

Abstract

We study the self-assembly of genetically engineered protein-based triblock copolymers consisting of a central pH-responsive silk-like middle block (SHn, where SH is a silk-like octapeptide, (GA)3GH and n is the number of repeats) flanked by hydrophilic random coil outer blocks (C2). Our previous work has already shown that triblocks with very long midblocks (n = 48) self-assemble into long, stiff protein filaments at pH values where the middle blocks are uncharged. Here we investigate the self-assembly behavior of the triblock copolymers for a range of midblock lengths, n = 8, 16, 24, 48. Upon charge neutralization of SHn by adjusting the pH, we find that C2SH8C2 and C2SH16C2 form spherical micelles, whereas both C2SH24C2 and C2SH48C2 form protein filaments with a characteristic beta-roll secondary structure of the silk midblocks. Hydrogels formed by C2SH48C2 are much stronger and form much faster than those formed by C2SH24C2. Enzymatic digestion of much of the hydrophilic outer blocks is used to show that with much of the hydrophilic outer blocks removed, all silk-midblocks are capable of self-assembling into stiff protein filaments. In that case, reduction of the steric repulsion by the hydrophilic outer blocks also leads to extensive fiber bundling. Our results highlight the opposing roles of the hydrophilic outer blocks and central silk-like midblocks in driving protein filament formation. They provide crucial information for future designs of triblock protein-based polymers that form stiff filaments with controlled bundling, that could mimick properties of collagen in the extracellular matrix.
Introduction
Designed recombinant protein-based polymers are a promising class of new polymer materials with potential applications in fields such as tissue engineering, drug- and gene delivery or self-healing biomaterials.1−8 A major advantage of recombinant protein-based polymers over polymers produced using synthetic chemistry is that the route of genetic engineering provides in principle a virtually perfect control over size, amino acid sequence and stereochemistry of the polymers. As a consequence, the final degree of control over the relevant physicochemical properties of materials made of these polymers is superior to that of any established chemical polymerization method. Being based on amino acids, a vast array of naturally occurring peptide sequences or domains can be used as inspiration for new designs. Domains that have been extensively explored in recent years include those with sequences inspired by, or based on, structural proteins known for their superior stimulus-responsive, mechanical, biocompatible, and structural properties. These include natural elastin,9−15 collagen,16−19 silk,9,19−24 and resilin.25−27
A key challenge in biomaterials is to mimic the extracellular matrix in order to make materials that can act as scaffolds for cell and tissue growth. Stiff collagen-like fibers are thought to be an important element in such materials. We have previously designed pH-responsive recombinant protein-based polymers that self-assemble into stiff fibers. Those polymers have a symmetric triblock structure and are composed of a silk-like midblock flanked on both sides by hydrophilic random coiling outer blocks.
The proteins in this study have a triblock conformation, with a silk-like middle block and random coiling hydrophilic outer blocks. The silk-like middle block consists of a number of repeats of the octapeptide GAGAGAGX (SX). This amino acid sequence is inspired by natural silk produced by the silk worm Bombyx mori(23) and is known to trigger self-assembly into a filamentous structure that most likely is a stack of so-called β-rolls.19,20,28 Charges on the residue X prevent the self-assembly, and by choosing amino acids with basic or acidic side chains, self-assembly of the protein filaments can be controlled by pH. The random coiling hydrophilic outer blocks are essential, since without them, the protein filaments aggregate and precipitate.29 Hence, their role is to provide colloidal stability by exposing a hydrophilic polymer brush on the outside of the filaments.30 The block is rich in hydrophilic amino acids glutamine, asparagine and serine and has a sequence that has similarities to natural collagen (GXY triplets). The basic repeating unit for this protein-based polymer is a 99 amino acid long “C”-block.19,30 The exact sequence can be found in the Supporting Information (Figure S1).
In previous studies, we have characterized protein filaments and gels formed by C2SX48C2 protein-based triblock copolymers, where C2 is a dimer of the “C” block, and SX48 is a 48-fold repetition of the silk-like octapeptide SX. pH-responsive residues in our previous studies have been glutamic acid (X = E), histidine (X = H), or lysine (X = K).19,20,31
For use of these and other protein filaments in applications, one ideally should have full and independent control over the relevant material properties such as gelling time after a pH adjustment, control of gel rheology independent from polymer concentration, and so on. This, in turn, requires full control over the properties of the protein filaments: growth kinetics, length and rigidity, and lateral association into fiber bundles. A key variable in controlling the self-assembly of our triblock copolymers into filaments obviously is the relative size of the various blocks. Therefore, we here study the role of the balance of self-assembling and random coiling domains for pH-responsive silk-collagen-like protein-based polymers.
We focus on the effect of changing the size of the central silk-like domain. As the X residue, we choose histidine, since this results in protein filament formation at physiological pH,32 which is most relevant for biomedical applications. A series of four protein-based polymers C2SHnC2 was constructed, produced, and characterized with a number n of octapeptide repeats of n = 8, 16, 24, and 48. As we will show, the silk-like blocks SHn with n = 8, 16, 24, and 48, all have a tendency to form protein filaments, but the driving force for doing so increases with the number of repeats n. Filament formation is opposed by the C2 side blocks, and below a certain critical number of repeats n of the silk block, C2SHnC2 polymers start forming micelles rather than filaments. Our study provides insights into the driving forces of filament formation of protein-based polymers that are crucial for future protein-based polymer designs with improved independent control over filament growth, lateral association of protein filaments and the resulting hydrogel properties.
