

Abstract
Most cases of systemic lupus erythematosus (SLE) are characterized by an impaired clearance of apoptotic cells in various tissues. Non-cleared apoptotic waste is considered an immunogen driving the autoimmune response in patients with SLE. During the execution of apoptosis, membrane blebs are formed and filled with cellular components. Here, we evaluate the cytoskeletal pathway(s) responsible for the loading of SLE prototypic nuclear autoantigens into the apoptotic cell-derived membranous vesicles (ACMV) generated during late phases of apoptosis. HeLa cells expressing a fusion protein of histone H2B with green fluorescent protein (GFP) were irradiated with ultraviolet (UV)-B to induce apoptosis. The appearance and trafficking of chromatin-derived material was monitored by fluorescence microscopy. Specific inhibitors of cytoskeletal pathways were employed to identify the motile elements involved in translocation and trafficking of the nuclear components. We observed that immediately after their appearance the ACMV did not contain histone H2BGFP; in this phase the fluorescence was contained in the nuclear remnants and the cytoplasm. Within consecutive minutes the ACMV were loaded with chromatin-derived material, whereas the loading of simultaneously created ACMV with histone H2BGFP was not uniform. Some ACMV were preferentially filled and, consequently, showed a remarkably higher histone H2BGFP accumulation. Inhibitors of the cytoskeleton revealed that functional microtubules and myosin light chain kinase are required for nuclear shrinkage and loading of nuclear material into the ACMV, respectively.
Keywords: apoptosis, blebbing, nuclear autoantigens, systemic lupus erythematosus
OTHER ARTICLES PUBLISHED IN THIS SERIES
Dying autologous cells as instructors of the immune system. Clinical and Experimental Immunology 2015, 179: 1–4.
Anti-dsDNA antibodies as a classification criterion and a diagnostic marker for systemic lupus erythematosus: critical remarks. Clinical and Experimental Immunology 2015, 179: 5–10.
The effect of cell death in the initiation of lupus nephritis. Clinical and Experimental Immunology 2015, 179: 11–16.
Desialylation of dying cells with catalytically active antibodies possessing sialidase activity facilitate their clearance by human macrophages. Clinical and Experimental Immunology 2015, 179: 17–23.
Instructive influences of phagocytic clearance of dying cells on neutrophil extracellular trap generation. Clinical and Experimental Immunology 2015, 179: 24–29.
Developmental regulation of p53-dependent radiation-induced thymocyte apoptosis in mice Clinical and Experimental Immunology 2015, 179: 30–38.
Low and moderate doses of ionizing radiation up to 2 Gy modulate transmigration and chemotaxis of activated macrophages, provoke an anti-inflammatory cytokine milieu, but do not impact upon viability and phagocytic function. Clinical and Experimental Immunology 2015, 179: 50–61.
Vessel-associated myogenic precursors control macrophage activation and clearance of apoptotic cells. Clinical and Experimental Immunology 2015, 179: 62–67.
Acetylated histones contribute to the immunostimulatory potential of neutrophil extracellular traps in systemic lupus erythematosus. Clinical and Experimental Immunology 2015, 179: 68–74.
Unconventional apoptosis of polymorphonuclear neutrophils (PMN): staurosporine delays exposure of phosphatidylserine and prevents phagocytosis by MΦ-2 macrophages of PMN. Clinical and Experimental Immunology 2015, 179: 75–84.
Introduction
Anti-nuclear autoantibodies appear in high titres in certain systemic rheumatic autoimmune diseases, such as systemic sclerosis [1], mixed connective tissue disease [2], juvenile idiopathic arthritis [3], chronic iridocyclitis [4] and, especially, systemic lupus erythematosus (SLE) [5]. These chronic inflammatory diseases are characterized by the presence of autoantibodies and immune complex deposition with activation of complement in various tissues causing acute or/and chronic inflammation and tissue damage [6,7]. This suggests an autoantigen-driven immune response to intracellular components altered and presented during cell death. It has been shown that the defective clearance of apoptotic cells and the accumulation of debris are crucial for the aetiopathogenesis of systemic autoimmunity [8].
Apoptosis is a regulated/programmed physiological process of cell death which plays an essential role in tissue turnover and homeostasis and has to be distinguished from the accidental, traumatic and un-regulated form of cell death usually referred to as necrosis [9]. The former occurs as a response to either noxious stimuli (extrinsic pathway) or to extracellular ligands activating the death receptors [Fas, tumour necrosis factor receptor (TNFR)-I], or is induced by the lack of survival signals, respectively [10].
Histones are basic nuclear chromatin proteins of eukaryotic cells. They operate like spools on which the cellular DNA is organized into nucleosomes; thus, they play a crucial role in condensing and stabilizing the chromatin and in regulating the transcription of genes. Five major classes of histones are known. H2A, H2B, H3 and H4 form a core structure referred to as nucleosome that serves as framework for the DNA double-strand helix. The linker histones H1/H5 are localized between the individual nucleosomes [11]. During apoptosis, the effector caspases 3, 6 and 7 are activated and cleave certain target proteins. This causes the breakdown of structures essential for the integrity of the cellular and subcellular architecture [12]. The cleavage of A- and B-type lamins, actin and ICAD (inhibitor of caspase activated DNAse, thereby freeing caspase activated DNase, CAD) causes the disassembly of the nucleus [13], the cytoskeleton [14] and chromatin [15], respectively. CAD initially generates high molecular weight DNA fragments by cleaving few internucleosomal sites of the chromatin, followed by the generation of mono- and oligonucleosomes, which occur as a so-called ‘DNA ladder’ in agarose gel electrophoresis [16,17]. The early fragmentation and extranuclear accumulation of chromatin fragments is a feature discriminating the apoptotic process from primary necrosis, where nuclei initially remain unaffected. A further characteristic of apoptosis is their swift non-phlogistic clearance by phagocytes, especially by macrophages [18]. These cells take up apoptotic cells and immediately degrade their macromolecules into small recyclable units such as nucleosides and amino acids. This highly efficient process avoids uncontrolled contact of the immune system with the corpses of dying cells, the challenge of tolerance and finally autoimmunity [19].
