

Abstract
Cell death and the release of chromatin have been demonstrated to activate the immune system producing autoantibodies against nuclear antigens in patients with systemic lupus erythematosus (SLE). Apoptosis, necrosis, necroptosis, secondary necrosis, autophagy and the clearance of dying cells by phagocytosis are processes believed to have a role in tolerance avoidance, activation of autoimmune lymphocytes and tissue damage by effector cells. The released chromatin not only activates the immune system; it also acts as antigen for the autoantibodies produced, including anti-dsDNA antibodies. The subsequent immune complex formed is deposited within the basement membranes and the mesangial matrix of glomeruli. This may be considered as an initiating event in lupus nephritis. The origin of the released chromatin is still debated, and the possible mechanisms and cell sources are discussed in this study.
Keywords: apoptosis, autophagy, necrosis, NETosis lupus nephritis
OTHER ARTICLES PUBLISHED IN THIS SERIES
Dying autologous cells as instructors of the immune system. Clinical and Experimental Immunology 2015, 179: 1–4.
Anti-dsDNA antibodies as a classification criterion and a diagnostic marker for systemic lupus erythematosus: critical remarks. Clinical and Experimental Immunology 2015, 179: 5–10.
Desialylation of dying cells with catalytically active antibodies possessing sialidase activity facilitate their clearance by human macrophages. Clinical and Experimental Immunology 2015, 179: 17–23.
Instructive influences of phagocytic clearance of dying cells on neutrophil extracellular trap generation. Clinical and Experimental Immunology 2015, 179: 24–29.
Developmental regulation of p53-dependent radiation-induced thymocyte apoptosis in mice Clinical and Experimental Immunology 2015, 179: 30–38.
Loading of nuclear autoantigens prototypically recognized by systemic lupus erythematosus sera into late apoptotic vesicles requires intact microtubules and myosin light chain kinase activity. Clinical and Experimental Immunology 2015, 179: 39–49.
Low and moderate doses of ionizing radiation up to 2 Gy modulate transmigration and chemotaxis of activated macrophages, provoke an anti-inflammatory cytokine milieu, but do not impact upon viability and phagocytic function. Clinical and Experimental Immunology 2015, 179: 50–61.
Vessel-associated myogenic precursors control macrophage activation and clearance of apoptotic cells. Clinical and Experimental Immunology 2015, 179: 62–67.
Acetylated histones contribute to the immunostimulatory potential of neutrophil extracellular traps in systemic lupus erythematosus. Clinical and Experimental Immunology 2015, 179: 68–74.
Unconventional apoptosis of polymorphonuclear neutrophils (PMN): staurosporine delays exposure of phosphatidylserine and prevents phagocytosis by MΦ-2 macrophages of PMN. Clinical and Experimental Immunology 2015, 179: 75–84.
Introduction
Chromatin or nucleosomes are the driving antigens in the induction of anti-double-stranded (ds)DNA antibodies. The presence of anti-dsDNA antibodies is a hallmark of systemic lupus erythematosus (SLE). The process is driven by special autoimmune T helper (Th) cells specific to epitopes in various DNA-binding nucleoproteins such as histones [1,2]. Anti-dsDNA antibodies form immune complexes (ICs) with nucleosomes that deposit within basement membranes in the body, e.g. skin and kidney, and may cause a systemic inflammation [3–6]. Lupus nephritis is characterized by the deposition of such ICs within the mesangial matrix and basement membranes of glomeruli, in addition to deposition within the basal membrane of the main renal arteries and microcapillaries surrounding the tubuli [7]. The binding of anti-dsDNA/nucleosomes immune complexes to basement membranes is mediated via nucleosomes. Nucleosomes show an affinity towards membrane components [8,9]. We have previously demonstrated that the production of anti-dsDNA antibodies, formation of ICs and subsequent deposition precedes the presence of infiltrating immune cells within the kidneys and the development of proteinuria of lupus-prone mice [10]. The release of nucleosomal antigens may therefore play a crucial role in the initiation of lupus nephritis.
Nucleosomes are complexes composed of dsDNA and histones. One nucleosome is composed of 180 base pairs of dsDNA and histone proteins organized as a protein octamer with the dsDNA wrapped in 1·65 turns of a superhelix [11]. An exterior linker histone stabilizes the structure together with a linker dsDNA connecting adjacent nucleosomes. Chromatin or nucleosomes contain protein complexes of DNA and histone binding proteins [12], and are normally located in the nucleus of the cell. Chromatin can be released during cell damage or death. However, a mechanism such as cell activation has also been shown to release nucleosomes in form of microparticles. The presence of nucleosomes has been detected in sera from normal individuals and in patients with SLE [13]. In mice, the levels of circulating nucleosomes decrease when anti-dsDNA antibody production increases during the progression of the disease, which may reflect formation and deposition of ICs [13]. Treatment with heparin prevented the deposition of ICs, probably by making the nucleosomes more accessible for degradation by nucleases [14]. The main source of nucleosomes in SLE is believed to originate from dead cells of apoptotic or necrotic origin. The possible mechanisms and cell sources of extracellular nucleosomes are discussed.
