

Abstract
Multiple sclerosis (MS) is an immune-mediated chronic central nervous system (CNS) disease affecting more than 400 000 people in the United States. Myelin-reactive CD4 T cells play critical roles in the formation of acute inflammatory lesions and disease progression in MS and experimental autoimmune encephalomyelitis (EAE), a well-defined mouse model for MS. Current MS therapies are only partially effective, making it necessary to develop more effective therapies that specifically target pathogenic myelin-specific CD4 T cells for MS treatment. While suppressing T-bet, the key transcription factor in T helper type 1 (Th1) cells, has been demonstrated to be beneficial in prevention and treatment of EAE, the therapeutic potential of retinoic acid-related orphan receptor gamma t (ROR)γt, the key transcription factor for Th17 cells, has not been well-characterized. In this study, we characterized the correlation between RORγt expression and other factors affecting T cell encephalitogenicity and evaluated the therapeutic potential of targeting RORγt by siRNA inhibition of RORγt. Our data showed that RORγt expression correlates with interleukin (IL)-17 production, but not with the encephalitogenicity of myelin-specific CD4 T cells. IL-23, a cytokine that enhances encephalitogenicity, does not enhance RORγt expression significantly. Additionally, granulocyte–macrophage colony-stimulating factor (GM-CSF) levels, which correlate with the encephalitogenicity of different myelin-specific CD4 T cell populations, do not correlate with RORγt. More importantly, inhibiting RORγt expression in myelin-specific CD4 T cells with an siRNA does not reduce disease severity significantly in adoptively transferred EAE. Thus, RORγt is unlikely to be a more effective therapeutic target for ameliorating pathogenicity of encephalitogenic CD4 T cells.
Keywords: EAE, multiple sclerosis, RORγt, T cell encephalitogenicity, transcription factor
Introduction
Multiple sclerosis (MS) is the leading cause of neurological disability in the United States in young adults after trauma, thus most patients suffer from the effects of MS for most of their adult life. Myelin-reactive CD4 T cells play an important role in the formation of acute inflammatory MS lesions and disease progression [1,2]. However, current MS therapies are only partially effective, making it necessary to develop more effective and specific therapies targeting pathogenic myelin-specific CD4 T cells for MS treatment. To this end, recent studies have focused on the identification of molecules that are specifically expressed in pathogenic myelin-specific CD4 T cells but not in other non-pathogenic cell populations, and on the evaluation of their therapeutic potential by suppressing those differentially expressed molecules during experimental autoimmune encephalomyelitis (EAE) development.
Naive CD4 T helper cells differentiate into different CD4 T effector lineages after antigen encounter, including T helper type 1 (Th1), Th2 and Th17 cells. When the environment is rich in interleukin (IL)-12 and/or interferon (IFN)-γ, naive CD4 T cells differentiate into IFN-γ-producing Th1 cells, driven by the transcription factor T-bet. Similarly, IL-4 induces IL-4-producing Th2 cells mediated by the transcription factor GATA binding protein 3 (GATA3) [3–8]. IFN-γ-producing Th1 CD4 cells were first shown to be pathogenic in EAE [3,9–12], while later IL-17-producing Th17 cells have been identified as a pathogenic population in EAE development [13]. However, IFN-γ, the Th1 signature cytokine, was shown subsequently to be dispensable for T cell encephalitogenicity and EAE development [14–17], while suppressing the Th1 transcription factor T-bet in myelin-specific CD4 T cells has been shown to ameliorate EAE [18–21], emphasizing the importance of lineage-specific transcription factors in regulating T cell encephalitogenicity and EAE development. Similarly, the contribution of IL-17, the Th17 signature cytokine, to EAE development is controversial, with one study showing that IL-17 plays a role in disease severity [22], while another study shows IL-17 does not contribute vitally [23]. Recently, retinoic acid-related orphan receptor gamma t (RORγt) has been explored as a therapeutic target for MS. Although RORγt–/– mice are resistant to EAE induction by immunization [24], the fact that RORγt–/– mice lack all peripheral lymph nodes (LNs) [24–26] compromises both the ability of those mice to prime T cells and the interpretation of this study. Furthermore, although RAG2–/– mice reconstituted with RORγt –/– CD4 T cells are resistant to EAE [24], they do not reveal the therapeutic potential of suppressing RORγt in treating MS patients. More recently, some small molecule compounds have been reported to suppress RORγt activity in EAE [27,28]; however, the preventative studies showed only minimal benefit in ameliorating EAE.
