

Abstract
Multiscale models are commonplace in cancer modeling, where individual models acting on different biological scales are combined within a single, cohesive modeling framework. However, model composition gives rise to challenges in understanding interfaces and interactions between them. Based on specific domain expertise, typically these computational models are developed by separate research groups using different methodologies, programming languages, and parameters. This paper introduces a graph-based model for semantically linking computational cancer models via domain graphs that can help us better understand and explore combinations of models spanning multiple biological scales. We take the data model encoded by TumorML, an XML-based markup language for storing cancer models in online repositories, and transpose its model description elements into a graph-based representation. By taking such an approach, we can link domain models, such as controlled vocabularies, taxonomic schemes, and ontologies, with cancer model descriptions to better understand and explore relationships between models. The union of these graphs creates a connected property graph that links cancer models by categorizations, by computational compatibility, and by semantic interoperability, yielding a framework in which opportunities for exploration and discovery of combinations of models become possible.
Keywords: tumor modeling, in silico oncology, model exploration, property graphs, neo4j
Introduction
Computational simulation of biology, and of cancer, is an active and increasingly rich area of research.1–3 In in silico environments, simulation-based studies and experiments can be relatively inexpensive in cost, time, and risk, where simulations can be used as tools for hypothesis testing and predictive treatments. The trend in cancer modeling in recent years has been toward the development of multiscale models. Such models allow us to capture the interdependence of biological phenomena that occur at different biological scales, for example combining models of subcellular processes with cell–cell interactions, as opposed to single-scale models that might operate at one of these scales in isolation. They offer a natural framework for studying phenomena, such as cancer, which are inherently multiscale in nature, and thus appear to offer the cutting edge with regard to potential predictive power and clinical applicability. There are generally two kinds of modeling approaches4: a bottom-up approach that looks at simulation from a functional and reductionist point of view, integrating multiple functional models of low-level processes; and a top-down approach that is formulated from holistic observations of biological phenomena to develop models that fit observed behaviors and outcomes. In cancer modeling, both of these approaches are used to investigate and simulate different aspects of cancer, and there is an increasing interest in combining mathematical modeling techniques in a hybrid fashion. Alas, as these models are typically created in isolation, interoperability is very rarely designed into the system.
One of the best hopes for developing novel models of cancer that span multiple biological scales is to reuse and extend existing models. However, the integration or extension of existing models of cancer (or elements of those models) currently represents a substantial technical challenge in the field.5 Composition of models or model components typically relies on specialist domain knowledge about model constructs, intended interactions, computational interfaces, as well as application domain knowledge, for example, the underlying biochemistry. Thus, a prerequisite for composing models is being able to reason semantically about commonalities or links between different models.
Efforts to relate data and models to domain knowledge are common in biology as the amount and diversity of data requires standards and structures in order to effectively manage it. Mature examples of open standards and ontologies include MicroArray and Gene Expression - Tabular format (MAGE-TAB),6 Biological Pathway Exchange (BioPAX),7 and the Gene Ontology (GO)8,9 to name but a few. Computational models can also be thought of as a kind of data, where the plethora of published models also need standards and structures. Mature standards for functional descriptions of computational models include markup languages such as CellML10,11 and the Systems Biology Markup Language (SBML).12,13 Dealing with the diversity of cancer models available has been discussed previously14 and is a continuing challenge in the wider context of biological modeling when considering numerous interoperability efforts ongoing in biology, where at the time of writing there are over 545 published standards.15
The de facto technologies for linking knowledge to data lie within the Semantic Web stack, which includes a set of specifications and languages including the Resource Description Framework (RDF),16 the Web Ontology Language (OWL),17 SPARQL Protocol and RDF Query Language (SPARQL),18 and the Semantic Web Rule Language (SWRL).19 These standards have been developed based on the philosophy of open data over the World Wide Web, and properties yielded by developing computational engines on data structures for logical inference. Advanced software has been developed for logical reasoning over linked data using these standards, such as HermiT20 and Pellet.21
While the Semantic Web technology stack is mature and its standards fully supported by the World Wide Web Consortium (W3C), the primary aim of such a technology is to facilitate machine processing and interoperation in a distributed fashion across the Web. The approach presented in this paper makes the assumption that all of the data, both domain-specific knowledge and model descriptions, lie within the scope of a single database, where in silico cancer models are semantically linked within this context. The overheads levied by the Semantic Web technology stack no longer restrict performance for querying data and for within-database analytics.
Our work in this paper builds on TumorML, a domain-specific XML-based markup language for computational cancer model description22 based on our experiences and requirements23 from the European Commission’s Transatlantic Tumor Model Repositories (TUMOR) project.24 The aim of the project was to develop a European-based digital cancer model repository to link and interoperate with a similar established digital model repository (DMR) based in the United States, and developed by the Center for the Development of a Virtual Tumor (CViT).25 TumorML was developed to act as the standard communication format between elements of the TUMOR infrastructure, and to facilitate model exportation. Its schema was designed to allow marked-up cancer model descriptions to hold essential metadata for search and retrieval of models from online repositories, as well as the linking of models via their computational interfaces. We extend and apply TumorML in this work with a property graph-based data model 26 and corresponding database implementation that semantically links model descriptions to each other via domain knowledge stored in a graph database.
