent with an increased proportion of CD25+Foxp3+ Tregs among colonic CD4 positive cells in nicotine-treated mice with oxazolone-, but not TNBS-induced colitis (Fig. 3B).
Contribution of the cytokine milieu to the dichotomous effect of nicotine on pro- and anti-inflammatory cytokine production
Next, we asked whether the reciprocal effects of IL-4 and IL-12 on nicotine-dependent changes in Foxp3 expression correlate with production of the pro- and anti-inflammatory cytokines IL-17/IFN-γ and IL-10, respectively. First, we sought to ascertain that the cytokine milieu of each form of experimental IBD correspond to the expected profile. Indeed, we found significantly (p<0.05) increased colonic levels of IL-12, IL-23 in TNBS colitis, and IL-4 in oxazolone colitis (Fig. 5A). Treatment with nicotine further elevated IL-23 and IFN-γ levels (p<0.05) in TBS colitis. Although the differences in IL-10 levels did not significnatly differ among tested groups (p>0.05), we detected a tendency for elevation of IL-10 in the oxazolone+nicotine group (not shown).
Figure 5. The influence of IL-4 and IL-12 on nicotinergic regulation IL-10, IL-17 and IFN-γ secretion.

A. Colons from mice with oxazolone- or TNBS-induced colitides were collected on day 5 after hapten treatment. Total protein was extracted, and tissue levels of IL-4, IL-12, IL-23 and IFN-γ were detected using mouse ELISA Immunoassay kits, as described in Materials and Methods. Asterisk = p<0.05 compared to control mice; pound sign = p<0.05 compared to untreated mice with the respective form of experimental colitis.
B. The naïve CD4+CD62L+ T cells from intact BALB/c mice were stimulated with anti-CD3 and anti-CD28 in the presence or absence of 100 μM of nicotine ± 10 ng/ml of IL-4 or IL-12 for 5 days, after which the cell culture supernatants were collected and analyzed by ELISA for the presence of IL-10, IL-17 and IFN-γ, as described in Materials and Methods. Asterisk = p<0.05 compared to naïve CD4+CD62L+ T cells stimulated by TCR/CD3 cross-linking without any additions; pound sign = p<0.05 compared to cells stimulated by anti-CD3/anti-C28 in the presence of nicotine given alone; and plus sign = p<0.05 compared to cells stimulated with anti-CD3/anti-C28 in the presence of relevant cytokine without nicotine.
C. The naïve CD4+CD62L+ T cells from intact BALB/c mice were stimulated with anti-CD3 and anti-CD28 in the presence or absence of 100 μM of nicotine ± 10 ng/ml of IL-4 or IL-12 for 5 days, after which the expression of IL-4, IL-10, IL-17 and IFN-γ was analyzed by FCM, as described in Materials and Methods.
To identify pharmacologic effects of nicotine on cytokine production by CD4+ T cells, we performed in vitro experiments. As expected, stimulation of naïve CD4+CD62L+ T cells with anti-CD3/CD28 antibodies upregulated secretion of IL-17 (Fig. 5B)—an effector cytokine of the Th17 lineage that plays a pathogenic role in IBD (56). This effect was blocked in the presence of nicotine (p<0.05). IL-4 did not change this effect of nicotine (p>0.05), which is consistent with the anti-inflammatory action of nicotine in mice with oxazolone colitis. On the contrary, IL-12 totally abolished nicotine-dependent inhibition of IL-17 production. Furthermore, in the presence of nicotine, IL-12 significantly (p<0.05) raised IL-17 above control levels (Fig. 5B). A similar pattern of IL-4 and IL-12-dependent modulation of nicotine effects was observed for IFN-γ production (Fig. 5B), another pro-inflammatory cytokine characteristic of TNBS-induced gut inflammation (57). As was the case with IL-17 production, IFN-γ production was maximal in the presence of both nicotine and IL-12 (p<0.05). These observations help explain how nicotine treatment exacerbates gut inflammation in mice with TNBS colitis.
CD3/CD28 stimulation of naïve T cells also induced very low levels of IL-10 secretion (Fig. 5B). When the cells were stimulated with anti-CD3/CD28 antibodies in the presence of nicotine, the concentration of IL-10 in culture supernatant rose approximately 5-fold (Fig. 5B). Simultaneous exposure to IL-4 did not alter the nicotine-dependent upregulation of IL-10 secretion (Fig. 5B). In marked contrast, stimulation with anti-CD3/CD28 antibodies in the presence of both nicotine and IL-12 not only abolished the stimulatory effect of nicotine on IL-10 secretion, but also decreased it below control levels (p<0.05). The latter phenomenon suggested that worsening of TNBS colitis by nicotine could be explained by its pro-inflammatory action in the presence of IL-12.
We also performed FCM analyses of cytokine expression in CD4+CD62L+ cells stimulated with anti-CD3/CD28 +/− nicotine. The results shown in Fig. 5C confirmed that nicotine can enhance IFN-γ and IL-10, but inhibit IL-17 expression. About 85% of IL-10 producing cells expressed Foxp3 (data not shown), which is consistent with a finding that nicotine enhances Foxp3 expression (Fig. 4B). Production of IL-4 was not affected.