Indeed, studies employing monocyte-derived macrophages of patients with SLE revealed an impaired clearance of apoptotic cell material in several patients [20]. This observation, the highly increased incidence of SLE in complement-deficient individuals, and several animal models of clearance deficiency (MFG-E8KO [21], DNase 2KO [22], sIgMKO [23], lupus-prone mouse strains [24]), suggest that a deficient or impaired clearance of apoptotic cells is causally involved in the aetiology and the pathogenesis of SLE [7,25]. The major autoimmunogen of SLE is discussed to be secondary necrotic material evolved from apoptotic cells that had escaped proper clearance.
Late apoptosis and secondary necrosis are rare events in healthy subjects and the metabolism of nuclear autoantigens in late apoptotic cells remains elusive. Thus, to assess the fate of nuclear material during the stages of apoptosis, we employed HeLa cells expressing the fluorescent histone H2BGFP fusion protein and monitored localization and trafficking of nuclear material in the dying cells. We analysed the cellular and nuclear remnants and various apoptotic cell-derived membranous vesicles (ACMV) during late apoptosis in the presence and absence of inhibitors of cell dynamics in vitro.
Here we report that the small (< 1 μm) ACMVS and medium-sized (1–3 μm) ACMVM formed during the early stages of apoptosis never contained histone H2BGFP. In contrast, in some of the large (> 3 μm) ACMVL formed during later stages of cell death, histone H2BGFP can be detected readily. Importantly, the vesicles do not contain histone H2BGFP in statu nascendi, but some vesicles are loaded within a couple of minutes. Although the ACMVL are seemingly morphologically homogeneous, they differ in their load of nuclear autoantigens.
These findings corroborate the hypothesis that the loading of cleaved nuclear material into the ACMVL during late stages of apoptosis is not just a passive random process, but an active and regulated process. Experiments with inhibitors for cytoskeletal remodelling revealed that microtubules and myosin light chain kinase (MLCK) are required for nuclear shrinkage and for the sorting of degraded chromatin into the ACMVL, respectively.
Materials and methods
Stable transfection
Human HeLa cells were transfected with the pBOS-H2BGFP plasmid for expression of a histone H2B-GFP fusion protein (BD Pharmingen, Heidelberg, Germany) using FuGENE® transfection reagent (Promega, Mannheim, Germany), according to the manufacturer's instructions. Limiting dilution was performed twice consecutively in 96-well plates for 2 weeks each in culture medium supplemented with the antibiotic blasticidin (2 μg/ml) (Invivogen, Toulouse, France), against which pBOS-H2BGFP confers resistance. Selection of stable single clones was carried out by fluorescence microscopy.
Cell culture and induction of apoptosis
Cell culturing of the H2BGFP positive HeLa cell line was performed routinely at 37°C and 5·5% CO2 in R10 medium, consisting of RPMI-1640 medium supplemented with 10% fetal calf serum (FCS), 10% glutamine, 1% penicillin–streptomycin (Gibco/Invitrogen, Karlsruhe, Germany) and 1% HEPES (10 mM, pH 7·2) (Calbiochem/Merck, Darmstadt, Germany). To restrain the growth of HeLa cells not containing the H2BGFP expression plasmid, blasticidin (2 μg/ml) was added to the R10-medium. Apoptosis was induced by irradiation with ultraviolet-B (UV-B) (900 mJ/cm2).
Inhibition of cytoskeletal plasticity and induction of apoptosis
Adherent HeLa cells were cultured in six-well plates in R10 medium to a density of 3 × 104 cells/cm2. Ten min after the addition of one of the inhibitors for cytoskeletal plasticity displayed in Table 1 (all from Calbiochem/Merck, Darmstadt, Germany) cells were irradiated with UV-B and prepared for fluorescence microscopy.
Table 1.
Ultraviolet (UV)-B-induced apoptosis in the presence of inhibitors of cellular dynamic and motility
| Target | Inhibitor | Action | Inhibitor (conc.) | Observations |
|---|---|---|---|---|
| F-actin | Jasplakinolide | Stabilizer of F-actin | 50 nM | Cells disaggregate immediately into small exocytotic vesicles. Weak contraction of cell body. Late ACMVL contain H2BGFP |
| G-actin | Latrunculin A | Inhibitor of G-actin | 5 μM | Intermediate exocytotic vesicles. Plastic nuclei, plane periphery with small motile branches. Late ACMVL contain H2BGFP |
| p160ROCK | (R)-(+)-trans-N-(4-Pyridyl)-4-(1-aminoethyl)-cyclohexanecarboxamide) (Y27632) | Inhibitor of Rho-associated protein kinase (p160ROCK) | 5 μM | Some cells: strong contraction, extensive blebbing and chromatin condensation. Other cells cannot contract properly, sporadic late ACMVL contain H2BGFP |
| Myosin S1 ATPase | N-Benzyl-p-toluenesulphonamide (BTS) | Inhibitor of myosin S1-ATPase | 125 μM | Cells bring their morphology into a round figure: strong contraction and condensation of chromatin. Late ACMVL contain H2BGFP |
| MLCK | 1-(5-chloronaphthalene-1-sulphonyl)homopiperazine, HCl (ML-9) | Inhibitor of myosin light chain kinase (MLCK) | 10 μM | No characteristic morphological changes during apoptosis. Strong condensation of chromatin and shrinkage of nuclei. No H2BGFP in late ACMVL |
| Tubulin (β-subunit) | Baccatin III N-benzyl-β-phenylisoserine Ester, Taxol® (Paclitaxel) | Stabilizer of microtubules | 25 μM | No characteristic morphological changes during apoptosis. Faint condensation of chromatin, no/weak shrinkage of nuclei. No H2BGFP in late ACMVL |
For each condition a total of approximately 50 cells was observed and analysed accurately in fluorescence microscopy. ACMV = apoptotic cell-derived membranous vesicles; H2BGFP = histone 2-green fluorescent protein; ML-9 = 1-(5-chloronaphthalene-1-sulphonyl)-1H-hexahydro-1,4-diazepine. MLCK = myosin light chain kinase; ROCK = Rho-associated protein kinase.