A property graph is a simple graph that consists of nodes and edges (representing relationships), where each node and edge can possess properties that store specific values. A traversal is how you query a property graph, navigating from starting nodes to related nodes according to an underlying algorithm. In contrast to traditional relational databases, queries can be run on graph data that map more conventionally to real-world questions, as many queries deal with how entities are related rather than finding or filtering on individual properties of entities. For example, social networks are commonly expressed as graphs, where typical queries might map to questions such as “Who are Alice’s friends?” or “Does Alice have any friends within 2-degrees of separation from Bob?”. While directed graphs have been used in data management in biology, graph models are typically used in describing biological data, such as metabolic and signaling pathways, taxonomies of terms, and structural and sequence data.27–29 Our approach is to combine metadata sets with representations of computational models, rather than with biological data itself.
Implementation
We store our graphs and queries in Neo4j, an open-source graph database written in the Java programming language.30 Neo4j was chosen as the graph database as it is a mature, open source, general-purpose NoSQL database system that implements the property graph data model. Neo4j uses its own query language called Cypher, which was designed to be expressive of the domain at hand rather than of the data structures. Cypher queries are declarative statements that allow querying and updating of graphs. They can be formulated to create and delete nodes and relationships, update properties in the graph, as well as traverse the graph and match certain sub-graph patterns. For example, given a graph consisting of nodes representing people, and relationships linking people as ‘friend of’, the Cypher query to ask the above question, “Who are Alice’s friends?”, may look like that shown in Listing 1.
Listing 1.

An example Cypher query to find friends of Alice.
In Listing 1, line 1 specifies to iterate through all nodes in the graph. Line 2 adds a constraint as only nodes that are connected to nodes containing the property name equating to the value “Alice” connected by the relationship FRIEND_OF. Line 3 returns each matching node based on lines 1 and 2. For brevity’s sake, we will not describe Cypher’s syntax and functionality here; a full introduction to the Cypher query language can be found in a book on Neo4j.31
Before loading any in silico cancer models into our graph database, we define how our models should link together via some domain-specific knowledge. This provides the context for our investigation, where our semantic queries utilize domain knowledge in order to return relevant information. For example, given a model, A, we may formulate a query to ask the question, “What other models are classified in the same categories as A?” In a relational model, this query would match values in a particular column to the category value, whereas in a property graph model, this query traverses the database graphs to look for models that are connected to A via a node representing the category of A. In other words, from A we traverse to a node representing the category of that cancer model, then return the set of all other model nodes connected to that category node.
As a starting point, we took the data model used in the TumorML XML schema produced out of the TUMOR project.24 The schema allows the recording of metadata relating to cancer model descriptions on a number of levels. Firstly, TumorML stores metadata relating to the model description documents themselves. This enables basic curation using Dublin Core,32 linking with relevant people and organizations using xCard,33 as well as referencing/citation metadata using BibTeXML.34 Also, a TUMOR-specific taxonomy of cancer models was developed allowing for categorization of cancer models stored within the project’s infrastructure.22 Secondly, abstract model descriptions are used to describe the functional interfaces to modularized cancer models in a black box fashion where information flows through parameter descriptors. The markup is inspired by xMML.35 Within the model descriptions, implementation metadata such as hardware and software environment requirements to run associated code or binary implementations are included in TumorML model descriptions. This allows computational engines to interpret and execute stored models, where appropriate. The entity-relationship diagram in Figure 1 illustrates this data model. Meanwhile, Tables 1 and 2 summarize our mappings from the concepts outlined in Figure 1 to property graph concepts.
Figure 1.
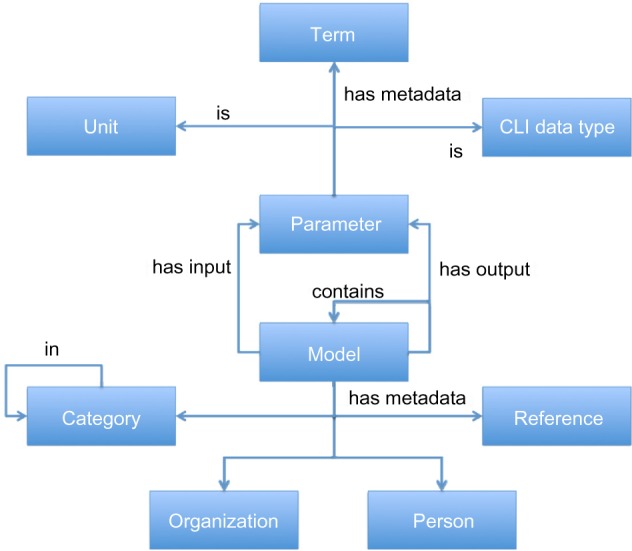
Entity-relationship diagram showing the TumorML data model. Here, we can see that a Model has input and output Parameters. Parameters are classified by Unit and Command Line Interface (CLI) data types, and also have metadata Terms attached to them. Other metadata includes bibliographic References, People, and Organizations, as well as Categories to classify the Model. Models can also be compositions and contain other models.
