

Abstract
Antibody–drug conjugates (ADCs) constitute a category of anticancer targeted therapy that has gathered great interest during the last few years because of their potential to kill cancer cells while causing significantly fewer side effects than traditional chemotherapy. In this paper, a process of computational construction of ADCs is described, using the surface lysines of an antibody and a non-covalent linker molecule, as well as a cytotoxic substance, as files in Protein Data Bank format. Also, aspects related to the function, properties, and development of ADCs are discussed.
Keywords: cancer, targeted therapy, antibody–drug conjugate
Introduction
Cancer is a term used for a group of diseases in which abnormal cells divide uncontrollably, having the ability to invade other tissues and spread to other parts of the body through the blood and the lymph systems. It is one of the dominating causes of death worldwide, being responsible for 8.2 million deaths in 2012, while annual cancer cases are expected to rise from 14 millions in 2012 to 22 millions during the next two decades. There are a number of cancer treatment approaches, with surgery, chemotherapy, and radiotherapy being some of the most common ones. Traditional chemotherapy drugs act against all rapidly dividing cells, including the non-cancerous ones, a disadvantage that led to an increased interest in the development of targeted anticancer therapies (TATs). TAT refers to the systemic administration of drugs or other substances with particular mechanisms that interfere with specific molecules involved in cancer cell growth and survival, and have minimized adverse effects on healthy cells.1–7
Antibody–drug conjugates (ADCs) constitute a class of TAT. An ADC molecule consists of three components: a cytotoxic drug, also called payload; a tumor-targeting monoclonal antibody or antibody fragment; and a molecule that connects the previous two, called linker. Its mechanism of action can be described in general terms as the binding of the antibody to its antigen and the subsequent release of the cytotoxin, which results in the death of the cancer cell. The choice of the antibody depends on the target antigen, usually a molecule present in cancer cells at a much higher concentration than in normal cells. Therefore, the expected performance from an ADC upon its administration to the patient is to remain stable in circulation and deliver the cytotoxic substance selectively to cancer cells, maximizing their exposure to the drug, while minimizing the exposure of healthy cells. Some ADCs are internalized by the cell upon the binding of the antibody to the target, although other ADCs reduce the blood supply of the tumor by targeting endothelial cells within the tumor vasculature. There are three internalization routes: clathrin-mediated endocytosis, caveolae-mediated uptake, and pinocytosis.8–18
The most common antibody form incorporated in ADCs is the full monoclonal antibody, or immunoglobulin (IgG). IgGs have a long half-life in blood, usually spanning from days to weeks, which enables them to travel in the blood vessels for a time long enough to locate the tumor cells.19–22 The second part of the ADC, the linker, is required to be stable during the circulation of the ADC in the bloodstream in order to avoid the premature release of the drug, as well as able to discharge the drug after the binding of the antibody to its antigen. Linkers currently explored can be categorized into cleavable and non-cleavable linkers. Cleavable linkers include lysosomal protease-sensitive linkers, acid-sensitive linkers, and glutathione-sensitive linkers.23–25 Regarding the cytotoxic part of the ADC, there are two main classes of ADC payloads that have been explored, the first one being drugs that disrupt microtubule assembly, such as auristatins and maytansinoids, and the second one being drugs that target DNA structure, such as calicheamicins and duocarmycins.8,9,25–29
Since the hypervariable regions of the antibody are expected to bind to the cancer-specific antigen, they are not advisable as conjugation sites. Instead, the conjugation of a linker to an antibody takes place at the more preserved regions of the antibody, at solvent-accessible reactive amino acids.30,31 Two amino acids typical for conjugation with drugs are lysines and cysteines, the latter being exposed after the reduction of the interchain disulfide bonds of the antibody. Even though the number of drugs linked to an antibody is usually 0–8, conjugation can occur at 40 different lysines and at 8 different cysteines per antibody. The numerous possible conjugation sites, in combination with the fact that the drug-to-antibody ratio (DAR) can be larger than 1, means that numerous different ADCs can be generated from the same antibody, drug, and linker. In addition, controlling the site and stoichiometry of drug conjugation to the antibody is not easy and typically results in heterogeneous mixtures of ADCs. Heterogeneous ADCs do not have well-defined in vivo pharmacokinetic properties, which is a disadvantage in terms of their therapeutic effect and manufacturing process, and they cannot be easily optimized. Therefore, the production of homogeneous ADCs is a necessity and a lot of effort is being made toward that direction. Part of that effort is the accomplishment of site-specific conjugation with a number of methods, such as cysteine engineering, glycoengineering, and enzymatic conjugation, as well as use of non-native amino acids such as selenocysteine, acetylphenylalanine, para-acetylphenylalanine, and para-azidophenylalanine.12,19,32–37
Drug development is a complicated, time-consuming, and expensive process. Bioinformatics is a multidisciplinary scientific field that aids this process, giving the opportunity to model biologically active molecules and make estimations about their properties. For example, computational drug design and molecular mechanics methods, although imperfect, aim to predict whether a given molecule will bind to a target and with what binding affinity. The information obtained through those computational methods, even though it is not exact, accelerates the process of drug discovery. However, even though a number of computational tools for drug design exist, the specialized field of ADC computational design has been explored to a much smaller degree.38–45
The in silico study of biological molecules requires their computational representation, usually in the Protein Data Bank (PDB) format. The PDB format provides a standard representation for three-dimensional structures of biological macromolecules, derived experimentally with X-ray diffraction and NMR studies. A PDB file includes information about the primary, secondary, and tertiary structure of the molecule described. The atomic coordinates of the molecule, as well as the bonds between its atoms are some of the most essential data contained. However, additional information can be included, such as crystallographic structure factors, NMR experimental data, sequence database references, and bibliographic citations.46 In this paper, a method of computational construction of ADCs using data from established databases is described. The three PDB files of the antibody, the linker, and the drug are processed and merged into a final PDB file of an ADC molecule. Specifically, the configuration of the linker and the drug molecules is changed so that they are aligned with the antibody, and hydrogen bonding occurs between the successive molecules in the ADC triplet. The amino acids of the antibody that were chosen to be conjugated with the linker are lysines in the surface of the antibody. The change in the configuration of the linker and the drug is accomplished via translation and rotation. The data used are antibodies from the RCSB Protein Data Bank and anticancer drugs from the Open National Cancer Institute Database, as well as the molecule C15N, which represents a non-cleavable linker, all as PDB files. The computational processes were executed in the C++ programming language. Molecular graphics and hydrogen addition were performed with the UCSF Chimera package. Chimera is developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIGMS P41-GM103311).41