To determine if RORγt could be a better therapeutic target for MS, we first compared RORγt expression in encephalitogenic and non-encephalitogenic myelin-specific CD4 T cells. We then determined whether IL-23, the cytokine that increases T cell encephalitogenicity and enhances EAE development, up-regulates RORγt expression. Furthermore, we defined the relationship between RORγt and granulocyte–macrophage colony-stimulating factor (GM-CSF) expression. Moreover, we used a siRNA specific for RORγt to achieve RORγt inhibition in myelin-specific CD4 T cells, followed by adoptive transfer, to determine whether suppressing RORγt in myelin-specific CD4 T cells reduces their encephalitogenicity.
Materials and methods
Animals
B6/wild-type (WT), B6/IFN-γ–/– and B6/T-bet–/– mice were purchased from the Jackson Laboratory and bred in a specific pathogen-free animal facility at Ohio State University (OSU) Wexner Medical Center. B10.PL mice transgenic for the myelin basic protein (MBP) Ac1-11-specific T cell receptor (TCR) chains Vα2·3 or Vβ8·2 were a gift from J. Goverman [29]. All animal protocols were approved by the OSU Institutional Animal Care and Use Committee.
In-vitro transfection with siRNA
Synthetic siRNAs were purchased from ThermoFisher Scientific (Fremont, CA, USA), and stocks were prepared in the RNase-free H2O at 160 μM. Splenocytes from naive Vα2·3/Vβ8·2 TCR transgenic mice or IFN-γ–/– Vα2·3/Vβ8·2 TCR transgenic mice were transfected with siRNA-NS, siRNA-RORγt (5′-GGUAGAUGGGAUAGAGAUAUU-3′) or siRNA-Tbet, as described previously [19,20]. After overnight transfection, the cells were washed and stimulated with 2 μg/ml of MBP Ac1-11 in the presence of WT, non-transfected and irradiated splenocytes at a ratio of 1 : 5 for 1–3 days.
In-vitro culture of splenocytes from TCR transgenic mice
Splenocytes were prepared from naive 5–10-week-old Vα2·3/Vβ8·2 TCR transgenic mice and cultured in 24-well plates at 2 × 106 cells/well with irradiated B10.PL splenocytes (6 × 106 cells/well). Cells were activated with MBP Ac1-11 (2 μg/ml) and different combinations of cytokines or neutralizing antibodies for cytokines to differentiate effector T helper cells. Cytokines and antibody concentrations were as follows: 0·5 ng/ml IL-12, 25 ng/ml IL-6, 1 ng/ml transforming growth factor (TGF)-β1, 2 μg/ml anti-IFN-γ, 1 μg/ml anti-IL-12, 2 μg/ml anti-IL-4 and 0·35 μg/ml anti-TGF-β [20].
EAE induction
Immunization
Eight–10-week-old B6/IFN-γ–/– mice were injected subcutaneously (s.c.) over four sites in the flank with 200 μg myelin oligodendrocyte glycoprotein (MOG) 35–55 (C S bio) in an emulsion with complete Freund's adjuvant (CFA) (Difco, Becton Dickinson Co., Franklin Lakes, NJ, USA). Pertussis toxin (200 ng) (List) per mouse in phosphate-buffered saline (PBS) was injected intraperitoneally (i.p.) at the time of immunization and 48 h later.
Adoptive transfer
Splenocytes were isolated from naive 5–10-week-old Vα2·3/Vβ8·2 TCR transgenic mice or IFN-γ–/– Vα2·3/Vβ8·2 TCR transgenic mice. The cells were first transfected with siRNA-NS, siRNA-RORγt or siRNA-T-bet overnight and activated with 2 μg/ml of MBP Ac1-11 in 24-well plates at 2 × 106 cells/well with irradiated B10.PL splenocytes (6 × 106 cells/well). After 72 h, the cells were washed with PBS and 5 × 106 cells were injected i.p. into naive B10.PL mice. The mice were evaluated daily for clinical signs of EAE. Mice were scored on scale of 0 to 6: 0, no clinical disease; 1, limp/flaccid tail; 2, moderate hind limb weakness; 3, severe hind limb weakness; 4, complete hind limb paralysis; 5 quadriplegia or premoribund state; and 6, death.