Table 1.
Details of relationships in our property graph model that map to the entity-relationship representation shown in Figure 1. These relationships roughly translate to those shown in the entity-relationship diagram of the TumorML data model shown in Figure 1. For example, the relationship HAS_INPUT links a Model to an input Parameter. We have extended the TumorML data model slightly, for example introducing the SYNONYM_OF_TERM relationship that links synonymous Terms together.
| RELATIONSHIP TYPE | DESCRIPTION |
|---|---|
| HAS_INPUT | Connects a model with its input parameters |
| HAS_OUTPUT | Connects a model with its output parameters |
| HAS_METADATA | Connects models and parameters to metadata |
| HAS_CATEGORY | Connects categories with other categories (ie, subcategories) |
| CONTAINS | Connects a model with other submodels (to show model composition) |
| SYNONYM_OF_TERM | Connects terms with synonymous terms |
| CREATED_BY | Connects a model with a creator (or author) of the model |
| CONTRIBUTED_BY | Connects a model with a publisher of the model description (ie, the database record) |
Table 2.
Details of node types in our property graph model that map to the entity-relationship representation of the TumorML data model shown in Figure 1. The node types in our graphs map directly to the entities in the TumorML data model. Note, we have not included CLI data types as the aim is not to enable the discovery of computational compatibilities, but rather for compatible models beyond implementation.
| NODE TYPE | DESCRIPTION |
|---|---|
| MODEL | Represents an abstract model description |
| PARAMETER | Represents a model interface parameter |
| CATEGORY | Metadata representing a categorization of a cancer model |
| TERM | Metadata representing a controlled vocabulary term |
| UNIT | Metadata representing a unit of measurement |
| PERSON | Represents a person |
| ORGANISATION | Represents an organization |
| REFERENCE | Represents a bibliographic reference linked to the model |
We initially loaded in three different domain- specific property graphs taken from within the TumorML schema: a Tumor Model Metadata model; a data model of computational types (eg, Strings, Integers, etc.); and a data model representing units (SI units and additional relevant units). Figure 2 shows a visualization of our Tumor Model Metadata model that is based on the TUMOR model taxonomy. What we can see here is a hierarchy where the endpoints of the graph represent taxa relating to each classifier. For example, the mathematical technique utilized, labeled Math, can take one of three values – Continuous, Discrete, or Hybrid, and SingleScale models may be representative of phenomena acting on specific scales, such as Organ or Subcellular. When new model descriptions are added to the graph database, they are categorized by connecting them to the relevant taxon. The edges connecting models to this domain-specific portion of the graph allow us to group related models together.
Figure 2.

TUMOR Taxonomy transposed to a property graph model and visualized in the Neo4j browser application. In this graph, we can see a hierarchy of categorizations. For example, the node Cancer has subcategories corresponding to Glioma, Nephroblastoma, Breast, Lung, and Generic.
We also load into the database domain property graphs to represent model parameter types and their units. The purpose of these is particularly relevant in the context of componentized cancer models, where linking model parameters to type and unit metadata allows us to describe how components may communicate with each other based on their computational interfaces and semantic compatibility. For example, the output of one component model may be a double-precision floating-point number, where its value represents a chemical concentration level of the phosphorolase-53 growth factor, expressed as molar concentration in μM (M = moles/liter). Therefore, it should only be able to connect to another model via an input with the same parameter profile (type = double-precision floating-point number; unit = μM). Where types and units are imprecise, for example, if the unit were in nM rather than μM, a computational execution environment, such as a workflow engine or simulation software, might still allow these parameters to be connected by identifying and normalizing these differences in scales. Likewise, type conversions and other unit conversions could also take place. With our graph database populated with three kinds of domain property graphs (model categorizations, computational types, units), we can load in our cancer model descriptions. Where in TumorML, metadata fields are used to annotate portions of models with domain-specific ontology or controlled vocabulary terms, in our graph database, we now create direct links to nodes that exist in our domain property graphs to annotate the models.
Our approach described so far relies on the graph database being pre-loaded with relevant domain information in order to link our cancer models. However, we can also add domain information to our domain-specific graphs on the fly by making use of external services to semantically enrich the graph database, without having to convert and load entire controlled vocabularies or ontologies into our graph database. As an example, we use NCBO’s BioPortal,36 an open repository of biomedical ontologies that provides access via Web services and Web browsers to ontologies developed in OWL, RDF, OBO format,37 and Protégé38 frames. Instead of loading an entire ontology or vocabulary downloaded from Bio-Portal, we query BioPortal for one particular term to annotate a model. In our case, we may seek to annotate a model input or output parameter. An appropriate BioPortal query allows us to retrieve a relevant term along with its synonyms and then store these as new property graphs in Neo4j. For this work, we have limited our scope to the NCI thesaurus39 (NCIt), as it is a rich and established collection of terms with good coverage for cancer research domains, which includes community standards, such as CDISC.40 This way, models are linked via NCIt codes, or by terms deemed synonymous with each other (including abbreviations), allowing property graph queries to traverse between models linked by common terminology. This could be easily extended to also relate coded terms together via BioPortal’s user-submitted mappings that would allow graph traversals between terms taken from a variety of different vocabularies and ontologies.