Methods
Conjugation process
In this section, the process of the computational conjugation of the antibody, the linker, and the drug is described in more detail. As previously explained, the goal of the program developed is to produce the PDB file of an ADC molecule, given the PDB files of an antibody, a linker, and a drug. The drug and the linker are reconfigured via rotation and translation in order to be brought in positions appropriate for the hydrogen bonding to occur between the linker and the drug, as well as between the linker and a surface lysine of the antibody. The change in the configuration of the antibody, the linker, and the drug was executed computationally by changing their atomic coordinates. First, the drug was rotated and translated in relation to the linker, while the linker remained stable. The two files were subsequently merged into a linker–drug conjugate PDB file. Second, the linker–drug conjugate was rotated and translated in relation to the selected surface lysine of the antibody, while the antibody remained stable. Similarly, the two files were merged to produce the final PDB file of the antibody–drug conjugate.
In order for the reconfiguration of the molecules to be correct, the changes in their atomic coordinates had to preserve the initial lines and distances between the atoms, which was accomplished with the use of an affine transformation. An affine transformation is any transformation that preserves collinearity, which means that all points placed on a line before the transformation still lie on a line after the transformation. It also conserves the ratios of distances, which means that the midpoint of a line segment remains the midpoint after the application of the transformation.47,48 According to the homogeneous transformation matrix defined in Ref. 49, the change in the position of an atom can be described with the following equations,
where x, y, z are the initial coordinates of an atom, x’, y’, z’ are the new coordinates of the atom, a is the scaling factor, set at the value of 1 since in this project scaling was not necessary, rx, ry, rz are the rotation angles of the atom around the x, y, z axis, respectively, and Tx, Ty, Tz are the distances according to which the atom will be translated in the x, y, z direction, respectively.
In order to rotate a molecule in relation to another molecule, an axis was defined for each of them. The axis of the linker was defined as the line connecting its two most distant non-hydrogen atoms, in order to be representative of the shape of the molecule. For that purpose, the distance between every pair of heavy atoms of the linker was calculated, and the pair with the largest distance was chosen. These two atoms also participated in the connections of the linker with the drug and the antibody, as will be explained in the following paragraph. The axis of the drug was defined as the line connecting the hydrogen atom of the drug that took part in the hydrogen bond with the linker and the electronegative atom of the drug covalently bonded to it. The axis of the linker–drug conjugate was defined as the line connecting the two most distant non-hydrogen atoms of the linker–drug conjugate, similarly to the axis of the linker. Finally, the axis of the lysine was defined as the line connecting the alpha carbon of the lysine and the nitrogen atom of its side chain, since this line is directed toward the exterior of the antibody, which is the desired direction for the linker–drug conjugate.
Similarly, in order to translate a molecule in relation to another molecule, a distance between them had to be defined. The distance between the drug and the linker was defined as the distance between the hydrogen atom of the drug that participates in the hydrogen bond with the linker and the linker atom that participates in the hydrogen bond with the drug, which was selected to be one of the two most distant atoms of the linker. The distance between the linker–drug conjugate and the antibody is defined as the distance between the linker atom that participates in the hydrogen bond with the antibody, which was selected to be one of the two most distant atoms of the linker–drug conjugate, the one that belongs to the linker, and a hydrogen atom covalently bonded to the nitrogen of the lysine side chain.
As a molecule (ie, the drug or the linker–drug conjugate) rotates in relation to a stable molecule (ie, the linker or the antibody, respectively), or equivalently as the values of the variables rx, ry, rz change, the angle between its axis and the axis of the stable molecule is calculated. The values of rx, ry, rz for which this angle becomes minimal are selected, and finally the molecule is rotated according to those angles. Similarly, as a molecule (ie, the drug or the linker–drug conjugate) is translated in relation to a stable molecule (ie, the linker or the antibody, respectively), or equivalently as the values of the variables Tx, Ty, Tz change, the distance between those two molecules is calculated as described in the previous paragraph. The values of Tx, Ty, Tz for which that distance becomes minimal are chosen, and the molecule is translated according to those values.
According to the Steiner–Saenger definition of the hydrogen bond, a hydrogen bond is any cohesive interaction X–H•••A where H carries a positive and A a negative (partial or full) charge and the charge on X is more negative than on H. The molecule C15N, which represents a common non-cleavable linker, can form hydrogen bonds with a drug as well as a lysine amino acid of an antibody. In particular, every hydrogen atom of the drug that is connected to a heavy atom more electronegative than hydrogen can form a hydrogen bond with a heavy atom of the linker, such as carbon. Similarly, the nitrogen atom of the lysine side chain and the hydrogen atom covalently bonded to it can form a hydrogen bond with a carbon atom of the linker. After a molecule has been rotated and translated in relation to the stable molecule, the hydrogen bond is simulated computationally by deleting the hydrogen atom of the hydrogen donor from the PDB file, before the two files are merged into one PDB file.50
The final ADC molecule obtains a realistic conformation, as ensured by a final minimization step using the molecular dynamics package Gromacs, and specifically the GROMOS bonding library.51
Results
The application of the process described in the Methods section resulted in the generation of a number of ADC PDB files. Since the antibody and the drug, as well as the conjugation sites of the antibody and the drug, can vary, the number of different combinations of initial molecules generating distinct ADCs is quite large.
