

Abstract
The identification of modifiable risk factors for the development of rheumatic conditions and their sequelae is crucial for reducing the substantial worldwide burden of these diseases. However, the validity of such research can be threatened by sources of bias, including confounding, measurement and selection biases. In this Review, we discuss potentially major issues of selection bias—a type of bias frequently overshadowed by other bias and feasibility issues, despite being equally or more problematic—in key areas of rheumatic disease research. We present index event bias (a type of selection bias) as one of the potentially unifying reasons behind some unexpected findings, such as the ‘risk factor paradox’—a phenomenon exemplified by the discrepant effects of certain risk factors on the development versus the progression of osteoarthritis (OA) or rheumatoid arthritis (RA). We also discuss potential selection biases owing to differential loss to follow-up in RA and OA research, as well as those due to the depletion of susceptibles (prevalent user bias) and immortal time bias. The lesson remains that selection bias can be ubiquitous and, therefore, has the potential to lead the field astray. Thus, we conclude with suggestions to help investigators avoid such issues and limit the impact on future rheumatology research.
Introduction
Rheumatic and musculoskeletal conditions, and their sequelae, constitute a tremendous disease burden worldwide. unbiased research that accurately and reliably determines modifiable risk factors for the development of rheumatic conditions and their sequelae is critical to reduce this burden. Among the major sources of bias that threaten the validity of research findings, confounding and measurement biases have generally received their due attention from investigators and clinicians. However, selection bias, which tends to be insidious (yet equally or more problematic), is frequently overshadowed by other bias and feasibility issues. In this article, we review potentially major selection bias issues in key areas of rheumatic disease research. As most of these issues are not unique to rheumatic conditions, we also describe notable examples from nonrheumatic conditions to help crystallize our discussions.
Disease burden of arthritic conditions
In the US alone, arthritis affected approximately 43 million people in 1997 and is projected to affect 60 million people by 2020.1 The burden of disease involves not only the morbidity from arthritis, but also its associated comorbidities, sequelae events, and premature mortality. For example, osteoarthritis (OA), the most common joint disorder among adults in the US, causes pain and decreased mobility, and OA progression leads to disability, joint failure, and total joint replacement. Rheumatoid arthritis (RA), a chronic and systemic inflammatory condition, leads to joint pain and deformity, as well as premature cardiovascular events and mortality. Sequelae events of these conditions play a major part in the disease burden among affected individuals, as well as in the burden to society in general; thus, these complications represent a compelling target for secondary or tertiary prevention. Our ability to prevent these potentially debilitating and costly disease sequelae depends on an accurate understanding of modifiable risk factors for these events. Ultimately, unbiased determination of risk factors for disease progression or sequelae events holds the promise of improving our ability to prevent these outcomes through risk factor modification in clinical care and public health practice.
The risk factor paradox
In rheumatic diseases
Despite substantial research progress over the past few decades enhancing our knowledge of the risk factors for the incidence of musculoskeletal conditions (for primary prevention), evidence regarding the risk factors for disease progression or sequelae events among individuals with musculoskeletal conditions (for secondary or tertiary prevention) has often been inconsistent, or sometimes even paradoxical (Table 1).2–6 For example, over the past few decades, a number of risk factors for incident knee OA have been consistently identified, including female sex, obesity, high bone mineral density, knee injury, repetitive occupational stress on joints, and certain sports.7,8 By contrast, a systematic review of 36 articles concluded that sex, knee pain, radiological severity, joint injury, quadriceps strength, and regular sport activities are not associated with the risk of OA progression.9 Furthermore, these studies have failed to find a consistent association even between obesity or ageing (two well-established risk factors) and the risk of knee OA progression.9 interestingly, high bone mineral density (another risk factor for the development of OA) was associated with a reduced risk of radiographic OA progression.10–13
Table 1. Examples of risk factor paradoxes in rheumatic disease contexts.
| Risk factor | Associations in the general population | Associations in the rheumatic disease (index) population |
|---|---|---|
| OA | ||
| Bone mineral density | Risk of incident OA | ↓ Risk of OA progression9 |
| Obesity | Risk of incident OA | ↔ Risk of OA progression9 |
| Low vitamin C levels | Risk of incident OA | ↓ Risk of OA progression9 |
| Female sex | Risk of incident OA | ↔ Risk of OA progression9 |
| RA | ||
| Smoking | Risk of incident RA | ↓ or ↔ Risk of RA progression14–16 |
| Risk of incident CVD | ↔ Risk of CVD among patients with RA17–18 | |
| Obesity | Risk of mortality | ↓ Mortality among patients with RA20 |
| PsA | ||
| Smoking | Risk of psoriasis | ↓ Risk of psoriatic arthritis among patients with psoriasis4 |
| HLA-Cw*0602 | Risk of psoriasis | ↓ Risk of psoriatic arthritis among patients with psoriasis26,27 |
Abbreviations: CVD, cardiovascular disease; OA, osteoarthritis; PsA, psoriatic arthritis; RA, rheumatoid arthritis.
Similarly, while smoking is a well-established risk factor for the development of incident RA, several cohort studies have found that smoking has an inverse or null association with radiological RA progression (the ‘smoking paradox’).14–16 For example, a study based on a prospective early RA cohort (n = 813) reported that current smokers had a 50% lower risk of structural disease progression compared with nonsmokers (multi variate odds ratio 0.50, 95% CI 0.27–0.93).14 Another RA cohort study (n = 2004) found that smoking intensity is associated with an inverse dose-response (P <0.001); heavy smokers progressed significantly less than moderate smokers or nonsmokers (average progression of the maximum damage score, 1.21%, 2.71%, and 2.86%, respectively).16 Furthermore, according to the rochester epidemiology Project, established cardiovascular risk factors, such as male sex, current smoking, past cardiac history, family cardiac history, and dyslipidemia, are not associated with the risk of cardiovascular disease (CVD) in patients with RA, implying a substantially weaker impact (if any) than in the general population.3,17 For example, the magnitude of association between current smoking and CVD risk among patients with ra was 32% (statistically insignificant), as compared with a 219% increased risk among current smokers without RA (P = 0.008).17 The results from another RA cohort study (n = 4,363) indicated a 20% decreased risk of cardio vascular morbidity among patients with this condition who are current smokers.18 Furthermore, unlike the threefold increased risk of cardiovascular mortality associated with obesity in the general population,19 obese patients with RA had a 70% decreased risk of cardio vascular mortality compared with those in the normal BMI range (the ‘obesity paradox’).20 Similarly, unlike in the general population,21 high levels of total or low-density lipoprotein (LDL) cholesterol were not associated with an increased cardio vascular risk among patients with RA, whereas low levels of total or LDL cholesterol were associated with an increased risk of cardiovascular events (the ‘lipid paradox’).22 Thus, the current evidence regarding the effects of smoking, obesity, and dyslipidemia on the risk of cardiovascular events among patients with RA does not provide support for the current practice guidelines regarding cardiovascular risk management among these patients.23,24
Likewise, the smoking paradox has been observed in studies of psoriatic arthritis (PsA). Current smoking has been found to increase the risk of incident PsA by threefold in the general population,25 but is associated with a 40% decreased risk of PsA in those with psoria-sis.4 Moreover, the genetic allele that is strongly associated with the risk of psoriasis (HLA-Cw*0602) predicts a lower risk of PsA among patients with psoriasis.26,27
In nonrheumatic diseases
Such inconsistent or paradoxical phenomena in the risk factors for sequelae events have also been observed in many nonrheumatic disease contexts (Table 2). For example, a smoking paradox has been observed among patients who undergo isolated coronary artery bypass grafting, with current smokers having a 30% lower risk for arrhythmia than nonsmokers.28 Similarly, among patients with a history of coronary heart disease, those who are overweight or obese (BMI ≥25) have a 20% lower risk of cardiovascular-specific mortality than individuals who are of normal weight (BMI <25) (the obesity paradox).29,30 In addition, in 2011, Canto et al.6 showed that, among people with myocardial infarction, well-established cardiovascular risk factors (such as hypertension, smoking, dyslipidemia, diabetes, and family history of coronary heart disease) had protective effects on the risk of hospital mortality. Furthermore, in the same year, Dahabreh and Kent31 discussed the study findings that patent foramen ovale (PFO; a type of atrial septal defect) doubles the risk of incident cryptogenic stroke,32 but does not increase the risk of recurrent events.31
Table 2. Examples of risk factor paradoxes in nonrheumatic disease contexts.