Results
To illustrate how we apply our property graph model, and also how to link together models expressed as graphs, we present two examples: one for describing a single model translated from TumorML and the other for mapping a domain-specific modeling framework’s modules into individual property graphs that are linked via domain-specific semantics.
Example 1: EGFR-ERK pathway module
For our first example, we took an EGFR-ERK pathway module,41 which describes the Ras/Raf/mitogen-activated protein kinase (MEK)/extracellular-signal-regulated kinase (ERK) pathway that is a key signaling network, governing proliferation, differentiation, and cell survival.42 Briefly, binding of epidermal growth factor (EGF) to EGF receptor (EGFR) produces a series of downstream effects through the activation of cell decision-making components.43 Pathway dynamics are regulated by material balance and kinetic equations as well as by reaction rates that are dependent on the changes in concentrations of pathway components over time, as done in many other modeling studies.44–46 Readers are encouraged to refer to the original article to have a further understanding of the model.
Here, the pathway module was modeled using TumorML, as set out in previous work,22 and we transformed the TumorML description into our graph-based representation. In Listing 2, we show the TumorML description of the EGFR module. In Listing 3, we show the same description expressed in a Cypher query that creates relevant nodes and edges to form a sub graph. Lines 19–24 of the Cypher query shown in Listing 3 create direct links to domain-specific graph nodes, in particular to those nodes in our TUMOR model taxonomy depicted in Figure 2. The EGFR-ERK module loaded into Neo4j using the Cypher query in Listing 3 yields the graph shown in Figure 3.
Listing 2.

TumorML description of the EGFR-ERK pathway module.
Listing 3.

Create: a Cypher query for creating the EGFR-ERK pathway module in Neo4j.
Figure 3.

The EGFR-ERK pathway module as a property graph and visualized in the Neo4j browser application.
When we have multiple models stored in the same database, we gradually build up connected property graphs of models clustered around metadata nodes that are part of the domain graphs. This enables us to formulate Cypher queries that ask questions about the connectedness of nodes. For example, a query that returns the EGFR-ERK Pathway depicted in Figure 3 might ask the question, “What models use continuous mathematics?”, where the graph database query would be “What model nodes are connected to the continuous node in our database?”. We express this query in Cypher in Listing 4.
Listing 4.

Pattern matching: a Cypher query to find model nodes connected to both Imageable and continuous nodes.
Line 1 of this query seeks to set the context for the pattern machine, iterating through all nodes. Line 2 attempts to match sub-graphs that consist of any node, n, connected by a relationship HAS_METADATA to a Continuous node. The query then specifies to return the ID and Title properties of any matching nodes as a list. A selection of matching nodes is shown in Table 3 (this is not exhaustive for brevity).
Table 3.
List of model node properties output from the example query in Listing 4.
| ID | TITLE |
|---|---|
| urn:miriam:tumor:000001 | EGFR-ERK Pathway model |
| urn:miriam:tumor:000004 | Alarcón 2005 Subcellular model |
| urn:miriam:tumor:000005 | Owen 2011 Subcellular model |
| urn:miriam:tumor:000007 | Alarcón 2005 VEGF calculator |
| urn:miriam:tumor:000008 | Alarcón 2006 VEGF calculator |
Example 2: Vascular tumor growth models
Our second example deals with angiogenesis, an essential process in normal tissue evolution and maintenance. This physiological process provides blood, which brings with it oxygen and nutrients, to many tumors allowing them to grow and spread. For many years angiogenesis has been a focus of intensive research, with several effective angiogenesis-related antitumor therapies developed by the pharmaceutical industry.47 Here, we focus on a family of models developed by Thomas Alarcón and collaborators that has been developed into an object-oriented (OO) modeling framework for implementing hybrid and multiscale models of vascular tumor growth by the University of Oxford’s Department of Computer Science, in conjunction with the Wolfson Centre for Mathematical Biology. The focus of the framework development has been to apply software engineering techniques that allow its elements to be highly reusable and extensible. Models published by Alarcón et al, in particular the family of models discussed and extended in Ref. 48, have been reverse-engineered in order to extract and abstract the common methodologies and data structures involved in the development of vascular tumor growth models. The OO framework has been developed based on these abstractions, and a functioning implementation of the framework has been developed in C++. The framework is presented fully by Connor et al in Ref. 49.
To demonstrate the use of our graph representation for exploring model composition, we loaded into our graph database model descriptions of components of the tumor growth modeling framework described above. The reason for using such a framework for our exemplar is that it contains a coherent, modular set of models that interoperate with each other in a predictable way. Thus, we are able to show how single-scale models can be linked together semantically to form multiscale models within the context of existing multiscale models. Moreover, through the use of our graph-based data schema, model developers may explore the modeling framework with different kinds of queries that would not normally be straightforward without metadata and a means to compute over it.