Mechanisms of programmed cell death as the origin for release of nucleosomes and activation of autoimmune cells
Apoptosis, or programmed cell death (PCD), is essential for embryonic development and renewal of tissue by eliminating cells that are abnormal and potentially dangerous [15]. PCD maintains homeostasis of the immune system, e.g. after massive expansion of reactive T cells and B cells in response to infection [16]. This is important in order to sustain immune tolerance and is a key process in the positive and negative selection of B and T lymphocytes eliminating potential self-reactive cells [17]. Apoptosis can be initiated by several death receptors on the cell surface or from signals within the cell in response to stress, DNA damage and defective cell cycle, etc. [15]. Apoptosis is characterized by activation of caspases, DNA fragmentation and membrane bleb formation [18]. Importantly, apoptosis does not normally activate the immune system, and apoptotic cells are cleared by phagocytes without the release of nucleosomes and with little consequent inflammation. However, nucleosomes are exposed at the cell surface of apoptotic blebs [19]. Inefficient clearance of dying cells may result in the accumulation of apoptotic cell remnants leading to a process called secondary necrosis [20]. This will lead to degeneration of the cell remnants and release of nucleosomes and other damage-associated molecular patterns (DAMPs) such as high-mobility group protein B1 (HMGB1), interleukin (IL)-1a, uric acid, mitochondrial content and adenosine triphosphate (ATP), like those observed during necrosis or necroptosis (see below) [21]. Incomplete chromatin digestion by phagocytes may also lead to the release of particles containing nucleic acids [20]. Chromatin modifications such as acetylation and methylation of histones have been demonstrated during apoptosis (reviewed in [22]), and autoantibodies specific for acetylated and methylated histone epitopes have been found in SLE patients and in mouse experimental models [23,24]. Because the apoptotic process may modify the nucleic acids to appear more like foreign antigen, e.g. a viral particle, the antigen-presenting cells [APCs, dendritic cells (DC) and B cells] may take it up, recognize it as foreign and activate it [25]. Apoptotic blebs with chromatin modifications have been demonstrated to activate DC with subsequent IL-17 production of co-cultured T cells [26].
In contrast to apoptosis, necrotic cell death is characterized by cellular organ and cytoplasma swelling, which leads to rupture of the plasma membrane and subsequent cell lysis [27]. It was originally considered a non-controlled death mechanism, but recent data have pointed out a tightly controlled type of necrosis called necroptosis [28]. This process is dependent upon the serine–threonine kinase RIP1 (RIPK1), and the RIP1 family member RIP3 has been identified as a key mediator of caspase-independent cell death [29]. The interplay between apoptosis and necroptosis is important for the elimination of excess T cells after an immune response [30]. Interestingly, the proinflammatory cytokine tumour necrosis factor (TNF) can induce apoptosis in certain cell types and necrosis on others [31]. Many other signals of the immune system, such as ligation of Fas, engagement of Toll-like receptors (TLR) or TCR by dsDNA and anti-CD3 antibodies, respectively, have been shown to trigger necroptosis [32]. The induction of necrosis or necroptosis by proinflammatory cytokines and other triggers may increase the release of nucleosomes. Necroptosis has not been investigated in SLE, but extensive studies on TNF-like weak inducer of apoptosis (TWEAK) indicate a central role for the TNF pathway in the induction of lupus nephritis [33].
Authophagy is a third mechanism that could lead to PCD. Autophagy is an intracellular degradation system that allows the cell to consume itself for energy. It is triggered by stress conditions such as starvation, or could be a part of the normal turnover of cellular contents [34]. Autophagy has been shown to be involved in the elimination of microorganisms, control of proinflammatory signalling, adaptive immunity and secretion of immune mediators (reviewed in [35]). Autophagy can rescue cells from the apoptotic process by interfering with the apoptotic signalling cascade. Inflammasome activation and secretion of immune mediators are regulated by autophagy through regulatory interactions with innate immune signalling pathways [35]. Autophagy has been demonstrated to be important in both T and B cell immunity by contributing to antigen presentation, T cell homeostasis and polarization, as well as to the survival of mature B cells. It is also required for plasmablast formation [35]. The impact of autophagy in SLE has been reviewed recently and the process has been shown to be activated in the disease [36]. Autophagy is increased in SLE patients and lupus-prone mice [36]. It is involved in the survival of autoreactive B cells and required for the differentiation of plasmablasts [37]. An increased amount of autophagic compartments was observed in T cells from lupus-prone mice and SLE patents, indicating a deregulation of autophagy in SLE [38].