| Risk factor paradox | Associations in the general population | Associations in the index population |
|---|---|---|
| Smoking paradox | Risk of incident CAD | ↓ Risk of hospital mortality in patients with CAD28 |
| Obesity paradox | Risk of incident CAD | ↓ Risk of cardiovascular-specifc mortality in patients with CAD29,30 |
| Aspirin paradox | Risk of incident COPD | ↓ Mortality in patients with COPD65 |
| Thrombophilia paradox | Risk of incident CHD | ↓ Risk of recurrent CHD events in patients with CHD66 |
| PFO paradox | Risk of incident VTE | ↔ Risk of recurrent VTE in patients with incident VTE35 |
| Low birth-weight paradox | Risk of incident stroke Risk of low-birth weight baby | ↔ Risk of recurrent stroke in patients with incident stroke31,32↓ Mortality in low-birth weight babies36 |
| Apolipoprotein E4 allele | Risk of incident Alzheimer disease | ↓ Risk of Alzheimer disease progression33,34 |
Abbreviations: CAD, coronary artery disease; CHD, coronary heart disease; COPD, chronic obstructive pulmonary disease; PFO, patent foramen ovale; VTE, venous thrombotic embolism.
Another commentary comprehensively discussed a prominent neurology example of paradoxical gene effects, in which the apolipoprotein E gene was associated with a risk of mild cognitive impairment (the onset of Alzheimer disease), but not with the progression of Alzheimer disease.33,34 Similarly, in the field of haematology, thrombophilia increases the risk of incident deep vein thrombosis, but not of recurrent events (the ‘thrombophilia paradox’).35 Finally, in paediatrics, smoking increases the risk of low birth weight, but new-borns exposed to maternal smoking have been shown to have a 20% reduced risk of mortality compared with low-birth-weight infants not exposed to maternal smoking (the ‘birth weight paradox’).36
These apparently paradoxical phenomena have been puzzling to investigators and clinicians, as current knowledge does not readily explain their biological mechanisms. Nevertheless, despite these contradictory findings that challenge conventional wisdom, clinicians continue to advocate consensus-based (as opposed to evidence-based) recommendations. For example, patients with coronary heart disease have been advised to control their weight, cholesterol and blood pressure, and to quit smoking.28,37
Apart from potential differences in underlying biology, and possibly a diminished role of established risk factors in the presence of competing disease-specific risk factors in index disease populations, an enticing alternative methodological explanation for these unexpected results is a type of selection bias known as index event bias. This type of bias is discussed in the next section.
Index event bias and risk factor paradox
A compelling explanation for the numerous paradoxes discussed above is index event bias (also known as collider stratification bias), a type of selection bias.31,36,38 This bias can affect research on the risk of disease sequelae when multiple risk factors for sequelae are also risk factors for having the disease in the first place. Examples of such research scenarios include studies of the effect of obesity on the risk of OA progression among those with OA (in which OA is considered the index event), the effect of smoking on the risk of sequelae events among those with RA (the index event), or the effect of PFO on the risk of recurrent stroke. In epidemiological terms, index event bias occurs because conditioning on the outcome (that is, considering the conditional situation in which a given event, the ‘outcome’, occurs)—for example, restricting the study population to those with an index event—induces dependence between these risk factors, even when they are not associated in the general population (that is, the unconditioned entire population). This effect thus creates a spurious association among those risk factors with an index event.39
As discussed in their 2011 JAMA commentary, Dahabreh and Kent demonstrated that these spurious inverse associations between PFO and other risk factors (such as age, hypertension, diabetes, and smoking) among patients with incident stroke were created by index event bias, and that such associations were not present in the general population.31 The spurious negative associations between these risk factors occur because patients with PFO (a strong risk factor by itself) require less of a contribution from other risk factors to have the first stroke as compared with those without PFO.31 Thus, among patients with incident stroke, the presence of PFO becomes inversely associated with other risk factors, including unknown or unmeasured ones that cannot be adjusted. These negative associations, particularly those with unadjusted risk factors, would bias the effect of PFO towards the null for recurrent stroke, as observed in multiple studies, because in individuals with PFO the effect of the PFO would be negatively confounded by those risk factors.31
To help general readers understand the causal framework and underlying logic of index event bias in graphical form, we have depicted an intuitive example in Figure 1. This classic, simple experiment of a coin toss (cause) and a ringing bell (effect) demonstrates the mechanism by which conditioning on a common effect (a ‘downstream variable’) induces a negative correlation between two causes (or risk factors) that were independent before conditioning.
Figure 1.
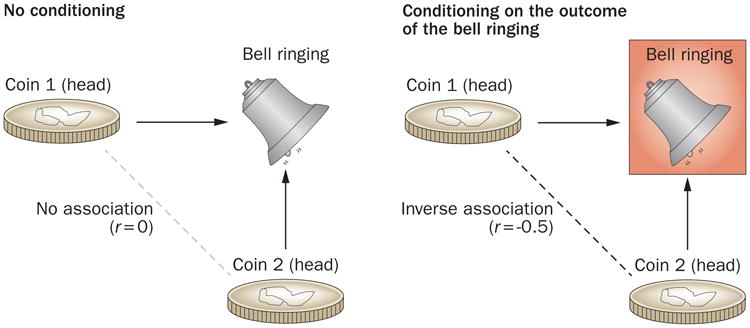
A causal diagram illustration of index event bias, also known as collider stratification bias. A causal diagram consists of a set of relevant variables (for example, exposures, potential confounders, and outcomes) and arrows to indicate the flow of causation between those variables. When there are multiple independent causes for an effect (i.e. a common effect), conditioning on this common effect (i.e. selecting only scenarios in which the effect is observed) leads to a spurious association between those causes. A classic, simple example of a coin toss (cause) and a ringing bell (effect) can illustrate the logic behind this phenomenon. In this experiment involving two coins and a bell, the bell rings whenever either coin comes up heads on a toss of both coins. Thus, the bell ringing is a common effect of heads appearing on the toss of either coin. In causal diagrams, this is depicted as colliding causal arrows on a given common effect variable (which gives the name ‘collider’). Obviously, heads appearing from one coin toss is independent of heads appearing from the other coin toss; thus, these two causes are mutually independent with a correlation coefficient between the two of 0. However, if we calculate the correlation from only the events when the bell rings (i.e. we condition on the common effect of the bell ringing), the appearances of heads on the two coins are no longer independent, resulting in a correlation coefficient of −0.5. This discrepancy occurs because if coin A came up tails, then that must mean that coin B came up heads (and vice versa), as we know that the bell rang. This simple experiment demonstrates that conditioning on a common effect induces a negative correlation between two causes or ‘risk factors’. Conditioning is marked by a box around the variable name, and the spurious association is marked by a dotted line between variables, as per causal diagram convention.