Here, four different ADCs are demonstrated as results of this process. The two conjugates depicted in Figure 2 contain the antibody with PDB id 4GAG. In the left conjugate, the linker has formed a hydrogen bond with the surface lysine with residue sequence number 147, located at the antibody chain with id L, while the drug with sequence number 600 (nci_600.pdb) has formed a hydrogen bond with the linker. In the right conjugate, the linker has been connected with a hydrogen bond to the surface lysine with residue sequence number 115, located at the antibody chain with id H, while the drug with sequence number 450 (nci_450.pdb) has formed a hydrogen bond with the linker. The conjugation areas of these two ADCs are depicted in Figure 3, from a different angle and a smaller distance.
Figure 2.
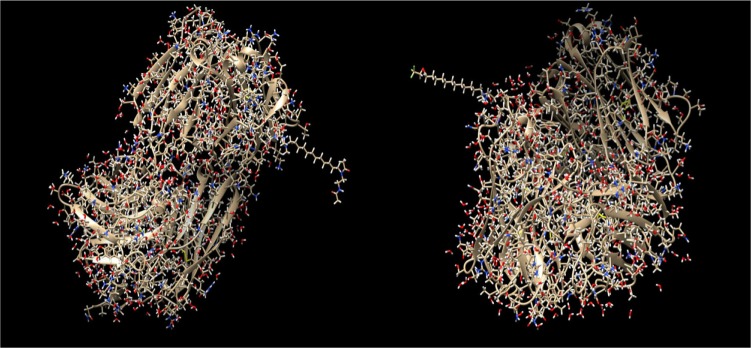
ADCs composed by the antibody with pdb id 4GAG, the linker molecule C15N, and a drug from the Open NCI database. Left: the drug with sequence number 600 (nci_600.pdb) has been conjugated through the linker molecule C15N to the surface lysine with residue sequence number 147, at the chain with id L. Right: the drug with sequence number 450 (nci_450.pdb) has been conjugated through the linker molecule C15N to the surface lysine with residue sequence number 115, at the chain with id H.
Figure 3.
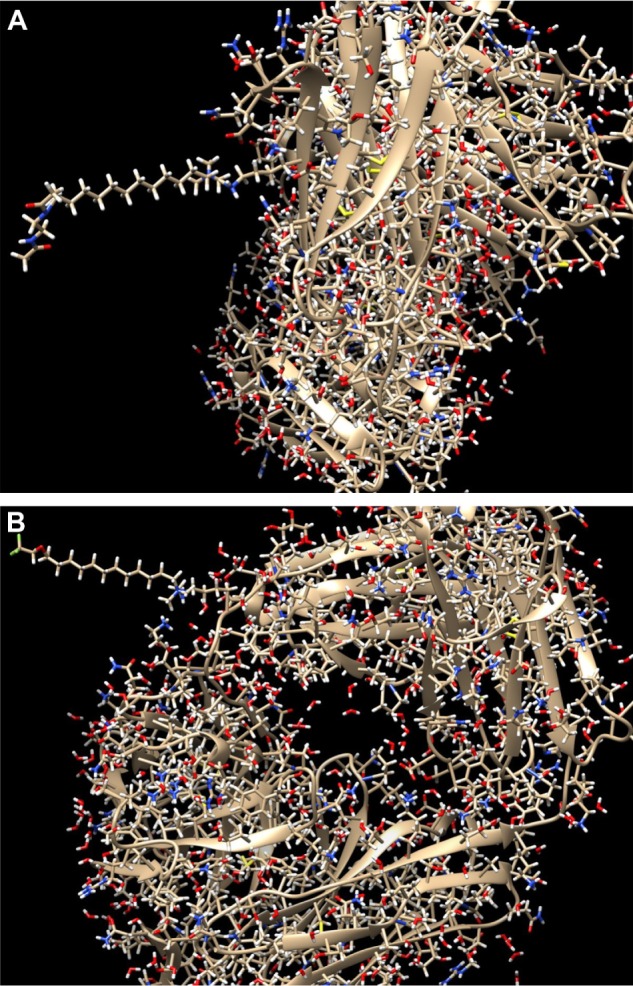
Zoomed images of the ADCs depicted in Figure 2, from different angles for better view of the conjugation. (A) Zoom in the conjunction of the drug with sequence number 600 (nci_600.pdb) to the surface lysine with residue sequence number 147 and chain id L of the antibody with pdb id 4GAG, through the linker molecule C15N. (B) Zoom in the conjunction of the drug with sequence number 450 (nci_450.pdb) to the surface lysine with residue sequence number 115 and chain id H of the antibody with pdb id 4GAG, through the linker molecule C15N.
In Figure 4, two different ADCs are illustrated, both containing the antibody with PDB id 4GAJ. In the left conjugate, the linker has formed a hydrogen bond with the surface lysine with residue sequence number 75, located at the antibody chain with id H, while the drug with sequence number 700 (nci_700.pdb) has formed a hydrogen bond with the linker. In the right conjugate, the linker has been connected to the surface lysine with residue sequence number 209, located at the chain with id H, with a hydrogen bond. Also, the drug with sequence number 14 (nci_14.pdb) has formed a hydrogen bond with the linker. The conjugation areas of these two ADCs are depicted in Figure 5, from a different angle and a smaller distance (Figs. 2–6).