Enzyme-linked immunosorbent assay (ELISA)
ELISA was performed to detect the expression of GM-CSF and IL-3 in supernatant. Supernatants were collected from B6/WT, B6/IFN-γ–/– or B6/T-bet–/– splenocytes cultured at 4 × 106 cells/well in 24-well plates. Purified anti-mouse GM-CSF primary antibody (R&D Systems, Minnealpolis, MN, USA) was diluted in 0·1 M NaHCO3 (pH 8·2) at 2 μg/ml. Immunolon II plates (Dynatech Laboratories, Chantilly, VA, USA) were coated with 50 μl of primary antibodies per well and incubated overnight at 4°C. The plates were washed twice with PBS/0·05% Tween 20, and were then blocked with 200 μl of 1% bovine serum albumin (BSA) in PBS per well for 2 h. The plates were washed twice with PBS/0·05% Tween 20, and 100 μl of supernatants were added in duplicate. The plates were incubated overnight at 4°C and washed four times with PBS/0·05% Tween 20. Biotinylated rat anti-mouse secondary antibody (R&D Systems) were diluted in PBS/1% BSA, 100 μl of 1 μg/ml biotinylated antibody was added to each well, and plates were incubated at room temperature for 1 h. The plates were washed six times with PBS/0·05%Tween 20, and 100 μl avidin–peroxidase was added at 2·5 μg/ml and incubated for 30 min. The plates were washed eight times with PBS/0·05% Tween 20 and 100 μl ABTS (2,2′-azino-bis(3-ethylbenzothiazoline-6-sulphonic acid) substrate containing 0·03% H2O2 was added to each well. The plate was monitored for 10–20 min for colour development and read at A 405. A standard curve was generated from GM-CSF standard, and the GM-CSF concentration in the samples was calculated.
Intracellular staining and flow cytometric analysis
Flow cytometric analysis was performed to evaluate cytokine production, T-bet and RORγt expression in CD4 T cells, as described previously [20]. Briefly, splenocytes were activated with antigen or αCD3/CD28. For the last 4–5 h of the incubation, 50 ng/ml phorbol myristate acetate (PMA) and 750 ng/ml ionomycin were added to cells for cytokine staining. 1 μl/ml GolgiPlug was added to each well to block cytokine secretion 4 h before staining. Cells were then collected, washed and resuspended in staining buffer (1% BSA in PBS). The cells were incubated with monoclonal antibodies (mAbs) to the cell-surface markers for 30 min at 4°C. After washing twice with staining buffer, cells were fixed and permeabilized using Cytofix/Cytoperm solution for 20 min (BD buffer for cytokine staining and T-bet staining; BD Biosciences, San Jose, CA, USA) or 60 min (eBioscience buffer for RORγt staining; eBioscience, San Diego, CA, USA) at 4°C. Cells were stained for intracellular cytokines, T-bet or RORγt with mAb diluted in PermWash solution for 30 min at 4°C; 80 000–100 000 live cell events were acquired on a fluorescence activated cell sorter (FACS)Canto (BD) and analysed using FlowJo software (Tree Star, Inc., Ashland, OH, USA). Phycoerythrin (PE)-anti-IL-17, allophycocyanin (APC)-anti-IFN-γ, peridinin chlorophyll (PerCP)-anti-CD4 and Pacific Blue-anti-CD44 were purchased from BD Biosciences. Fluorescein isothiocyanate (FITC)-anti-T-bet was purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA); PE-anti-RORγt was purchased from eBioscience.
Statistical analysis
A statistically significant difference in EAE clinical scores was considered to be P < 0·05, as determined by Mann–Whitney U-test. The Mann–Whitney U-test is non-parametric, and therefore accounts for the fact that EAE scores are ordinal and not interval-scaled.