Apoptosis in the absence of the inhibitors served as control. The cells were monitored for 24 h employing fluorescence microscopy (Axiovert 200 microscope; Zeiss, Göttingen, Germany; the ORCA-HRC4742-95 camera was supplied by Hamamatsu Photonics, Hamamatsu, Japan) and morphometry. The latter was performed using Adobe Photoshop CS5 to measure ACMV, nuclear areas and mean fluorescence intensity (MFI). Because several factors (e.g. photograph bleaching, exposure time, etc.) may affect the MFIs, all fluorescence microscopy pictures were taken under the same conditions. For morphometric analyses, a total of 25 cells were analysed for each condition. Before performing the experiment, it was ensured that the inhibitors had no effect on the cell viability during the observation period.
Results
ACMV are formed and loaded differentially with nuclear material during apoptosis
When we followed apoptosis in UV-B-irradiated H2BGFP transfected HeLa cells, we observed the formation of early small (ACMVS, < 1 μM), early intermediate (ACMVM, 1 μM–3 μM) and late large (ACMVL, > 3 μM) ACMV starting at approximately 180, 270 and 300 min after induction of apoptosis, respectively (Fig. 1a–d); we analysed approximately 50 cells by fluorescence microscopy.
Fig. 1.
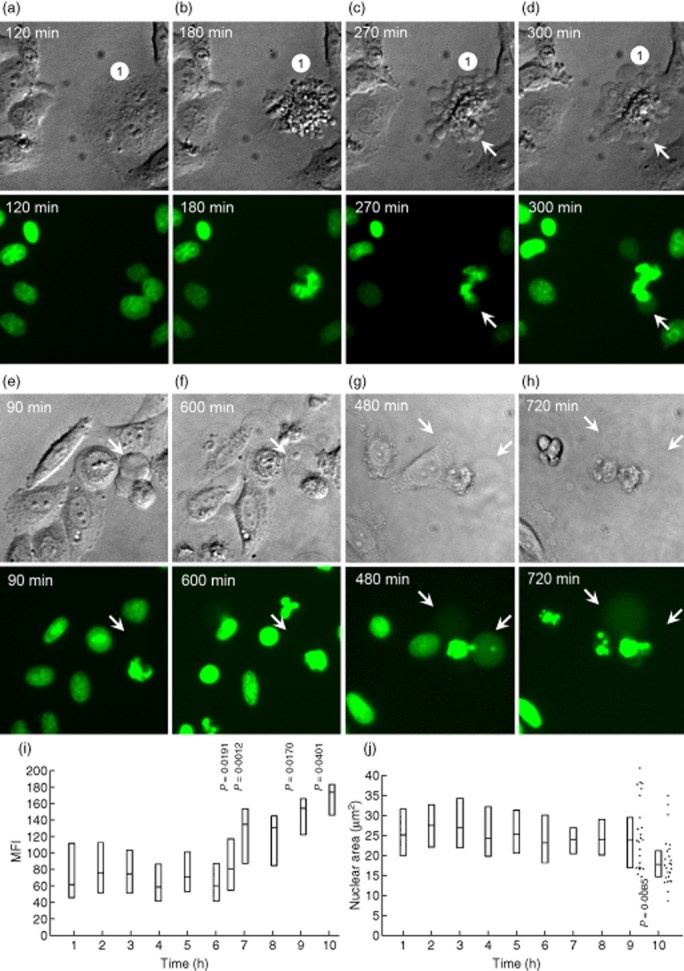
Redistribution of histone 2B-green fluorescent protein (H2BGFP) during ultraviolet (UV)-B-induced apoptosis in HeLa cells. Adherent HeLa cells expressing fluorescent H2BGFP histone were cultured to a density of 3 × 104 cells/cm2. After UV-B irradiation with 900 mJ/cm2 the distribution of H2BGFP was monitored by video-microscopy. (a–d) The characteristic nuclear changes that had been described for adherent cells undergoing apoptosis. After 270 min the cell marked with ‘1’ had undergone waves of formation of apoptotic cell-derived membranous vesicles (ACMV). None of these contained detectable amounts of H2BGFP; 30 min later two of the vesicles were filled with substantial amounts of H2BGFP (arrows). Note that (1) not all ACMV contain degraded chromatin (H2BGFP); (2) ACMV are formed initially without nuclear material, and are loaded in a second step (arrows). (e,f) Some large ACMVL remain unloaded even 10 h after their formation. (g,h) In very late phases of apoptosis some ACMVL which had been loaded with H2BGFP disintegrate and release their load into the culture supernatant, most probably during secondary necrosis. (i) mean fluorescence intensity (MFI) of the nuclei during the 10 h after irradiation. Note a significant increase of the MFI starting 6 h after irradiation (Student's t-test of cellular MFI at t = 0 versus t = × hours after irradiation). (j) nuclear area during the 10 h after irradiation. Note a significant shrinkage of the nuclei 9 h after irradiation (Student's t-test of cellular nuclear area at t = 0 versus t = × hours after irradiation).
Whereas at time-point 270 min none of the ACMV contained detectable H2BGFP (Fig. 1c), 30 min later few ACMV were loaded with chromatin fragments generated from the degraded nucleus (marked with arrows in Fig. 1c,d). Even after prolonged monitoring for up to 10 h, most of the ACMV did not contain visible amounts of H2BGFP (Fig. 1e,f). Observations of late cell death phases showed that some of the ACMVL which had been filled with H2BGFP lost their content by disintegration (Fig. 1g,h).
Morphometric measurements of H2BGFP-transfected HeLa cells revealed that the chromatin contracted significantly 6–7 h after induction of cell death, reflected by increased MFI values (Fig. 1i). Significant shrinkage of the nuclear area was to be observed after 9–10 h (Fig. 1j).
Cytoskeletal rearrangements are required for the morphological changes of apoptosing cells
Next we studied the morphological changes of apoptotic HeLa cells in the presence of inhibitors for cytoskeletal dynamics.