Impact on OA progression studies
Index event bias could have also contributed to many null or paradoxical findings that have been observed in OA progression studies, as we have previously discussed using causal diagrams.5 For example, we have depicted a typical design for an OA progression study in relation to prior obesity in Figure 2, in which we have indicated the study population of patients with OA at study baseline (this conditioning is marked by a box around ‘OA incidence’ as per causal diagram convention [see Figure 1 and its legend for details]). In these studies, obesity (the exposure of interest) is a known cause of OA incidence and is often assessed before or at the time of OA incidence. Thus, as in the earlier PFO discussion, restricting the study population to patients with OA (those with an index event) introduces index event bias by creating a spurious negative association (marked by a dotted line) between obesity and other risk factors (including unknown or unmeasured risk factors [URFs]) for OA progression (Figure 2).5 Heuristically, the spurious negative association between these risk factors occurs because patients with obesity (a strong risk factor by itself) require less of a contribution from other risk factors to develop incident OA, as compared with those who are not obese.5 Thus, among patients already with incident OA, the presence of obesity becomes inversely associated with other risk factors, including URFs that cannot be adjusted. This spurious negative confounding, particularly by unadjusted risk factors, would bias the effect of obesity towards the null for the risk of OA progression, as observed in previous studies.9
Figure 2.

A causal diagram of a typical observational study showing the assessment of the effect of obesity on OA progression among patients with (incident) OA. Conditioning on (or restricting to) those with OA incidence (i.e. conditioning on a common effect, as explained in Figure 1) results in obesity and the URFs becoming negatively associated, as indicated by a dotted line between obesity and URFs, even though these two factors were not associated before OA incidence. This artificially-generated negative confounding results in a biased association between obesity and OA progression (represented as obesity—URF→OA progression), leading to effect estimation biased towards the null (see Figure 1 legend for details). Abbreviations: OA, osteoarthritis; URFs, unknown or unmeasured risk factors.
Impact on RA progression studies
The index event bias mechanism could also explain the inverse or null association between smoking and RA progression14–16 (or CVD, a sequelae event of RA3,17–18) among patients with RA (Figure 3). Similarly to in our earlier discussion, we consider an example using risk factors that are mutually independent but are associated with RA incidence, namely smoking and URFs. As URFs are not associated with smoking before individuals develop RA, they are not confounders in a study for RA incidence (or CVD) in the general population. However, when studying patients with RA, smoking and these URFs are no longer independent, owing to index event bias (indicated by a dotted line linking the two factors in Figure 3). As smoking becomes inversely associated with URFs,16 the resulting effect measure becomes underestimated or reversed (that is, paradoxical) unless the URF is appropriately adjusted for. However, not all risk factors are measured, or even known, in many observational studies, leading to biased effect estimates. The magnitude and direction of the bias would vary according to the direction and strength of associations between smoking and URFs, and between URFs and RA progression (or CVD).5,31 Thus, index event bias can explain the apparently diminished or paradoxical role of established risk factors among those with index rheumatic conditions (Table 1), as well as in a number of nonrheumatic disease contexts (Table 2).6,29,30,38
Figure 3.

A causal diagram of a typical observational study showing the assessment of the effect of smoking on RA progression (or CVD complications) among patients with RA. Similarly to in Figures 1 and 2, we consider independent risk factors (specifically, smoking and URFs) that are associated with both RA and RA progression (or CVD). Note that URFs are not associated with smoking (as indicated by the absence of a line between the two factors) before individuals develop RA. Thus, URFs would not be a confounder in a study of smoking and RA progression (or CVD) in the general population. However, smoking and URFs are no longer independent (as indicated by a dotted line between them) following conditioning on a common effect (in this case, restriction of the study sample to patients with RA, as denoted by a box around RA). Consequently, a biased association occurs between smoking and RA progression (or CVD) (represented as smoking—URFs → RA progression [or CVD]). As the study design leads this spurious association with URFs to operate as a negative confounder, the resulting effect measure becomes underestimated or reversed (that is, paradoxical) unless the study appropriately adjusts for URFs. Abbreviations: CvD, cardiovascular disease; RA, rheumatoid arthritis; URFs, unknown or unmeasured risk factors.
The quantitative impact of index event bias has been reported by a simulation study using numerical examples that was published in 2013.40 The authors found that index event bias can be substantial, reducing the impact of a given risk factor for an incident event with a relative risk (RR) of 9.0 all the way down to a null effect for recurrent events.40 Using the same framework,40 we have simulated an example that reflects a rheumatic disease context for interested readers (see Supplementary Information). In this example, when comparing the impact of a given risk factor (such as smoking) on RA incidence versus progression, an RR of 2.5 for RA incidence becomes protective for RA progression (RR = 0.64). These simulation data provide a numerical display for index event bias as an explanation for the smoking paradox in the risk of RA progression (or CVD sequelae) in patients with RA, as also depicted in Figure 3.
Total effects of risk factors
Even beyond the index event bias issue discussed earlier, obtaining the total effect of a particular risk factor for a subsequent or sequelae event among individuals with index events can be challenging. As an example, we consider the total effect of smoking on the risk of CVD among patients with RA (Figure 4). To contrast our discussion of this issue with risk factor investigation (smoking in this case) in the general population, we have depicted the associations in a ‘general’ epidemiology context using a causal diagram in Figure 4a. To simplify our discussion, we assume that smoking increases the risk of CVD through two causal pathways relevant to RA: one pathway mediated through RA (that is, smoking increases the risk of RA, which in turn increases the risk of CVD —an indirect or mediated effect), and the other through a mechanism that does not involve RA (that is, smoking increases the risk of CVD—a direct effect). In this case, the total effect of smoking on the risk of CVD is the net causal effect through both pathways. In the general population, this total effect can be estimated using conventional confounding adjustment (indicated by the box around ‘confounders’ in Figure 4a), fulfilling the primary aim of such general-population studies.
Figure 4.