Experimental Section
Construction of Recombinant Strains and Protein Biosynthesis
The cloning of the triblock C2SH48C2 has been described by us previously.32,33 The DNA fragment encoding the midblock in this protein consists of 24 repeats of a [(GAGAGAGH]2-encoding BsaI/BanI fragment. The DNA fragments encoding the shorter mid blocks studied here, SHn (n = 8, 16, and 24), were constructed in the same manner. These fragments consist of 4, 8, and 12 repeats of the BsaI/BanI fragment, respectively, and were released from their vector by digestion with AccI/BanI. Vector pMTL23-C232 was opened with AccI/BsaI, after which the SHn fragments were inserted. The resulting plasmids were openened with AccI/BsaI, after which the second C2-encoding DNA fragment was inserted. This fragment had been obtained by digestion of pMTL23-C232 with AccI/BanI. The final C2SHnC2-encoding genes were cloned into expression vector pPIC9 (Invitrogen) via EcoRI/NotI. Transformation of P. pastoris and protein production in bioreactors were as before.34
Purification
The purification of the three smallest proteins (C2SH8C2, C2SH16C2, C2SH24C2) was performed by first selectively precipitating the protein polymers from cell-free fermentation broth in a similar way as for C2SH48C2.32 This was done by adding ammonium sulfate up to 45% saturation. After an incubation time of 30 min at room temperature, the solution was centrifuged (16000g, 40 min, 4 °C). The protein polymer pellet was resuspended in 60% of the original volume of 50 mM formic acid. The precipitation step with ammonium sulfate (45% saturation) was repeated once. After the centrifugation step the protein polymers were resuspended in 100 mL 50 mM formic acid and extensively dialyzed against 10 mM formic acid at 4 °C. Finally the proteins were freeze-dried for storage.
MALDI-TOF
Matrix-assisted laser desorption/ionization (MALDI) mass spectrometry was performed using an ultrafleXtreme mass spectrometer (Bruker). Samples were prepared by the dried droplet method on a 600 μm AnchorChip target (Bruker), using 5 mg/mL 2,5-dihydroxyacetophenone, 1.5 mg/mL diammonium hydrogen citrate, 25% (v/v) ethanol, and 1% (v/v) trifluoroacetic acid as matrix. Spectra were derived from 10 500-shot (1000 Hz) acquisitions taken at nonoverlapping locations across the sample. Measurements were made in the positive linear mode, with ion source 1, 25.0 kV; ion source 2, 23.3 kV; lens, 6.5 kV; pulsed ion extraction, 680 ns. Protein Calibration Standard II (Bruker) was used for external calibration.
SDS-PAGE
Electrophoresis (SDS-PAGE) was performed using the NuPAGE Novex system with 10% Bis-Tris gels, MES-SDS running buffer, and Novex Sharp Protein Standard prestained molecular mass markers. Gels were stained with Coomassie SimplyBlue SafeStain (all Invitrogen).
Dynamic Light Scattering (DLS)
DLS measurements were performed using a Zetasizer NanoZS (Malvern Instruments, U.K.), equipped with a He–Ne laser (4 mW), operating at a wavelength of 633 nm. Each measurement was performed at an angle of 173° and a temperature of 25 °C. Measurements at pH 2 were performed by dissolving protein in 10 mM HCl at a concentration of 1 g/L. Solutions were filtrated (200 nm, Millipore). Measurements at pH 8 were performed by diluting the former solutions a factor 2, using filtrated 100 mM phosphate buffer (pH 8). Reported hydrodynamic radii are z-averaged values determined by DTS Software, version 5.10. Reversibility was examined by adding an excess of 1 M filtered HCl to solutions containing protein micelles or fibers.
Atomic Force Microscopy
AFM samples were made by applying a drop of protein solution on a 10 × 10 mm hydrophilic silicon wafer (Siltronic Corp.) bearing a thin oxide layer, rinsing the wafer with milli-Q water to remove any nonadsorbed material, and drying it under a stream of nitrogen. The samples were analyzed using a Digital Instruments Nanoscope V in ScanAsyst mode and NP-10 silicon nitride tips with a spring constant of 0.350 N/m and a 10 nm tip radius (Bruker, CA, U.S.A.). Images were processed using NanoScope Analysis 1.40. All samples contained 1 g/L of protein and a 50 mM phosphate buffer (pH 8).
Circular Dichroism
CD measurements were performed on a Jasco J-715 spectropolarimeter at 298 K. The spectra were recorded between 190 and 260 nm with a resolution of 0.2 nm and a scanning speed of 1 nm/s. Each spectrum was an average of 20 measurements. A quartz cuvette with a path length of 0.5 mm was used. Protein concentration was 0.25 g/L and the solvent was 10 mM HCl (pH 2) or 50 mM phosphate buffer (pH 8). For the kinetic study, 1 g/L solutions in 50 mM phosphate buffer (pH 8) were used, which were diluted 4× with the same buffer prior to measuring. Ellipticity was measured at a wavelength of 198 nm.
Rheology
Rheological measurements were performed on an Anton Paar MCR 301 rheometer with Couette CC10/TI geometry. Cup and bob radii were 5.420 mm and 5.002 mm, respectively. A solvent trap was used to prevent evaporation. Samples containing 25 g/L of protein were adjusted to pH 8 in a 50 mM phosphate buffer. Immediately after adjusting the pH the storage modulus was measured using oscillatory deformation (f = 1 Hz and γ = 0.1%) until a plateau value was reached. Temperature was controlled by a Peltier element at 298 K during measurements.
Enzymatic Digestion
Trypsin from bovine pancreas (Sigma-Aldrich) was used to digest the C2-block of C2SH8C2, C2SH16C2, and C2SH24C2. Samples contained 1 g/L of protein and 0.02 g/L of trypsin. After mixing protein and enzyme, we adjusted the pH to 8 in 50 mM phosphate buffer. Samples were incubated for 72 h at 310 K before measuring them with AFM and SDS-PAGE.
Results and Discussion
Protein Characterization
The four proteins described in this study include three new constructs and one described previously.32 The molecular weight (MW) of each newly constructed protein was measured with MALDI-TOF MS and compared to the theoretical mass predicted from the amino acid composition. As shown in Table 1, experimentally determined masses match those expected theoretically within the experimental uncertainty. Additionally, Dynamic Light Scattering (DLS) was used to determine the size of the proteins when fully charged at pH 2. Hydrodynamic sizes of the four proteins C2SHnC2 with n = 8, 16, 24, and 48 in solution at pH 2 are shown in Table 1 and are typical for molecularly dissolved nonglobular proteins of these molar masses.