In response to pathogens, neutrophils may die in a process called NETosis [39]. Upon stimulation or engagement with pathogens, neutrophils release extracellular traps (NETs) that consist of nucleosomes and neutrophil proteins. These may surround the pathogen and increase the likelihood of phagocytosis by macrophages [40]. NETs released by dying neutrophils are normally degraded by DNase1 within the circulation [40]. Impaired degradation of NETs has been seen in sera from SLE patients, and this is associated with lupus nephritis [41]. Recently published studies demonstrate that NETs can induce innate plasmacytoid DC (pDC) activation through TLR-9 and TLR-7 [42,43]. Increased infiltration and activation of neutrophils within the kidney with subsequent release of NETs may be harmful in SLE. The NETs may be targeted by anti-dsDNA antibodies which may promote IC deposition within the mesangial matrix and glomerular basement membrane (GBM), therefore representing a potentially crucial factor in the development of lupus nephritis (see below).
Dying cells as source of released chromatin in lupus nephritis
Whether or not apoptotic kidney cells are the source of released renal chromatin is controversial. Some groups demonstrated increased apoptosis of residential kidneys cells [3,4,44,45], while others claimed that there was no increase in apoptotic kidney cells of lupus patients and in mouse models of lupus nephritis [46,47]. Looking at immune complex deposition, there are several kidney cell types and infiltrating immune cells that might be a possible source of chromatin.
Glomerular mesangial cells, endothelial cells and podocytes
The first deposition of ICs is observed within the glomerular mesangial matrix. It is therefore likely that the source of released chromatin may come from dying mesangial cells. One of the theories of anti-dsDNA antibody deposition is the ‘planted antigen’ theory [48]. Chromatin released from mesangial cells into the mesangial matrix or glomerular basement membrane may bind circulating anti-dsDNA antibodies and thereby form the ICs observed as electron dense structures [48]. However, released chromatin without autoantibodies has never been observed in the mesangial matrix or the GBM of lupus-prone mice or in biopsies of SLE patients. ICs may be actively cleared by mesangial cells into the mesangial matrix [49]. Due to the affinity of nucleosomes towards the matrix components, a consequent accumulation of ICs are a more likely mechanism and may be named the ‘planted immune complex’ theory. The ICs may activate the mesangial cells through FcRs or TLRs to produce cytokines and chemokines that will attract infiltrating cells [7] (see below). We have recently demonstrated that nucleosomes alone can activate mesangial cells to produce cytokines and chemokines [50]. Other cells nearby, such as glomerular endothelial cells and podocytes, are believed to contribute to the subendothelial and subepithelial deposits observed within the GBM. This theory has never been proved, and it is speculated that it is more likely that the subendothelial and subepithelial depositions are due to the pressure of the blood flow forcing the ICs increasingly towards the epithelial side of the GBM, where they are stopped by the podocytes.
Tubular cells and microcapillary endothelial cells
Other renal cells include the different tubular cells and endothelial cells in arteries and capillaries within the renal circulation. Apoptosis of tubular cells has been detected in human kidney biopsies, but the levels were found to be too low to be considered as a reasonable quantitative source for autoantibody binding and IC formation [46]. Capillary endothelial cells are the most likely candidates for the source of released chromatin fragments of the residential kidney cells. They are in direct contact with the circulating anti-dsDNA antibodies/nucleosomes and the activated effector cells. Nucleosomes have been shown to activate endothelial cells to produce proinflammatory cytokines such as IL-6 and IL-8 [51]. In addition, endothelial bound nucleosomes and C1q are targets for autoantibodies and lead to activation of complement [52]. Endothelial cells are activated during inflammatory processes and may release microparticles containing chromatin fragments that bind anti-dsDNA antibodies [53,54]. Staining kidney sections of New Zealand black/white F1 (NZB/W F1) mice with anti-Meca32 and endoglin (CD105) antibodies revealed that IC deposits in the mesangial matrix contained Meca32 and endoglin (Fig. 1) during different stages of the disease. Endoglin is known to be expressed on activated endothelial cells, thereby explaining the staining within the glomeruli. Meca32 is not expressed on glomerular endothelial cells. This may indicate dead endothelial cells or released endothelial microparticles as a source of the chromatin found in IC depositions [55]. However, the release of microparticles into the circulation is not restricted to the kidney, and may originate from endothelial cells outside the kidneys.