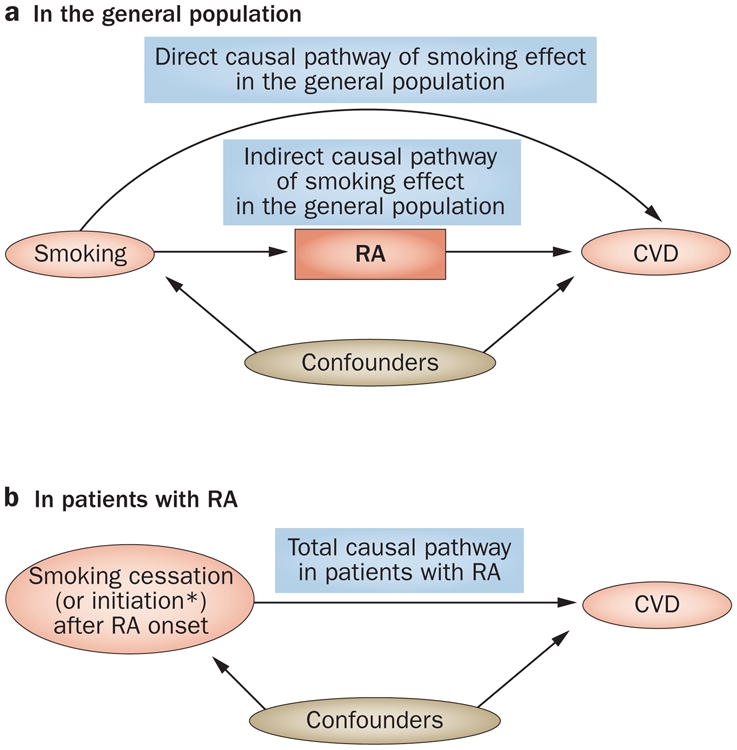
Causal diagrams displaying the effect of smoking on CVD a | A causal diagram displaying two causal pathways (direct and indirect) for the total effect of smoking on CVD complications in the general (unselected) population. The box around ‘confounders’ denotes adjustments. The total effect of smoking on the risk of CvD in this population is the net combined causal effect through both pathways. b | A causal diagram of the total causal effect of smoking on CVD complications among patients with RA (i.e. a restricted population). *Theoretically, smoking initiation after RA onset would be equivalent to smoking exposure in the general population in part a; however, in practice, this would be unusual after RA onset. Alternatively, the impact of smoking cessation can be evaluated in these studies. Abbreviations: CVD, cardiovascular disease; RA, rheumatoid arthritis.
By contrast, in research studies that evaluate the risk factors for sequelae events among individuals with an index event, even the correction of index event bias (as discussed) might not generate an estimate of the total effect of smoking on the risk of CVD among patients with RA, which is usually the intended aim of these studies (Figure 4b). This problem occurs because smoking status is often assessed before or at the time of RA onset, or it is mostly unchanged before and after RA onset, even if it was measured at or after the study baseline. Thus, although investigators think that they are evaluating the impact of smoking status on CVD among patients with RA, they are actually evaluating the effect of continued smoking since before the study baseline (and RA onset), reflecting the scenario depicted in Figure 3. In the end, most of these studies estimate the direct effect of smoking (not mediated through RA) in the general population, not the total effect of smoking among patients with RA as intended. As both the direct and indirect effects of smoking on the risk of CVD are expected to occur in the same direction, one would expect the direct effect of smoking to be smaller than its total effect measured in the general population for the risk of incident CVD, which has been the case in such studies.41,42,43
Differential loss to follow-up
Classical selection bias issues are often raised in the selection of controls for case–control studies. However, cohort studies, including randomized controlled trials (RCTs), are subject to the same type of selection bias as a result of differential loss to follow-up.
Impact on OA progression studies
This type of selection bias has been raised in the OA research context as another potential reason underlying the null associations between obesity and OA progression (as determined by radiography) in several well-established OA cohort studies.5 These studies have all used serial knee radiographs at multiple follow-up time points to document disease progression. However, in most of these studies, a substantial proportion of subjects were lost to follow-up, particularly when the time between the radiographic assessments was long. For example, the average follow-up time between repeated knee radiographs in the Framingham OA study was 9 years, and of 1,473 individuals with knee radiographs taken at baseline, 40% did not have knee radiographs at the follow-up visit.44 A similar proportion of loss to follow-up (40%) was also reported in a UK study.45 As obese individuals are less likely to complete follow-up owing to poor health, and patients whose OA has worsened are less likely to return for the last study visit owing to loss of mobility, this differential loss to follow-up can lead to a selection bias, ‘diluting’ the effect of obesity on OA progression.5 The comparison of baseline characteristics between individuals who completed the follow-up with those who did not, a frequently used approach, does not guarantee protection against selection bias, as the determinants for loss to follow-up might not be the same between the exposed and unexposed groups, and they might even share common causes with the disease outcomes.
Impact on RA therapy studies
Major loss to follow-up has also been observed in a series of pharmaco-epidemiological studies that investigated comparative safety profiles of anti-TNF DMARDs.46,47 Although these studies addressed the issue of confounding (by baseline covariates) by active comparator design or propensity score analyses, they experienced considerable loss to follow-up during a relatively short period of time (≤1 year), which is within the range of contemporary RA RCT durations.48–50 For example, as acknowledged by its accompanying editorial,51 a pharmaco-epidemiological study that investigated the risk of hospitalized infection among biologic agent users over a 1-year period showed exposure retention rates of 82% in the anti-TNF agent group and 60% in the nonbiologic DMARD comparison group at 60 days of follow-up (Figure 5a).46 These high and differential loss rates between groups continued during further follow-up: retention rates fell to 53% and 29%, respectively, by 180 days of follow-up (the study mid-point), and only 31% and 14% of participants were retained by 360 days (the end of the study) (Figure 5a).46 Despite effectively controlling for confounders to a level similar to that achieved in RCTs through state-of-the-art methods, such a high level of differential loss to follow-up threatens the embedded assumption that loss to follow-up is completely random (that is, not associated with an outcome, or mediators of an outcome), leaving the study design open to potential selection bias. If patients tend to discontinue TNF-inhibitor treatment following a low-grade infection (for example, an infection not requiring hospitalization), the patients remaining on TNF inhibitors would have a lower rate of hospitalized infection.51 However, relatively insufficient efficacy of the nonbiologic DMARD agents could have led to glucocorticoid use, which would have contributed to both higher rates of infection and discontinuation of the corresponding drug (thus resulting in loss to follow-up). Unfortunately, no data are provided on the reasons for this high rate of loss to follow-up,46 and it remains impossible to determine the direction or extent of this potential bias.51
Figure 5.
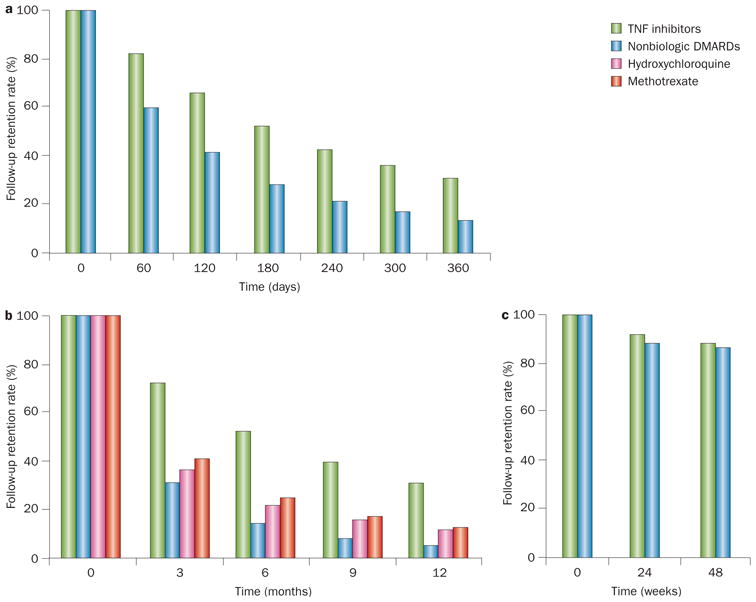
Differential loss to follow-up in studies of RA therapy. a,b | Two observational pharmaco-epidemiological studies46,47 showed high and differential loss rates between groups. c | By contrast, much lower levels of loss to follow-up were observed in a randomized trial46 of a biologic agent in RA at similar time points. Despite effectively controlling for confounders in the observational studies,46,47 such a high level of differential loss to follow-up threatens the embedded assumption that loss to follow-up is completely random (i.e. not associated with an outcome, or mediators of an outcome), leaving the study design open to potential selection bias. Abbreviation: RA, rheumatoid arthritis.