Table 1. Characterization of Protein–Polymers Described in This Study; Theoretical and Measured (MALDI-TOF MS) Mass, and hydrodynamic radius (Rh), as Measured by DLS at pH 2 (10 mM HCl).
| protein | theoretical MW (Da) | measured MW (Da) | Rh at pH 2 (nm) |
|---|---|---|---|
| C2SH8C2 | 42992 | 42952 | 4.9 ± 0.2 |
| C2SH16C2 | 47621 | 47617 | 5.6 ± 0.6 |
| C2SH24C2 | 52249 | 52242 | 6.2 ± 0.2 |
| C2SH48C2 | 66135 | 6607632 | 6.8 ± 0.6 |
Protein–polymers are secreted in the medium by the production organism, Pichia pastoris, and simple ammonium sulfate precipitation suffices to obtain highly pure protein polymers. SDS-PAGE gels for the newly constructed proteins C2SH8C2, C2SH16C2, and C2SH24C2 after purification using ammonium sulfate precipitation are shown in Figure 1.
Figure 1.
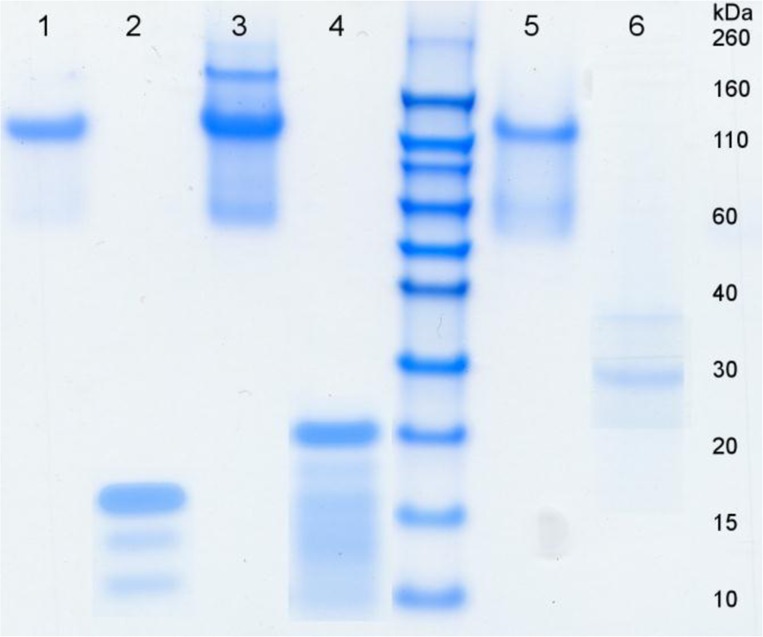
SDS-PAGE gel of purified and trypsin treated protein–polymers. Lane 1: C2SH8C2; lane 2: C2SH8C2 after trypsin digestion; lane 3: C2SH16C2; lane 4: C2SH16C2, C2SH16C2 after trypsin digestion; lane 5: C2SH24C2; lane 6: C2SH24C2 after trypsin digestion.
For each of the three proteins, there was a clear main band corresponding to the protein polymer. Note that the migration of the protein-based polymers is anomalously slow due to the poor SDS-binding capacity of the hydrophilic C2-blocks, as has been described before.19,30 This leads to an apparent mass of approximately 120 kDa. The band that is visible for all proteins migrating to an apparent mass of 60 kDa is similar to the band found in purified C2SH48C2. This band represents an SDS-PAGE artifact, as N-terminal sequencing combined with MALDI-TOF showed it was the intact protein.35 We attribute the band at 200 kDa to multimers of the intact protein. The high purity of the protein samples is also confirmed with MALDI-TOF (Figure S2). Figure 1 also shows SDS-PAGE of purified proteins treated with trypsin to remove most of the outer blocks. These digested protein–polymers are also used in our physical studies and will be discussed in detail later on.
AFM
First we study the self-assembly of the protein–polymers after a pH shift from pH 2 to pH 8 using Atomic Force Microscopy (AFM) imaging. As is shown by the AFM images in Figure 2a,b, after prolonged incubation at pH 8 (72 h) the proteins with the longest silk-like midblocks, C2SH48C2 and C2SH24C2, form long, stiff filaments. For both proteins, the filaments have a height of approximately 2 nm and lengths up to many micrometers. We did not find significant differences in the final filament lengths for the two proteins. The average width of the C2SH24C2 is 7 nm smaller than that of the C2SH48C2 filaments, which is close to half of the expected width of the folded S48 blocks.19,28 In contrast, the proteins with the shorter silk-like midblocks, C2SH16C2 and C2SH8C2, did not form filaments after a pH shift from pH 2 to pH 8, after prolonged incubation. Instead, these proteins appear to form micelles, as suggested by the pancake-like structures found with AFM and shown in Figure 2c,d.
Figure 2.

AFM images of self-assembled protein–polymers (1 g/L), adsorbed to silica 72 h after a pH quench from pH 2 to pH 8 (50 mM phosphate buffer): (a) C2SH48C2, (b) C2SH24C2, (c) C2SH16C2, (d) C2SH8C2. Images are 5 × 5 μm (a, b) or 2 × 2 μm (c, d).
DLS
Dynamic light scattering confirms the appearance of micelles at pH 8 in samples of C2SH16C2 and C2SH8C2. While at pH 2 both proteins are present as single molecules with Rh = 5.6 and 4.9 nm, at pH 8 they assemble into micelles with hydrodynamic radii more than doubled: 12.6 and 11.2 nm, respectively.
When the pH of a solution containing micelles or fibers was lowered well below the pKa of histidine by the addition of an excess HCl, we observed an immediate drop in scattered intensity and observed molecularly dissolved protein polymers. This shows that the self-assembly of all four protein polymers is fully reversible.
Circular Dichroism
The very different self-assembled structures of the proteins with the longest silk-like midblocks versus those with the shorter ones raises the question whether their secondary structure is also different. In order to assess changes in secondary structure after the pH shift from pH 2 to pH 8, we have performed circular dichroism (CD) spectroscopy of all proteins, both in their fully charged, monomeric form at pH 2, and in their neutralized and self-assembled form at pH 8.