Jasplakinolide is a macrocyclic cyclodepsipeptide, extracted from the marine sponge Jaspis johnstoni [26]. It competes with phalloidin for the binding to F-actin. Induction of actin polymerization and stabilization of pre-existing actin filaments cause cytotoxic and anti-proliferative effects [27]. The effect of jasplakinolide on HeLa cell apoptosis is the almost immediate formation of motile ACMVS 10 min after the addition of jasplakinolide (Fig. 2a). The cells lose volume and shrink. However, the loading of H2BGFP material into late apoptotic ACMVL remains unaffected (Fig. 2b). Compared with control cells and cells treated with alternative inhibitors of cytoskeletal dynamics [Y27632, 1-(5-chloronaphthalene-1-sulphonyl)-1H-hexahydro-1,4-diazepine (ML-9), latrunculin A, N-benzyl-p-toluene sulphonamide (BTS)], we detected a significantly greater amount of ACMV in cells incubated with jasplakinolide 90 min after apoptosis induction (Fig. 2c).
Fig. 2.

F- and G-actin, p160ROCK and myosin are not required for loading into apoptotic cell-derived membranous vesicles (ACMV)L of histone 2B-green fluorescent protein (H2BGFP). Culturing and induction of apoptosis in HeLa cells was performed as described in Fig. 1. (a,b) HeLa cells were incubated with the F-actin inhibitor jasplakinolide. (a) Ten min after addition of the inhibitor, the cells start to form large numbers of ACMVS/M. (b) This leads to a loss of cell volume followed by shrinkage of the cell corpses. Whereas the inhibition of F-actin causes quick and dramatic changes in plasma membrane appearance, the creation and the loading of ACMVL with H2BGFP remains unaffected as to be observed 360 min after irradiation (arrows). (c) ACMV count 90 min after induction of apoptosis. HeLa cells incubated with jasplakinolide (Jasp) show a significant higher number of released ACMVS/M. HeLa cells treated with latrunculin A (Lat A) show no significant change in the ACMV count (most ACMV were observed transiently during the first 10–60 min (see d). (d,e,f) HeLa cells were incubated with the G-actin inhibitor latrunculin A. (d) Ten min after addition of the inhibitor, the cells start to form small ACMVS/M. (e) After 120 min the nuclei appear plastically with a flat cytoplasmic rim. (f) Note that loading of ACMVL with histone H2BGFP fusion protein remains unaffected as to be observed 720 min after irradiation (arrows). (g,h) HeLa cells were incubated with the p160ROCK inhibitor Y-27632; (g) 390 min after addition of the inhibitor, some cells display abnormalities in uropod protrusion (arrow). (g,h) Note that loading of ACMVL with H2BGFP remains unaffected (1). (i,j) HeLa cells were incubated with the myosin inhibitor N-benzyl-p-toluene sulphonamide (BTS); (i) 210 min after addition of BTS, most cells adopted a round shape but preserved their capabilities to move. (j) Note that ACMVL were still loaded with H2BGFP. Small, medium-sized and large apoptotic cell-derived membranous vesicles are marked ACMVS/M/l, respectively.
Latrunculin A is a macrolide toxin of the Red Sea sponge Latrunculia magnifica. It creates an equimolar complex with G-actin affecting the actin polymerization and interferes with microfilament organization. Latrunculin A has no impact on microtubules [28]. Within 10 min after addition of latrunculin A, HeLa cells generate ACMVS and ACMVM transiently and form membranous tubular extensions (Fig. 2d). After 90 min, HeLa cells treated with latrunculin A do not release newly generated ACMVS/M, and therefore show similar amounts of released ACMV compared with the control cells (Fig. 2c). The nuclei of latrunculin A-treated cells appear plastic, whereas the cytoplasm of the cell is somewhat flat (Fig. 2e). This plasticity disappears during progression of apoptosis. Inhibition of G-actin has no impact on nuclear shrinkage and on the redistribution of H2BGFP into ACMVL (Fig. 2f).
The pyridine derivate Y-27632 inhibits the functions of Rho-associated protein kinase (ROCK)-I and ROCK-II by binding to the catalytic site of these protein kinases. During the apoptotic process, ROCK-I is cleaved by caspase-3 at the carboxy-terminal region, thus becoming a constitutively activated kinase inducing membrane blebbing [29–31]. Treatment with Y-27632 inhibits uropod protrusion (Fig. 2g; arrow). Some cells (marked with ‘1’ in Fig. 2g,h) still performed the usual morphological changes with cell shrinkage and the formation of ACMVS. However, formation and loading of ACMVL was not affected (Fig. 2h); 90 min after apoptosis induction HeLa cells treated with Y-27632 have released similar amounts of ACMV compared with the control cells (Fig. 2c).
BTS is an aryl sulphonamide, which specifically inhibits the Ca2+-stimulated myosin S1 ATPase and compromises gliding motility reversibly. BTS does not compete for the nucleotide-binding site of myosin II; rather, it handicaps myosin's interaction with F-actin. The primary target of BTS is Ca2+-stimulated S1 ATPase and the secondary target is actin-stimulated ATPase [32]. If HeLa cells are incubated with BTS, most of them contract and adopt a round shape. The cells are not ‘paralyzed’, as they still maintain their ability to move (Fig. 2i). ACMVL were formed and loaded with H2BGFP in the presence of BTS (Fig. 2j). BTS-treated HeLa cells show normal numbers of ACMV after 90 min (Fig. 2c).
Microtubules are required for the nuclear shrinkage of apoptosing cells
Paclitaxel is an anti-proliferative drug isolated from the bark of the pacific yew tree (Taxus brevifolia). It stabilizes microtubule formation by interacting with tubulin on its β-tubulin subunit [33] and prevents disassembly of the microtubule polymer. We observed that paclitaxel-treated H2BGFP-transfected HeLa cells formed ACMVS and ACMVM within 30 min after irradiation (not shown); 60 min later the cells adopted a unique phenotype. They remained adherent, formed ACMVL, lost their mobility and did not condense and degrade their chromatin and nuclei, respectively (Fig. 3a). This appearance lasted for at least 960 min. At this time-point, most nuclei displayed a viable morphology with no signs of condensation and shrinkage. Faint ACMVL were visible; however, none of these were loaded with H2BGFP (Fig. 3b). Morphometric measurements of the area of nuclei during a period of 10 h revealed that cells incubated with paclitaxel, an inhibitor of microtubules, stopped the shrinkage process at an early point of cell death (Fig. 4). The cells are not able to degrade and condense their nuclear material. Thus, they cannot translocate the histone H2BGFP fusion protein into ACMV (Fig. 3b).