A similarly designed pharmaco-epidemiology study that investigated the risk of type-2 diabetes also showed a large difference in the follow-up rate between the anti-TNF agent group and other DMARD groups47 as depicted in Figure 5b. Interestingly, this particular study conducted an intent-to-treat (ITT) analysis (typically used in RCTs) up to a half of their follow-up period. Although this approach might have helped to estimate the effects of the baseline exposures without potential post-baseline confounding,52 this method would not guard against potential selection bias owing to differential loss to follow-up.52 Notably, the first pharmaco-epidemiology study showed no increased risk of hospitalized infection associated with anti-TNF agents,46 which conflicts with the findings of a previous meta-analysis of randomized trials53 and a recently conducted RCT published in 2013 (Figure 5c displays the high follow-up retention rate of this RCT over 48 weeks).48 We are not aware of any RCT data on the impact of biologic DMARDs on the risk of type-2 diabetes to cross-validate the results of the latter pharmaco-epidemiology study.47 Of note, a summary of the US National Research Council guidelines for missing values (including loss to follow-up rates) stated the following as part of their key findings: “Substantial instances of missing data are a serious problem that undermines the scientific credibility of causal conclusions from clinical trials. The assumption that analysis methods can compensate for such missing data are not justified, so aspects of trial design that limit the likelihood of missing data should be an important objective.”54 This conclusion emphasizes the importance of minimizing loss to follow-up in design and execution of clinical trials, as well as the associated difficulties of dealing with missing data at the analysis phase.54 Furthermore, in an editorial published in 2013, Hernan and colleagues outlined the limitations of the widely popular ITT approach, even in randomized trials (particularly for safety trials), and suggested advanced methods to overcome these limitations.52 These principles should also be applicable to observational studies, as they are clearly not immune to this type of bias; if anything, observational studies ought to be subject to a higher level of scrutiny for any type of bias.
Depletion of susceptibles
Differential depletion of susceptible participants can bias effect estimates of hazardous exposures towards the null or in a protective direction owing to attrition of study participants based on their susceptibility to the exposure. A classical example is the age-stratified effects of smoking on coronary heart disease mortality in the British Physician's study (n = 34,439).55 With ageing, participants who were susceptible to coronary heart disease (particularly that related to smoking) tended to die of smoking-related causes, and only those who were less susceptible remained. Thus, both the RRs and the risk differences for coronary heart disease in that study showed a graded decline with ageing from the youngest age category (35–44 years), and smoking became protective in the oldest category of 75–84 years (RRs 5.7 and 0.9, respectively).55
Similar phenomena have been observed in rheumatic disease contexts. For example, downward trends in the associations between obesity and mortality among patients with RA have been reported, ranging from an RR of 1.6 (positive) for those <50 years of age to 0.9 (negative) for those >70 years of age.56 A simple methodological explanation is that those who were most susceptible to obesity-related complications died before the age at which they would have been eligible for study enrolment.57 Furthermore, the same differential depletion of susceptible individuals between compared groups can explain the decreasing RR of complications (such as venous thrombotic embolism) in inflammatory rheumatic conditions over time following the onset of the underlying inflammatory conditions.58,59 This phenomenon highlights the superiority of using incident exposures (as opposed to prevalent exposures), particularly in a setting in which the induction time (the time between RA onset and the incidence of venous thrombotic embolism) is short. Nevertheless, when the induction time associated with chronic exposures is relatively long (for example, in the case of the impact of smoking on the risk of lung cancer), this selection bias manifests after the peak effect age of the exposure, thus primarily among elderly individuals, as described earlier.55,57
The most notable example is that of the impact of hormonal replacement therapy (HRT) on CVD among postmenopausal women. Prominent observational studies that investigated the effects of a mixture of both prevalent and incident use of HRT have shown a 40–50% protective effect against the risk of myocardial infarction.60,61 However, a large subsequent RCT (the Women's Health initiative [n = 16,608]) showed hazardous effects, particularly during the early stages of HRT use.62 When the same previous observational study was analysed by emulating an RCT study design (using incident exposure of HRT and an ITT-equivalent approach), the results converged with those of the RCT.63 The lesson remains that selection bias can be ubiquitous and could, therefore, potentially lead the field astray.
Immortal time bias
Immortal time bias occurs owing to periods of follow-up time that are protected from deaths or end points by virtue of the study design.64 Such bias can either lead to misclassification bias through the incorrect classification of unexposed ‘immortal time’ (before initial exposure) as part of the follow-up period of the exposed group or result in selection bias when periods of immortal time are differentially excluded from the analysis.64 Both types of bias lead to a downward bias of effect estimates (that is, towards the null or in a protective direction). This differential exclusion can occur from the use of a hierarchical approach to determination of treatment status or a predetermined prescription pattern of compared treatment options in the study population.64 For example, in a study of the potential survival impact of biologic agents in patients with RA (compared with nonbiologic agents), the start of follow-up (defined as the date of the first use of the biologic agent or nonbiologic therapy) would come considerably later following RA diagnosis for biologic agent users than for nonbiologic agent users (the comparator), because a large proportion of biologic agent users (if not all, as in some countries or regions) had been previously treated with nonbiologic agents (and survived these previous treatments) (Figure 6). Thus, unless the excluded period of nonbiologic agent use before biologic agent use (the unexposed immortal time) is appropriately assigned to the nonbiologic treatment group in a time-varying manner,64 the immortal time-induced selection bias could lead to a major survival advantage for biologic agent users.
Figure 6.
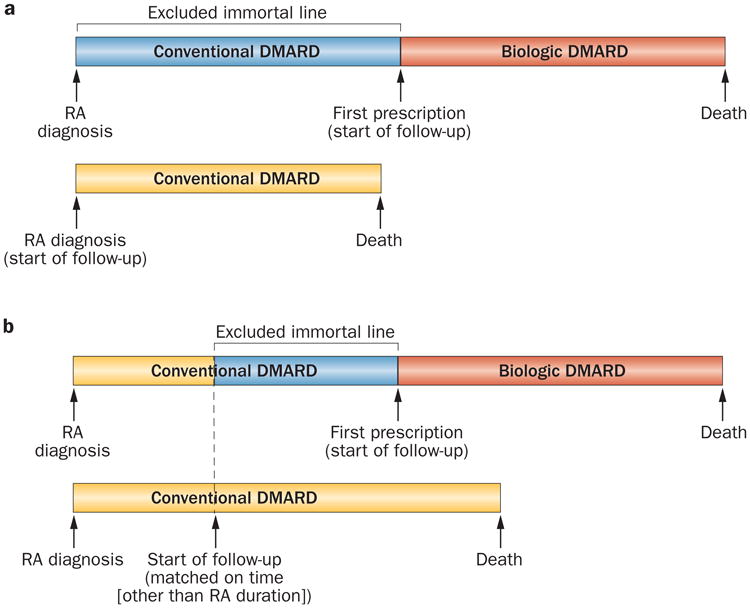
Immortal time bias as a form of selection bias. Immortal time bias is introduced as a form of selection bias in cohort studies when a period of ‘immortal time’ is excluded from the analysis. This exclusion occurs because the start of follow-up for the group receiving treatment (a biologic DMARD in this example) is defined by the start of treatment and is, by design (or by practice pattern), later than that for the comparison group (receiving a conventional DMARD). a | A depiction of the comparison group's follow-up starting at the time of RA diagnosis. b | A depiction of the comparison group's follow-up starting sometime after RA diagnosis (matched on certain time factors other than RA duration), but before biologic DMARD use. In both cases, unless the excluded period of nonbiologic agent use before biologic agent use (i.e. the unexposed immortal time) is appropriately assigned to the nonbiologic group in a time-varying manner,64 the immortal time-induced selection bias could lead to a major survival advantage for biologic agent users. Abbreviation: RA, rheumatoid arthritis.