Differential expression of α7 nAChR by colonic CD4 T cells in two forms of experimental IBD
Inflammatory cytokines can modify expression levels of distinct nAChR subtypes in immune cells (18, 58), and specific nAChR subtypes exhibit diverse effects on inflammatory cytokines, inhibiting production of pro-inflammatory cytokines via both α4 and α7 nAChRs (17, 59). Therefore, the dichotomous effects of nicotine in oxazolone- and TNBS-induced colitis might reflect differences in the repertoire of nAChRs expressed by colonic T cells in each form of colitis. To test this hypothesis, we quantified the proportion of CD4 T cells expressing α4 vs. α7 in inflammatory infiltrates of oxazolone and TNBS colitis. α4 expression in colonic CD4 T cells in both forms of experimental IBD was similarly low, but the level of α7 expression differed dramatically (Fig. 6A). Fewer than 3% of colonic CD4 T cells in TNBS-induced colitis expressed α7, whereas approximately 2/3 of CD4 T cells expressed this receptor in oxazolone colitis, i.e., the frequency of α7 expression in oxazolone colitis exceeded that in TNBS colitis by more than 30-fold (Fig. 6A). Hence, the therapeutic action of nicotine on oxazolone colitis could be explained by its ability to inhibit the pro-inflammatory function of the majority of CD4 T cells by activating their α7 nAChR. The pivotal role of α7 nAChR in mediating the therapeutic activity of nicotine in oxazolone-induced colitis was confirmed by the absence of such activity in knockout mice lacking α7 (Fig. 6B).
Figure 6. Variations in the expression of α4 and α7 nAChRs by CD4 T cells.

A. The expression of α4 and α7 nAChRs by colonic CD4 T cells in different forms of experimental IBD. The percentages of CD4 T cells expressing α4 or α7 in the colonic inflammatory infiltrates of BALB/c mice with oxazolone (Ox)- and TNBS (T)-induced colitides were computed in ten microscopic fields of double stained cryostat sections of colons from three mice in each group (n = 3). Asterisk = p<0.05 compared to mice with oxazolone-induced colitis.
B. Clinical evaluation of nicotine treatment on the severity of oxazolone colitis in α7 knockout (KO) mice (B6.129S7-Chrna7tm1Bay/J; stock# 003232). The DAI values were computed as described in detail in Materials and Methods. Each point represents the mean ± SD of data obtained in 5 mice. The differences between the nicotine treated and not treated groups are not significant (p>0.05).
C. The effects of IL-4 and IL-12 on α7 nAChR expression. The enriched naïve CD4+CD62L+ T cells from intact BALB/c mice were stimulated with anti-CD3/anti-CD28 antibodies alone (control) or in the presence of 10 ng/ml of either IL-4 or IL-12 for 5 days in culture, and the expression of α7 mRNA and protein was detected by qPCR and ICW, respectively, as detailed in Materials and Methods. The results were expressed as means ± SD of relative receptor mRNA or protein levels in cells exposed to cytokines compared to that in controls, taken as 1. Asterisk = p<0.05 compared to control group, and pound sign = p<0.05 compared to cells exposed to IL-12.
Alterations of α7 nAChR expression in normal murine CD4 T cells by IL-4 and IL-12
To elucidate possible causes for differential expression of α7 nAChR by colonic CD4 T cells in the two forms of experimental IBD, we measured changes in α7 expression under influence of IL-4 and IL-12—the principal inflammatory cytokines characteristic of oxazolone- and TNBS-induced gut inflammation, respectively. α7 gene expression at the mRNA level was analyzed by qPCR and at the protein level by ICW. Following 5 days of anti-CD3/CD28 stimulation of naïve CD4+CD62L+ T cells from intact BALB/c mice in the presence of IL-4 or IL-12. qPCR revealed that IL-12 reduced the α7 mRNA expression to undetectable levels. In marked contrast, IL-4 increased α7 mRNA by approximately 1.5-fold (Fig. 6C). By ICW, IL-12 reduced the protein level of α7 by 70% (p<0.05), whereas IL-4 increased it by approximately 1.3-fold (Fig. 6C). Neither cytokine altered the expression level of α9 nAChR (data not shown). Thus, IL-4 and IL-12 could enhance the anti- and pro-inflammatory effects of nicotine in oxazolone- and TNBS-indices colitides, respectively, by selectively up- and downregulating expression of the anti-inflammatory α7 nAChR in colonic CD4 T cells.
Discussion
In this study, we demonstrated for the first time that the dichotomous effect of smoking that improves UC and worsens CD can be reproduced by nicotine treatment in mice with oxazolone and TNBS colitides. We also showed that the differences in the repertoire of nAChRs expressed by colonic CD4 T cells play an important role in modifying the outcome of nicotinergic treatment of gut inflammation. The inflammatory milieu characteristic of UC/oxazolone colitis facilitated expression of the anti-inflammatory α7 nAChR by colonic CD4 T cells, thus improving colitis, whereas the milieu in CD/TNBS colitis abolished α7 expression, which could exacerbate gut inflammation. These findings underscore the potential for nicotinergic drugs in regulating colonic inflammation.
It is well known that smoking affects both UC and CD by ameliorating UC and worsening CD. Since both smoking and pure nicotine can alter production of Th1- and Th2-type cytokines (60–62), we hypothesized that the nicotinergic agonist nicotine would mimic the effects of smoking in the mouse models of UC- and CD-like colitides. As expected, oxazolone and TNBS induced acute forms of UC- or CD-like colonic inflammation, respectively, and mice with each form of experimental IBD demonstrated directly opposing responses to nicotine treatment. The analysis of peripheral and colonic T cell subpopulations revealed that clinical and histological features of experimental IBD types correlated well with the immunopathologic characteristics of each form of colitis (19–27). Clinical and histological improvement of mice with oxazolone colitis was associated with a relative increase of CD25+Foxp3+ Tregs and a reciprocal decrease of CD4 IL-17+ cells among LPMCs, indicating that in this form of IBD the therapeutic effect of nicotine stems from skewing the Treg/Th17 balance toward the immunosuppressive Tregs. The opposite was observed in mice with nicotine-treated TNBS colitis, indicating that the Treg/Th17 balance was skewed toward pro-inflammatory Th17 cells.
