

Abstract
Arginine methylation has emerged as a widespread post-translational modification with influence over myriad cellular processes. However, the molecular mechanisms underlying such methylarginine-dependent phenomena remain unclear. To aid in this research, a facile method was developed to install methylarginine analogues on recombinant protein for use in biochemical, biophysical, and structural studies. Through chemical conjugation of novel α,β-unsaturated amidine precursors with proteins, methylarginine mimics can be displayed with control of methylation site, extent, and regiospecificity. Analogue installation into histones using this strategy produced modified proteins that were recognized by antibodies specific to endogenous methylarginine, and these histones retained the capacity to form mononucleosomes. Moreover, a native methylarginine-specific binding domain was shown to interact with methylarginine analogue-modified substrates. This chemical conjugation method for installing methylarginine analogues provides an efficient route to produce homogeneous modified proteins for subsequent investigations of methylarginine-dependent processes.
A large and expanding group of proteins, with diverse cellular functions, are known to harbor post-translational arginine methylation.1 Because the guanidino group of arginine can participate in polydentate ionic and hydrogen bonding interactions, arginine serves as a critical component of certain protein–protein2,3 and protein-nucleic acid4 interfaces. Arginine-mediated interactions are modulated by methylation, which exists in three major physiological forms: ω-N-methylarginine (monomethylarginine, MMA), ω-N,N-dimethylarginine (asymmetric dimethylarginine, ADMA), and ω-N,N′-dimethylarginine (symmetric dimethylarginine, SDMA).1 In vivo, arginine methylation is installed by protein arginine methyltransferase (PRMT) enzymes. The PRMT paralogues are classified as either Type I or Type II based on product selectivity: Type I PRMTs produce ADMA, Type II PRMTs produce SDMA, and both classes produce MMA. For example, the Type I enzyme PRMT1 methylates histone H4 at residue R3 to yield both H4R3Me1 (monomethyl) and H4R3Me2a (dimethyl, asymmetric). Likewise, the Type II paralogue PRMT5 deposits H4R3Me1 and H4R3Me2s (dimethyl, symmetric). Ultimately, these marks interact with cellular machinery to elicit a physiological output.5,6
While arginine methylation is implicated in many cellular processes—such as transcriptional regulation,7 DNA repair8 and RNA trafficking9—there remains much to be elucidated about the underlying biochemical mechanisms. For example, methylarginine-specific binding modules are present among diverse classes of proteins;2 yet, in many cases, the biological significance of methylarginine-dependent interactions is unclear. At the protein level, methylarginine-specific association can affect localization,9 complex participation,10 and may also influence catalytic function.6 Therefore, methylated substrate proteins are necessary reagents for the study of methylarginine-dependent activity. However, current methods to access homogeneous recombinant protein with prescribed site, extent and regiospecificity of arginine methylation suffer from technical challenges that include demanding biochemical manipulations11 and limited control of methylation status.12 Presented here is a facile method to install methylarginine analogues on recombinant protein with defined methylation status.
Akin to the chemical conjugation strategies employed for methyl-,13 acetyl-,14,15 and ubiquityl-lysine analogues;16 the site of methylarginine analogue installation is directed by chemo-selective reaction with a mutant cysteine residue. As such, methylarginine analogue conjugation is ideal for modifying proteins that lack cysteine residues, other than at the site of analogue installation. Methylarginine analogue precursors are based on a novel α,β-unsaturated amidine scaffold with various degrees of N-methylation, which reacts with cysteinyl thiols via conjugate addition (Scheme 1). Once appended to protein, the amidine moiety serves as a guanidino group side chain mimic with shape and pKa (acetamidine pKa(DMSO) = 27.1, guanidine pKa(DMSO) = 28.5)17 similar to native arginine—features shared by amidine-based inhibitors for arginine-specific proteases,18 nitric oxide synthases,19 dimethylarginine dimethylamino-hydrolases,19 arginine deiminases,20 and PRMTs.21 Unlike the guanidino side chain of arginine, amidine analogues possess an ε-methylene group in place of nitrogen that introduces nonplanar geometry and precludes polar interactions at this atom. However, structural studies suggest that at least a subset of methylarginine-dependent interactions is primarily mediated via the terminal nitrogen atoms (Figure S1), which are conserved between the guanidino and amidino side chains.
Scheme 1.