Figure 3 shows that at pH 2 all proteins have nearly identical spectra. These spectra clearly have the signature of a random coil and are very similar to that of a pure C4 block, for which it was previously shown that it behaves as a random coil over a wide range of solution conditions.30 The similarity of the spectra over the entire series of triblocks leads us to conclude that at pH 2, both the hydrophilic outer blocks and the silk-like middle blocks have a random coil conformation.
Figure 3.

Molar ellipticity per amino acid of protein–polymer solutions of C2SH8C2 (a), C2SH16C2 (b), C2SH24C2 (c), and C2SH48C2 (d) in 10 mM HCl (pH 2) and 50 mM phosphate buffer (pH 8). Samples at pH 8 have been measured 96 h after adjusting the pH from pH 2 to pH 8.
Figure 3 also shows the CD spectra for the proteins at pH 8. The micelle-forming proteins with the shortest silk-like middle blocks only show a minor spectral shift as compared to the spectra at pH 2. The spectrum at pH 8 still mostly resembles that of a random coil. Note, however, that this could still simply be a consequence of the relatively small contributions of the rather short silk-like middle-blocks to the total spectra. In contrast, the spectra of the filament forming proteins with the longer silk-like midblocks at pH 8 show a very clear spectral shift as compared to pH 2. For this case, it is clear that a significant change of secondary structure occurs upon adjusting the pH.
In order to isolate the contribution of the silk-like midblocks to the total CD spectra, we have also acquired the spectra of a pure C4 polymer, that should be identical to the combined spectrum of the two C2 outer-blocks. Difference spectra pertaining to the isolated silk-like midblocks obtained by subtracting the spectra of the outer blocks are shown, for all four proteins, in Figure 4. For each protein, the mass fraction of the outer blocks was determined and the spectrum of the corresponding concentration of C2 blocks was subtracted from the spectrum of the whole protein. It is clear that the absence of a change in secondary structure for the two proteins with the shortest silk-like midblocks is real, and is not caused by the signal of the outer blocks overwhelming that of the silk-like midblocks: for this case, the difference spectrum still has the signature characteristic of a random coil. Difference spectra for the SH24 and SH48 midblocks at pH 8 are also very similar but have a distinctly different CD spectrum suggesting that both have a secondary structure that is very different from a random coil. Molecular Dynamics simulations have indicated that the neutralized and folded silk-like block SE48 obtains a β-roll structure in solution.28 This structure consists of two interconnected parallel β-sheets and is consistent with fiber dimensions found with AFM and SAXS.19 The CD spectra at pH 8 of C2SH24C2 and C2SH48C2 are very similar to that of neutralized C2SE48C2,19 leading us to conclude the same β-roll structure is present in the folded C2SH24C2 and C2SH48C2.
Figure 4.

Molar ellipticity per amino acid of the isolated SH8 and SH16 (a) and SH24 and SH48 (b) after subtraction of the signal of the C2 side chains from the signal of the triblock protein–polymers. All samples contained a total of 0.25 g/L of protein and were measured at pH 8 (50 mM phosphate buffer) 96 h after a pH adjustment from pH 2.
Moreover, from the fact that after extensive incubation at pH 8 the ellipticities per amino acid estimated for the SH24 and SH48 midblocks are very nearly equal in magnitude, we conclude that, most likely, in both cases virtually all protein molecules self-assemble into filaments.
Time Resolved AFM
For the two proteins that self-assemble into filaments (C2SH48C2 and C2SH24C2), we have also elucidated the kinetics of filament formation using time-resolved AFM imaging. This was achieved by taking aliquots after different times of incubation at pH 8, after the pH adjustment from acidic pH. Immediately after taking the aliquot, it was deposited on a silica wafer, to quench the filament growth. For each aliquot, the length of a fair number of filaments (50–90) was determined and used to estimate the average filament length and its standard deviation. Results of this analysis are shown in Figure 5.
Figure 5.
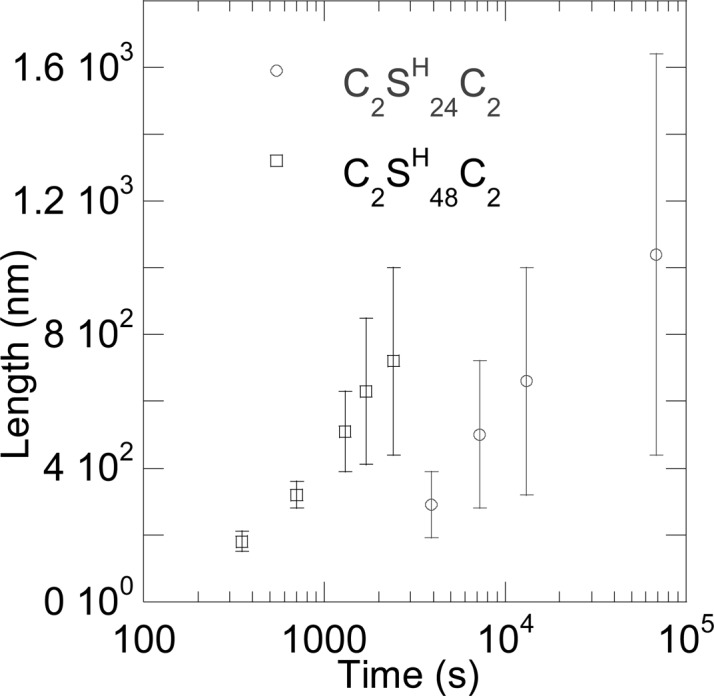
Average fiber length (n = 50–90) as a function of the time after shift from pH 2 to pH 8 (50 mM phosphate buffer) for 1 g/L solutions of C2SH24C2 and C2SH48C2, as measured by AFM. Error bars represent standard deviations.