To elucidate the mechanisms responsible for the dichotomous outcomes following nicotine treatment of mice with oxazolone vs. TNBS-induced colitides, we investigated the direct effect of nicotine on freshly isolated murine naïve CD4+CD62L+ T cells. In vitro nicotine stimulation of naïve T cells activated via TCR/CD3 cross-linking resulted in upregulated expression of Foxp3+, indicating that facilitated development of Tregs from naïve T cells is a normal outcome of nicotinergic stimulation. Furthermore, nicotine upregulated production of the immunosuppressive cytokine IL-10 and inhibited that of IL-17—an effector cytokine of Th17 cells. Upregulated production of IL-10 provided indirect evidence that the Foxp3+ Tregs induced by nicotine were functional cells that could produce and respond to IL-10, long known to suppress intestinal inflammation in mice with experimental colitis (63). These observations are in keeping with earlier reports that treatment with nicotine can induce “suppressor” T cells (64, 65), inhibit IL-18-induced T cell proliferation (66), and influence cytokine profiles and subsequent cell cycling/apoptotic responses of peripheral blood mononuclear cells from IBD patients (37). These results, taken together, suggest that the anti-inflammatory action of nicotine in mice with oxazolone-induced colitis resulted from stimulating production of Tregs. This observation, however, did not explain why nicotine worsened TNBS colitis, and prompted additional mechanistic studies.
The induction and function of Foxp3+ T cells can both affect and be affected by inflammatory cytokines (67, 68). Therefore, differences between nicotine effects on mice with oxazolone vs. TNBS colitis might reflect differences in the gut inflammatory environment produced by each haptenating agent. Since it is well-known that IL-4 is an effector cytokine in oxazolone colitis (53), and that IL-12 plays a crucial role in TNBS colitis, these cytokines could modulate the nicotinergic effects on the T cells mediating colonic inflammation in mice with each form of colitis. To test this hypothesis, we determined whether IL-4 and IL-12 alter the ability of nicotine to upregulate Foxp3 expression by naïve CD4+CD62L+ T cells stimulated with anti-CD3/CD28 antibodies. While IL-4 did not alter the nicotine effect, IL-12 completely abolished the nicotine-dependent upregulation of Tregs. Decreased number/function of Tregs skews the dynamic equilibrium within the Treg/Th17 axis toward Th17 cells (69). Hence, worsening of colonic inflammation by nicotine/smoking may be explained by a loss of control of Th17 cells playing the pathogenic role in both TNBS-induced colitis in mice (70) and patients with CD (71).
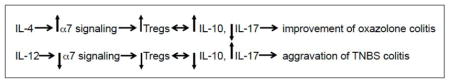
To elucidate the molecular mechanisms allowing cytokines to diversify the immunopharmacologic action of nicotine, we investigated the role of T cell nAChRs. Immunologic stimulation has been shown to alter the expression and function of nAChRs in T cells, and different nAChR subtypes are known to exhibit diverse immunoregulatory effects due to differential regulation of pro- and inflammatory cytokines through the signaling pathway couples by distinct nAChR subtypes (16, 17, 72–78). In keeping with the well-established immunosuppressive/anti-inflammatory function of α7 nAChR (reviewed in (79)), we observed a dramatic difference in the percentages of colonic CD4 T cells expressing α7 in oxazolone vs. TNBS colitis, 67% vs. 3%, respectively, and confirmed the abilities of IL-4 and IL-12 to exhibit reciprocal effects on α7 gene expression in murine CD4 T cells at the mRNA and protein level. The pivotal role of α7 in mediating therapeutic effects of nicotine in oxazolone colitis was confirmed in experiments with α7 nAChR knockout mice that showed no improvement due to nicotine tretment. The pro-inflammatory action of nicotine that aggravated TNBS colitis could be mediated via the alternative nicotinergic signaling pathways activated in colonic CD4 T cells that lacked α7 nAChR. The reported pro-inflammatory/immunostimulatory effects of nicotine include increased secretion of IL-12 by Th1 T cells (80), enhanced Con A-induced production of IFN-γ (62), and protection of lymphocytes from cortisol-induced apoptosis (81). It has been proposed that the nicotinergic pro-inflammatory cascade is mediated by α9 nAChR (82), whose expression was not affected by either IL-4 or IL-12. Thus, upregulated expression of α7 by colonic T cells, facilitated by IL-4, could mediate an anti-inflammatory effect of nicotine on oxazolone colitis and, vice versa, downregulation of this receptor by IL-12 could worsen TNBS colitis. These chains of events can be summarized as follows:
A recent discovery that the efferent vagus nerve modulates the immune response and controls inflammation through nAChRs expressed by inflammatory cells gave rise to a concept of a “cholinergic (nicotinic) anti-inflammatory pathway” (83), and opened a novel avenue for treatment of inflammatory diseases with nicotinic agonists (79, 84, 85). The goal is to avoid the undesired side effects of the therapeutic doses of nicotine (86), which, unfortunately, is not effective in UC at low doses (12). The following nicotinic agonists that avoid the toxicity of nicotine are currently under investigation: AR-R17779 ((−)-spiro[1-azabicyclo[2.2.2]octane-3,5′-oxazolidin-2′-one]), GTS21 (3-[(2,4-dimethoxy)benzylidene]-anabaseine), CAP55, Exo2 (exo-2-(2-pyridyl)-7-azabicyclo[2.2.1] heptane), PNU-282987 ([N-[(3R)-1-Azabicyclo[2.2.2]oct-3-yl]-4-chlorobenzamide hydrochloride]) and 4OHGTS (3-(4-hydroxy,2-methoxybenzylidene) anabaseine), some of which have already been successfully used in various in vitro and in vivo models of inflammation (reviewed in (84, 87)). In IBD associated with elevated levels of IL-4, such as UC, the therapeutic effect may be achieved from α7 agonists, whereas α7 antagonists may be therapeutic in the IBD type associated with elevated IL-12, like CD. By now, both nicotinic agonists and antagonists (35, 36) have been demonstrated to be therapeutic in experimental IBD, and it also has been shown that administration of α7 agonists to the IBD type caused by IL-12, such as DSS-induced colitis (88), worsen gut inflammation (38).