Results
RORγt is expressed in both encephalitogenic and non-encephalitogenic myelin-specific CD4 T cells
We first determined whether RORγt is expressed differentially in encephalitogenic and non-encephalitogenic myelin-specific CD4 T cells. The combination of TGF-β1 and IL-6 in vitro induces the expression of the transcription factor RORγt, leading to IL-17 production [24,30–34]. However, we and others demonstrated that myelin-specific Th17 cells differentiated with TGF-β1 and IL-6 are not encephalitogenic, as they fail to transfer disease following adoptive transfer [20,35–37]. However, IL-6, in the absence of Th1 and Th2 differentiating signals, differentiates naive CD4 T cells into encephalitogenic Th17 cells, which transfer disease as efficiently as conventional Th1 cells [20,35,36]. We previously identified an encephalitogenic CD4 T cell population that produces minimal amounts of IL-17, and no IFN-γ, when anti-TGF-β is added with IL-6 + αIL-12/IFN-γ/IL-4 [20]. This population is highly encephalitogenic when utilized in adoptively transferred EAE. Using splenocytes from naive TCR transgenic mice expressing TCR genes Vα2·3/Vβ8·2 that recognize MBP Ac1-11, these three Th cell populations were generated, as well as IL-12-differentiated Th1 cells, and IL-17, RORγt and T-bet expression was analysed. As we expected, the non-encephalitogenic Th17 cells activated with MBP Ac1-11 plus TGF-β1/IL-6 had the highest IL-17-producing population (25%), the encephalitogenic Th17 cells activated with MBP Ac1-11 plus IL-6 + αIL-12/IFN-γ/IL-4 had a modest but significant IL-17-producing population (14%), while the encephalitogenic CD4 T cells activated with MBP Ac1-11 plus IL-6 + αIL-12/IFN-γ/IL4/TGF-β had a minimal IL-17-producing population (4%), and Th1 cells differentiated with IL-12 had no IL-17-producing cells (Fig. 1a). T-bet expression was highest in Th1 cells and modest in encephalitogenic Th17 cells differentiated with IL-6 + αIL-12/IFN-γ/IL-4 and encephalitogenic CD4 T cells differentiated with IL-6 + αIL-12/IFN-γ/IL-4/TGF-β, but almost undetectable in non-encephalitogenic Th17 cells differentiated with TGF-β1/IL-6. In contrast, RORγt was expressed in all four myelin-specific CD4 T cell populations, with the non-encephalitogenic Th17 cells differentiated with TGF-β1/IL-6 having the highest RORγt expression (57%), and Th1 cells having the lowest expression (5%), while encephalitogenic Th17 cells differentiated with IL-6 + αIL-12/IFN-γ/IL-4 and encephalitogenic CD4 T cells differentiated with IL-6 + αIL-12/IFN-γ/IL-4/TGF-β, having modest levels of RORγt, 13 and 6% respectively (Fig. 1b). These data indicate that RORγt expression is expressed in both encephalitogenic and non-encephalitogenic myelin-specific CD4 T cells, and RORγt expression level does not correlate with encephalitogenicity (Table 1).
Fig. 1.