Conclusions
Unbiased research that accurately determines modifiable risk factors for the development of rheumatic and musculo skeletal conditions and their sequelae is critical to reduce their disease burden. As reviewed here, the evidence on risk factors for disease sequelae among patients with rheumatic diseases has been inconsistent or paradoxical, unlike the research findings on risk factors for incident conditions. Beyond potential biological explanations for these counterintuitive findings, an enticing methodological explanation could be a type of selection bias known as index event bias, which can affect research on disease sequelae (as shown in many nonrheumatic conditions). Furthermore, mismatches between the study question of interest and study design could have led to the many apparent paradoxes in the field because many of these studies have not investigated the total impact of those risk factors that occur after the index events. Many powerful confounding control methods (such as propensity score methods or active comparator analysis) have been effective in pharmaco-epidemiological research in rheumatic conditions; however, they do not address selection bias caused by potential differential loss to follow-up. Depletion of susceptibles (another type of selection bias) can explain the decreasing impact of risk factors on mortality with ageing in many rheumatic conditions, including RA, and the null (or inverse) associations of prevalent exposure studies. Immortal time bias as a form of selection bias (as opposed to as a type of misclassification bias) can also create strong spurious inverse associations (for example, resulting in an apparent highly protective effect of a particular drug).
The key lesson from these experiences is that selection bias can be ubiquitous, and holds the potential to mislead the field. Therefore, researchers and clinicians should pay attention to this issue in all phases of research, including its design, execution and interpretation. Several practical measures can be taken. First, when planning a research study, a well-formed and testable hypothesis should be developed that considers potential selection bias issues (at least to the same extent as confounding and measurement bias issues) from the start. Study design tends to be more important than analytical choices in avoiding selection bias, and involving methodologists with relevant expertise from the beginning of the study design process can help greatly to avoid these biases. Second, the specific intent of the research question and its corresponding effect measures (for example, total, direct, or indirect effects) should be clearly determined to avoid mismatches between the two. We find constructing plausible causal diagrams (as in Figures 1–4) to be helpful, particularly in facilitating explicit display and discussion of causal questions and the potential role of involved variables. Third, an appropriate study design and analytical method should be used to fit the specific research question that was intended. This measure is crucial when assessing the direct and mediated effects of risk factors, as potential selection bias (for example, index event bias) is likely to occur. Fourth, in general, use of incident exposure (as opposed to prevalent exposure) helps to avoid depletion of susceptibles, particularly if outcomes occur early after the initiation of exposure and cumulative exposure is less relevant (although chronic exposures, such as obesity and smoking, are less or not subject to these recommendations). This approach further helps to avoid selection bias by ensuring that the exposure precedes the occurrence of any mediators and the outcome, and that any confounders precede the occurrence of the exposure. Fifth, more studies about the natural history of rheumatic conditions and their sequelae are needed to clarify the time sequence of relevant biological stages and to avoid adjusting for causal mediators. With such knowledge often incomplete or unavailable, investigators are encouraged to perform sensitivity analyses under various causal assumptions, to assess the impact of such assumptions on their results. Finally, more methodological research on selection bias issues specifically in rheumatic conditions would help to refine these recommendations towards more comprehensive and practical guidelines, as well as help investigators avoid the pitfalls posed by this crucial type of bias and ensure the validity of future rheumatology research.
Supplementary Material
Key points.
Unlike research findings on risk factors for incident conditions, the evidence on risk factors for disease sequelae among patients with rheumatic diseases have often been inconsistent or paradoxical
Although biological explanations for these counterintuitive results might exist, an enticing methodological explanation is a type of selection bias called index event bias, which can affect research on disease sequelae
Propensity score methods or active comparator analysis in pharmaco-epidemiological research helps to address confounding issues in observational studies, but does not address selection bias owing to potential differential loss to follow-up
The depletion of susceptibles can explain the decreasing impact of risk factors on mortality with ageing in rheumatic conditions, as well as explain the null (or inverse) associations of prevalent exposure studies
To avoid these issues, investigators should carefully specify the research question of interest and clarify the time sequence of exposures, mediators, and outcome variables
Furthermore, investigators should use incident exposures whenever possible, minimize loss to follow-up, and exercise proper inference
Review criteria.
A search for original articles published between 1965 and 2013 and focusing on the risk factors for key rheumatic conditions as well as those for their sequela conditions was performed in MEDLINE and PubMed. The search terms included “osteoarthritis”, “rheumatoid arthritis”, “psoriasis”, “psoriatic arthritis”, “risk factors”, “smoking”, “obesity”, “gene”, “genetic”, “loci”, “progression”, “myocardial infarction”, “cardiovascular outcomes”, and “death” alone and in combination. A few selected key reviews and seminal papers were also included. Papers cited in this Review were selected according to their relevance to the subject.
Footnotes
Competing interests: The authors declare no competing interests.
Author contributions All authors contributed equally to researching the data for the article, discussions of the content, writing the article and editing of the manuscript before submission.
Supplementary information is linked to the online version of this paper at www.nature.com/nrrheum.