In conclusion, our data indicate that the dichotomous clinical and immunopharmacologic effects of smoking/nicotine in UC and CD can be explained, in part, by the ability of the cytokine milieu characteristic of each type of IBD to modify in a unique fashion the nicotinergic signaling by altering the repertoire of T cell nAChRs. Although there is significant potential for developing selective immunomodulation using nicotinergic agents that target nAChRs in T cells, an equally broad and specific range of nAChR-related outcomes can be expected, which may be both beneficial and harmful to the host. Identification of the nAChR subtypes coupled to specific functions of a particular T cell subset involved in each type of IBD should allow selective immunomodulation. Thus, future in depth analysis of cholinergic immunopharmacology of distinct forms of gut inflammation should be rewarded with novel therapeutic strategies.
References
- 1.Motley RJ, Rhodes J, Kay S, Morris TJ. Late presentation of ulcerative colitis in ex-smokers. Int J Colorectal Dis. 1988;3:171–175. doi: 10.1007/BF01648362. [DOI] [PubMed] [Google Scholar]
- 2.Koutroubakis I, Manousos ON, Meuwissen SGM, Pena AS. Environmental risk factors in inflammatory bowel disease. Hepato-Gastroenterology. 1996;43:381–393. [PubMed] [Google Scholar]
- 3.Rubin DT, Hanauer SB. Smoking and inflammatory bowel disease. Eur J Gastroenterol Hepatol. 2000;12:855–862. doi: 10.1097/00042737-200012080-00004. [DOI] [PubMed] [Google Scholar]
- 4.Eliakim R, Karmeli F, Cohen P, Heyman SN, Rachmilewitz D. Dual effect of chronic nicotine administration: augmentation of jejunitis and amelioration of colitis induced by iodoacetamide in rats. Int J Colorectal Dis. 2001;16:14–21. doi: 10.1007/s003840000262. [DOI] [PubMed] [Google Scholar]
- 5.Harries AD, Baird A, Rhodes J. Non-smoking: a feature of ulcerative colitis. Br Med J (Clin Res Ed) 1982;284:706. doi: 10.1136/bmj.284.6317.706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Logan RF, Edmond M, Somerville KW, Langman MJ. Smoking and ulcerative colitis. Br Med J (Clin Res Ed) 1984;288:751–753. doi: 10.1136/bmj.288.6419.751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Motley RJ, Rhodes J, Ford GA, Wilkinson SP, Chesner IM, Asquith P, Hellier MD, Mayberry JF. Time relationships between cessation of smoking and onset of ulcerative colitis. Digestion. 1987;37:125–127. doi: 10.1159/000199478. [DOI] [PubMed] [Google Scholar]
- 8.de Castella H. Non-smoking: a feature of ulcerative colitis. Br Med J (Clin Res Ed) 1982;284:1706. doi: 10.1136/bmj.284.6330.1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Birtwistle J, Hall K. Does nicotine have beneficial effects in the treatment of certain diseases? Br J Nurs. 1996;5:1195–1202. doi: 10.12968/bjon.1996.5.19.1195. [DOI] [PubMed] [Google Scholar]
- 10.Wolf JM, Lashner BA. Inflammatory bowel disease: sorting out the treatment options. Cleve Clin J Med. 2002;69:621–626. 629–631. doi: 10.3949/ccjm.69.8.621. [DOI] [PubMed] [Google Scholar]
- 11.Hilsden RJ, Hodgins DC, Timmer A, Sutherland LR. Helping patients with Crohn’s disease quit smoking. Am J Gastroenterol. 2000;95:352–358. doi: 10.1111/j.1572-0241.2000.t01-1-01789.x. [DOI] [PubMed] [Google Scholar]
- 12.Thomas GA, Rhodes J, Ingram JR. Mechanisms of disease: nicotine--a review of its actions in the context of gastrointestinal disease. Nat Clin Pract Gastroenterol Hepatol. 2005;2:536–544. doi: 10.1038/ncpgasthep0316. [DOI] [PubMed] [Google Scholar]
- 13.Coulie B, Camilleri M, Bharucha AE, Sandborn WJ, Burton D. Colonic motility in chronic ulcerative proctosigmoiditis and the effects of nicotine on colonic motility in patients and healthy subjects. Aliment Pharmacol Ther. 2001;15:653–663. doi: 10.1046/j.1365-2036.2001.00959.x. [DOI] [PubMed] [Google Scholar]
- 14.McGrath J, McDonald JW, Macdonald JK. Transdermal nicotine for induction of remission in ulcerative colitis. Cochrane Database Syst Rev. 2004:CD004722. doi: 10.1002/14651858.CD004722.pub2. [DOI] [PubMed] [Google Scholar]
- 15.Pullan RD, Rhodes J, Ganesh S, Mani V, Morris JS, Williams GT, Newcombe RG, Russell MA, Feyerabend C, Thomas GA, et al. Transdermal nicotine for active ulcerative colitis. N Engl J Med. 1994;330:811–815. doi: 10.1056/NEJM199403243301202. [DOI] [PubMed] [Google Scholar]
- 16.Fujii T, Takada-Takatori Y, Kawashima K. Basic and clinical aspects of non-neuronal acetylcholine: expression of an independent, non-neuronal cholinergic system in lymphocytes and its clinical significance in immunotherapy. J Pharmacol Sci. 2008;106:186–192. doi: 10.1254/jphs.fm0070109. [DOI] [PubMed] [Google Scholar]
- 17.Chernyavsky AI, Arredondo J, Galitovskiy V, Qian J, Grando SA. Structure and function of the nicotinic arm of acetylcholine regulatory axis in human leukemic T cells. Int J Immunopathol Pharmacol. 2009;22:461–472. doi: 10.1177/039463200902200223. [DOI] [PubMed] [Google Scholar]