In Figure 4 we have illustrated a portion of the vascular tumor growth framework, highlighting a simple case of inter facing with a model, using the Unified Modeling Language (UML) where the diagram was produced using Visual Paradigm for UML,50 a computer-aided software engineering tool. In this case, objects of type Alarcon2005SubCellularModel interact with Cell objects that are collectively contained in a CellPopulation object. The parameters that are passed between the objects include chemical concentrations, and information such as cell state, mass, and cycle times. Listing 5 shows the corresponding Cypher query to create the property graph describing the Alarcón 2005 subcellular model given in Ref. 51. After loading all of the framework’s model descriptions into the graph database, we can begin to explore our semantically linked models with Cypher queries.
Figure 4.

UML class diagram illustrating parameters passed between subcellular models and cell objects.
Listing 5.

Create: a Cypher query to describe the Alarcón 2005 sub cellular model shown in Figure 4.
We demonstrated earlier how we could link models by categorizations of cancer models. Now, using the example of the vascular tumor growth modeling framework, we can show how models can be linked less trivially through annotated model inputs and outputs. For example, queries involving cancer model parameters might ask, “What cancer models have input parameters that are compatible with another model’s output parameters?”. Based on how we have linked our model parameters and variables to domain-specific data, such as using our controlled vocabulary terms, standard units, and command-line data types, we can determine the most compatible single-scale models that could be used to compose a multiscale compound model. This can be expressed in Cypher as shown in Listing 6.
Listing 6.

Recommend compatible models: a Cypher query to find model nodes that have parameters that are compatible using metadata terms.
Line 1 seeks to iterate through all patterns of nodes, where the pattern consists of a model node n connected to an input parameter p, which is in turn connected to a metadata term meta that is shared with an output parameter q of a model m. Next, line 2 specifies that node n should not be the same as node m (to omit finding itself as a compatible model in the query). Finally, line 3 specifies to return all matches as a list of pairs of model titles under two headings, where ModelA has outputs that match inputs for ModelB, along with what terms are matched as NCIt codes. An example output to the query is shown in Table 4.
Table 4.
Output of query on matching model outputs to inputs.
In this example, we can see that, based on the metadata terms with which model inputs and outputs are annotated, the outputs of the subcellular models are compatible with the inputs of the VEGF calculator models. Although the model para meters have different labels (under table headings Output A and Input B), the annotated NCI terms ensure specific and identical meaning. C1272 corresponds to the NCIt code for Recombinant Vascular Endothelial Growth Factor (VEGF), and C28217 to the term for Intracellular. Explicitly, then, the combination of these terms indicates that the VEGF calculator components can be connected via intracellular VEGF to the Alarcón 2005 sub-cellular model. Here, importantly, by capturing compositional relationships such as those that we know to exist between the subcellular models and VEGF calculators in an existing family of multiscale models, we validate our approach.
Finally, another kind of query we may ask involves reasoning on compositional relationships between existing multiscale models. While the previous example uses Cypher to recommend a composition, we may have stored models that we know are already composed of sub-models. Such compositions are denoted in our database by the CONTAINS relationship, as shown in Figure 5. This property graph expresses the fact that the Alarcón 2003 model is composed of a subcellular model, a cell proliferation model, a vascular structural adaptation model, and an oxygen calculator. Each of these sub-models is associated with a specific single scale via appropriate metadata nodes. Questions such as “Over what biological scales does the Alarcon 2003 model extend?” are now possible. Clearly, an appropriately phrased query would return a list containing the Cell, Tissue, and Subcellular scales.
Figure 5.
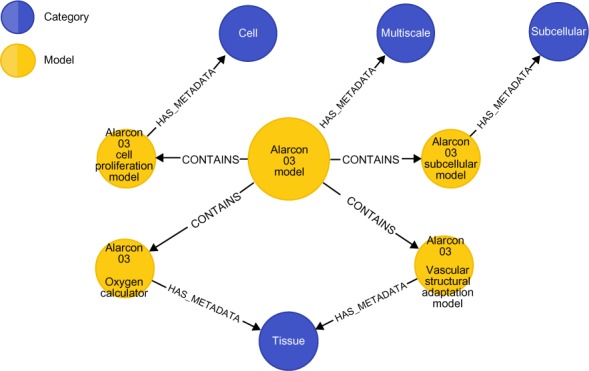
The Alarcón 2003 model and its component models and biological scale metadata, represented as a property graph and visualized in the Neo4j browser application.
Discussion
To date, many computational models of cancer have been developed to account for phenomena occurring at individual biological scales. One of the major challenges we now face is to integrate and extend these models into a fully multiscale computational framework. However, model composition and extension is non-trivial, requiring specialist domain knowledge. Our approach intends to facilitate both the integration of single-scale models across multiple scales and the extension of existing multiscale models. We achieve this by enabling the annotation of cancer models and the formation of semantic links between them through the use of a property graph database. Our approach, by making in silico models of cancer more understandable, also has the potential to close the gap between experimentalists and modelers and to encourage greater collaboration between them.
We believe that graph databases have a very relevant place in informatics systems for cancer study, and also more broadly in biomedical informatics. While semantic technologies are mature and well supported by organizations such as the W3C, much of the philosophy behind their development is based on interoperating systems for linked data across the Internet. With our tumor model graphs, we have taken the approach where metadata is stored alongside model descriptions, removing much of the overhead that comes with systems built on RDF and OWL. We, however, should make it clear that we do believe that the Semantic Web stack has its place and its uses. For importing and exporting data and metadata from a tumor model repository, we would certainly expect to leverage some or all of these technologies, and this is why the XML markup in TumorML was originally developed – to transmit such data in a standardized and interoperable format, which could be combined with the Semantic Web’s XML-based tools and formats.