Clearly, the average length of the C2SH48C2 filaments increases at a much higher rate than the average length of the C2SH24C2 filaments. For both proteins, the size distribution of the filaments quite dramatically broadens with incubation time. This must mean that there is continuous nucleation of filaments, with existing fibers elongating by the attachment of additional proteins, and new filaments being formed at the same time. Such a continuous nucleation is very different from the self-assembly of proteins of an inverted silk-collagen triblocks SE24C2C2SE24 that we have studied before. For that polymer, we observed fast nucleation immediately after the pH induced charge neutralization, followed by elongation of existing fibers without the formation of many new ones.20 Such a mechanism obviously leads to a much narrower size distribution than the continuous nucleation mechanism that we observe for C2SH48C2 and C2SH24C2. The inverted sequence of SE24C2C2SE24 results in an extra complicating factor for nucleation, namely the meeting of the two ends of one molecule. This can slow down homogeneous nucleation of new fibers severely. We anticipate that the occurrence of heterogeneous nucleation (possibly initiated by a small fraction of irreversibly folded protein, partially degraded protein or impurities that bind protein) leads to a fast nucleation step, followed by elongation of growing fibers. During this elongation, homogeneous nucleation is almost nonexisting. C2SH48C2 and C2SH24C2 do not require this extra step during homogeneous nucleation and can therefore combine a quick heterogeneous nucleation with a continuous homogeneous one.
Time Resolved CD
While time resolved AFM is a powerful tool to obtain kinetic data on the growth of individual protein filaments, it does not provide information on the total conversion of protein monomers into filaments. To obtain such data, we have used time-resolved CD. As the spectrum of this type of proteins only changes when they assemble into filaments,20 one can use the magnitude of this spectral shift as a measure for the total fraction of proteins that have self-assembled. At a wavelength of 198 nm, where the change in ellipticity (θ) between pH 2 and pH 8 is the largest, we have followed the change in ellipticity over time, for both C2SH48C2 and C2SH24C2. The fraction f of unfolded (and thus molecularly dissolved) proteins at time t is estimated from
| 1 |
The result of the analysis of the time-resolved CD experiment for the fraction f of unfolded protein as a function of incubation time is shown in Figure 6 for both C2SH48C2 and C2SH24C2.
Figure 6.
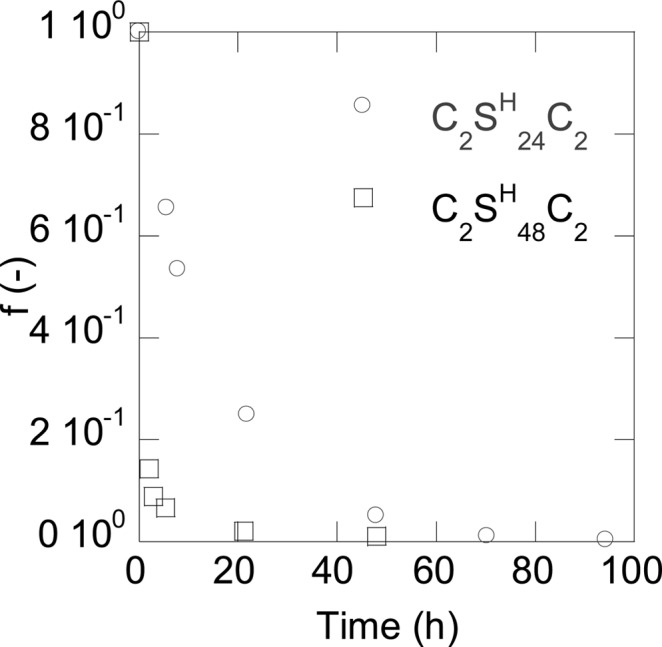
Fraction f of unfolded protein of C2SH24C2 and C2SH48C2 in time after incubation in 50 mM phosphate buffer (pH 8). Both solutions contained 1 g/L of protein.
The CD data fully confirm the conclusion from the AFM data that under the same conditions (pH and weight concentration), the self-assembly of C2SH48C2 into filaments is significantly faster than that of C2SH24C2, This must mean that the folding of the silk-like block is not the rate-determining step. The fact that C2SH48C2 has twice the hydrophobic surface area compared to C2SH24C2, must be a key factor in the docking of a new protein onto a growing end of an existing fiber. Next we consider implications of the differences in filament formation and filament properties for gels that form when letting the proteins self-assemble into filaments at much higher concentrations.
Rheology
C2SH48C2 is already known to form hydrogels at neutral or higher pH,32 at weight concentrations exceeding 10 g/L. Here we have shown that the C2SH24C2 protein also self-assembles into protein filaments, and that after prolonged incubation, essentially all protein is incorporated in protein filaments. Next, we follow the gelation of 25 g/L solutions of both proteins by online rheometry, as a function of the incubation time at pH 8, for a time period of up to 2 days. Figure 7 shows the development of the storage modulus of both solutions in time. There are two distinct differences between the curves for the two proteins. First, gelation of C2SH24C2 is very much slower than that of C2SH48C2. The graph shows a lag time of several hours before the storage modulus starts increasing, while C2SH48C2 starts gelling virtually instantaneously. This observation is in line with our findings with Time Resolved AFM of much slower filament growth rates. Apparently, filaments of C2SH24C2 grow so slowly that it takes a significant time to reach the overlap concentration, while this transition point is reached much faster for the case of C2SH48C2. Second, the limiting value of the storage modulus (after 48 h of incubation time at pH 8) differs by almost an order of magnitude. Since it appears that all protein is eventually incorporated into protein filaments, at identical weight concentrations, we anticipate that the total length of protein filament should be roughly equal, and the difference observed must be due to differences in either the length or structural organization of the fibers in the network structure. For dilute samples we have observed that final filament lengths are comparable for the two proteins. Assuming that this also holds for more concentrated samples, a possible cause could be a difference in filament–filament interactions, that lead to a different structural organization of the fibers in the network structure. The C2SH48C2 fibers have twice the exposed histidine rich (hydrogen bonding and aromatic character) surface area as compared to C2SH24C2 fibers, and this might leads to a stronger attractive force between fibers. The difference in size of the tightly packed silk-like domain in the protein filaments may result in a difference in stiffness of the filaments. This might contribute to the difference in gel properties as well. If these hypotheses are true, a further increase of the silk-like domain, or a decrease of the random coiling domain (facilitating contact between the silk-like domains of neighboring filaments) should lead to stronger hydrogels, at even lower concentrations than those we observe here for C2SH48C2.
Figure 7.