- 18.Qian J, Galitovskiy V, Chernyavsky AI, Marchenko S, Grando SA. Plasticity of the murine spleen T-cell cholinergic receptors and their role in in vitro differentiation of naive CD4 T cells toward the Th1, Th2 and Th17 lineages. Genes Immun. 2011;12:222–230. doi: 10.1038/gene.2010.72. [DOI] [PubMed] [Google Scholar]
- 19.Uhlig HH, Powrie F. Dendritic cells and the intestinal bacterial flora: a role for localized mucosal immune responses. J Clin Invest. 2003;112:648–651. doi: 10.1172/JCI19545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Monteleone G, Biancone L, Marasco R, Morrone G, Marasco O, Luzza F, Pallone F. Interleukin 12 is expressed and actively released by Crohn’s disease intestinal lamina propria mononuclear cells. Gastroenterology. 1997;112:1169–1178. doi: 10.1016/s0016-5085(97)70128-8. [DOI] [PubMed] [Google Scholar]
- 21.Sartor RB. Mechanisms of disease: pathogenesis of Crohn’s disease and ulcerative colitis. Nat Clin Pract Gastroenterol Hepatol. 2006;3:390–407. doi: 10.1038/ncpgasthep0528. [DOI] [PubMed] [Google Scholar]
- 22.Fuss IJ, Neurath M, Boirivant M, Klein JS, de la Motte C, Strong SA, Fiocchi C, Strober W. Disparate CD4+ lamina propria (LP) lymphokine secretion profiles in inflammatory bowel disease. Crohn’s disease LP cells manifest increased secretion of IFN-gamma, whereas ulcerative colitis LP cells manifest increased secretion of IL-5. J Immunol. 1996;157:1261–1270. [PubMed] [Google Scholar]
- 23.Parronchi P, Romagnani P, Annunziato F, Sampognaro S, Becchio A, Giannarini L, Maggi E, Pupilli C, Tonelli F, Romagnani S. Type 1 T-helper cell predominance and interleukin-12 expression in the gut of patients with Crohn’s disease. Am J Pathol. 1997;150:823–832. [PMC free article] [PubMed] [Google Scholar]
- 24.Mannon PJ, Fuss IJ, Mayer L, Elson CO, Sandborn WJ, Present D, Dolin B, Goodman N, Groden C, Hornung RL, Quezado M, Yang Z, Neurath MF, Salfeld J, Veldman GM, Schwertschlag U, Strober W. Anti-interleukin-12 antibody for active Crohn’s disease. N Engl J Med. 2004;351:2069–2079. doi: 10.1056/NEJMoa033402. [DOI] [PubMed] [Google Scholar]
- 25.Fuss IJ, Heller F, Boirivant M, Leon F, Yoshida M, Fichtner-Feigl S, Yang Z, Exley M, Kitani A, Blumberg RS, Mannon P, Strober W. Nonclassical CD1d-restricted NK T cells that produce IL-13 characterize an atypical Th2 response in ulcerative colitis. J Clin Invest. 2004;113:1490–1497. doi: 10.1172/JCI19836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schmidt C, Giese T, Ludwig B, Mueller-Molaian I, Marth T, Zeuzem S, Meuer SC, Stallmach A. Expression of interleukin-12-related cytokine transcripts in inflammatory bowel disease: elevated interleukin-23p19 and interleukin-27p28 in Crohn’s disease but not in ulcerative colitis. Inflamm Bowel Dis. 2005;11:16–23. doi: 10.1097/00054725-200501000-00003. [DOI] [PubMed] [Google Scholar]
- 27.Fichtner-Feigl S, Fuss IJ, Preiss JC, Strober W, Kitani A. Treatment of murine Th1- and Th2-mediated inflammatory bowel disease with NF-kappa B decoy oligonucleotides. J Clin Invest. 2005;115:3057–3071. doi: 10.1172/JCI24792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Elson CO, Cong Y, Brandwein S, Weaver CT, McCabe RP, Mahler M, Sundberg JP, Leiter EH. Experimental models to study molecular mechanisms underlying intestinal inflammation. Ann N Y Acad Sci. 1998;859:85–95. doi: 10.1111/j.1749-6632.1998.tb11113.x. [DOI] [PubMed] [Google Scholar]
- 29.Neurath MF, Fuss I, Kelsall BL, Stuber E, Strober W. Antibodies to interleukin 12 abrogate established experimental colitis in mice. J Exp Med. 1995;182:1281–1290. doi: 10.1084/jem.182.5.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kitani A, Fuss IJ, Nakamura K, Schwartz OM, Usui T, Strober W. Treatment of experimental (Trinitrobenzene sulfonic acid) colitis by intranasal administration of transforming growth factor (TGF)-beta1 plasmid: TGF-beta1-mediated suppression of T helper cell type 1 response occurs by interleukin (IL)-10 induction and IL-12 receptor beta2 chain downregulation. J Exp Med. 2000;192:41–52. doi: 10.1084/jem.192.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fuss IJ, Marth T, Neurath MF, Pearlstein GR, Jain A, Strober W. Anti-interleukin 12 treatment regulates apoptosis of Th1 T cells in experimental colitis in mice. Gastroenterology. 1999;117:1078–1088. doi: 10.1016/s0016-5085(99)70392-6. [DOI] [PubMed] [Google Scholar]
- 32.Strober W, Fuss IJ, Blumberg RS. The immunology of mucosal models of inflammation. Annu Rev Immunol. 2002;20:495–549. doi: 10.1146/annurev.immunol.20.100301.064816. [DOI] [PubMed] [Google Scholar]
- 33.Heller F, Fuss IJ, Nieuwenhuis EE, Blumberg RS, Strober W. Oxazolone colitis, a Th2 colitis model resembling ulcerative colitis, is mediated by IL-13-producing NK-T cells. Immunity. 2002;17:629–638. doi: 10.1016/s1074-7613(02)00453-3. [DOI] [PubMed] [Google Scholar]