Fig. 1.
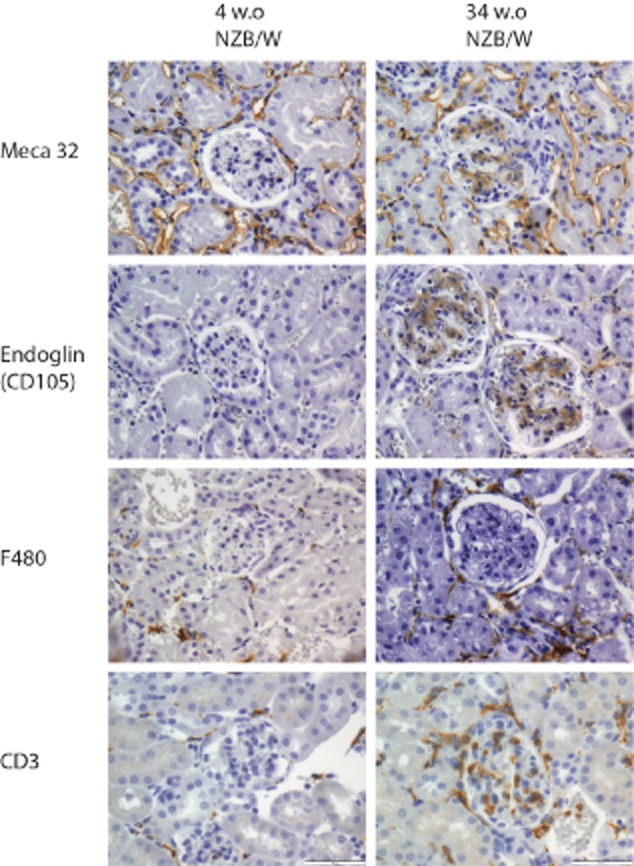
Glomerular detection of autologous cells as source of released chromatin. Kidney sections of lupus prone New Zealand black/white F1 (NZB/WF1) mice at different ages (4 and 34 weeks) were stained with anti-Meca32, anti-endoglin (CD105), anti-F480 (all from BioLegend, San Diego, CA, USA) and anti-CD3 (Dako, Glostrup, Denmark). Antibodies were detected by immunohistochemistry using Polink-2 Plus horseradish peroxidase detection kits for tissue (Golden Bridge International, Inc., Mukilteo, WA, USA) according to the protocol described in detail in [50]. All pictures are taken at ×400, scale bar = 50 μm.
Infiltrating granulocytes, macrophages, T and B cells
Polymorphonuclear leucocytes (granulocytes) are normally the first cells recruited to sites of inflammation [56]. Nucleosomes have been shown to induce activation and secretion of proinflammatory cytokines by polymorphonuclear neutrophils (PMN) [57]. Netting neutrophils have been demonstrated recently to induce endothelial damage, infiltrate tissue and expose immunological stimulatory molecules in SLE patients [58]. However, their role in lupus nephritis was not investigated [58]. Impairment in clearance of nets by reduced degradation is associated with proteinuria in lupus patients [41]. An imbalance in pro- and anti-apoptotic factors was shown in both neutrophils and sera from patients with juvenile-onset SLE, leading to increased neutrophil apoptosis in these patients [59]. Mouse studies have not revealed the role of neutrophils, as the mice have small amounts of these cells in the circulation in comparison to humans [56]. However, we found an increase in infiltrating granulocytes within the glomeruli and interstitia between tubuli in B/W mice with anti-dsDNA antibodies and proteinuric mice compared with young mice, although no indication that these cells are the origin of deposited chromatin [50].
Macrophages are essential for the clearance of apoptotic cell debris and ICs. Studies have shown that induction of apoptosis in circulating macrophages by chlodronate-filled liposomes accelerated the development of lupus nephritis in lupus-prone mice [60]. This was due possibly to a subsequent decrease in the clearance of apoptotic cells, thereby increasing the amount of circulating chromatin [60]. There are no known studies on the role of increased cell death of T cells in lupus nephritis. There are, however, enormous numbers of infiltrating T cells and macrophages within the kidney of lupus-prone mice and SLE patients. The T cells infiltrating glomeruli in lupus-prone mice are CD3+ T cells (Fig. 1) of all classes, Th1, Th2 and Th17 cells [61]. The fates of these cells during the course of the disease have not been investigated, but considering the amount and the life span of these helper and effector cells, they may be a source of released chromatin. Apoptosis of B cells has been demonstrated to accelerate lupus nephritis in lupus-prone mice [62]. Studies of B cell infiltration have revealed that this happens only after IC deposition and is located mainly In tertiary lymphoid structures within the kidney, and not in glomeruli and interstitia between tubules. However, B cells are essential for driving disease progression from silent mesangial nephritis to end-stage disease [63].