Fig. 3.

Microtubules and myosin light chain kinase (MLCK) are required for nuclear shrinkage and loading of apoptotic cell-derived membranous vesicles (ACMV)L with histone 2B-green fluorescent protein (H2BGFP), respectively. (a,b) HeLa cells incubated with an agent stabilizing microtubules (paclitaxel) are arrested at an early stage of apoptosis. (b) Nuclear shrinkage is impaired as well as the sorting of nuclear autoantigens into late ACMVL. (c,d) The formation of ACMVL is not affected during apoptosis in the presence of 1-(5-chloronaphthalene-1-sulphonyl)-1H-hexahydro-1,4-diazepine (ML-9), an inhibitor of myosin light chain kinase (MLCK). (d) Nuclear shrinkage and chromatin condensation is performed regularly. However, sorting of nuclear autoantigens into late ACMVL cannot be observed. (e) In the absence of inhibitors, and in the presence of jasplakinolide, latrunculin A, Y-27632 and N-benzyl-p-toluene sulphonamide (BTS) histone H2BGFP-positive ACMVL are readily observed (arrows). Paclitaxel conserves the cell at an early state of cell death. Nuclear shrinkage and chromatin condensation is arrested. Cells incubated with ML-9 regularly generate ACMVL; translocation of nuclear autoantigens into these was not detected.
Fig. 4.
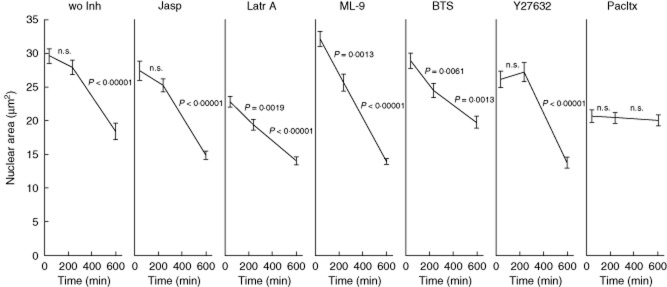
Morphometric measurement of nuclear size 30, 240 and 600 min after irradiation. Y-27632 and jasplakinolide do not have any influence on nuclear shrinkage. 1-(5-chloronaphthalene-1-sulphonyl)-1H-hexahydro-1,4-diazepine (ML-9) and N-benzyl-p-toluene sulphonamide (BTS) significantly accelerate and decelerate nuclear shrinkage, respectively. Cells treated with latrunculin A or paclitaxel (Pacltx) already display a significantly smaller nuclear area 30 min after treatment. Whereas cells treated with latrunculin A continue shrinkage, the nuclei of paclitaxel-treated cells maintain their size for at least 10 h.
MLCK activity is required for the loading of H2B into late ACMVL
ML-9 is a synthetic compound that inhibits the catalytic site of MLCK with high selectivity. It competes with ATP and acts as an inhibitor of the phosphate acceptor in the catalytic centre [34]. Employing ML-9, we observed a somewhat normal execution of apoptosis in HeLa cells; 270 min after irradiation most cells harboured condensed chromatin and regularly formed ACMVM/l (Fig. 3c). Up to 960 min after induction of apoptosis the nuclei appeared shrunken and displayed condensed amounts of H2BGFP. However, we never observed loading into ACMVL of H2BGFP (Fig. 3d). ML-9-treated cells showed normal numbers of ACMV compared with the apoptotic control cells 90 min after apoptosis induction (Fig. 2c).
Table 1 and Figs 3e and 4 summarize the results of the inhibitor study. Whereas in the apoptotic control cells and in the presence of the inhibitors jasplakinolide, latrunculin A, Y-27632 and BTS H2BGFP-loaded ACMVL are readily seen (arrows), paclitaxel and ML-9 inhibited the formation and loading, respectively, of ACMVL (Figs 3e and 4).
Discussion
In this study, we analysed the fate of a prototypic nuclear autoantigen recognized by the humoral immune response in patients with SLE in the execution phase of apoptotic cell death in vitro. We observed that the histone H2BGFP fusion protein is loaded into ACMVL that are generated in the late stages of apoptosis. Pharmacological inhibition studies revealed that microtubules and MLCK are required for nuclear shrinkage and for packing nuclear autoantigens into membranous apoptotic vesicles, respectively (Fig. 5).
Fig. 5.
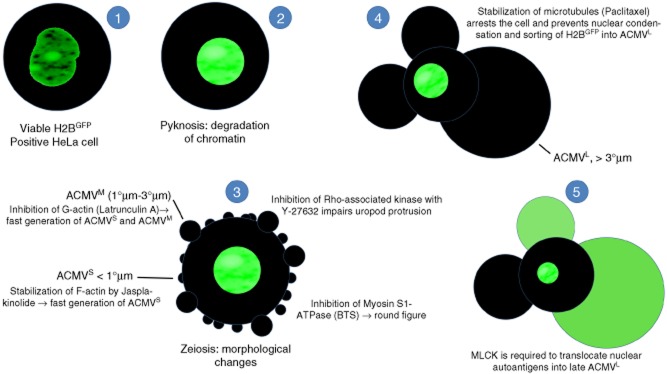
Synopsis: schematic display of the translocation in apoptotic cells of nuclear autoantigens. HeLa cells were transfected with pBOS-histone 2B-green fluorescent protein (H2BGFP) to label histone H2B as a representative of nuclear proteins serving as autoantigens in patients with systemic lupus erythematosus (SLE). After induction of apoptosis via irradiation with ultraviolet (UV)-B, processing and translocation of H2B was monitored employing fluorescence microscopy. The transfected cells were incubated with inhibitors of cell dynamics to identify the cytoskeletal proteins involved in the trafficking of H2B. (1) Viable H2BGFP expressing cell with a regularly configured nucleus. (2) Apoptosis starts with chromatin condensation and degradation (pyknosis). (3) In early apoptosis, the cells undergo dramatic morphological changes. They shrink and generate apoptotic cell-derived membranous vesicles (ACMV), zeiosis. Initially small (< 1 μm) and intermediate (1–3 μm) ACMVS/M are generated. These ACMV do not contain substantial amounts of H2BGFP. Several inhibitors of cell dynamics have impact on the morphology of the dying cell. p160ROCK (Y-27632) is required for uropod protrusion. Inhibition of F-actin (jasplakinolide) or G-actin (latrunculin A) leads to the early generation of ACMVS and ACMVM. Inhibition of myosin S1 ATPase [N-benzyl-p-toluene sulphonamide (BTS)] causes rounding of the cell. (4) However, all the above listed inhibitors do not have any impact on formation of late ACMVL and the translocation of H2B into these. Inhibition of microtubules (paclitaxel) arrests the apoptotic process at an early time-point. It impairs nuclear shrinkage as well as formation of ACMVL and translocation of H2BGFP. (5) Inhibition of myosin light chain kinase (MLCK) by 1-(5-chloronaphthalene-1-sulphonyl)-1H-hexahydro-1,4-diazepine (ML-9) does not preclude the formation of ACMVL, but impairs their loading with histone H2BGFP.