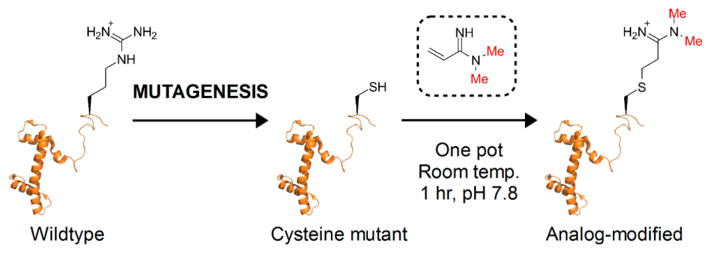
Chemical Conjugation of Methylarginine Analogues with Recombinant Histone
Analogue precursor compounds 1–4 (Figure 1A) were synthesized with predefined methylation extent and regio-specificity at the amidine group to mimic various forms of endogenous methylarginine. Precursor 1 was prepared by addition of methylchloroaluminum amide to acrylonitrile followed by hydrolysis.22 The MMA and ADMA analogue precursors (compounds 2 and 3, respectively) were produced by aminolysis of ethyl 3-hydroxypropionimidate hydrochloride (5), prepared by the Pinner reaction (Scheme 2A). Subsequently, chlorination of the hydroxyl group allowed for base-promoted β-elimination to yield the desired α,β-unsaturated amidines. Access to the symmetric amidine precursor 4 was achieved via methylamine addition to an activated N-methylamide obtained by reaction of 3-hydroxy-N-methylpropanamide tert-butyldimethylsilyl ether (10) with triethyloxonium tetrafluoroborate (Scheme 2B). Deprotection, halogenation, and elimination yielded the target unsaturated symmetric amidine 4.
Figure 1.

Site-specific and regiospecific installation of arginine analogues into histones. (A) Alkylation of histone mutants with analogue precursors 1–4. Observed m/z values for analogue-modified histones are in agreement with expected values. (B) Deconvoluted intact mass (inset) and ETD mass spectrum of H4R3C-ADMAA yielding product ions consistent with site-specific modification.
Scheme 2. Preparation of Analogue Precursorsa.
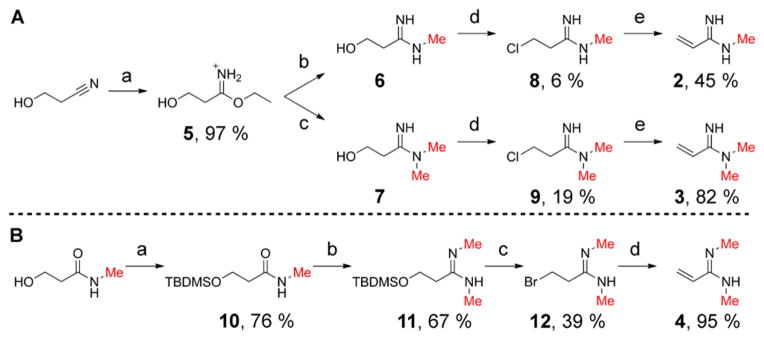
a(A) Synthesis of monomethyl and asymmetric dimethyl analogue precursors: (a) HCl, EtOH, Et2O, 3 h; (b) MeNH2, MeOH, 19 h; (c) Me2NH, MeOH, 16 h; (d) PPh3, CCl4, DMF, 22–24 h; (e) triethylamine, acetonitrile, 1–5 min. (B) Synthesis of symmetric dimethyl analogue precursor (a) TBDMSCl, imidazole, DMF, 24 h; (b) i. Et3OBF4, DCM, 1 h; ii. MeNH2, DCM/THF, 1 h; (c) PPh3Br2, DCM, 20 h; (d) triethylamine, acetonitrile, 1 min. All reactions were performed at room temperature.
The analogue precursors were reacted with recombinant Xenopus histones H3 and H4 containing cysteine substitutions: H3R2C (possesses an additional C110A mutation that is functionally akin to the wild-type protein13) and H4R3C, respectively. The reactions were performed at room temperature under denaturing and reducing conditions for ~1 h. The following analogue-modified histone H4 derivatives were produced by reaction with precursors 1–4, respectively (Figure 1A): H4R3C-AA (arginine analogue), H4R3C-MMAA (mono-methyl arginine analogue), H4R3C-ADMAA (asymmetric dimethyl arginine analogue), and H4R3C-SDMAA (symmetric dimethyl arginine analogue). Likewise, histone H3R2C was reacted with 1 and 3 to yield H3R2C-AA and H3R2C-ADMAA, respectively. Formation of the analogue-modified histones was monitored by ESI-MS, and observed m/z values were in accordance with those expected (Figure 1A). Mass spectrometric analysis of alkylation reactions showed complete loss of starting protein masses concomitant with the emergence of corresponding alkylated product ions, which indicates highly efficient analogue conjugation (Figure S2). Analysis of intact H4R3C-ADMAA by tandem mass spectrometry (MS/MS) confirmed site-specific installation of the methylarginine analogue. The predominant precursor ion at m/z 11281.4 corresponding to H4R3C-ADMAA (Figure 1B, inset) was subjected to electron-transfer dissociation (ETD), and the resultant product ion series was consistent with analogue installation at Cys 3 (Figure 1B).