Figure 4.
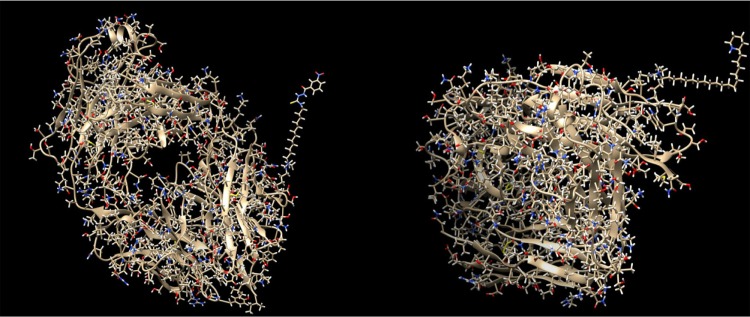
ADCs composed by the antibody with pdb id 4GAJ, the linker molecule C15N, and a drug from the Open NCI database. Left: the drug with sequence number 700 (nci_700.pdb) has been conjugated through the linker molecule C15N to the surface lysine with residue sequence number 75, at the chain with id H. Right: the drug with sequence number 14 (nci_14.pdb) has been conjugated through the linker molecule C15N to the surface lysine with residue sequence number 209, at the chain with id H.
Figure 5.
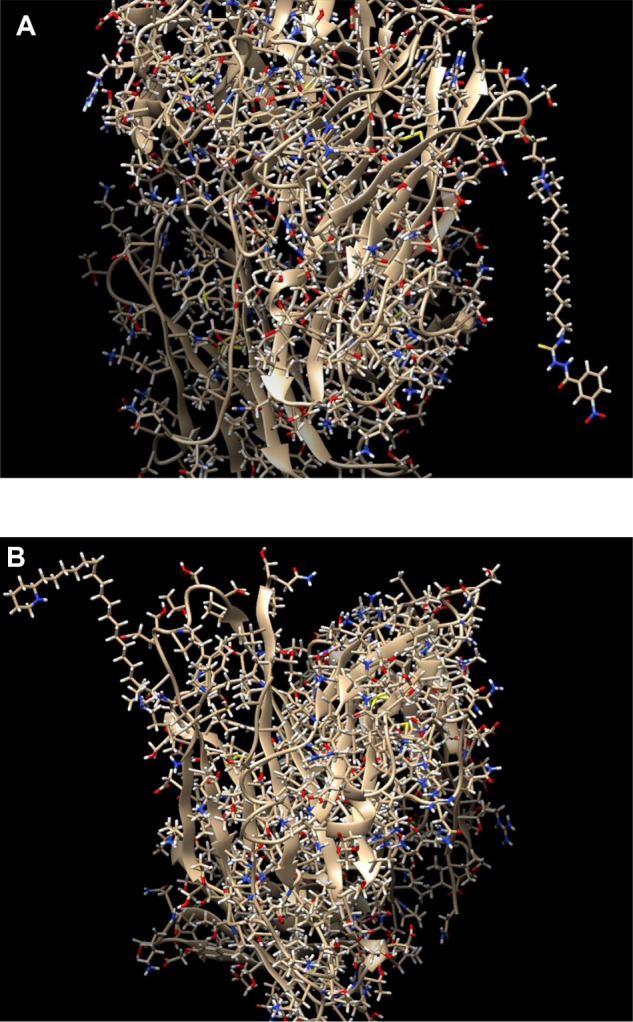
Zoomed images of the ADCs depicted in Figure 4, from different angles for better view of the conjugation. (A) Zoom in the conjunction of the drug with sequence number 700 (nci_700.pdb) to the surface lysine with residue sequence number 75 and chain id H of the antibody with pdb id 4GAJ, through the linker molecule C15N. (B) Zoom in the conjunction of the drug with sequence number 14 (nci_14.pdb) to the surface lysine with residue sequence number 209 and chain id H of the antibody with pdb id 4GAJ, through the linker molecule C15N.
Figure 6.
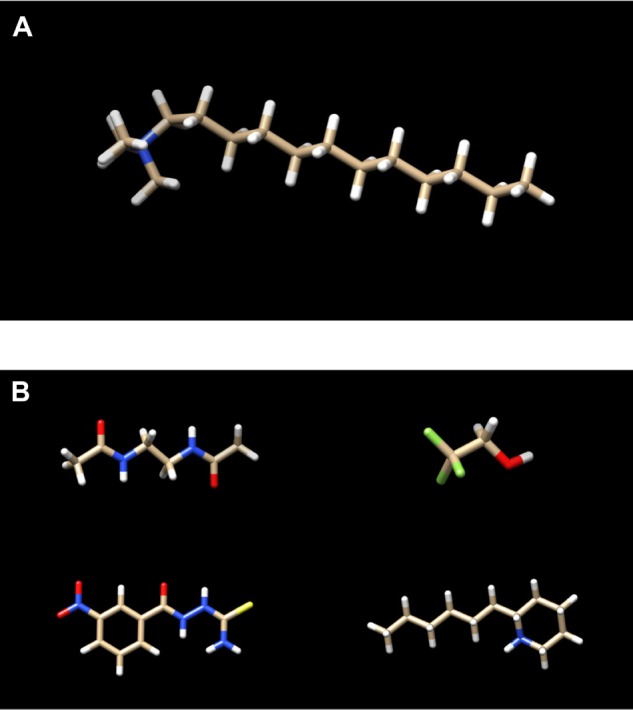
(A) The linker molecule (C15N) participating in the conjugations shown in Figures 2 and 3. (B) The cytotoxic substances participating in the conjugations shown in Figures 2 and 3. Top left: nci_600.pdb. Top right: nci_450.pdb. Bottom left: nci_700.pdb. Bottom right: nci_14.pdb.