References
- 1.Reginster JY. The prevalence and burden of arthritis. Rheumatology (Oxford) 2002;41(Suppl. 1):3–6. [PubMed] [Google Scholar]
- 2.Symmons DP, Gabriel SE. Epidemiology of CVD in rheumatic disease, with a focus on RA and SLE. Nat Rev Rheumatol. 2011;7:399–408. doi: 10.1038/nrrheum.2011.75. [DOI] [PubMed] [Google Scholar]
- 3.Gabriel SE. Heart disease and rheumatoid arthritis: understanding the risks. Ann Rheum Dis. 2010;69(Suppl. 1):i61–i64. doi: 10.1136/ard.2009.119404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eder L, et al. The association between smoking and the development of psoriatic arthritis among psoriasis patients. Ann Rheum Dis. 2012;71:219–224. doi: 10.1136/ard.2010.147793. [DOI] [PubMed] [Google Scholar]
- 5.Zhang Y, et al. Methodologic challenges in studying risk factors for progression of knee osteoarthritis. Arthritis Care Res (Hoboken) 2010;62:1527–1532. doi: 10.1002/acr.20287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Canto JG, et al. Number of coronary heart disease risk factors and mortality in patients with first myocardial infarction. JAMA. 2011;306:2120–2127. doi: 10.1001/jama.2011.1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang Y, Jordan JM. Epidemiology of osteoarthritis. Rheum Dis Clin North Am. 2008;34:515–529. doi: 10.1016/j.rdc.2008.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Felson DT, et al. Osteoarthritis: new insights. Part 1: the disease and its risk factors. Ann Intern Med. 2000;133:635–646. doi: 10.7326/0003-4819-133-8-200010170-00016. [DOI] [PubMed] [Google Scholar]
- 9.Belo JN, Berger MY, Reijman M, Koes BW, Bierma-Zeinstra SM. Prognostic factors of progression of osteoarthritis of the knee: a systematic review of observational studies. Arthritis Rheum. 2007;57:13–26. doi: 10.1002/art.22475. [DOI] [PubMed] [Google Scholar]
- 10.Zhang Y, et al. Bone mineral density and risk of incident and progressive radiographic knee osteoarthritis in women: the Framingham Study. J Rheumatol. 2000;27:1032–1037. [PubMed] [Google Scholar]
- 11.Hart DJ, et al. The relationship of bone density and fracture to incident and progressive radiographic osteoarthritis of the knee: the Chingford Study. Arthritis Rheum. 2002;46:92–99. doi: 10.1002/1529-0131(200201)46:1<92::AID-ART10057>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 12.Lane NE et al. Wnt signaling antagonists are potential prognostic biomarkers for the progression of radiographic hip osteoarthritis in elderly Caucasian women. Arthritis Rheum. 2007;56:3319–3325. doi: 10.1002/art.22867. [DOI] [PubMed] [Google Scholar]
- 13.McAlindon TE, et al. Do antioxidant micronutrients protect against the development and progression of knee osteoarthritis? Arthritis Rheum. 1996;39:648–656. doi: 10.1002/art.1780390417. [DOI] [PubMed] [Google Scholar]
- 14.Vesperini V, et al. Tobacco exposure reduces radiographic progression in early rheumatoid arthritis. Results from the ESPOIR cohort. Arthritis Care Res (Hoboken) 2013;65:1899–1906. doi: 10.1002/acr.22057. [DOI] [PubMed] [Google Scholar]
- 15.Harrison BJ, Silman AJ, Wiles NJ, Scott DG, Symmons DP. The association of cigarette smoking with disease outcome in patients with early inflammatory polyarthritis. Arthritis Rheum. 2001;44:323–330. doi: 10.1002/1529-0131(200102)44:2<323::AID-ANR49>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 16.Finckh A, Dehler S, Costenbader KH, Gabay C. Cigarette smoking and radiographic progression in rheumatoid arthritis. Ann Rheum Dis. 2007;66:1066–1071. doi: 10.1136/ard.2006.065060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gonzalez A, et al. Do cardiovascular risk factors confer the same risk for cardiovascular outcomes in rheumatoid arthritis patients as in non-rheumatoid arthritis patients? Ann Rheum Dis. 2008;67:64–69. doi: 10.1136/ard.2006.059980. [DOI] [PubMed] [Google Scholar]
- 18.Naranjo A, et al. Cardiovascular disease in patients with rheumatoid arthritis: results from the QUEST-RA study. Arthritis Res Ther. 2008;10:R30. doi: 10.1186/ar2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Manson JE, et al. A prospective study of obesity and risk of coronary heart disease in women. N Engl J Med. 1990;322:882–889. doi: 10.1056/NEJM199003293221303. [DOI] [PubMed] [Google Scholar]
- 20.Escalante A, Haas RW, del Rincon I. Paradoxical effect of body mass index on survival in rheumatoid arthritis: role of comorbidity and systemic inflammation. Arch Intern Med. 2005;165:1624–1629. doi: 10.1001/archinte.165.14.1624. [DOI] [PubMed] [Google Scholar]
- 21.Wilson PW, et al. Prediction of coronary heart disease using risk factor categories. Circulation. 1998;97:1837–1847. doi: 10.1161/01.cir.97.18.1837. [DOI] [PubMed] [Google Scholar]
- 22.Myasoedova E, et al. Lipid paradox in rheumatoid arthritis: the impact of serum lipid measures and systemic inflammation on the risk of cardiovascular disease. Ann Rheum Dis. 2011;70:482–487. doi: 10.1136/ard.2010.135871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Peters MJ, et al. EULAR evidence-based recommendations for cardiovascular risk management in patients with rheumatoid arthritis and other forms of inflammatory arthritis. Ann Rheum Dis. 2009;69:325–331. doi: 10.1136/ard.2009.113696. [DOI] [PubMed] [Google Scholar]
- 24.Solomon DH, Peters MJ, Nurmohamed MT, Dixon W. Unresolved questions in rheumatology: motion for debate: the data support evidence-based management recommendations for cardiovascular disease in rheumatoid arthritis. Arthritis Rheum. 2013;65:1675–1683. doi: 10.1002/art.37975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li W, Han J, Qureshi AA. Smoking and risk of incident psoriatic arthritis in US women. Ann Rheum Dis. 2011;71:804–808. doi: 10.1136/annrheumdis-2011-200416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bowcock AM, Cookson WO. The genetics of psoriasis, psoriatic arthritis and atopic dermatitis. Hum Mol Genet. 2004;13(Suppl. 1):R43–R55. doi: 10.1093/hmg/ddh094. [DOI] [PubMed] [Google Scholar]
- 27.Duffin KC, et al. Genetics of psoriasis and psoriatic arthritis: update and future direction. J Rheumatol. 2008;35:1449–1453. [PMC free article] [PubMed] [Google Scholar]
- 28.Aune E, Roislien J, Mathisen M, Thelle DS, Otterstad JE. The “smoker's paradox” in patients with acute coronary syndrome: a systematic review. BMC Med. 2011;9:97. doi: 10.1186/1741-7015-9-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Romero-Corral A, et al. Association of bodyweight with total mortality and with cardiovascular events in coronary artery disease: a systematic review of cohort studies. Lancet. 2006;368:666–678. doi: 10.1016/S0140-6736(06)69251-9. [DOI] [PubMed] [Google Scholar]
- 30.Lavie CJ, De Schutter A, Patel D, Artham SM, Milani RV. Body composition and coronary heart disease mortality—an obesity or a lean paradox? Mayo Clin Proc. 2011;86:857–864. doi: 10.4065/mcp.2011.0092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dahabreh IJ, Kent DM. Index event bias as an explanation for the paradoxes of recurrence risk research. JAMA. 2011;305:822–823. doi: 10.1001/jama.2011.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kent DM, Thaler DE. Is patent foramen ovale a modifiable risk factor for stroke recurrence? Stroke. 2010;41:S26–S30. doi: 10.1161/STROKEAHA.110.595140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tyas SL, et al. Transitions to mild cognitive impairments, dementia, and death: findings from the Nun Study. Am J Epidemiol. 2007;165:1231–1238. doi: 10.1093/aje/kwm085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Glymour MM. Invited commentary: when bad genes look good—APOE*E4, cognitive decline, and diagnostic thresholds. Am J Epidemiol. 2007;165:1239–1246. doi: 10.1093/aje/kwm092. author reply 1247. [DOI] [PubMed] [Google Scholar]