During apoptosis, cells undergo a plethora of morphological changes, including cellular shrinkage, condensation of chromatin (pyknosis), nuclear fragmentation (karyorrhexis) and development of bubble-like surface blebs (zeiosis), and break down into subcellular membranous vesicles (referred to in this paper as ACMV) (Fig. 5). Apoptotic cells usually release ‘find-me’ signals, expose ‘eat me’ signals and orchestrate their swift and early clearance accompanied by the release of the tolerogenic cytokines interleukin (IL)-10 and transforming growth factor (TGF)-β [35,36].
Consequently, apoptotic cells are rarely seen in healthy tissues, and even sites of massive apoptosis do not show any signs of inflammation. Indeed, apoptotic cells have been reported to dampen inflammation [36]. For rare cases of apoptotic cells that had escaped this primary, phosphatidylserine-dependent clearance, back-up mechanisms exist that opsonize late apoptotic cells that expose immature glycoproteins [37,38].
A proper clearance, as found in most subjects, means that apoptotic cells are phagocytosed before they lose their membrane integrity without induction of inflammatory responses. In many patients with SLE the clearance process is impaired, as reflected by the occurrence in the affected tissues of post-apoptotic debris and inflammation. In the germinal centres of the lymph nodes, nuclear autoantigens released from secondary necrotic cells have been identified to decorate the surfaces of follicular dendritic cells and may there serve as selecting antigens for B cells that have accidentally gained autoreactivity during somatic diversification [39]. The molecular basis of this defect includes deficiencies for the complement components C1q [40], C2 [41] and C4 [42] and even a lupus-associated genetic variant in CR3 [43]. Furthermore, anti-dsDNA and other autoantibodies have been identified to shift the clearance of apoptotic cells towards inflammation [8,44]. Clearance deficiency, humoral autoimmunity and inflammation induced by antibody-bound late apoptotic/secondary necrotic cells fuel a vicious circle that precipitates tissue inflammation in patients with SLE [7].
The accumulation of cellular remnants is prone to challenge self-tolerance due to access of the immune system to post-apoptotic material in an inflammatory environment. Therefore, we investigated the fate of the prototypic nuclear autoantigen histone H2B in the late phases of apoptosis, which occur predominantly in individuals with a deficient clearance. We observed that nuclear histone H2BGFP is detectable in large membranous vesicles (ACMVL) formed in the late stages of apoptosis in vitro. In the early-occurring ACMVS/M we did not observe histone H2BGFP-related fluorescence (Fig. 5). However, this does not exclude the presence of chromatin in these vesicles; it only shows that the amount of DNA has not reached the level of detection, which requires other methods [45,46]. We used the H2BGFP distribution to follow the translocation of bulk chromatin for several hours and did not intend to detect minor traces of chromatin.
Coleman and colleagues described that ROCK I is necessary and sufficient for formation of membrane blebs and for relocalization of fragmented DNA into blebs and apoptotic bodies [29]. In our system, treatment with Y-27632 inhibits uropod protrusion and generation of ACMVS. However, loading of ACMVL was not prevented. In agreement with the above-mentioned paper, we observed that MLCK activity is required for loading with chromatin (reflected by H2BGFP translocation) of ACMVL [29].
In summary, we describe that translocation of the hallmark autoantigens addressed by the humoral autoimmune response in patients with SLE are loaded actively into large preformed membrane vesicles (ACMVL) in the late stages of apoptosis (Fig. 5). Although these stages are somewhat rare, and seldom occur in healthy subjects, they may become prominent in individuals with deficiency of apoptotic cell clearance. It may be speculated that this translocation of cleaved chromatin is involved in the atiopathogenesis of SLE. However, whether or not pharmacological interference with chromatin redistribution can be employed for the treatment of SLE needs further investigation.
Acknowledgments
This work was supported by the Masterswitch project of the European Union by SFB 643, the DFG training grant GK SFB 643, by the Emerging Fields Initiative of the Friedrich-Alexander-University Erlangen-Nuremberg, the Interdisciplinary Center of Clinical Research (IZKF) (grant J-20) at the University Hospital Erlangen and the K&R Wucherpfennig Stiftung. We thank Professor Andrew H. Wyllie (Department of Pathology, University of Cambridge, UK) for fruitful discussions during a conference on the clearance of apoptotic cells.
Author contributions
M. Z., C. J., L. M. and B. F. performed the experiments; R. V. provided the stably transfected HeLa cells. C. G., G. S. and M. H. designed the study and wrote the manuscript.
Disclosures
The authors declare no conflicts of interest.