Discussion
Computational prospects in the field of antibody– drug conjugation
In this paper, a process of computational construction of ADCs has been described, choosing surface lysines as the antibody conjugation sites. For this purpose, drug PDB files from the Open National Cancer Institute Database, antibody PDB files from the RCSB Protein Data Bank, and the PDB file of the non-cleavable linker molecule C15N were used. Between the linker and the drug, as well as between the antibody lysine and the linker–drug conjugate, hydrogen bonds were formed. The change in the configuration of the molecules was accomplished using the affine transformation.
The method described in this paper represents one of the many ways a drug can be conjugated with an antibody. Parameters such as the amino acid participating in the conjugation, the DAR, the type of linker and drug used, as well as the chemical bonds formed between the components of the conjugate can vary. Besides lysine, other natural or synthetic amino acids such as cysteine, selenocysteine, or acetylphenylalanine can be connected with a linker–drug conjugate. In addition, other non-cleavable or cleavable linkers can be used, while maytansinoids, auristatins, calicheamicins, and duocarmycins can be used as cytotoxic substances.
An example of a different conjugation technique is the formation of a covalent bond between an amino acid and a reactive functional group pendent to the linker–drug conjugate. The direct conjugation of a lysine with a linker–drug conjugate through amide bonds is also possible, using an N-hydroxysuccinimide (NHS) ester appended to the linker–drug, a method applied in the commercial ADC Mylotarg. Surface lysines of the antibody can be modified to introduce a reactive group such as maleimide, and then conjugated to a linker–drug that contains an appropriate reactive handle such as thiol, a method applied in the commercial ADC Kadcyla. Regarding cysteine conjugation, the controlled reduction of existing disulfide bonds in an antibody liberates cysteine residues, which can react with a maleimide attached to the linker–drug, a method used in the commercial ADC Adcetris. However, the conjugation processes described above are rather random and produce heterogeneous mixtures of conjugates with variable DARs. In addition, the site of conjugation could be different for each ADC species containing even only one drug. Heterogeneity in overall charge can impact negatively the solubility, stability, and pharmacokinetic properties of the produced ADC.52–55
The need for the development of homogeneous ADCs led to an increasing interest in site-specific conjugation, through which the number of conjugated drugs and the site of conjugation can be controlled. Site-specific conjugation has been achieved with a number of methods, such as cysteine engineering, amino acid insertion, enzymatic conjugation, and glycoengineering. THIOMAB–drug conjugates (TDCs) are a class of engineered antibodies in which cysteines have been introduced into the amino acid sequence by single point mutations. The ideal sites for mutation are identified with phage display techniques, while the engineered antibody is reduced and reoxidized to present the thiols of the mutated cysteines for conjugation. Conjugation using the engineered cysteine site leaves the antigen-binding regions unaffected and is quite homogeneous, with greater than 92% of the engineered antibody (THIOMAB) conjugates containing two drugs. Another approach is the replacement of interchain cysteines with serines in order to reduce the number of potential conjugation sites. Site-specific conjugation has also been accomplished by inserting non-native amino acids into the antibody, such as selenocysteine, acetylphenylalanine, para-acetylphenylalanine, and para-azidophenylalanine, which can provide orthogonal conjugation chemistries that otherwise are not available from functional groups present in the 20 canonical amino acids. For example, site-specific ADCs have been produced using anti-5T4 and anti-Her2 antibodies, with oxime bond formation between ketones on the side chain of the incorporated non-native amino acid and hydroxylamine-functionalized monomethyl auristatin D with either protease-cleavable or non-cleavable linkers. An additional strategy is conjugation using the enzymes glycotransferase and transglutaminase. In particular, glycotransferase has been used to attach a chemically active sugar functional group to a glycosylation site on an antibody, which serves as a conjugation site. Transglutaminase, on the other hand, has been used to form a bond between an amine group on the linker–drug and an engineered glutamine residue on the antibody. Chemoenzymatic approaches have been explored with the aim to generate bioorthogonal reactive groups for selective conjugation. For example, the formylglycine-generating enzyme (FGE) has been utilized to recognize a CXPXR sequence and convert a cysteine to formylglycine, in order to generate antibodies with aldehyde tags. Finally, site-specific conjugation has also been achieved by conjugating drugs at the glycosylation site of antibodies, without changing their amino acid sequence or cell culture condition. In particular, the native glycans on an asparagine residue (Asn297) of antibodies have been enzymatically remodeled in vitro using galactosyl and sialyltransferases in order to introduce terminal sialic acids. These sialic acids were subjected to periodate oxidation, resulting in aldehyde groups that were used to conjugate cytotoxic agents with oxime ligation. The ADCs produced with site-specific conjugation proved to have improved pharmacokinetic and therapeutic properties in comparison with the randomly generated ADCs. Therefore, a future step regarding ADC-related computational operations can be the simulation of site-specific conjugation methods as those mentioned above.25,36,37,52,56–60
Another aspect that could be examined is the evaluation of the computationally produced ADCs, perhaps taking into account the existing analytical technologies that assess the physicochemical properties of real ADCs. Properties of real ADCs, such as the DAR, drug distribution, aggregation, and fragmentation, are evaluated with a variety of techniques. For instance, UV/VIS spectroscopic analysis and hydrophobic interaction chromatography (HIC) are two methods used to measure the DAR, while technologies such as mass spectroscopic analysis and chromatographic analysis are used to evaluate drug distribution. Therefore, computational techniques could be developed in order to perform the equivalent analytical evaluations on computational ADCs, in order to further aid the design and manufacturing processes of real ADCs.10,61 In addition, computational techniques that make estimations about the pharmacokinetic and therapeutic properties of a given ADC would be beneficial to the process of ADC design, since an ADC could not only be constructed, but also evaluated in silico, obtaining valuable information in relatively little time and cost prior to in vitro development.