Concluding hypothesis
Production of anti-dsDNA antibody, formation of ICs and their subsequent deposition within the mesangial matrix activate mesangial cells to produce cytokines and chemokines, thereby attracting immune cells. Circulating ICs activate endothelial cells and may release chromatin-containing microparticles that, if not properly cleared, may act as antigen for autoantibodies. This will contribute to the amount of ICs formed. The infiltration of effector cells accelerates the inflammation and tissue damage within the glomeruli and activates endothelial cells. This will recruit and maintain lymphocyte infiltration into the interstitium. Increased inflammation will increase the amount of dying cells within the kidney, irrespective of their origin, and lead to excess amounts of released chromatin that amplify the disease process. The exact mechanism(s) leading to breakage of tolerance and activation of autoantigen-specific lymphocytes are still unknown. Novel strategies for the prevention of lupus nephritis may depend upon the further investigation of programmed cell death mechanisms and the role of circulating chromatin in SLE.
Acknowledgments
This work was supported by the Foundation for Health and Rehabilitation through the Norwegian Rheumatology Organization (project 2008/2/0229).
Disclosure
There are no competing interests.
References
- 1.Kaliyaperumal A, Mohan C, Wu W, Datta SK. Nucleosomal peptide epitopes for nephritis-inducing T helper cells of murine lupus. J Exp Med. 1996;183:2459–2469. doi: 10.1084/jem.183.6.2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.DesaiMehta A, Mao CC, Rajagopalan S, Robinson T, Datta SK. Structure and specificity of T-cell receptors expressed by potentially pathogenic anti-DNA autoantibody-inducing T-cells in human lupus. J Clin Invest. 1995;95:531–541. doi: 10.1172/JCI117695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kalaaji M, Fenton KA, Mortensen ES, et al. Glomerular apoptotic nucleosomes are central target structures for nephritogenic antibodies in human SLE nephritis. Kidney Int. 2007;71:664–672. doi: 10.1038/sj.ki.5002133. [DOI] [PubMed] [Google Scholar]
- 4.Kalaaji M, Mortensen E, Jorgensen L, Olsen R, Rekvig OP. Nephritogenic lupus antibodies recognize glomerular basement membrane-associated chromatin fragments released from apoptotic intraglomerular cells. Am J Pathol. 2006;168:1779–1792. doi: 10.2353/ajpath.2006.051329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hedberg A, Fismen S, Fenton KA, Mortensen ES, Rekvig OP. Deposition of chromatin–IgG complexes in skin of nephritic MRL-lpr/lpr mice is associated with increased local matrix metalloprotease activities. Exp Dermatol. 2010;19:e265–e274. doi: 10.1111/j.1600-0625.2010.01064.x. [DOI] [PubMed] [Google Scholar]
- 6.Fismen S, Hedberg A, Fenton KA, et al. Circulating chromatin–anti-chromatin antibody complexes bind with high affinity to dermo-epidermal structures in murine and human lupus nephritis. Lupus. 2009;18:597–607. doi: 10.1177/0961203308100512. [DOI] [PubMed] [Google Scholar]
- 7.Lech M, Anders HJ. The pathogenesis of lupus nephritis. J Am Soc Nephrol. 2013;24:1357–1366. doi: 10.1681/ASN.2013010026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mjelle JE, Rekvig OP, Fenton KA. Nucleosomes possess a high affinity for glomerular laminin and collagen IV and bind nephritogenic antibodies in murine lupus-like nephritis. Ann Rheum Dis. 2007;66:1661–1668. doi: 10.1136/ard.2007.070482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mjelle JE, Rekvig OP, van der Vlag J, Fenton KA. Nephritogenic antibodies bind in glomeruli through interaction with exposed chromatin fragments and not with renal cross-reactive antigens. Autoimmunity. 2011;44:373–383. doi: 10.3109/08916934.2010.541170. [DOI] [PubMed] [Google Scholar]