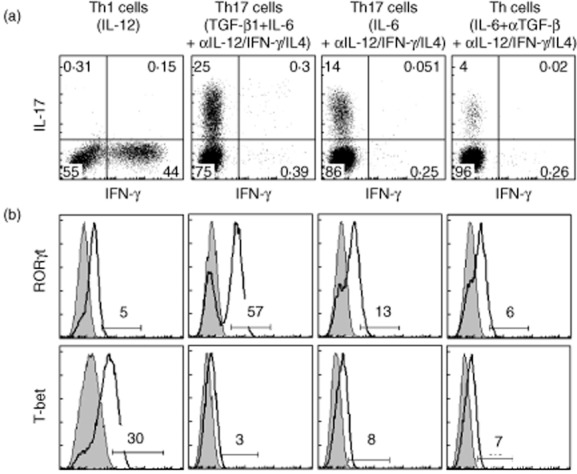
Retinoic acid-related orphan receptor gamma t (RORγt) is expressed in both encephalitogenic and non-encephalitogenic myelin-specific CD4 T cells. Splenocytes from naive T cell receptor (TCR) Vα2·3/Vβ8·2 transgenic mice were activated in vitro with myelin basic protein (MBP) Ac1-11 plus different cytokines for 3 days to differentiate different lineages of myelin-specific CD4 T cells. Transforming growth factor (TGF)-β1 and interleukin (IL)-6 were used to differentiate non-encephalitogenic T helper type 17 (Th17) cells, while IL-6 plus neutralizing antibodies to interferon (IFN)-γ, IL-4 and IL-12 were used to differentiate encephalitogenic Th17 cells. IL-6 plus neutralizing antibodies to IFN-γ, IL-4, IL-12 and TGF-β were used to differentiate encephalitogenic CD4 T cells with minimal levels of IL-17 but no IFN-γ production; and IL-12 was used to differentiate Th1 cells. Cells were harvested at 72 h after activation and analysed by flow cytometry for IL-17 and IFN-γ production (a), RORγt and T-bet expression (b). Cells were gated on CD4+ T cells. Data are representative of multiple independent experiments.
Table 1.
Retinoic acid-related orphan receptor gamma t (RORγt) expression and encephalitogenicity of CD4 T helper cells
| RORγt expression (%) | Incidence of EAE | Mean day of onset | Maximum clinical score | Mean clinical score on day of onset | |
|---|---|---|---|---|---|
| Th1 (IL-12) | 5 | 5/6(83%) | 7·2 | 4 | 2·4 |
| Th17 (TGF-β1 + IL-6) | 57 | 0/12(0%) | n.a. | n.a. | n.a. |
| Th17 (IL-6 + αIL-12/IFN-γ/IL-4) | 13 | 9/9(100%) | 6·6 | 6 | 2·2 |
| Th cells (IL-6 + αIL-12/IFN-γ/IL-4/TGF-β) | 6 | 4/5(80%) | 6·3 | 4 | 1·8 |
Th1 = T helper type 1; IL = interleukin; TGF = transforming growth factor; IFN = interferon; n.a. = not applicable.
RORγt expression is not IL-23-dependent
IL-23 is a critical cytokine for T cell encephalitogenicity and EAE development [13,38]. Exogenous IL-23, when added into the culture of myelin-specific CD4 T cells generated in immunized mice, favours expansion of Th17 cells which are highly encephalitogenic following adoptive transfer [39]. Therefore, we determined whether IL-23 enhances RORγt expression in pathogenic myelin-specific CD4 T cells from mice with spontaneous EAE. Splenocytes from TCR transgenic mice that developed spontaneous EAE were cultured with MBP Ac1-11, MBP Ac1-11 + IL-23 or MBP Ac1-11 + IL-12. Flow cytometric analysis demonstrated that there was no significant change in RORγt levels between the antigen-only group and antigen plus IL-23 group (Fig. 2a), while ELISA analysis showed that the addition of IL-23 significantly increased IL-17 production from myelin-specific CD4 T cells (Fig. 2b), suggesting that RORγt expression is not influenced by IL-23.
Fig. 2.

Exogenous interleukin (IL)-23 does not significantly enhance retinoic acid-related orphan receptor gamma t (RORγt) expression. (a,b) Splenocytes from T cell receptor (TCR) transgenic mice that developed spontaneous experimental autoimmune encephalomyelitis (EAE) were isolated and cultured with myelin basic protein (MBP) Ac1-11, MBP Ac1-11 + interleukin (IL)-23 or MBP Ac1-11 + IL-12 for 72 h. RORγt expression was analysed by flow cytometry (a) and IL-17 production was analysed by enzyme-linked immunosorbent assay (ELISA) (b). (c,d) B6/interferon (IFN)-γ–/– mice were immunized with myelin oligodendrocyte glycoprotein (MOG) 35–55 emulsified in complete Freund's adjuvant (CFA). Splenocytes were isolated on day 21 after immunization and stimulated with MOG 35–55, MOG 35–55 plus IL-12 or MOG 35–55 plus IL-23 for 3 days. IL-17 production (c) and RORγt expression (d) were analysed by flow cytometry. Cells were gated on CD4+ T cells. Data are representative of multiple independent experiments.