In conclusion, ADCs have emerged as a promising anticancer targeted therapy that aims to treat cancer by selectively attacking cancer cells while leaving normal cells unaffected for the most part. In comparison to conventional chemotherapy, this targeted mechanism of action has fewer side effects and enables the use of more potent drugs. The fact that ADCs are formed from three molecules instead of one makes their design and manufacturing even more complex than the already complicated discovery process of standalone drugs. However, it also results in an increased number of possible combinations of antibody conformation, linker, drug, and conjugation method. Therefore, the scientific field of ADCs provides vast research opportunities, not only in biology but also in bioinformatics, since specialized computational methods could benefit considerably the evolution of TAT.
Figure 1.
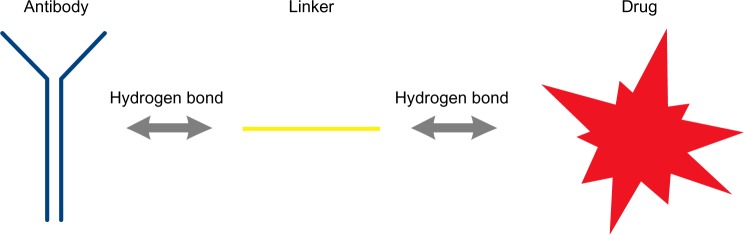
General scheme of the molecules and the bonds forming an ADC,9 according to the method described in this paper.
Footnotes
Author Contributions
Conceived and designed the experiments: AF, DV. Analyzed the data: AF, DV. Wrote the first draft of the manuscript: AF. Contributed to the writing of the manuscript: DV, GM, SK. Agree with manuscript results and conclusions: AF, DV, GM, SK. Jointly developed the structure and arguments for the paper: AF, DV. Made critical revisions and approved final version: AF, DV, GM, SK. All authors reviewed and approved of the final manuscript.
ACADEMIC EDITOR: JT Efird, Editor in Chief
FUNDING: Funding was received through an IKY Fellowship of Excellence for postgraduate studies in Greece – Siemens Program. The authors confirm that the funder had no influence over the study design, content of the article, or selection of this journal.
COMPETING INTERESTS: Authors disclose no potential conflicts of interest.
Paper subject to independent expert blind peer review by minimum of two reviewers. All editorial decisions made by independent academic editor. Upon submission manuscript was subject to anti-plagiarism scanning. Prior to publication all authors have given signed confirmation of agreement to article publication and compliance with all applicable ethical and legal requirements, including the accuracy of author and contributor information, disclosure of competing interests and funding sources, compliance with ethical requirements relating to human and animal study participants, and compliance with any copyright requirements of third parties. This journal is a member of the Committee on Publication Ethics (COPE).
REFERENCES
- 1.National Cancer Institute at the National Institutes of Health [Accessed August 1, 2014]. Available from: http://www.cancer.gov/. Updated in 2014.
- 2.World Health Organization [Accessed August 1, 2014]. Available from: http://www.who.int/. Updated in 2014.
- 3.American Cancer Society [Accessed August 2, 2014]. Available from: http://www.cancer.org/. Updated in 2014.
- 4.Cancer.net [Accessed August 2, 2014]. Available from: http://www.cancer.net/. Updated in 2014.
- 5.Cancer Research Institute [Accessed August 1, 2014]. Available from: http://www.cancerresearch.org/. Updated in 2014.
- 6.Cancer Research UK [Accessed August 2, 2014]. Available from: http://www.cancerresearchuk.org/. Updated in 2014.
- 7.Li J, Chen F, Cona MM, et al. A review on various targeted anticancer therapies. Target Oncol. 2012;7(1):69–85. doi: 10.1007/s11523-012-0212-2. [DOI] [PubMed] [Google Scholar]
- 8.Goldmacher VS, Chittenden T, Chari RVJ, Kovtun YV, Lambert JM. Antibody-drug conjugates for targeted cancer therapy. In: Desai MC, editor. Annu Rep Med Chem. Vol. 47. New York, NY: Academic Press; 2012. pp. 349–66. [Google Scholar]
- 9.Casi G, Neri D. Antibody-drug conjugates: basic concepts, examples and future perspectives. J Control Release. 2012;161(2):422–8. doi: 10.1016/j.jconrel.2012.01.026. [DOI] [PubMed] [Google Scholar]
- 10.Alley SC, Anderson KE. Analytical and bioanalytical technologies for characterizing antibody-drug conjugates. Curr Opin Chem Biol. 2013;17(3):406–11. doi: 10.1016/j.cbpa.2013.03.022. [DOI] [PubMed] [Google Scholar]
- 11.Kovtun YV, Goldmacher VS. Cell killing by antibody-drug conjugates. Cancer Lett. 2007;255(2):232–40. doi: 10.1016/j.canlet.2007.04.010. [DOI] [PubMed] [Google Scholar]
- 12.Conner SD, Schmid SL. Regulated portals of entry into the cell. Nature. 2003;422(6927):37–44. doi: 10.1038/nature01451. [DOI] [PubMed] [Google Scholar]
- 13.Schliemann C, Neri D. Antibody-based targeting of the tumor vasculature. Biochim Biophys Acta. 2007;1776:175–92. doi: 10.1016/j.bbcan.2007.08.002. [DOI] [PubMed] [Google Scholar]