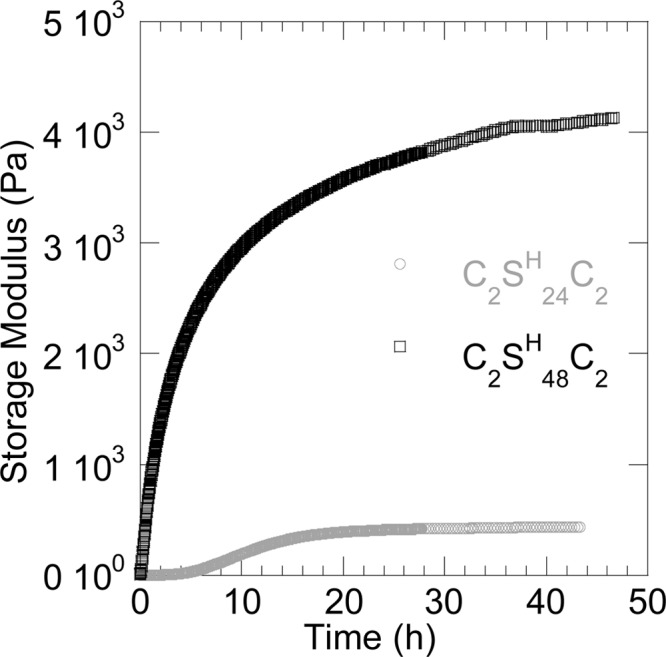
Storage modulus in time of 25 g/L solutions of C2SH24C2 and C2SH48C2 directly after quenching to pH 8 in 50 mM phosphate buffer at 298 K.
Enzymatic Digestion of the C2 Blocks
So far, our analysis of the series of triblocks has shown that decreasing the ratio of the self-assembling silk-like block to the random coiling blocks by reducing the length of the former, leads to a transition from fibers to micelles at pH 8. This raises the question whether the tendency of the triblocks to self-assemble into filaments is completely lost below a certain length of the silk-like midblocks, or that below this critical length, filament growth is simply opposed too much by the random coiling outer blocks. In order to distinguish between these cases, we have used enzymatic degradation by trypsin of the C2 blocks for the three smallest triblocks. Trypsin typically cleaves at sites immediately following a lysine or arginine, except when this amino acid is followed by proline.100 The C2 block has a total of eight putative cleavage sites, while the silk-like blocks have none. Hence, we expect the size of the C2 block can be reduced down to 42 amino acids on the N-terminus and 23 amino acids on the C-terminus using trypsin digestion. The enzyme works optimally at pH 8, corresponding to the conditions that the proteins self-assemble into either micelles or protein filaments. The presence of a clear main band in the SDS-PAGE gels in Figure 1 of digested C2SH8C2, C2SH16C2, and C2SH24C2, with highly increased mobilities compared to the intact protein polymers, confirms that indeed much of the hydrophilic C2 blocks was removed by the enzyme. Note that the C-fragments after digestion are smaller than 4 kDa and are therefore not visible on the gel. After extensive trypsin digestion, AFM imaging was used to check for changes in the self-assembled structures. Selected images are shown in Figure 8. For C2SH8C2, we find very few micellar structures, plus some short filaments. For C2SH16C2, there is a very clear transition from micelle formation to filament formation upon removal of much of the outer block by trypsin digestion. We also find that the filaments formed by trypsin-treated triblocks have a notable tendency to bundle. Finally, for C2SH24C2, we find that trypsin digestion leads to very strong filament bundling. Returning to the question posed at the beginning of this paragraph, it is now clear that even the shorter silk-like midblocks do have an intrinsic tendency to fold and stack into filaments, but that filament-formation can apparently be halted by the hydrophilic random coiling outer blocks, if these are sufficiently long. It is also clear that the precise length of the outer blocks not only determines whether micelles will be formed or filaments, but that it also determines the likelihood of the silk-like midblocks of neighboring filaments coming into contact, and leading to lateral filament-filament association and bundling, which is crucial in determining the final mechanical properties of hydrogels formed by our triblock protein-based polymers.
Figure 8.

Effect of trypsin digestion on fiber formation and fiber bundling. AFM pictures of C2SH8C2 (a), C2SH16C2 (b), and C2SH24C2 (c) adsorbed on silica after digestion by trypsin. All samples contained 1 g/L protein and 0.02 g/L trypsin. Samples were analyzed after 72 h of incubation at pH 8 (50 mM phosphate buffer) at 310 K. Image size is 5 × 5 μm (a, b) or 2 × 2 μm (c).
Conclusions
We have constructed a series of recombinant triblock protein polymers that consist of a hydrophilic inert random coiling block and a pH-responsive silk-like block. The number of octapeptide repeats in the silk-like midblock was varied over a broad range: 8, 16, 24, and 48. All proteins show pH-responsive self-assembly behavior. In each case there was a transition from molecularly dissolved charged proteins at pH 2 to self-assembled structures at pH 8. We observed a transition from spherical micelles (C2SH8C2 and C2SH16C2) to fiber formation (C2SH24C2 and C2SH48C2). The longest silk-like block yields the strongest and fastest forming hydrogels. Enzymatic digestion of the random coiling block triggered the micelle forming proteins into forming fibers. It also leads to more sticky fibers than the ones formed by intact C2SH24C2 and C2SH48C2.
In our previous work, we have described fiber-forming triblock protein–polymers with the structure C2SX48C2 that form dilute hydrogels, for some residues X (notably histidine) at physiological conditions (pH, temperature, ionic strength).20,31,32 Although the current dimensions of the two different domains are suitable for making hydrogels, they may not be ideal when aiming for strong hydrogels at extremely dilute concentrations, or for hydrogels with large pore sizes. Bundling of protein filaments can lead to both gelation at very low concentrations36 and to large pore sizes that may be desirable in applications such as tissue culture. This leads us to believe that a further increase of the silk-like block or a decrease of the hydrophilic random coiling block could give controlled bundling of the protein filaments, leading to extremely long and stiff fiber bundles, more faithfully mimicking the structure of collagen bundles in the extracellular matrix. Such control over bundling would very much broaden the range of moduli and pore sizes that can be acquired using our fiber based gels.