In summary, we have presented a property graph model for representing tumor models so that combinations of models can be explored, based on semantic compatibility. A single graph database can be used to store domain data, such as taxonomies, controlled terminologies, and ontologies alongside model descriptions. By annotating parameters with command-line interface data types, we can validate what might computationally fit together. Linking model descriptions to unit metadata allows us to reason about relative scalings between parameters, and by annotating with biological terms, as in the previous example using NCI codes, we can check the semantic compatibility of parameters based on biological knowledge (terms). We can then build relatively simple queries that propose links between models. In the future, we aim to build recommendation queries that could be quantified with compatibility metrics based on the proportions of common annotations, and also the distance of the metadata paths where terms might not match directly, but have common ancestry within ontologies or other relations between annotated terms.
To date, we have demonstrated the possibilities for exploration of model composition using Cypher queries and a number of existing multiscale cancer model descriptions as test data. Efforts are underway to use this work as the basis for developing a set of usable software tools for exploring cancer models. In particular, we believe that the effective utilization of several modeling approaches: continuum models, discrete models as well as fitting of model parameters via biological and clinical data may be optimized using this methodology. The work described in this paper is fully available as an interactive demo and can be downloaded as a Neo4j GraphGist at http://gist.neo4j.org/?6038a7b526bfa48da2c0.
Footnotes
Author Contributions
Wrote the first draft of the manuscript: DJ. Contributed to the writing of the manuscript: DJ, AJC, SM. Contributed to TumorML: DJ, SM, ZW, TSD. Developed the graph model: DJ. Developed the vascular tumor growth modeling framework: AJC. TQ, ES provided support and guidance for AJC’s contributions. Agree with manuscript results and conclusions: All authors. Jointly developed the structure and arguments for the paper: DJ, AJC. All authors made critical revisions and approved the final version.
ACADEMIC EDITOR: JT Efird, Editor in Chief
FUNDING: Contributions by DJ, SM, ZW, and TSD toward the development of TumorML were initially supported by the European Commission under the Transatlantic Tumor Model Repositories (TUMOR) project (Contract # FP7-ICT−2009.5.4–247754). Contributions by AJC are supported in part by the UK Engineering and Physical Sciences Research Council (EPSRC) and F. Hoffman la-Roche Ltd. Contributions by SM are supported in part by the Government of the Russian Federation (Grant 074-U01). The authors confirm that the funders had no influence over the study design, content of the article, or selection of this journal.
COMPETING INTERESTS: AJC discloses grants and non-financial support from Hoffman-La Roche, and grants from the Engineering and Physical Sciences Research Council, during the conduct of the study, and grants and non-financial support from Hoffman-La Roche outside the work presented here. All of the aforementioned disclosures were in support of AJC’s PhD research. Other authors disclose no competing interests.
Paper subject to independent expert blind peer review by minimum of two reviewers. All editorial decisions made by independent academic editor. Upon submission manuscript was subject to anti-plagiarism scanning. Prior to publication all authors have given signed confirmation of agreement to article publication and compliance with all applicable ethical and legal requirements, including the accuracy of author and contributor information, disclosure of competing interests and funding sources, compliance with ethical requirements relating to human and animal study participants, and compliance with any copyright requirements of third parties. This journal is a member of the Committee on Publication Ethics (COPE).
REFERENCES
- 1.Deisboeck TS, Wang Z, Macklin P, Cristini V. Multiscale cancer modeling. Annu Rev Biomed Eng. 2011;13:127–55. doi: 10.1146/annurev-bioeng-071910-124729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Southern J, Pitt-Francis J, Whiteley J, et al. Multi-scale computational modelling in biology and physiology. Prog Biophys Mol Biol. 2008;96:60–89. doi: 10.1016/j.pbiomolbio.2007.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tracqui P. Biophysical models of tumour growth. Rep Prog Phys. 2009;72:56701. [Google Scholar]
- 4.Walker DC, Southgate J. The virtual cell–a candidate co-ordinator for ‘middle-out’ modelling of biological systems. Brief Bioinform. 2009;10:450–61. doi: 10.1093/bib/bbp010. [DOI] [PubMed] [Google Scholar]
- 5.Wolkenhauer O, Auffray C, Brass O, et al. Enabling multiscale modeling in systems medicine. Genome Med. 2014;6:21–3. doi: 10.1186/gm538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rayner TF, Rocca-Serra P, Spellman PT, et al. A simple spreadsheet-based, MIAME-supportive format for microarray data: MAGE-TAB. BMC Bioinformatics. 2006;7:489. doi: 10.1186/1471-2105-7-489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Demir E, Cary MP, Paley S, et al. The BioPAX community standard for pathway data sharing. Nat Biotechnol. 2010;28:935–42. doi: 10.1038/nbt.1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.The Gene Ontology Consortium. Gene ontology: tool for the unification of biology. Nat Genet. 2000;25:25–9. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.The Gene Ontology Consortium. The gene ontology project in 2008. Nucleic Acids Res. 2008;36:D440–4. doi: 10.1093/nar/gkm883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cuellar AA, Lloyd CM, Nielsen P, Bullivant DP, Nickerson DP, Hunter PJ. An overview of CellML 1.1, a biological model description language. SIMUL. 2003;79(12):740–7. [Google Scholar]
- 11.Lloyd CM, Halstead MD, Nielsen P. CellML: its future, present and past. Prog Biophys Mol Biol. 2004;85(2–3):433–50. doi: 10.1016/j.pbiomolbio.2004.01.004. [DOI] [PubMed] [Google Scholar]
- 12.Hucka M, Finney A, Bornstein BJ, et al. Evolving a lingua franca and associated software infrastructure for computational systems biology: the Systems Biology Markup Language (SBML) project. IET Syst Biol. 2004;1(1):41–53. doi: 10.1049/sb:20045008. [DOI] [PubMed] [Google Scholar]
- 13.Hucka M, Finney A, Bornstein BJ, et al. The Systems Biology Markup Language (SBML): language specification for level 3 version 1 Core (release 1 candidate) Nature Precedings. 2010. [Accessed April 29, 2014]. Available from: http://dx.doi.org/10.1038/npre.2010.4123.1.