- 10.Fenton K, Fismen S, Hedberg A, et al. Anti-dsDNA antibodies promote initiation, and acquired loss of renal Dnase1 promotes progression of lupus nephritis in autoimmune (NZB×NZW)F1 mice. PLOS ONE. 2009;4:e8474. doi: 10.1371/journal.pone.0008474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 1997;389:251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- 12.Tan S, Davey CA. Nucleosome structural studies. Curr Opin Struct Biol. 2011;21:128–136. doi: 10.1016/j.sbi.2010.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jorgensen MH, Rekvig OP, Jacobsen RS, Jacobsen S, Fenton KA. Circulating levels of chromatin fragments are inversely correlated with anti-dsDNA antibody levels in human and murine systemic lupus erythematosus. Immunol Lett. 2011;138:179–186. doi: 10.1016/j.imlet.2011.04.006. [DOI] [PubMed] [Google Scholar]
- 14.Hedberg A, Fismen S, Fenton KA, et al. Heparin exerts a dual effect on murine lupus nephritis by enhancing enzymatic chromatin degradation and preventing chromatin binding in glomerular membranes. Arthritis Rheum. 2011;63:1065–1075. doi: 10.1002/art.30211. [DOI] [PubMed] [Google Scholar]
- 15.Taylor RC, Cullen SP, Martin SJ. Apoptosis: controlled demolition at the cellular level. Nat Rev Mol Cell Biol. 2008;9:231–241. doi: 10.1038/nrm2312. [DOI] [PubMed] [Google Scholar]
- 16.Opferman JT, Korsmeyer SJ. Apoptosis in the development and maintenance of the immune system. Nat Immunol. 2003;4:410–415. doi: 10.1038/ni0503-410. [DOI] [PubMed] [Google Scholar]
- 17.Fuchs Y, Steller H. Programmed cell death in animal development and disease. Cell. 2011;147:742–758. doi: 10.1016/j.cell.2011.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Siegel RM. Caspases at the crossroads of immune-cell life and death. Nat Rev Immunol. 2006;6:308–317. doi: 10.1038/nri1809. [DOI] [PubMed] [Google Scholar]
- 19.Radic M, Marion T, Monestier M. Nucleosomes are exposed at the cell surface in apoptosis. J Immunol. 2004;172:6692–6700. doi: 10.4049/jimmunol.172.11.6692. [DOI] [PubMed] [Google Scholar]
- 20.Munoz LE, Lauber K, Schiller M, Manfredi AA, Herrmann M. The role of defective clearance of apoptotic cells in systemic autoimmunity. Nat Rev Rheumatol. 2010;6:280–289. doi: 10.1038/nrrheum.2010.46. [DOI] [PubMed] [Google Scholar]
- 21.Kaczmarek A, Vandenabeele P, Krysko DV. Necroptosis: the release of damage-associated molecular patterns and its physiological relevance. Immunity. 2013;38:209–223. doi: 10.1016/j.immuni.2013.02.003. [DOI] [PubMed] [Google Scholar]
- 22.Fullgrabe J, Hajji N, Joseph B. Cracking the death code: apoptosis-related histone modifications. Cell Death Differ. 2010;17:1238–1243. doi: 10.1038/cdd.2010.58. [DOI] [PubMed] [Google Scholar]
- 23.van Bavel CC, Dieker JW, Kroeze Y, et al. Apoptosis-induced histone H3 methylation is targeted by autoantibodies in systemic lupus erythematosus. Ann Rheum Dis. 2011;70:201–207. doi: 10.1136/ard.2010.129320. [DOI] [PubMed] [Google Scholar]
- 24.van Bavel CC, Dieker JW, Tamboer WP, van der Vlag J, Berden JH. Lupus-derived monoclonal autoantibodies against apoptotic chromatin recognize acetylated conformational epitopes. Mol Immunol. 2010;48:248–256. doi: 10.1016/j.molimm.2010.08.003. [DOI] [PubMed] [Google Scholar]
- 25.Migliorini A, Anders HJ. A novel pathogenetic concept – antiviral immunity in lupus nephritis. Nat Rev Nephrol. 2012;8:183–189. doi: 10.1038/nrneph.2011.197. [DOI] [PubMed] [Google Scholar]
- 26.Fransen JH, Hilbrands LB, Ruben J, et al. Mouse dendritic cells matured by ingestion of apoptotic blebs induce T cells to produce interleukin-17. Arthritis Rheum. 2009;60:2304–2313. doi: 10.1002/art.24719. [DOI] [PubMed] [Google Scholar]
- 27.Golstein P, Kroemer G. Cell death by necrosis: towards a molecular definition. Trends Biochem Sci. 2007;32:37–43. doi: 10.1016/j.tibs.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 28.Linkermann A, Green DR. Necroptosis. N Engl J Med. 2014;370:455–465. doi: 10.1056/NEJMra1310050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ofengeim D, Yuan J. Regulation of RIP1 kinase signalling at the crossroads of inflammation and cell death. Nat Rev Mol Cell Biol. 2013;14:727–736. doi: 10.1038/nrm3683. [DOI] [PubMed] [Google Scholar]