- 35.Baglin T. Unraveling the thrombophilia paradox: from hypercoagulability to the prothrombotic state. J Thromb Haemost. 2010;8:228–233. doi: 10.1111/j.1538-7836.2009.03702.x. [DOI] [PubMed] [Google Scholar]
- 36.Hernandez-Diaz S, Schisterman EF, Hernan MA. The birth weight “paradox” uncovered? Am J Epidemiol. 2006;164:1115–1120. doi: 10.1093/aje/kwj275. [DOI] [PubMed] [Google Scholar]
- 37.Myers J, et al. The obesity paradox and weight loss. Am J Med. 2011;124:924–930. doi: 10.1016/j.amjmed.2011.04.018. [DOI] [PubMed] [Google Scholar]
- 38.VanderWeele TJ, Mumford SL, Schisterman EF. Conditioning on intermediates in perinatal epidemiology. Epidemiology. 2011;23:1–9. doi: 10.1097/EDE.0b013e31823aca5d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.VanderWeele TJ, Robins JM. Directed acyclic graphs, sufficient causes, and the properties of conditioning on a common effect. Am J Epidemiol. 2007;166:1096–1104. doi: 10.1093/aje/kwm179. [DOI] [PubMed] [Google Scholar]
- 40.Smits LJ, et al. Index event bias-a numerical example. J Clin Epidemiol. 2013;66:192–196. doi: 10.1016/j.jclinepi.2012.06.023. [DOI] [PubMed] [Google Scholar]
- 41.Westreich D, Greenland S. The Table 2 fallacy: presenting and interpreting confounder and modifier coefficients. Am J Epidemiol. 2013;177:292–298. doi: 10.1093/aje/kws412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Valeri L, Vanderweele TJ. Mediation analysis allowing for exposure-mediator interactions and causal interpretation: theoretical assumptions and implementation with SAS and SPSS macros. Psychol Methods. 2013;18:137–150. doi: 10.1037/a0031034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang Y, Neogi T, Hunter D et al. What effect is really being measured? An alternative explanation of paradoxical phenomenon in studies of osteoarthritis progression. Arthritis Care & Res (Hoboken) doi: 10.1002/acr.22213. http://dx.doi.org/10.1002/acr.22213. [DOI] [PMC free article] [PubMed]
- 44.Felson DT, et al. Risk factors for incident radiographic knee osteoarthritis in the elderly: the Framingham Study. Arthritis Rheum. 1997;40:728–733. doi: 10.1002/art.1780400420. [DOI] [PubMed] [Google Scholar]
- 45.Cooper C, et al. Risk factors for the incidence and progression of radiographic knee osteoarthritis. Arthritis Rheum. 2000;43:995–1000. doi: 10.1002/1529-0131(200005)43:5<995::AID-ANR6>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 46.Grijalva CG, et al. Initiation of tumor necrosis factor-alpha antagonists and the risk of hospitalization for infection in patients with autoimmune diseases. JAMA. 2011;306:2331–2339. doi: 10.1001/jama.2011.1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Solomon DH, et al. Association between disease-modifying antirheumatic drugs and diabetes risk in patients with rheumatoid arthritis and psoriasis. JAMA. 2011;305:2525–2531. doi: 10.1001/jama.2011.878. [DOI] [PubMed] [Google Scholar]
- 48.O'Dell JR, et al. Therapies for active rheumatoid arthritis after methotrexate failure. N Engl J Med. 2013;369:307–318. doi: 10.1056/NEJMoa1303006. [DOI] [PubMed] [Google Scholar]
- 49.O'Dell JR, et al. Treatment of rheumatoid arthritis with methotrexate and hydroxychloroquine, methotrexate and sulfasalazine, or a combination of the three medications: results of a two-year, randomized, double-blind, placebo-controlled trial. Arthritis Rheum. 2002;46:1164–1170. doi: 10.1002/art.10228. [DOI] [PubMed] [Google Scholar]
- 50.O'Dell JR, et al. Treatment of rheumatoid arthritis with methotrexate alone, sulfasalazine and hydroxychloroquine, or a combination of all three medications. N Engl J Med. 1996;334:1287–1291. doi: 10.1056/NEJM199605163342002. [DOI] [PubMed] [Google Scholar]
- 51.Dixon W, Felson DT. Is anti-TNF therapy safer than previously thought? JAMA. 2011;306:2380–2381. doi: 10.1001/jama.2011.1705. [DOI] [PubMed] [Google Scholar]
- 52.Hernan MA, Hernandez-Diaz S, Robins JM. Randomized trials analyzed as observational studies. Ann Intern Med. doi: 10.7326/0003-4819-159-8-201310150-00709. http://dx.doi.org/10.7326/0003-4819-159-8-201310150-00709. [DOI] [PMC free article] [PubMed]
- 53.Bongartz T, et al. Anti-TNF antibody therapy in rheumatoid arthritis and the risk of serious infections and malignancies: systematic review and meta-analysis of rare harmful effects in randomized controlled trials. JAMA. 2006;295:2275–2285. doi: 10.1001/jama.295.19.2275. [DOI] [PubMed] [Google Scholar]
- 54.Little RJ, et al. The prevention and treatment of missing data in clinical trials. N Engl J Med. 2012;367:1355–1360. doi: 10.1056/NEJMsr1203730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Doll R, Hill AB. Mortality of British doctors in relation to smoking: observations on coronary thrombosis. Natl Cancer Inst Monogr. 1966;19:205–268. [PubMed] [Google Scholar]
- 56.Wolfe F, Michaud K. Effect of body mass index on mortality and clinical status in rheumatoid arthritis. Arthritis Care Res (Hoboken) 2012;64:1471–1479. doi: 10.1002/acr.21627. [DOI] [PubMed] [Google Scholar]
- 57.Nguyen US, Niu J, Choi HK, Zhang Y. Body mass index and mortality: comment on article by Wolfe and Michaud. Arthritis Care Res (Hoboken) 2013;65:834–835. doi: 10.1002/acr.21910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Choi HK, et al. The risk of pulmonary embolism and deep vein thrombosis in rheumatoid arthritis: a UK population-based outpatient cohort study. Ann Rheum Dis. 2013;72:1182–1187. doi: 10.1136/annrheumdis-2012-201669. [DOI] [PubMed] [Google Scholar]
- 59.Zoller B, Li X, Sundquist J, Sundquist K. Risk of pulmonary embolism in patients with autoimmune disorders: a nationwide follow-up study from Sweden. Lancet. 2012;379:244–249. doi: 10.1016/S0140-6736(11)61306-8. [DOI] [PubMed] [Google Scholar]
- 60.Grodstein F, Stampfer M. The epidemiology of coronary heart disease and estrogen replacement in postmenopausal women. Prog Cardiovasc Dis. 1995;38:199–210. doi: 10.1016/s0033-0620(95)80012-3. [DOI] [PubMed] [Google Scholar]
- 61.Grady D, Rubin SM, Petitti DB, et al. Hormone therapy to prevent disease and prolong life in postmenopausal women. Ann Intern Med. 1992;117:1016–1037. doi: 10.7326/0003-4819-117-12-1016. [DOI] [PubMed] [Google Scholar]
- 62.Manson JE, et al. Estrogen plus progestin and the risk of coronary heart disease. N Engl J Med. 2003;349:523–534. doi: 10.1056/NEJMoa030808. [DOI] [PubMed] [Google Scholar]
- 63.Hernan MA, et al. Observational studies analyzed like randomized experiments: an application to postmenopausal hormone therapy and coronary heart disease. Epidemiology. 2008;19:766–779. doi: 10.1097/EDE.0b013e3181875e61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Levesque LE, Hanley JA, Kezouh A, Suissa S. Problem of immortal time bias in cohort studies: example using statins for preventing progression of diabetes. BMJ. 2010;340:b5087. doi: 10.1136/bmj.b5087. [DOI] [PubMed] [Google Scholar]
- 65.Tsai CL, Camargo CA., Jr Methodological considerations, such as directed acyclic graphs, for studying “acute on chronic” disease epidemiology: chronic obstructive pulmonary disease example. J Clin Epidemiol. 2009;62:982–990. doi: 10.1016/j.jclinepi.2008.10.005. [DOI] [PubMed] [Google Scholar]
- 66.Rich JD, et al. Prior aspirin use and outcomes in acute coronary syndromes. J Am Coll Cardiol. 2010;56:1376–1385. doi: 10.1016/j.jacc.2010.06.028. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.