- 14.Gerber HP, Senter PD, Grewal IS. Antibody drug-conjugates targeting the tumor vasculature. Current and future developments. MABS. 2009;1:247–53. doi: 10.4161/mabs.1.3.8515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Palumbo A, Hauler F, Dziunycz P, et al. A chemically modified antibody mediates complete eradication of tumours by selective disruption of tumour blood vessels. Br J Cancer. 2011;104:1106–15. doi: 10.1038/bjc.2011.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goldmacher VS, Blättler WA, Lambert JM, Chari RVJ. Immunotoxins and antibody–drug conjugates for cancer treatment. In: Muzykantov V, Torchilin V, editors. Biomedical Aspects of Drug Targeting. Boston, Dordrecht, London: Kluwer Academic Publishers; 2002. pp. 291–309. [Google Scholar]
- 17.Adler MJ, Dimitrov DS. Therapeutic antibodies against cancer. Hematol Oncol Clin North Am. 2012;26(3):447–81. doi: 10.1016/j.hoc.2012.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sapra P, Shor B. Monoclonal antibody-based therapies in cancer: advances and challenges. Pharmacol Ther. 2013;138(3):452–69. doi: 10.1016/j.pharmthera.2013.03.004. [DOI] [PubMed] [Google Scholar]
- 19.Wu AM, Senter PD. Arming antibodies: prospects and challenges for immunoconjugates. Nat Biotechnol. 2005;23(9):1137–46. doi: 10.1038/nbt1141. [DOI] [PubMed] [Google Scholar]
- 20.Wu AM, Yazaki PJ. Designer genes: recombinant antibody fragments for biological imaging. Q J Nucl Med. 2000;44:268–83. [PubMed] [Google Scholar]
- 21.Hulahov A, Chester KA. Engineered single chain antibody fragments for radioimmunotherapy. Q J Nucl Med Mol Imaging. 2004;48:279–88. [PubMed] [Google Scholar]
- 22.Neri D. Antibody-drug conjugates for cancer therapy. Nat Biotech. 2013;31(12) Available from: http://www.nature.com/nbt/extra/adc/pdf/adc.pdf. [Google Scholar]
- 23.LoRusso PM, Weiss D, Guardino E, Girish S, Sliwkowski MX. Trastuzumab emtansine: a unique antibody-drug conjugate in development for human epidermal growth factor receptor 2-positive cancer. Clin Cancer Res. 2011;17:6437–47. doi: 10.1158/1078-0432.CCR-11-0762. [DOI] [PubMed] [Google Scholar]
- 24.Younes A, Bartlett NL, Leonard JP, et al. Brentuximab vedotin (SGN-35) for relapsed CD30-positive lymphomas. N Engl J Med. 2010;363:1812–21. doi: 10.1056/NEJMoa1002965. [DOI] [PubMed] [Google Scholar]
- 25.Perez HL, Cardarelli PM, Deshpande S, et al. Antibody-drug conjugates: current status and future directions. Drug Discov Today. 2014;19(7):869–81. doi: 10.1016/j.drudis.2013.11.004. [DOI] [PubMed] [Google Scholar]
- 26.Doronina SO, Toki BE, Torgov MY, et al. Development of potent monoclonal antibody auristatin conjugates for cancer therapy. Nat Biotechnol. 2003;21:778–84. doi: 10.1038/nbt832. [DOI] [PubMed] [Google Scholar]
- 27.Chari RV, Martell BA, Gross JL, et al. Immunoconjugates containing novel maytansinoids: promising anticancer drugs. Cancer Res. 1992;52:127–31. [PubMed] [Google Scholar]
- 28.Hinman LM, Hamann PR, Wallace R, Menendez AT, Durr FE, Upeslacis J. Preparation and characterization of monoclonal antibody conjugates of the calicheamicins: a novel and potent family of antitumor antibiotics. Cancer Res. 1993;53:3336–42. [PubMed] [Google Scholar]
- 29.Coccia MA, Terrett JA, King DJ, et al. Human antibodies that bind CD70 and uses thereof. Medarex. Inc US Patent Appl. 20100150950:2007. [Google Scholar]
- 30.Iyer U, Kadambi VJ. Antibody drug conjugates – Trojan horses in the war on cancer. J Pharmacol Toxicol Methods. 2011;64(3):207–12. doi: 10.1016/j.vascn.2011.07.005. [DOI] [PubMed] [Google Scholar]
- 31.Carter PJ. Potent antibody therapeutics by design. Nat Rev Immunol. 2006;6:343–57. doi: 10.1038/nri1837. [DOI] [PubMed] [Google Scholar]
- 32.Wang L, Amphlett G, Blattler WA, Lambert JM, Zhang W. Structural characterization of the maytansinoidmonoclonal antibody immunoconjugate, huN901-DM1, by mass spectrometry. Protein Sci. 2005;14:2436–46. doi: 10.1110/ps.051478705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Junutula JR, Flagella KM, Graham RA, et al. Engineered thio-trastuzumab- DM1 conjugate with an improved therapeutic index to target human epidermal growth factor receptor2-positive breast cancer. Clin Cancer Res. 2010;16:4769–78. doi: 10.1158/1078-0432.CCR-10-0987. [DOI] [PubMed] [Google Scholar]
- 34.Dere R, Yi JH, Lei C, et al. PK assays for antibody-drug conjugates: case study with ado-trastuzumab emtansine. Bioanalysis. 2013;5:1025–40. doi: 10.4155/bio.13.72. [DOI] [PubMed] [Google Scholar]
- 35.Hamblett KJ, Senter PD, Chace DF, et al. Effects of drug loading on the antitumor activity of a monoclonal antibody drug conjugate. Clin Cancer Res. 2004;10:7063–70. doi: 10.1158/1078-0432.CCR-04-0789. [DOI] [PubMed] [Google Scholar]
- 36.Flemming A. Fine-tuning antibody-drug conjugates. Nat Rev Drug Discov. 2014;(13):178. doi: 10.1038/nrd4266. [DOI] [PubMed] [Google Scholar]