Because myelin-specific CD4 T cells from immunized B6/IFN-γ–/– mice produce large amounts of IL-17 and no IFN-γ, B6/IFN-γ–/– mice were immunized with MOG 35–55 to generate myelin-specific IL-17-producing cells. Splenocytes were isolated on day 21 after immunization and activated with MOG 35–55 in the presence or absence of IL-12 or IL-23. Similar to that observed in WT mice, exogenous IL-23 significantly expanded the IL-17-producing population in IFN-γ–/– myelin-specific CD4 T cells (from 10 to 24%), while IL-12 did not (from 10 to 12%) (Fig. 2c). However, IL-23 did not significantly enhance RORγt expression in IFN-γ–/– CD4 T cells (Fig. 2d). Interestingly, IL-12 treatment, which was shown previously to enhance the encephalitogenicity of myelin-specific CD4 T cells [12], reduced RORγt expression (Fig. 2a,d). These data indicate that in the absence of IFN-γ, RORγt expression in myelin-specific CD4 T cells is not influenced by IL-23.
RORγt expression does not correlate with GM-CSF production
To determine further whether the key molecules of the Th1 pathway, IFN-γ or T-bet, modulate RORγt expression, we compared RORγt expression in three mouse strains, B6/WT, B6/IFN-γ–/– and B6/T-bet–/–. Additionally, GM-CSF has been shown recently to be important for EAE development [40–43], and GM-CSF produced by autoreactive CD4 T cells is critical for T cell encephalitogenicity [42,43]. However, the role of RORγt in regulating GM-CSF production by CD4 T cells is debated. Thus, we also compared the GM-CSF expression in CD4 T cells from B6/WT, B6/IFN-γ–/– and B6/T-bet–/– mice, cultured with different cytokines to determine the relationship between RORγt expression and GM-CSF production. Splenocytes from naive B6/WT, B6/IFN-γ–/– and B6/T-bet–/– mice were activated with αCD3/CD28 alone (neutral), αCD3/CD28 plus IL-12 (Th1 condition), αCD3/CD28 plus TGF-β1 + IL-6 + αIL-12/IFN-γ/IL-4 (non-encephalitogenic Th17 condition), or αCD3/CD28 plus IL-6/αIL-12/IFN-γ/IL-4 (encephalitogenic Th17 condition). As shown in Fig. 3a, RORγt was expressed in CD4 T cells from all three strains of mice, albeit at different levels. Interestingly, B6/IFN-γ–/– and B6/T-bet–/– cells, which produce high levels of IL-17 and no IFN-γ, had substantially lower levels of RORγt expression compared to the WT mice that produce both IL-17 and IFN-γ. This suggests that RORγt expression is not positively regulated by the absence of Th1-associated transcription factors. Although TGF-β1 and IL-6 induced the highest levels of RORγt expression in CD4 cells from WT (85%), IFN-γ–/–(66%) and T-bet–/– (90%) mice (Fig. 3a), GM-CSF production was almost abolished in TGF-β1/IL-6-induced CD4 T cells from all three mice strains (Fig. 3b), suggesting that differentiation with TGF-β1 and IL-6, although inducing the highest levels of RORγt expression, significantly inhibits the production of the pathogenic cytokine GM-CSF by CD4 T cells. This inverse correlation between RORγt expression and GM-CSF production suggests that RORγt is not a positive regulator of GM-CSF production by CD4 T cells, as described previously [43].
Fig. 3.