- 30.Ch'en IL, Tsau JS, Molkentin JD, Komatsu M, Hedrick SM. Mechanisms of necroptosis in T cells. J Exp Med. 2011;208:633–641. doi: 10.1084/jem.20110251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Laster SM, Wood JG, Gooding LR. Tumor necrosis factor can induce both apoptic and necrotic forms of cell lysis. J Immunol. 1988;141:2629–2634. [PubMed] [Google Scholar]
- 32.Han J, Zhong CQ, Zhang DW. Programmed necrosis: backup to and competitor with apoptosis in the immune system. Nat Immunol. 2011;12:1143–1149. doi: 10.1038/ni.2159. [DOI] [PubMed] [Google Scholar]
- 33.Sanz AB, Izquierdo MC, Sanchez-Nino MD, et al. TWEAK and the progression of renal disease: clinical translation. Nephrol Dial Transplant. 2014;29(Suppl. 1):i54–i62. doi: 10.1093/ndt/gft342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147:728–741. doi: 10.1016/j.cell.2011.10.026. [DOI] [PubMed] [Google Scholar]
- 35.Deretic V, Saitoh T, Akira S. Autophagy in infection, inflammation and immunity. Nat Rev Immunol. 2013;13:722–737. doi: 10.1038/nri3532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhou XJ, Cheng FJ, Zhang H. Emerging view of autophagy in systemic lupus erythematosus. Int Rev Immunol. 2014 doi: 10.3109/08830185.2013.879711. doi: 10.3109/08830185.2013.879711. [DOI] [PubMed] [Google Scholar]
- 37.Clarke AJ, Ellinghaus U, Cortini A, et al. Autophagy is activated in systemic lupus erythematosus and required for plasmablast development. Ann Rheum Dis. 2014 doi: 10.1136/annrheumdis-2013-204343. doi: 10.1136/annrheumdis-2013-204343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gros F, Arnold J, Page N, et al. Macroautophagy is deregulated in murine and human lupus T lymphocytes. Autophagy. 2012;8:1113–1123. doi: 10.4161/auto.20275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brinkmann V, Reichard U, Goosmann C, et al. Neutrophil extracellular traps kill bacteria. Science. 2004;303:1532–1535. doi: 10.1126/science.1092385. [DOI] [PubMed] [Google Scholar]
- 40.Brinkmann V, Zychlinsky A. Beneficial suicide: why neutrophils die to make NETs. Nat Rev Microbiol. 2007;5:577–582. doi: 10.1038/nrmicro1710. [DOI] [PubMed] [Google Scholar]
- 41.Hakkim A, Furnrohr BG, Amann K, et al. Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc Natl Acad Sci USA. 2010;107:9813–9818. doi: 10.1073/pnas.0909927107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lande R, Ganguly D, Facchinetti V, et al. Neutrophils activate plasmacytoid dendritic cells by releasing self-DNA-peptide complexes in systemic lupus erythematosus. Sci Transl Med. 2011;3:73ra19. doi: 10.1126/scitranslmed.3001180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Garcia-Romo GS, Caielli S, Vega B, et al. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci Transl Med. 2011;3:73ra20. doi: 10.1126/scitranslmed.3001201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cui JH, Qiao Q, Guo Y, et al. Increased apoptosis and expression of FasL, Bax and caspase-3 in human lupus nephritis class II and IV. J Nephrol. 2012;25:255–261. doi: 10.5301/JN.2011.8451. [DOI] [PubMed] [Google Scholar]
- 45.Zheng L, Sinniah R, Hsu SI. Renal cell apoptosis and proliferation may be linked to nuclear factor-kappaB activation and expression of inducible nitric oxide synthase in patients with lupus nephritis. Hum Pathol. 2006;37:637–647. doi: 10.1016/j.humpath.2006.01.002. [DOI] [PubMed] [Google Scholar]
- 46.Faurschou M, Penkowa M, Andersen CB, Starklint H, Jacobsen S. Renal cell apoptosis in human lupus nephritis: a histological study. Lupus. 2009;18:994–999. doi: 10.1177/0961203309106175. [DOI] [PubMed] [Google Scholar]