The micelles formed by C2SH8C2 and C2SH16C2 might be worthwhile candidates for nanodelivery vehicles that release their contents in acidic environments such as the stomach. For example, this could be useful in taste-masking. It would also be interesting to aim for a much more precise control of the pH dependence of the self-assembly, in view of delivery to tumor cells exploiting the somewhat more acidic extracellular environment of tumor cells (6.5–6.9 compared to 7.2–7.4 around healthy cells).37
Finally, our work highlights how the familiar concept of the control of block copolymer self-assembly by tuning block lengths, translates to the case of protein-based polymers, with blocks that not merely self-assemble, but have well-defined folds into specific secondary structures. Specifically, our results point to the possibility to design self-assembling triblock protein–polymers with not only controlled fiber growth, but also controlled fiber bundling.
Acknowledgments
This research was funded by the Dutch Organization for Scientific Research (NWO) and by the European Research Council through Grant 267254 “Biomate”.
Supporting Information Available
Amino acid composition and MALDI-TOF spectra for the three newly produced protein polymers. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Huang J.; Foo C. W. P.; Kaplan D. L. Biosynthesis and applications of silk-like and collagen-like proteins. Polym. Rev. 2007, 47129–62. [Google Scholar]
- Meyer D. E.; Trabbic-Carlson K.; Chilkoti A. Protein purification by fusion with an environmentally responsive elastin-like polypeptide: Effect of polypeptide length on the purification of thioredoxin. Biotechnol. Prog. 2001, 174720–728. [DOI] [PubMed] [Google Scholar]
- Li M. Y.; Mondrinos M. J.; Gandhi M. R.; Ko F. K.; Weiss A. S.; Lelkes P. I. Electrospun protein fibers as matrices for tissue engineering. Biomaterials 2005, 26305999–6008. [DOI] [PubMed] [Google Scholar]
- Daamen W. F.; Veerkamp J. H.; van Hest J. C. M.; van Kuppevelt T. H. Elastin as a biomaterial for tissue engineering. Biomaterials 2007, 28304378–4398. [DOI] [PubMed] [Google Scholar]
- Nettles D. L.; Chilkoti A.; Setton L. A. Applications of elastin-like polypeptides in tissue engineering. Adv. Drug Delivery Rev. 2010, 62151479–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez-Garcia A.; Werten M. W. T.; Stuart M. C.; de Wolf F. A.; de Vries R. Coating of single DNA molecules by genetically engineered protein diblock copolymers. Small 2012, 8223491–3501. [DOI] [PubMed] [Google Scholar]
- Chilkoti A.; Dreher M. R.; Meyer D. E. Design of thermally responsive, recombinant polypeptide carriers for targeted drug delivery. Adv. Drug Delivery Rev. 2002, 5481093–1111. [DOI] [PubMed] [Google Scholar]
- van Hest J. C. M.; Tirrell D. A. Protein-based materials, toward a new level of structural control. Chem. Commun. 2001, 19, 1897–1904. [DOI] [PubMed] [Google Scholar]
- Golinska M. D.; Pham T. T. H.; Werten M. W. T.; de Wolf F. A.; Stuart M. A. C.; van der Gucht J. Fibril formation by pH and temperature responsive silk-elastin block copolymers. Biomacromolecules 2013, 14148–55. [DOI] [PubMed] [Google Scholar]
- Meyer D. E.; Chilkoti A. Genetically encoded synthesis of protein-based polymers with precisely specified molecular weight and sequence by recursive directional ligation: Examples from the elastin-like polypeptide system. Biomacromolecules 2002, 32357–367. [DOI] [PubMed] [Google Scholar]
- Koria P.; Yagi H.; Kitagawa Y.; Megeed Z.; Nahmias Y.; Sheridan R.; Yarmush M. L. Self-assembling elastin-like peptides growth factor chimeric nanoparticles for the treatment of chronic wounds. Proc. Natl. Acad. Sci. U.S.A. 2011, 10831034–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anumolu R.; Gustafson J. A.; Magda J. J.; Cappello J.; Ghandehari H.; Pease L. F. Fabrication of highly uniform nanoparticles from recombinant silk-elastin-like protein polymers for therapeutic agent delivery. ACS Nano 2011, 575374–5382. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- van Eldijk M. B.; Wang J. C. Y.; Minten I. J.; Li C. L.; Zlotnick A.; Nolte R. J. M.; Cornelissen J. J. L. M.; van Hest J. C. M. Designing two self-assembly mechanisms into one viral capsid protein. J. Am. Chem. Soc. 2012, 1344518506–18509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schipperus R.; Teeuwen R. L. M.; Werten M. W. T.; Eggink G.; de Wolf F. A. Secreted production of an elastin-like polypeptide by Pichia pastoris. Appl. Microbiol. Biotechnol. 2009, 852293–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schipperus R.; Eggink G.; de Wolf F. A. Secretion of elastin-like polypeptides with different transition temperatures by Pichia pastoris. Biotechnol. Prog. 2012, 281242–247. [DOI] [PubMed] [Google Scholar]
- Silva C. I. F.; Skrzeszewska P. J.; Golinska M. D.; Werten M. W. T.; Eggink G.; de Wolf F. A. Tuning of collagen triple-helix stability in recombinant telechelic polymers. Biomacromolecules 2012, 1351250–1258. [DOI] [PubMed] [Google Scholar]
- Skrzeszewska P. J.; Jong L. N.; de Wolf F. A.; Stuart M. A. C.; van der Gucht J. Shape-memory effects in biopolymer networks with collagen-like transient nodes. Biomacromolecules 2011, 1262285–2292. [DOI] [PubMed] [Google Scholar]