References
- 1.Hasegawa M, Sato S, Kikuchi K, Takehara K. Antigen specificity of antihistone antibodies in systemic sclerosis. Ann Rheum Dis. 1998;57:470–475. doi: 10.1136/ard.57.8.470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wayaku T, Hasegawa M, Kaji K, et al. Antigen specificity of antihistone antibodies in connective tissue disease patients with anti-U1RNP antibodies. Rheumatol Int. 2007;28:113–119. doi: 10.1007/s00296-007-0398-2. [DOI] [PubMed] [Google Scholar]
- 3.Ostensen M, Fredriksen K, Kass E, Rekvig OP. Identification of antihistone antibodies in subsets of juvenile chronic arthritis. Ann Rheum Dis. 1989;48:114–117. doi: 10.1136/ard.48.2.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Leak AM, Woo P. Juvenile chronic arthritis, chronic iridocyclitis, and reactivity to histones. Ann Rheum Dis. 1991;50:653–657. doi: 10.1136/ard.50.9.653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gompertz NR, Isenberg DA, Turner BM. Correlation between clinical features of systemic lupus erythematosus and levels of antihistone antibodies of the IgG, IgA, and IgM isotypes. Ann Rheum Dis. 1990;49:524–527. doi: 10.1136/ard.49.7.524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Munoz LE, Janko C, Schulze C, et al. Autoimmunity and chronic inflammation – two clearance-related steps in the etiopathogenesis of SLE. Autoimmun Rev. 2010;10:38–42. doi: 10.1016/j.autrev.2010.08.015. [DOI] [PubMed] [Google Scholar]
- 7.Munoz LE, Lauber K, Schiller M, Manfredi AA, Herrmann M. The role of defective clearance of apoptotic cells in systemic autoimmunity. Nat Rev Rheumatol. 2010;6:280–289. doi: 10.1038/nrrheum.2010.46. [DOI] [PubMed] [Google Scholar]
- 8.Munoz LE, Janko C, Grossmayer GE, et al. Remnants of secondarily necrotic cells fuel inflammation in systemic lupus erythematosus. Arthritis Rheum. 2009;60:1733–1742. doi: 10.1002/art.24535. [DOI] [PubMed] [Google Scholar]
- 9.Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26:239–257. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Green DR. Apoptotic pathways: paper wraps stone blunts scissors. Cell. 2000;102:1–4. doi: 10.1016/s0092-8674(00)00003-9. [DOI] [PubMed] [Google Scholar]
- 11.Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 1997;389:251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- 12.Slee EA, Adrain C, Martin SJ. Executioner caspase-3, -6, and -7 perform distinct, non-redundant roles during the demolition phase of apoptosis. J Biol Chem. 2001;276:7320–7326. doi: 10.1074/jbc.M008363200. [DOI] [PubMed] [Google Scholar]
- 13.Broers JL, Bronnenberg NM, Kuijpers HJ, Schutte B, Hutchison CJ, Ramaekers FC. Partial cleavage of A-type lamins concurs with their total disintegration from the nuclear lamina during apoptosis. Eur J Cell Biol. 2002;81:677–691. doi: 10.1078/0171-9335-00282. [DOI] [PubMed] [Google Scholar]
- 14.Charras GT, Hu CK, Coughlin M, Mitchison TJ. Reassembly of contractile actin cortex in cell blebs. J Cell Biol. 2006;175:477–490. doi: 10.1083/jcb.200602085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Enari M, Sakahira H, Yokoyama H, Okawa K, Iwamatsu A, Nagata S. A caspase-activated DNase that degrades DNA during apoptosis, and its inhibitor ICAD. Nature. 1998;391:43–50. doi: 10.1038/34112. [DOI] [PubMed] [Google Scholar]
- 16.Herrmann M, Lorenz HM, Voll R, Grunke M, Woith W, Kalden JR. A rapid and simple method for the isolation of apoptotic DNA fragments. Nucleic Acids Res. 1994;22:5506–5507. doi: 10.1093/nar/22.24.5506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Newell MK, Haughn LJ, Maroun CR, Julius MH. Death of mature T cells by separate ligation of CD4 and the T-cell receptor for antigen. Nature. 1990;347:286–289. doi: 10.1038/347286a0. [DOI] [PubMed] [Google Scholar]
- 18.Fadeel B, Xue D, Kagan V. Programmed cell clearance: molecular regulation of the elimination of apoptotic cell corpses and its role in the resolution of inflammation. Biochem Biophys Res Commun. 2010;396:7–10. doi: 10.1016/j.bbrc.2010.02.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Geske FJ, Monks J, Lehman L, Fadok VA. The role of the macrophage in apoptosis: hunter, gatherer, and regulator. Int J Hematol. 2002;76:16–26. doi: 10.1007/BF02982714. [DOI] [PubMed] [Google Scholar]
- 20.Herrmann M, Voll RE, Zoller OM, Hagenhofer M, Ponner BB, Kalden JR. Impaired phagocytosis of apoptotic cell material by monocyte-derived macrophages from patients with systemic lupus erythematosus. Arthritis Rheum. 1998;41:1241–1250. doi: 10.1002/1529-0131(199807)41:7<1241::AID-ART15>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 21.Hanayama R, Tanaka M, Miyasaka K, et al. Autoimmune disease and impaired uptake of apoptotic cells in MFG-E8-deficient mice. Science. 2004;304:1147–1150. doi: 10.1126/science.1094359. [DOI] [PubMed] [Google Scholar]
- 22.Kitahara Y, Kawane K, Nagata S. Interferon-induced TRAIL-independent cell death in DNase II–/– embryos. Eur J Immunol. 2010;40:2590–2598. doi: 10.1002/eji.201040604. [DOI] [PubMed] [Google Scholar]
- 23.Ogden CA, Kowalewski R, Peng Y, Montenegro V, Elkon KB. IGM is required for efficient complement mediated phagocytosis of apoptotic cells in vivo. Autoimmunity. 2005;38:259–264. doi: 10.1080/08916930500124452. [DOI] [PubMed] [Google Scholar]
- 24.Potter PK, Cortes-Hernandez J, Quartier P, Botto M, Walport MJ. Lupus-prone mice have an abnormal response to thioglycolate and an impaired clearance of apoptotic cells. J Immunol. 2003;170:3223–3232. doi: 10.4049/jimmunol.170.6.3223. [DOI] [PubMed] [Google Scholar]