In studies of methylarginine-dependent gene expression, antibodies specific for histones harboring arginine methylation have been used to map the distribution of methylarginine along chromatin.23 These antibodies were employed to assess recognition of analogue-modified histones by Western blot analysis. As expected, α-H4R3Me2a antibody recognized wild-type histone H4 treated with PRMT1—which enzymatically installs native ADMA—whereas the untreated histone exhibited no signal (Figure 2A). The same antibody also recognized H4R3C-ADMAA but failed to detect both H4R3C and H4R3C-AA. Similar results were observed using α-H4R3Me1 with H4R3C-MMAA (Figure 2B), α-H4R3Me2s with H4R3C-SDMAA (Figure 2C), and α-H3R2Me2a with H3R2C-ADMAA (Figure 2D). In addition, the analogue-modified histones displayed minimal antigen cross-reactivity among the respective antibodies (Figure S3). Together, these antibody recognition results indicate that analogue-modified histones are reasonable mimics of native arginine and methylarginine. Next, the analogue-modified histones were used to reconstitute nucleosomes, protein:DNA complexes that form chromatin. Mononucleosomes were assembled from octamers, composed of histones H3, H2A, H2B, and either wild-type H4 or analogue-modified H4, wrapped by the 601 DNA sequence (Figure S4). Native polyacrylamide gel electrophoresis (PAGE) analysis of the WT, AA-modified, and ADMAA-modified nucleosome reconstitution experiments shows nearly equal extent of retarded DNA migration upon assembly, indicating that the process of analogue installation does not affect nucleosome reconstitution efficiency. Thus, incorporation of methylarginine analogues into nucleosome complexes provides access to homogeneous modified chromatin substrates. The ease of analogue installation makes this strategy amenable to large-scale preparation of nucleosomes.
Figure 2.

Analogue-modified histones were recognized by methylarginine-specific antibodies in Western blot analyses. Antibody specificities: (A) H4R3Me2a, (B) H4R3Me1, (C) H4R3Me2s, and (D) H3R2Me2a. Lower row in each panel shows protein loading control.
The ability of methylarginine analogues to mimic native arginine methylation was further tested by investigating analogue association with the putative methylarginine-specific effector protein TDRD3. The Tudor domain-containing protein TDRD3 localizes to transcription start sites of many highly expressed genes and binds to H4R3Me2a; therefore, TDRD3 was postulated to be a methylarginine-specific effector protein with transcriptional co-activator function.24 In contrast to these findings, an isothermal titration calorimetry (ITC) binding study indicated no detectable association between the Tudor domain of TDRD3 and an 8-mer H4R3Me2a peptide.25 To gain further insight into TDRD3 binding specificity, equilibrium dissociation constants (Kd) were determined for the Tudor domain with various peptide substrates using a fluorescence polarization anisotropy saturation binding assay. Under conditions similar to those of the aforementioned ITC study, the Tudor domain of TDRD3 demonstrated affinity for 10-mer and 15-mer peptide substrates with Kd values in the low- to mid-micromolar range (Figures 3A and S5A). The observed increase in affinity as a function of peptide length may explain the reported lack of binding with a minimal 8-mer substrate.25,26 Furthermore, the 15-mer peptide displayed the most pronounced methylation-dependent binding, demonstrating a ~1.6-fold decrease in Kd attributable to methylation. Site-directed mutagenesis (Y566A) of the aromatic cage binding pocket within TDRD3 markedly decreased peptide binding affinity; further indicating that substrate interactions are specific to the Tudor domain (Figure S6).2,25 Taken together, these results show that the Tudor domain of TDRD3 is a binding module that discriminates, albeit weakly, between non-methylated and asymmetrically dimethylated arginine in histone H4.
Figure 3.

The Tudor domain of TDRD3 binds native asymmetric dimethylarginine and the corresponding analogue-modified substrate. (A) Measured equilibrium dissociation constants from fluorescence polarization saturation binding assays. (B) Western blot analysis of GST-TDRD3 precipitated on peptide-coated beads. (C) Fluorescence polarization competition assay. N.B. = no binding. N.D. = not determined. * = from ref 25.