- Skrzeszewska P. J.; de Wolf F. A.; Werten M. W. T.; Moers A. P. H. A.; Stuart M. A. C.; van der Gucht J. Physical gels of telechelic triblock copolymers with precisely defined junction multiplicity. Soft Matter 2009, 5102057–2062. [Google Scholar]
- Martens A. A.; Portale G.; Werten M. W. T.; de Vries R. J.; Eggink G.; Stuart M. A. C.; de Wolf F. A. Triblock protein copolymers forming supramolecular nanotapes and pH-responsive gels. Macromolecules 2009, 4241002–1009. [Google Scholar]
- Beun L. H.; Beaudoux X. J.; Kleijn J. M.; de Wolf F. A.; Stuart M. A. C. Self-assembly of silk-collagen-like triblock copolymers resembles a supramolecular living polymerization. ACS Nano 2012, 61133–140. [DOI] [PubMed] [Google Scholar]
- Smeenk J. M.; Otten M. B. J.; Thies J.; Tirrell D. A.; Stunnenberg H. G.; van Hest J. C. M. Controlled assembly of macromolecular β-sheet fibrils. Angew. Chem., Int. Ed. 2005, 44131968–1971. [DOI] [PubMed] [Google Scholar]
- Krejchi M. T.; Atkins E. D. T.; Fournier M. J.; Mason T. L.; Tirrell D. A. Observation of a silk-like crystal structure in a genetically engineered periodic polypeptide. J. Macromol. Sci., Part A: Pure Appl.Chem. 1996, A33101389–1398. [Google Scholar]
- Krejchi M. T.; Cooper S. J.; Deguchi Y.; Atkins E. D. T.; Fournier M. J.; Mason T. L.; Tirrell D. A. Crystal structures of chain-folded antiparallel β-sheet assemblies from sequence-designed periodic polypeptides. Macromolecules 1997, 30175012–5024. [Google Scholar]
- Krejchi M. T.; Atkins E. D. T.; Waddon A. J.; Fournier M. J.; Mason T. L.; Tirrell D. A. Chemical sequence control of β-sheet assembly in macromolecular crystals of periodic polypeptides. Science 1994, 26551771427–1432. [DOI] [PubMed] [Google Scholar]
- Li L. Q.; Tong Z. X.; Jia X. Q.; Kiick K. L. Resilin-like polypeptide hydrogels engineered for versatile biological function. Soft Matter 2013, 93665–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elvin C. M.; Carr A. G.; Huson M. G.; Maxwell J. M.; Pearson R. D.; Vuocolo T.; Liyou N. E.; Wong D. C. C.; Merritt D. J.; Dixon N. E. Synthesis and properties of crosslinked recombinant pro-resilin. Nature 2005, 4377061999–1002. [DOI] [PubMed] [Google Scholar]
- Charati M. B.; Ifkovits J. L.; Burdick J. A.; Linhardt J. G.; Kiick K. L. Hydrophilic elastomeric biomaterials based on resilin-like polypeptides. Soft Matter 2009, 5183412–3416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schor M.; Martens A. A.; de Wolf F. A.; Stuart M. A. C.; Bolhuis P. G. Prediction of solvent dependent β-roll formation of a self-assembling silk-like protein domain. Soft Matter 2009, 5132658–2665. [Google Scholar]
- Werten M. W. T.; Moers A. P. H. A.; Vong T.; Zuilhof H.; van Hest J. C. M.; de Wolf F. A. Biosynthesis of an amphiphilic silk-like polymer. Biomacromolecules 2008, 971705–1711. [DOI] [PubMed] [Google Scholar]
- Werten M. W. T.; Wisselink W. H.; van den Bosch T. J. J.; de Bruin E. C.; de Wolf F. A. Secreted production of a custom-designed, highly hydrophilic gelatin in Pichia pastoris. Protein Eng. 2001, 146447–454. [DOI] [PubMed] [Google Scholar]
- Martens A. A.; van der Gucht J.; Eggink G.; de Wolf F. A.; Stuart M. A. C. Dilute gels with exceptional rigidity from self-assembling silk-collagen-like block copolymers. Soft Matter 2009, 5214191–4197. [Google Scholar]
- Golinska M. D.; Wlodarczyk-Biegun M. K.; Werten M. W. T.; Stuart M. A. C.; de Wolf F. A.; de Vries R. Dilute self-healing hydrogels of silk-collagen-like block copolypeptides at neutral pH. Biomacromolecules 2014, 153699–706. [DOI] [PubMed] [Google Scholar]
- Yan Y.; Martens A. A.; Besseling N. A. M.; de Wolf F. A.; de Keizer A.; Drechsler M.; Stuart M. A. C. Nanoribbons self-assembled from triblock peptide polymers and coordination polymers. Angew. Chem., Int. Ed. 2008, 47224192–4195. [DOI] [PubMed] [Google Scholar]
- Werten M. W. T.; Van den Bosch T. J.; Wind R. D.; Mooibroek H.; De Wolf F. A. High-yield secretion of recombinant gelatins by Pichia pastoris. Yeast 1999, 15111087–1096. [DOI] [PubMed] [Google Scholar]
- Włodarczyk-Biegun M. K.; Werten M. W. T.; de Wolf F. A.; van den Beucken J. J. J. P.; Leeuwenburgh S. C. G.; Kamperman M.; Stuart M. A. C. Genetically engineered silk–collagen-like copolymer for biomedical applications: Production, characterization and evaluation of cellular response. Acta Biomater. 2014, 1083620–3629. [DOI] [PubMed] [Google Scholar]
- ExPASy Bioinformatics Resource Portal Peptide Cutter. http://web.expasy.org/peptide_cutter/, accessed August 22, 2013.
- Kouwer P. H. J.; Koepf M.; Le Sage V. A. A.; Jaspers M.; van Buul A. M.; Eksteen-Akeroyd Z. H.; Woltinge T.; Schwartz E.; Kitto H. J.; Hoogenboom R.; Picken S. J.; Nolte R. J. M.; Mendes E.; Rowan A. E. Responsive biomimetic networks from polyisocyanopeptide hydrogels. Nature 2013, 4937434651–655. [DOI] [PubMed] [Google Scholar]
- Estrella V.; Chen T. A.; Lloyd M.; Wojtkowiak J.; Cornnell H. H.; Ibrahim-Hashim A.; Bailey K.; Balagurunathan Y.; Rothberg J. M.; Sloane B. F.; Johnson J.; Gatenby R. A.; Gillies R. J. Acidity generated by the tumor microenvironment drives local invasion. Cancer Res. 2013, 7351524–1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.