- 14.Johnson D, McKeever S, Stamatakos G, et al. Dealing with diversity in computational cancer modeling. Cancer Inform. 2013;12:115–24. doi: 10.4137/CIN.S11583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.BioSharing BioSharing Standards: a catalogue of reporting standards and organizations that develop these. [Accessed April 29, 2014]. 2014. Available from: http://www.biosharing.org/standards.
- 16.Selçuk Candan K, Liu H, Suvarna R. Resource description framework: metadata and its applications. SIGKDD Explor. 2001;3(1):6–19. [Google Scholar]
- 17.Smith MK, Welty C, McGuinness DL, editors. OWL Web Ontology Language Guide, W3C Recommendation 10 February 2004. 2004. [Accessed April 29, 2014]. Available from: http://www.w3.org/TR/2004/REC-owl-guide-20040210/
- 18.Prud’hommeaux E, Seaborne A. SPARQL Query Language for RDF, W3C Recommendation 15 January 2008. 2008. [Accessed April 29, 2014]. Available from: http://www.w3.org/TR/2008/REC-rdf-sparql-query-20080115/
- 19.Horrocks I, Patel-Schneider PF, Boley H, et al. SWRL: A Semantic Web Rule Language Combining OWL and RuleML, W3C Member Submission 21 May 2004. 2014. [Accessed April 29, 2014]. Available from: http://www.w3.org/Submission/2004/SUBM-SWRL-20040521/
- 20.Shearer R, Motik B, Horrocks I. In: Ruttenberg A, Sattler U, Dolbear C, editors. HermiT: A Highly-Efficient OWL Reasoner; Proceedings of 5th Inter national Workshop on OWL: Experiences and Directions (OWLED 2008 EU); October 26–27, 2008; Karlsruhe, Germany. [Accessed April 29, 2014]. Available from: http://webont.org/owled/2008/papers/owled2008eu_submission_12.pdf. [Google Scholar]
- 21.Sirin E, Parsia B, Cuenca Grau B, Kalyanpur A, Katz Y. Pellet: a practical OWL-DL reasoner. Web Semant. 2007;5(2):51–3. [Google Scholar]
- 22.Johnson D, McKeever S, Deisboeck TS, Wang Z. Connecting digital cancer model repositories with markup: introducing TumorML version 1.0. ACM SIGBio Record. 2013;3(3):5–11. [Google Scholar]
- 23.Johnson D, Cooper J, McKeever S. TumorML: Concept and requirements of an in silico cancer modelling markup language. Conf Proc IEEE Eng Med Biol Soc. 2011;2011:441–4. doi: 10.1109/IEMBS.2011.6090060. [DOI] [PubMed] [Google Scholar]
- 24.Sakkalis V, Sfakianakis S, Marias K, et al. The TUMOR project: integrating cancer model repositories for supporting predictive oncology; Abstract Booklet for VPH2012 Integrative Approaches to Computational Biomedicine; September 18–20, 2012; London, UK. [Google Scholar]
- 25.Deisboeck TS, Zhang L, Martin S. Advancing cancer systems biology: introducing the Center for the Development of a Virtual Tumor, CViT. Cancer Inform. 2007;5:1–8. [PMC free article] [PubMed] [Google Scholar]
- 26.Parastatidis S. On graph data model design – relationships. 2013. [Accessed July 17, 2014]. Available from: http://savas.me/2013/03/on-graph-data-model-design-relationships/
- 27.Graves M, Bergeman ER, Lawrence CB. Graph database systems for genomics. IEEE Eng Med Biol Mag. 1995;14(6):737–45. [Google Scholar]
- 28.Olken F. Graph data management for molecular biology. OMICS. 2003;7(1):75–8. doi: 10.1089/153623103322006652. [DOI] [PubMed] [Google Scholar]
- 29.Eckman BA, Brown PG. Graph data management for molecular and cell biology. IBM J Res Dev. 2006;50(6):545–60. [Google Scholar]
- 30.Neo Technology inc Neo4j: the world’s leading graph database. 2014. [Accessed April 29, 2014]. Available from: http://www.neo4j.org.