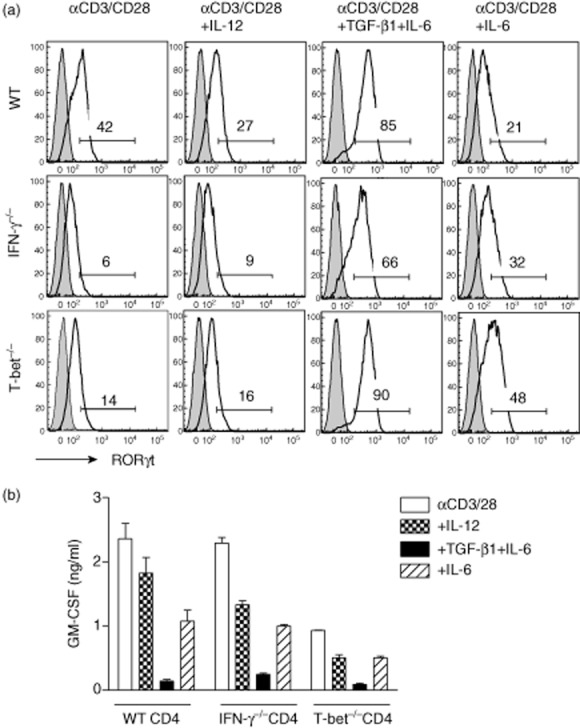
Retinoic acid-related orphan receptor gamma t (RORγt) is expressed in CD4 T cells from wild-type (WT), interferon (IFN)γ–/– and T-bet–/– mice, but does not correlate with granulocyte-macrophage colony-stimulating factor (GM-CSF) production. Splenocytes from naive B6/WT, B6/IFN-γ–/– and B6/T-bet–/– mice were activated with αCD3/CD28, αCD3/CD28 plus interleukin (IL)-12, αCD3/CD28 plus transforming growth factor (TGF)-β1 + IL-6 + αIFN-γ/IL-12/IL-4 or αCD3/CD28 plus IL-6 + αIFN-γ/IL-12/IL-4 for 2 days. RORγt expression was determined by flow cytometry (a). Cells were gated on CD4+ T cells. Supernatant was collected 48 h after activation and GM-CSF production was analysed by enzyme-linked immunosorbent assay (ELISA) (b). Data are representative of multiple independent experiments.
Suppressing RORγt expression does not reduce T cell encephalitogenicity significantly
siRNA is an efficient way of suppressing gene expression and exogenous siRNA has been used extensively to inhibit/silence target gene expression and explored recently in clinical trials for human diseases [44,45]. To evaluate the suppressive efficiency of siRNA-RORγt, a siRNA specifically targeting Rorc, the gene encoding for RORγt, siR-RORγt was transfected into splenocytes of WT/B6 mice and then differentiated with αCD3/CD28 plus TGF-β1 and IL-6. As expected, cells transfected with a control non-sense siRNA (siRNA-NS) and activated with TGF-β1 and IL-6 had the highest RORγt expression with a geometric mean of 465, while siRNA-NS transfected cells activated with αCD3/CD28 alone have the lowest RORγt expression with a geometric mean of 68. siRNA-RORγt efficiently inhibited RORγt expression, as shown by a significant decrease of the geometric mean in siRNA-RORγt-transfected CD4 T cells differentiated with TGF-β1 and IL-6, compared to siRNA-NS-treated cells under the same conditions (geometric mean: from 465 to 175) (Fig. 4a), indicating that siRNA-RORγt effectively suppresses RORγt expression in Th17 cells.
Fig. 4.
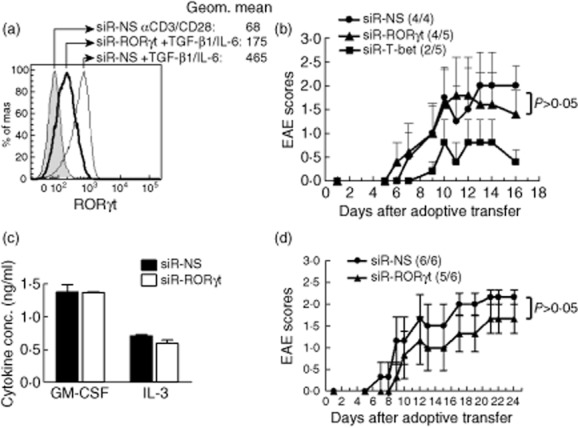