- 25.Kruse K, Janko C, Urbonaviciute V, et al. Inefficient clearance of dying cells in patients with SLE: anti-dsDNA autoantibodies, MFG-E8, HMGB-1 and other players. Apoptosis. 2010;15:1098–1113. doi: 10.1007/s10495-010-0478-8. [DOI] [PubMed] [Google Scholar]
- 26.Scott VR, Boehme R, Matthews TR. New class of antifungal agents: jasplakinolide, a cyclodepsipeptide from the marine sponge, Jaspis species. Antimicrob Agents Chemother. 1988;32:1154–1157. doi: 10.1128/aac.32.8.1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bubb MR, Senderowicz AM, Sausville EA, Duncan KL, Korn ED. Jasplakinolide, a cytotoxic natural product, induces actin polymerization and competitively inhibits the binding of phalloidin to F-actin. J Biol Chem. 1994;269:14869–14871. [PubMed] [Google Scholar]
- 28.Spector I, Shochet NR, Kashman Y, Groweiss A. Latrunculins: novel marine toxins that disrupt microfilament organization in cultured cells. Science. 1983;219:493–495. doi: 10.1126/science.6681676. [DOI] [PubMed] [Google Scholar]
- 29.Coleman ML, Sahai EA, Yeo M, Bosch M, Dewar A, Olson MF. Membrane blebbing during apoptosis results from caspase-mediated activation of ROCK I. Nat Cell Biol. 2001;3:339–345. doi: 10.1038/35070009. [DOI] [PubMed] [Google Scholar]
- 30.Riento K, Ridley AJ. Rocks: multifunctional kinases in cell behaviour. Nat Rev Mol Cell Biol. 2003;4:446–456. doi: 10.1038/nrm1128. [DOI] [PubMed] [Google Scholar]
- 31.Sebbagh M, Renvoize C, Hamelin J, Riche N, Bertoglio J, Breard J. Caspase-3-mediated cleavage of ROCK I induces MLC phosphorylation and apoptotic membrane blebbing. Nat Cell Biol. 2001;3:346–352. doi: 10.1038/35070019. [DOI] [PubMed] [Google Scholar]
- 32.Cheung A, Dantzig JA, Hollingworth S, et al. A small-molecule inhibitor of skeletal muscle myosin II. Nat Cell Biol. 2002;4:83–88. doi: 10.1038/ncb734. [DOI] [PubMed] [Google Scholar]
- 33.Lowe J, Li H, Downing KH, Nogales E. Refined structure of alpha beta-tubulin at 3.5 A resolution. J Mol Biol. 2001;313:1045–1057. doi: 10.1006/jmbi.2001.5077. [DOI] [PubMed] [Google Scholar]
- 34.Saitoh M, Ishikawa T, Matsushima S, Naka M, Hidaka H. Selective inhibition of catalytic activity of smooth muscle myosin light chain kinase. J Biol Chem. 1987;262:7796–7801. [PubMed] [Google Scholar]
- 35.Lauber K, Blumenthal SG, Waibel M, Wesselborg S. Clearance of apoptotic cells: getting rid of the corpses. Mol Cell. 2004;14:277–287. doi: 10.1016/s1097-2765(04)00237-0. [DOI] [PubMed] [Google Scholar]
- 36.Voll RE, Herrmann M, Roth EA, Stach C, Kalden JR, Girkontaite I. Immunosuppressive effects of apoptotic cells. Nature. 1997;390:350–351. doi: 10.1038/37022. [DOI] [PubMed] [Google Scholar]
- 37.Bilyy RO, Shkandina T, Tomin A, et al. Macrophages discriminate glycosylation patterns of apoptotic cell-derived microparticles. J Biol Chem. 2012;287:496–503. doi: 10.1074/jbc.M111.273144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Franz S, Herrmann K, Furnrohr BG, et al. After shrinkage apoptotic cells expose internal membrane-derived epitopes on their plasma membranes. Cell Death Differ. 2007;14:733–742. doi: 10.1038/sj.cdd.4402066. [DOI] [PubMed] [Google Scholar]
- 39.Baumann I, Kolowos W, Voll RE, et al. Impaired uptake of apoptotic cells into tingible body macrophages in germinal centers of patients with systemic lupus erythematosus. Arthritis Rheum. 2002;46:191–201. doi: 10.1002/1529-0131(200201)46:1<191::AID-ART10027>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 40.Berkel AI, Birben E, Oner C, Oner R, Loos M, Petry F. Molecular, genetic and epidemiologic studies on selective complete C1q deficiency in Turkey. Immunobiology. 2000;201:347–355. doi: 10.1016/S0171-2985(00)80089-3. [DOI] [PubMed] [Google Scholar]
- 41.Jonsson G, Truedsson L, Sturfelt G, Oxelius VA, Braconier JH, Sjoholm AG. Hereditary C2 deficiency in Sweden: frequent occurrence of invasive infection, atherosclerosis, and rheumatic disease. Medicine. 2005;84:23–34. doi: 10.1097/01.md.0000152371.22747.1e. [DOI] [PubMed] [Google Scholar]
- 42.Ripoche J, Fontaine M, Godin M, Hauptmann G, Goetz J. Partial deficiency of the fourth component of human complement (C4) and autoantibody directed against C4 in a patient with SLE. Ann Immunol (Paris) 1983;134D:233–245. doi: 10.1016/s0771-050x(83)80089-8. [DOI] [PubMed] [Google Scholar]
- 43.Rhodes B, Furnrohr BG, Roberts AL, et al. The rs1143679 (R77H) lupus associated variant of ITGAM (CD11b) impairs complement receptor 3 mediated functions in human monocytes. Ann Rheum Dis. 2012;71:2028–2034. doi: 10.1136/annrheumdis-2012-201390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Grossmayer GE, Munoz LE, Weber CK, et al. IgG autoantibodies bound to surfaces of necrotic cells and complement C4 comprise the phagocytosis promoting activity for necrotic cells of systemic lupus erythaematosus sera. Ann Rheum Dis. 2008;67:1626–1632. doi: 10.1136/ard.2007.081828. [DOI] [PubMed] [Google Scholar]
- 45.Schiller M, Bekeredjian-Ding I, Heyder P, Blank N, Ho AD, Lorenz HM. Autoantigens are translocated into small apoptotic bodies during early stages of apoptosis. Cell Death Differ. 2008;15:183–191. doi: 10.1038/sj.cdd.4402239. [DOI] [PubMed] [Google Scholar]
- 46.Schiller M, Parcina M, Heyder P, et al. Induction of type I IFN is a physiological immune reaction to apoptotic cell-derived membrane microparticles. J Immunol. 2012;189:1747–1756. doi: 10.4049/jimmunol.1100631. [DOI] [PubMed] [Google Scholar]