Having confirmed TDRD3 Tudor domain specificity for native methylarginine, binding of analogue-modified peptides was tested. Histone peptides (15-mers) possessing a biotin tag were conjugated to streptavidin-coated beads, to which a solution of TDRD3 Tudor domain fusion protein (GST-TDRD3) was added. The extent of Tudor domain binding was assessed by Western blot analysis of precipitated protein. For the native H4R3Me2a peptide, adhesion of GST-TDRD3 was evident; in contrast, the non-methylated peptide (H4R3) gave a comparatively diminished signal (Figure 3B). These results were mirrored by the corresponding analogue-modified peptides: H4R3C-ADMAA and H4R3C-AA. In addition, free H4R3Me2a antagonized the association of GST-TDRD3 with bead-conjugated H4R3C-ADMAA (Figure S5B), indicating specific interaction between the Tudor domain and the analogue-modified substrate.
A fluorescence polarization competition assay was used to assess TDRD3 Tudor domain binding specificity under homogeneous solution-phase conditions. The association between His6-MBP-TDRD3 (a high-yielding soluble fusion construct of the Tudor domain) and a tetramethylrhodamine-tagged 15-mer H4R3Me2a peptide produces polarized fluorescence, which was antagonized by competitor peptides (10-mer, untagged). The displacement of fluorescent peptide due to competition causes a decrease in polarized emission inversely proportional to competitor ligand affinity. As expected, the H4R3Me2a ligand was the most potent competitor and the corresponding H4R3C-ADMAA variant was ~1.5-fold less effective at competitor concentrations between 1.7 and 3.3 mM (Figure 3C). Non-methylated H4R3 and H4R3C-AA competitor peptides demonstrated no affinity for TDRD3. Taken together, methylarginine analogues functionally mimic the native mark and TDRD3 exhibits only a mild preference for methylated over non-methylated arginine. Despite the subtle substrate specificity of TDRD3, the observed methylation-dependent binding affinity differences may represent a physiological mechanism to fine-tune its proposed transcriptional co-activation function within a cellular context.
The site-specific incorporation of methylarginine analogues into recombinant protein offers a facile method to attain homogeneous substrates for biochemical, biophysical, and structural investigations into the functions of arginine methylation. As demonstrated, analogue precursors 1–4 quantitatively modify histone proteins containing a single cysteine mutation that directs site-specific conjugation. This method provides control of methylation extent (non-, mono-, and dimethylarginine) in addition to regiospecificity (symmetric and asymmetric dimethylarginine). Chemically modified histones displaying methylarginine analogues were recognized by antibodies specific for native methylation states. Additionally, such histones were capable of forming mononucleosomes, demonstrating that methylarginine analogues can provide access to modified chromatin substrates. Experiments with both endogenous asymmetric dimethylarginine and the corresponding analogue variant indicated that the methylarginine recognition module of TDRD3 preferentially binds the asymmetric dimethylated mark in histone H4. Taken together, this work demonstrates that methylarginine analogue installation into recombinant protein by chemical conjugation is an effective method to produce substrates for in vitro investigations of methylarginine-dependent processes.
Supplementary Material
Acknowledgments
We thank Lauren Buckley (formerly of UW Madison) for assistance with alkylation condition optimization, the laboratories of James Wells (UCSF) and Kevan Shokat (UCSF) for instrument usage, in addition to Steven Clarke (UCLA) for expression plasmids of PRMT paralogues. This work was supported by the UCSF Program for Breakthrough Biomedical Research, the Searle Scholars Program (D.G.F.), the V Scholar Award (D.G.F.), and the NSF (graduate research fellowship to D.D.L.). Mass spectrometry was provided by the Bio-Organic Biomedical Mass Spectrometry Resource at UCSF (A.L.B., Director), supported by NIH NIGMS 8P41GM103481 and the Howard Hughes Medical Institute.
Footnotes
The authors declare no competing financial interest.