- 37.Tian F, Lu Y, Manibusan A, et al. A general approach to site-specific antibody drug conjugates. Proc Natl Acad Sci. 2014;111(5):1766–71. doi: 10.1073/pnas.1321237111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Filntisi A, Papangelopoulos N, Bencurova E, et al. State-of-the-art neural networks applications in biology. IIJSBBT. 2013;2(4):63–85. [Google Scholar]
- 39.Filntisi A, Vlachakis D, Matsopoulos G, Kossida S. 3D structural bioinformatics of proteins and antibodies: state of the art perspectives and challenges. IJSBBT. 2013;2(3):67–74. [Google Scholar]
- 40.Vlachakis D, Tsagkrasoulis D, Megalooikonomou V, Kossida S. Introducing Drugster: a comprehensive drug design, lead and structure optimization toolkit. Bioinformatics. 2013;29(1):126–8. doi: 10.1093/bioinformatics/bts637. [DOI] [PubMed] [Google Scholar]
- 41.Pettersen EF, Goddard TD, Huang CC, et al. UCSF Chimera – A visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–12. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 42.Chemical Computing Group Inc . Molecular Operating Environment (MOE), 2013.08. Montreal, QC, Canada: Chemical Computing Group Inc; 2014. [Google Scholar]
- 43.Toshimoto K, Wakayama N, Kusama M, Maeda K, Sugiyama Y, Akiyama Y. Drug Metab Dispos; In silico prediction of major drug clearance pathways by support vector machines with feature-selected descriptors. ; 2014. pp. 1811–9. [DOI] [PubMed] [Google Scholar]
- 44.Mitra A, Kesisoglou F, Dogterom P. Application of Absorption Modeling to Predict Bioequivalence Outcomme of Two Batches of Etoricoxib Tablets. AAPS PharmSciTech. 2014:1–9. doi: 10.1208/s12249-014-0194-8.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vlachakis D, Kossida S. Antibody drug conjugate bioinformatics: drug delivery through the letterbox. Comput Math Methods Med. 2013;2013:4. doi: 10.1155/2013/282398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.RCSB Protein Data Bank [Accessed June 4, 2014]. Available from: http://www.rcsb.org/. Updated in 2014.
- 47.Wolfram Mathworld [Accessed April 5, 2014]. Available from http://mathworld.wolfram.com/AffineTransformation.html. Updated in 2010.
- 48.Michigan Technical University, Department of Computer Science [Accessed April 5, 2014]. Available from: http://www.cs.mtu.edu/~shene/COURSES/cs3621/NOTES/geometry/geo-tran.html. Updated in 2002.
- 49.Planning Algorithms, Cambridge University [Accessed April 5, 2014]. Available from: http://planning.cs.uiuc.edu/node104.html. Updated in 2006.
- 50.Steiner T, Saenger W. Role of C-H.cntdot.cntdot.cntdot.O hydrogen bonds in the coordination of water molecules. Analysis of neutron diffraction data J Am Chem Soc. 1993;115(11):4540–7. [Google Scholar]
- 51.Van Der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJ. GROMACS: fast, flexible, and free. J Comput Chem. 2005;26(16):1701–18. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- 52.Junutula JR, Raab H, Clark S, et al. Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nat Biotechnol. 2008;26:925–32. doi: 10.1038/nbt.1480. [DOI] [PubMed] [Google Scholar]
- 53.Bross PF, Beitz J, Chen G, et al. Approval summary: gemtuzumab ozogamicin in relapsed acute myeloid leukemia. Clin Cancer Res. 2001;17:1490–6. [PubMed] [Google Scholar]
- 54.Senter PD. Potent antibody drug conjugates for cancer therapy. Curr Opin Chem Biol. 2009;13:235–44. doi: 10.1016/j.cbpa.2009.03.023. [DOI] [PubMed] [Google Scholar]
- 55.Boylan NJ, Zhou W, Proos RJ, Tolbert TJ, Wolfe JL, Laurence JS. Conjugation site heterogeneity causes variable electrostatic properties in Fc conjugates. Bioconjugate Chem. 2013;24:1008–16. doi: 10.1021/bc4000564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Panowski S, Bhakta S, Raab H, Polakis P, Junutula JR. Site-specific antibody drug conjugates for cancer therapy. MABS. 2014;6:34–45. doi: 10.4161/mabs.27022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhou Q, Stefano JE, Manning C, et al. Site-specific antibody-drug conjugation through glycoengineering. Bioconjugate Chem. 2014;25(3):510–20. doi: 10.1021/bc400505q. [DOI] [PubMed] [Google Scholar]
- 58.McDonagh CF, Turcott E, Westendorf L, et al. Engineered antibody-drug conjugates with defined sites and stoichiometries of drug attachment. Protein Eng Des Sel. 2006;19:299–307. doi: 10.1093/protein/gzl013. [DOI] [PubMed] [Google Scholar]
- 59.Carrico IS, Carlson BL, Bertozzi CR. Introducing genetically encoded aldehydes into proteins. Nat Chem Biol. 2007;3:321–2. doi: 10.1038/nchembio878. [DOI] [PubMed] [Google Scholar]
- 60.Rabuka D, Rush JS, deHart GW, Wu P, Bertozzi CR. Site-specific chemical protein conjugation using genetically encoded aldehyde tags. Nat Protoc. 2012;7:1052–67. doi: 10.1038/nprot.2012.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wakankar A, Chen Y, Gokarn Y, Jacobson FS. Analytical methods for physicochemical characterization of antibody drug conjugates. MABS. 2011;3:161–72. doi: 10.4161/mabs.3.2.14960. [DOI] [PMC free article] [PubMed] [Google Scholar]