- 34.Boirivant M, Fuss IJ, Chu A, Strober W. Oxazolone colitis: A murine model of T helper cell type 2 colitis treatable with antibodies to interleukin 4. J Exp Med. 1998;188:1929–1939. doi: 10.1084/jem.188.10.1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ghia JE, Blennerhassett P, Kumar-Ondiveeran H, Verdu EF, Collins SM. The vagus nerve: a tonic inhibitory influence associated with inflammatory bowel disease in a murine model. Gastroenterology. 2006;131:1122–1130. doi: 10.1053/j.gastro.2006.08.016. [DOI] [PubMed] [Google Scholar]
- 36.Ghia JE, Blennerhassett P, El-Sharkawy RT, Collins SM. The protective effect of the vagus nerve in a murine model of chronic relapsing colitis. Am J Physiol Gastrointest Liver Physiol. 2007;293:G711–718. doi: 10.1152/ajpgi.00240.2007. [DOI] [PubMed] [Google Scholar]
- 37.Aldhous MC, Prescott RJ, Roberts S, Samuel K, Waterfall M, Satsangi J. Does nicotine influence cytokine profile and subsequent cell cycling/apoptotic responses in inflammatory bowel disease? Inflamm Bowel Dis. 2008;14:1469–1482. doi: 10.1002/ibd.20523. [DOI] [PubMed] [Google Scholar]
- 38.Snoek SA, Verstege MI, van der Zanden EP, Deeks N, Bulmer DC, Skynner M, Lee K, Te Velde AA, Boeckxstaens GE, de Jonge WJ. Selective alpha7 nicotinic acetylcholine receptor agonists worsen disease in experimental colitis. Br J Pharmacol. 2010;160:322–333. doi: 10.1111/j.1476-5381.2010.00699.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kojima R, Kuroda S, Ohkishi T, Nakamaru K, Hatakeyama S. Oxazolone-induced colitis in BALB/C mice: a new method to evaluate the efficacy of therapeutic agents for ulcerative colitis. J Pharmacol Sci. 2004;96:307–313. doi: 10.1254/jphs.fp0040214. [DOI] [PubMed] [Google Scholar]
- 40.Boirivant M, Strober W, Fuss IJ. Regulatory cells induced by feeding TNP-haptenated colonic protein cross-protect mice from colitis induced by an unrelated hapten. Inflamm Bowel Dis. 2005;11:48–55. doi: 10.1097/00054725-200501000-00007. [DOI] [PubMed] [Google Scholar]
- 41.Kihara N, de la Fuente SG, Fujino K, Takahashi T, Pappas TN, Mantyh CR. Vanilloid receptor-1 containing primary sensory neurones mediate dextran sulphate sodium induced colitis in rats. Gut. 2003;52:713–719. doi: 10.1136/gut.52.5.713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ekstrom GM. Oxazolone-induced colitis in rats: effects of budesonide, cyclosporin A, and 5-aminosalicylic acid. Scand J Gastroenterol. 1998;33:174–179. doi: 10.1080/00365529850166914. [DOI] [PubMed] [Google Scholar]
- 43.Iijima H, Neurath MF, Nagaishi T, Glickman JN, Nieuwenhuis EE, Nakajima A, Chen D, Fuss IJ, Utku N, Lewicki DN, Becker C, Gallagher TM, Holmes KV, Blumberg RS. Specific regulation of T helper cell 1-mediated murine colitis by CEACAM1. J Exp Med. 2004;199:471–482. doi: 10.1084/jem.20030437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ndoye A, Buchli R, Greenberg B, Nguyen VT, Zia S, Rodriguez JG, Webber RJ, Lawry MA, Grando SA. Identification and mapping of keratinocyte muscarinic acetylcholine receptor subtypes in human epidermis. J Invest Dermatol. 1998;111:410–416. doi: 10.1046/j.1523-1747.1998.00299.x. [DOI] [PubMed] [Google Scholar]
- 45.Weigmann B, Tubbe I, Seidel D, Nicolaev A, Becker C, Neurath MF. Isolation and subsequent analysis of murine lamina propria mononuclear cells from colonic tissue. Nat Protoc. 2007;2:2307–2311. doi: 10.1038/nprot.2007.315. [DOI] [PubMed] [Google Scholar]
- 46.Arredondo J, Chernyavsky AI, Jolkovsky DL, Webber RJ, Grando SA. SLURP-2: A novel cholinergic signaling peptide in human mucocutaneous epithelium. J Cell Physiol. 2006;208:238–245. doi: 10.1002/jcp.20661. [DOI] [PubMed] [Google Scholar]
- 47.Wang X, Ouyang Q, Luo WJ. Oxazolone-induced murine model of ulcerative colitis. Chin J Dig Dis. 2004;5:165–168. doi: 10.1111/j.1443-9573.2004.00173.x. [DOI] [PubMed] [Google Scholar]
- 48.Eri R, Kodumudi KN, Summerlin DJ, Srinivasan M. Suppression of colon inflammation by CD80 blockade: evaluation in two murine models of inflammatory bowel disease. Inflamm Bowel Dis. 2008;14:458–470. doi: 10.1002/ibd.20344. [DOI] [PubMed] [Google Scholar]
- 49.Himmel ME, Hardenberg G, Piccirillo CA, Steiner TS, Levings MK. The role of T-regulatory cells and Toll-like receptors in the pathogenesis of human inflammatory bowel disease. Immunology. 2008;125:145–153. doi: 10.1111/j.1365-2567.2008.02939.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yu QT, Saruta M, Avanesyan A, Fleshner PR, Banham AH, Papadakis KA. Expression and functional characterization of FOXP3+ CD4+ regulatory T cells in ulcerative colitis. Inflamm Bowel Dis. 2007;13:191–199. doi: 10.1002/ibd.20053. [DOI] [PubMed] [Google Scholar]