- 47.Seredkina N, Zykova SN, Rekvig OP. Progression of murine lupus nephritis is linked to acquired renal Dnase1 deficiency and not to up-regulated apoptosis. Am J Pathol. 2009;175:97–106. doi: 10.2353/ajpath.2009.080943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.de Zubiria SA, Herrera-Diaz C. Lupus nephritis: an overview of recent findings. Autoimmune Dis. 2012;2012:849684. doi: 10.1155/2012/849684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schlondorff D, Banas B. The mesangial cell revisited: no cell is an island. J Am Soc Nephrol. 2009;20:1179–1187. doi: 10.1681/ASN.2008050549. [DOI] [PubMed] [Google Scholar]
- 50.Kanapathippillai P, Hedberg A, Fenton CG, Fenton KA. Nucleosomes contribute to increase mesangial cell chemokine expression during the development of lupus nephritis. Cytokine. 2013;62:244–252. doi: 10.1016/j.cyto.2013.03.016. [DOI] [PubMed] [Google Scholar]
- 51.Tanner JE. Nucleosomes activate NF-kappaB in endothelial cells for induction of the proangiogenic cytokine IL-8. Int J Cancer. 2004;112:155–160. doi: 10.1002/ijc.20390. [DOI] [PubMed] [Google Scholar]
- 52.O'Flynn J, Flierman R, van der Pol P, et al. Nucleosomes and C1q bound to glomerular endothelial cells serve as targets for autoantibodies and determine complement activation. Mol Immunol. 2011;49:75–83. doi: 10.1016/j.molimm.2011.07.020. [DOI] [PubMed] [Google Scholar]
- 53.Ostergaard O, Nielsen CT, Iversen LV, et al. Unique protein signature of circulating microparticles in systemic lupus erythematosus. Arthritis Rheum. 2013;65:2680–2690. doi: 10.1002/art.38065. [DOI] [PubMed] [Google Scholar]
- 54.Nielsen CT, Ostergaard O, Stener L, et al. Increased IgG on cell-derived plasma microparticles in systemic lupus erythematosus is associated with autoantibodies and complement activation. Arthritis Rheum. 2012;64:1227–1236. doi: 10.1002/art.34381. [DOI] [PubMed] [Google Scholar]
- 55.Pisetsky DS, Gauley J, Ullal AJ. Microparticles as a source of extracellular DNA. Immunol Res. 2011;49:227–234. doi: 10.1007/s12026-010-8184-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol. 2013;13:159–175. doi: 10.1038/nri3399. [DOI] [PubMed] [Google Scholar]
- 57.Lindau D, Ronnefarth V, Erbacher A, Rammensee HG, Decker P. Nucleosome-induced neutrophil activation occurs independently of TLR9 and endosomal acidification: implications for systemic lupus erythematosus. Eur J Immunol. 2011;41:669–681. doi: 10.1002/eji.201040593. [DOI] [PubMed] [Google Scholar]
- 58.Villanueva E, Yalavarthi S, Berthier CC, et al. Netting neutrophils induce endothelial damage, infiltrate tissues, and expose immunostimulatory molecules in systemic lupus erythematosus. J Immunol. 2011;187:538–552. doi: 10.4049/jimmunol.1100450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Midgley A, McLaren Z, Moots RJ, Edwards SW, Beresford MW. The role of neutrophil apoptosis in juvenile-onset systemic lupus erythematosus. Arthritis Rheum. 2009;60:2390–2401. doi: 10.1002/art.24634. [DOI] [PubMed] [Google Scholar]
- 60.Denny MF, Chandaroy P, Killen PD, et al. Accelerated macrophage apoptosis induces autoantibody formation and organ damage in systemic lupus erythematosus. J.Immunol. 2006;176:2095–2104. doi: 10.4049/jimmunol.176.4.2095. [DOI] [PubMed] [Google Scholar]
- 61.Apostolidis SA, Crispin JC, Tsokos GC. IL-17-producing T cells in lupus nephritis. Lupus. 2011;20:120–124. doi: 10.1177/0961203310389100. [DOI] [PubMed] [Google Scholar]
- 62.Trebeden-Negre H, Weill B, Fournier C, Batteux F. B cell apoptosis accelerates the onset of murine lupus. Eur J Immunol. 2003;33:1603–1612. doi: 10.1002/eji.200323665. [DOI] [PubMed] [Google Scholar]
- 63.Teichmann LL, Schenten D, Medzhitov R, Kashgarian M, Shlomchik MJ. Signals via the adaptor MyD88 in B Cells and DCs make distinct and synergistic contributions to immune activation and tissue damage in lupus. Immunity. 2013;38:528–540. doi: 10.1016/j.immuni.2012.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]