Precursor synthesis and characterization, full experimental methods, and supplemental figures. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Bedford MT, Clarke SG. Mol Cell. 2009;33:1. doi: 10.1016/j.molcel.2008.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen C, Nott TJ, Jin J, Pawson T. Nat Rev Mol Cell Biol. 2011;12:629. doi: 10.1038/nrm3185. [DOI] [PubMed] [Google Scholar]
- 3.Trievel RC, Shilatifard A. Nat Struct Mol Biol. 2009;16:678. doi: 10.1038/nsmb0709-678. [DOI] [PubMed] [Google Scholar]
- 4.Luscombe NM, Laskowski RA, Thornton JM. Nucleic Acids Res. 2001;29:2860. doi: 10.1093/nar/29.13.2860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhao Q, Rank G, Tan YT, Li H, Moritz RL, Simpson RJ, Cerruti L, Curtis DJ, Patel DJ, Allis CD, Cunningham JM, Jane SM. Nat Struct Mol Biol. 2009;16:304. doi: 10.1038/nsmb.1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.An W, Kim J, Roeder RG. Cell. 2004;117:735. doi: 10.1016/j.cell.2004.05.009. [DOI] [PubMed] [Google Scholar]
- 7.Migliori V, Muller J, Phalke S, Low D, Bezzi M, Mok WC, Sahu SK, Gunaratne J, Capasso P, Bassi C, Cecatiello V, De Marco A, Blackstock W, Kuznetsov V, Amati B, Mapelli M, Guccione E. Nat Struct Mol Biol. 2012;19:136. doi: 10.1038/nsmb.2209. [DOI] [PubMed] [Google Scholar]
- 8.Guo Z, Zheng L, Xu H, Dai H, Zhou M, Pascua MR, Chen QM, Shen B. Nat Chem Biol. 2010;6:766. doi: 10.1038/nchembio.422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McBride AE, Cook JT, Stemmler EA, Rutledge KL, McGrath KA, Rubens JA. J Biol Chem. 2005;280:30888. doi: 10.1074/jbc.M505831200. [DOI] [PubMed] [Google Scholar]
- 10.Gao X, Zhao X, Zhu Y, He J, Shao J, Su C, Zhang Y, Zhang W, Saarikettu J, Silvennoinen O, Yao Z, Yang J. J Biol Chem. 2012;287:18130. doi: 10.1074/jbc.M111.311852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Akahoshi A, Suzue Y, Kitamatsu M, Sisido M, Ohtsuki T. Biochem Biophys Res Commun. 2011;414:625. doi: 10.1016/j.bbrc.2011.09.137. [DOI] [PubMed] [Google Scholar]
- 12.Kölbel K, Ihling C, Bellmann-Sickert K, Neundorf I, Beck-Sickinger AG, Sinz A, Kühn U, Wahle E. J Biol Chem. 2009;284:8274. doi: 10.1074/jbc.M809547200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Simon MD, Chu F, Racki LR, de la Cruz CC, Burlingame AL, Panning B, Narlikar GJ, Shokat KM. Cell. 2007;128:1003. doi: 10.1016/j.cell.2006.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang R, Holbert MA, Tarrant MK, Curtet S, Colquhoun DR, Dancy BM, Dancy BC, Hwang Y, Tang Y, Meeth K, Marmorstein R, Cole RN, Khochbin S, Cole PA. J Am Chem Soc. 2010;132:9986. doi: 10.1021/ja103954u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li F, Allahverdi A, Yang R, Lua GB, Zhang X, Cao Y, Korolev N, Nordenskiold L, Liu CF. Angew Chem, Int Ed. 2011;50:9611. doi: 10.1002/anie.201103754. [DOI] [PubMed] [Google Scholar]
- 16.Valkevich EM, Guenette RG, Sanchez NA, Chen YC, Ge Y, Strieter ER. J Am Chem Soc. 2012;134:6916. doi: 10.1021/ja300500a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bordwell FG, Ji GZ. J Am Chem Soc. 1991;113:8398. [Google Scholar]
- 18.Nar H. Trends Pharmacol Sci. 2012;33:279. doi: 10.1016/j.tips.2012.03.004. [DOI] [PubMed] [Google Scholar]
- 19.Wang Y, Monzingo AF, Hu S, Schaller TH, Robertus JD, Fast W. Biochemistry. 2009;48:8624. doi: 10.1021/bi9007098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Luo Y, Arita K, Bhatia M, Knuckley B, Lee YH, Stallcup MR, Sato M, Thompson PR. Biochemistry. 2006;45:11727. doi: 10.1021/bi061180d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Obianyo O, Causey CP, Jones JE, Thompson PR. ACS Chem Biol. 2011;6:1127. doi: 10.1021/cb2001473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Garigipati RS. Tetrahedron Lett. 1990;31:1969. [Google Scholar]
- 23.Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K. Cell. 2007;129:823. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 24.Yang Y, Lu Y, Espejo A, Wu J, Xu W, Liang S, Bedford MT. Mol Cell. 2010;40:1016. doi: 10.1016/j.molcel.2010.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu K, Guo Y, Liu H, Bian C, Lam R, Liu Y, Mackenzie F, Rojas LA, Reinberg D, Bedford MT, Xu RM, Min J. PLoS One. 2012;7:e30375. doi: 10.1371/journal.pone.0030375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Voigt P, Reinberg D. Chembiochem. 2011;12:236. doi: 10.1002/cbic.201000493. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.