- 51.Walker MR, Kasprowicz DJ, Gersuk VH, Benard A, Van Landeghen M, Buckner JH, Ziegler SF. Induction of FoxP3 and acquisition of T regulatory activity by stimulated human CD4+CD25- T cells. J Clin Invest. 2003;112:1437–1443. doi: 10.1172/JCI19441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Moon C, Kim SH, Park KS, Choi BK, Lee HS, Park JB, Choi GS, Kwan JH, Joh JW, Kim SJ. Use of epigenetic modification to induce FOXP3 expression in naive T cells. Transplant Proc. 2009;41:1848–1854. doi: 10.1016/j.transproceed.2009.02.101. [DOI] [PubMed] [Google Scholar]
- 53.Tozawa K, Hanai H, Sugimoto K, Baba S, Sugimura H, Aoshi T, Uchijima M, Nagata T, Koide Y. Evidence for the critical role of interleukin-12 but not interferon-gamma in the pathogenesis of experimental colitis in mice. J Gastroenterol Hepatol. 2003;18:578–587. doi: 10.1046/j.1440-1746.2003.03024.x. [DOI] [PubMed] [Google Scholar]
- 54.Wessler I, Kirkpatrick CJ. Acetylcholine beyond neurons: the non-neuronal cholinergic system in humans. Br J Pharmacol. 2008;154:1558–1571. doi: 10.1038/bjp.2008.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kurzen H, Wessler I, Kirkpatrick CJ, Kawashima K, Grando SA. The non-neuronal cholinergic system of human skin. Horm Metab Res. 2007;39:125–135. doi: 10.1055/s-2007-961816. [DOI] [PubMed] [Google Scholar]
- 56.Fujino S, Andoh A, Bamba S, Ogawa A, Hata K, Araki Y, Bamba T, Fujiyama Y. Increased expression of interleukin 17 in inflammatory bowel disease. Gut. 2003;52:65–70. doi: 10.1136/gut.52.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sun Y, Wu XX, Yin Y, Gong FY, Shen Y, Cai TT, Zhou XB, Wu XF, Xu Q. Novel immunomodulatory properties of cirsilineol through selective inhibition of IFN-gamma signaling in a murine model of inflammatory bowel disease. Biochem Pharmacol. 2010;79:229–238. doi: 10.1016/j.bcp.2009.08.014. [DOI] [PubMed] [Google Scholar]
- 58.Kondo Y, Tachikawa E, Ohtake S, Kudo K, Mizuma K, Kashimoto T, Irie Y, Taira E. Inflammatory cytokines decrease the expression of nicotinic acetylcholine receptor during the cell maturation. Mol Cell Biochem. 2010;333:57–64. doi: 10.1007/s11010-009-0204-4. [DOI] [PubMed] [Google Scholar]
- 59.Chernyavsky AI, Arredondo J, Skok M, Grando SA. Auto/paracrine control of inflammatory cytokines by acetylcholine in macrophage-like U937 cells through nicotinic receptors. Int Immunopharmacol. 2010;10:308–315. doi: 10.1016/j.intimp.2009.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kalra R, Singh SP, Savage SM, Finch GL, Sopori ML. Effects of cigarette smoke on immune response: chronic exposure to cigarette smoke impairs antigen-mediated signaling in T cells and depletes IP3-sensitive Ca(2+) stores. J Pharmacol Exp Ther. 2000;293:166–171. [PubMed] [Google Scholar]
- 61.Phaybouth V, Wang SZ, Hutt JA, McDonald JD, Harrod KS, Barrett EG. Cigarette smoke suppresses Th1 cytokine production and increases RSV expression in a neonatal model. Am J Physiol Lung Cell Mol Physiol. 2006;290:L222–231. doi: 10.1152/ajplung.00148.2005. [DOI] [PubMed] [Google Scholar]
- 62.Hallquist N, Hakki A, Wecker L, Friedman H, Pross S. Differential effects of nicotine and aging on splenocyte proliferation and the production of Th1- versus Th2-type cytokines. Proc Soc Exp Biol Med. 2000;224:141–146. doi: 10.1046/j.1525-1373.2000.22412.x. [DOI] [PubMed] [Google Scholar]
- 63.Murai M, Turovskaya O, Kim G, Madan R, Karp CL, Cheroutre H, Kronenberg M. Interleukin 10 acts on regulatory T cells to maintain expression of the transcription factor Foxp3 and suppressive function in mice with colitis. Nat Immunol. 2009;10:1178–1184. doi: 10.1038/ni.1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Richman DP, Arnason BG. Nicotinic acetylcholine receptor: evidence for a functionally distinct receptor on human lymphocytes. Proc Natl Acad Sci U S A. 1979;76:4632–4635. doi: 10.1073/pnas.76.9.4632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Menard L, Rola-Pleszczynski M. Nicotine induces T-suppressor cells: modulation by the nicotinic antagonist D-tubocurarine and myasthenic serum. Clin Immunol Immunopathol. 1987;44:107–113. doi: 10.1016/0090-1229(87)90056-0. [DOI] [PubMed] [Google Scholar]
- 66.Takahashi HK, Iwagaki H, Hamano R, Kanke T, Liu K, Sadamori H, Yagi T, Yoshino T, Tanaka N, Nishibori M. The immunosuppressive effects of nicotine during human mixed lymphocyte reaction. Eur J Pharmacol. 2007;559:69–74. doi: 10.1016/j.ejphar.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 67.Zhao Z, Yu S, Fitzgerald DC, Elbehi M, Ciric B, Rostami AM, Zhang GX. IL-12R beta 2 promotes the development of CD4+CD25+ regulatory T cells. J Immunol. 2008;181:3870–3876. doi: 10.4049/jimmunol.181.6.3870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zheng Y, Chaudhry A, Kas A, deRoos P, Kim JM, Chu TT, Corcoran L, Treuting P, Klein U, Rudensky AY. Regulatory T-cell suppressor program co-opts transcription factor IRF4 to control T(H)2 responses. Nature. 2009;458:351–356. doi: 10.1038/nature07674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Heo YJ, Joo YB, Oh HJ, Park MK, Heo YM, Cho ML, Kwok SK, Ju JH, Park KS, Cho SG, Park SH, Kim HY, Min JK. IL-10 suppresses Th17 cells and promotes regulatory T cells in the CD4+ T cell population of rheumatoid arthritis patients. Immunol Lett. 2010;127:150–156. doi: 10.1016/j.imlet.2009.10.006. [DOI] [PubMed] [Google Scholar]