- 31.Robinson I, Webber J, Eifrém E. Graph Databases. Sebastopol, CA: O’Reilly Media; 2014. [Google Scholar]
- 32.Kunze J, Baker T. The Dublin Core Metadata Element Set. RFC 5013 (Informational) 2007. [Accessed October 10, 2014]. Available from: http://www.ietf.org/rfc/rfc5013.txt.
- 33.Perreault S. xCard: vCard XML Representation. RFC 6351 (Proposed Standard) 2011. [Accessed October 10, 2014]. Updated by RFC 6868. Available from: http://tools.ietf.org/rfc/rfc6351.txt.
- 34.Gundersen VB, Hendrikse ZW. BibTeX as XML markup. [Accessed online October 10, 2014]. 2007. Available from: http://bibtexml.sf.net/
- 35.Borgdorff J, Lorenz E, Hoekstra AG, Falcone J, Chopard B. A principled approach to distributed multiscale computing, from formalization to execution; Proceedings of IEEE eScience Workshops; Stockholm, Sweden. December 5–8, 2011. [Google Scholar]
- 36.Noy NF, Shah NH, Whetzel PL, et al. BioPortal: ontologies and integrated data resources at the click of a mouse. Nucleic Acids Res. 2009;37(2):W170–3. doi: 10.1093/nar/gkp440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Osumi-Sutherland D. OBO Format. Ontogenesis. 2010. [Accessed October 10, 2014]. Available from: http://ontogenesis.knowledgeblog.org/245.
- 38.Stanford Center for Biomedical Informatics Research Protégé – a free open-source ontology editor and framework for building intelligent systems. 2014. [Accessed May 1, 2014]. Available from: http://protege.stanford.edu/
- 39.Sioutos N, de Coronado S, Haber MW, Hartel FW, Shaiu WL, Wright LW. NCI thesaurus: a semantic model integrating cancer-related clinical and molecular information. J Biomed Inform. 2007;40(1):30–43. doi: 10.1016/j.jbi.2006.02.013. [DOI] [PubMed] [Google Scholar]
- 40.de Montjoie AJ. Introducing the CDISC Standards: New Efficiencies for Medical Research. Austin, TX: CDISC Publications; 2009. [Google Scholar]
- 41.Wang Z, Zhang L, Sagotsky J, Deisboeck TS. Simulating non-small cell lung cancer with a multiscale agent-based model. Theor Biol Med Model. 2007;4:50. doi: 10.1186/1742-4682-4-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kolch W. Meaningful relationships: the regulation of the Ras/Raf/MEK/ERK pathway by protein interactions. Biochem J. 2000;351(2):289–305. [PMC free article] [PubMed] [Google Scholar]
- 43.Friedl P, Wolf K. Tumour-cell invasion and migration: diversity and escape mechanisms. Nat Rev Cancer. 2003;3:362–74. doi: 10.1038/nrc1075. [DOI] [PubMed] [Google Scholar]
- 44.Chen WW, Schoeberl B, Jasper PJ, et al. Input-output behavior of ErbB signaling pathways as revealed by a mass action model trained against dynamic data. Mol Syst Biol. 2009;5:239. doi: 10.1038/msb.2008.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang Z, Birch CM, Sagotsky J, Deisboeck TS. Cross-scale, cross-pathway evaluation using an agent-based non-small cell lung cancer model. Bioinformatics. 2009;25:2389–96. doi: 10.1093/bioinformatics/btp416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schoeberl B, Pace EA, Fitzgerald JB, et al. Therapeutically targeting ErbB3: a key node in ligand-induced activation of the ErbB receptor-PI3K axis. Sci Signal. 2009;2:ra31. doi: 10.1126/scisignal.2000352. [DOI] [PubMed] [Google Scholar]
- 47.Kerbel RS. Tumor angiogenesis. N Engl J Med. 2008;358(19):2039–49. doi: 10.1056/NEJMra0706596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Owen MR, Alarcón T, Maini PK, Byrne HM. Angiogenesis and vascular remodelling in normal and cancerous tissues. J Math Biol. 2009;58(4–5):689–721. doi: 10.1007/s00285-008-0213-z. [DOI] [PubMed] [Google Scholar]
- 49.Connor AJ, Cooper J, Byrne HM, Maini PK, McKeever S. Object-oriented paradigms for modelling vascular tumour growth: a case study; Proceedings of The Fourth International Conference on Advances in System Simulation (SIMUL 2012); 2012; Lisbon, Spain. pp. 74–83. [Google Scholar]
- 50.Visual Paradigm for UML. [Accessed October 10, 2014]. 2014. Available from: http://www.visual-paradigm.com/
- 51.Alarcón T, Byrne HM, Maini PK. A multiple scale model for tumor growth. Multiscale Model Simul. 2005;3(2):440–75. [Google Scholar]