- 70.Alex P, Zachos NC, Nguyen T, Gonzales L, Chen TE, Conklin LS, Centola M, Li X. Distinct cytokine patterns identified from multiplex profiles of murine DSS and TNBS-induced colitis. Inflamm Bowel Dis. 2009;15:341–352. doi: 10.1002/ibd.20753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Brand S. Crohn’s disease: Th1, Th17 or both? The change of a paradigm: new immunological and genetic insights implicate Th17 cells in the pathogenesis of Crohn’s disease. Gut. 2009;58:1152–1167. doi: 10.1136/gut.2008.163667. [DOI] [PubMed] [Google Scholar]
- 72.Kawashima K, Fujii T. Extraneuronal cholinergic system in lymphocytes. Pharmacol Ther. 2000;86:29–48. doi: 10.1016/s0163-7258(99)00071-6. [DOI] [PubMed] [Google Scholar]
- 73.Fujii T, Watanabe Y, Inoue T, Kawashima K. Upregulation of mRNA encoding the M5 muscarinic acetylcholine receptor in human T- and B-lymphocytes during immunological responses. Neurochem Res. 2003;28:423–429. doi: 10.1023/a:1022840416292. [DOI] [PubMed] [Google Scholar]
- 74.Kawashima K, Yoshikawa K, Fujii YX, Moriwaki Y, Misawa H. Expression and function of genes encoding cholinergic components in murine immune cells. Life Sci. 2007;80:2314–2319. doi: 10.1016/j.lfs.2007.02.036. [DOI] [PubMed] [Google Scholar]
- 75.Kawashima K, Fujii T. Expression of non-neuronal acetylcholine in lymphocytes and its contribution to the regulation of immune function. Front Biosci. 2004;9:2063–2085. doi: 10.2741/1390. [DOI] [PubMed] [Google Scholar]
- 76.Kawashima K, Fujii T. The lymphocytic cholinergic system and its contribution to the regulation of immune activity. Life Sci. 2003;74:675–696. doi: 10.1016/j.lfs.2003.09.037. [DOI] [PubMed] [Google Scholar]
- 77.Paldi-Haris P, Szelenyi JG, Nguyen TH, Hollan SR. Changes in the expression of the cholinergic structures of human T lymphocytes due to maturation and stimulation. Thymus. 1990;16:119–122. [PubMed] [Google Scholar]
- 78.Nizri E, Hamra-Amitay Y, Sicsic C, Lavon I, Brenner T. Anti-inflammatory properties of cholinergic up-regulation: A new role for acetylcholinesterase inhibitors. Neuropharmacology. 2006;50:540–547. doi: 10.1016/j.neuropharm.2005.10.013. [DOI] [PubMed] [Google Scholar]
- 79.de Jonge WJ, Ulloa L. The alpha7 nicotinic acetylcholine receptor as a pharmacological target for inflammation. Br J Pharmacol. 2007;151:915–929. doi: 10.1038/sj.bjp.0707264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Aicher A, Heeschen C, Mohaupt M, Cooke JP, Zeiher AM, Dimmeler S. Nicotine strongly activates dendritic cell-mediated adaptive immunity: potential role for progression of atherosclerotic lesions. Circulation. 2003;107:604–611. doi: 10.1161/01.cir.0000047279.42427.6d. [DOI] [PubMed] [Google Scholar]
- 81.De Rosa MJ, del Esandi MC, Garelli A, Rayes D, Bouzat C. Relationship between alpha 7 nAChR and apoptosis in human lymphocytes. J Neuroimmunol. 2005;160:154–161. doi: 10.1016/j.jneuroim.2004.11.010. [DOI] [PubMed] [Google Scholar]
- 82.Vincler M, Vetter DE, McIntosh M. a9a10 and a7 nicotinic acetylcholine receptors have opposite roles in the immune response to peripheral nerve injury. Biochem Pharmacol. 2007;74:SMA49. (45.41) [Google Scholar]
- 83.Borovikova LV, Ivanova S, Zhang M, Yang H, Botchkina GI, Watkins LR, Wang H, Abumrad N, Eaton JW, Tracey KJ. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature. 2000;405:458–462. doi: 10.1038/35013070. [DOI] [PubMed] [Google Scholar]
- 84.Ulloa L. The vagus nerve and the nicotinic anti-inflammatory pathway. Nat Rev Drug Discov. 2005;4:673–684. doi: 10.1038/nrd1797. [DOI] [PubMed] [Google Scholar]
- 85.Ulloa L, Wang P. The neuronal strategy for inflammation. Novartis Found Symp. 2007;280:223–233. discussion 233–227. [PubMed] [Google Scholar]
- 86.Tapper AR, McKinney SL, Nashmi R, Schwarz J, Deshpande P, Labarca C, Whiteaker P, Marks MJ, Collins AC, Lester HA. Nicotine activation of alpha4* receptors: sufficient for reward, tolerance, and sensitization. Science. 2004;306:1029–1032. doi: 10.1126/science.1099420. [DOI] [PubMed] [Google Scholar]
- 87.Bonaz B. The cholinergic anti-inflammatory pathway and the gastrointestinal tract. Gastroenterology. 2007;133:1370–1373. doi: 10.1053/j.gastro.2007.08.061. [DOI] [PubMed] [Google Scholar]
- 88.Sadakane C, Koseki J, Inagaki Y, Hasegawa Y, Shindo S, Maruyama H, Takeda S, Takeda H, Hattori T. TJN-419 improves dextran sulfate sodium-induced colitis via inhibition of interleukin-12 release. Biol Pharm Bull. 2010;33:84–90. doi: 10.1248/bpb.33.84. [DOI] [PubMed] [Google Scholar]