Suppressing retinoic acid-related orphan receptor gamma t (RORγt) expression does not significantly reduce T cell encephalitogenicity. (a) Splenocytes from naive B6 mice were transfected with either siRNA-NS or siRNA-RORγt for 18 h. The cells were then activated with αCD3/CD28 alone or in the presence of transforming growth factor (TGF)-β1 and interleukin (IL)-6 for 2 days. RORγt expression was determined by intracellular staining (filled grey histogram represents siRNA-NS treated cells activated with αCD3/CD28; thin-line open histogram represents siRNA-NS treated cells activated with αCD3/CD28 plus TGF-β1 and IL-6; bold-line open histogram represents siRNA-RORγt-treated cells activated with αCD3/CD28 and TGF-β1 and IL-6). Cells were gated on CD4+ T cells. (b) Splenocytes from naive TCR Vα2·3/Vβ8·2 transgenic mice were transfected with siRNA-NS, siRNA-RORγt or siRNA-T-bet for 18 h. The cells were then activated with myelin basic protein (MBP) Ac1-11 for 3 days and transferred into naive B10 PL recipient mice by intraperitoneal (i.p.) injection. The mice were monitored for experimental autoimmune encephalomyelitis (EAE) development. (c,d) Splenocytes from naive IFN-γ–/– TCR Vα2·3/Vβ8·2 transgenic mice were transfected with siRNA-NS or siRNA-RORγt for 18 h. The cells were then activated with MBP Ac1-11 for 3 days and transferred into naive B10 PL recipient mice by i.p. injection. Supernatant was collected 72 h after activation and granulocyte-macrophage colony-stimulating factor (GM-CSF) and IL-3 production were analysed by enzyme-linked immunosorbent assay (ELISA) (c). The recipient mice were monitored for EAE development (d). Data are representative of two independent experiments.
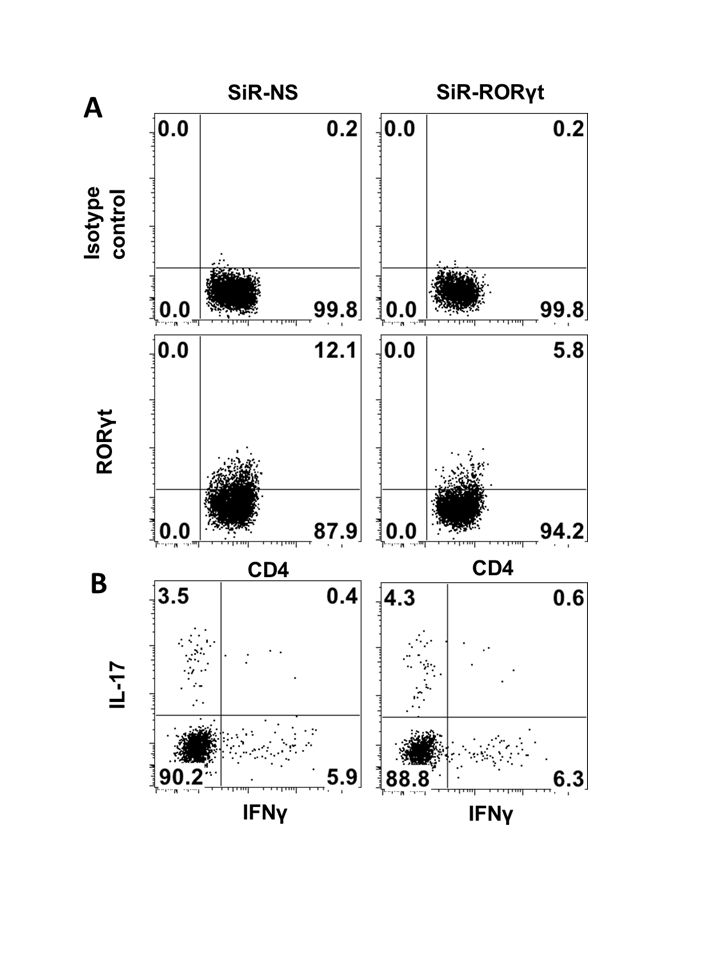
To determine the extent to which inhibiting RORγt expression alters the encephalitogenicity of myelin-specific CD4 T cells, splenocytes from naive myelin-specific TCR transgenic mice were transfected with siRNA-NS, siRNA-RORγt or siRNA-Tbet, activated with MBP Ac1-11 for 3 days, and transferred into naive B10.PL recipient mice. Although RORγt expression is significantly lower in CD4 T cells activated under Th neutral conditions than Th17 differentiating conditions, the transfection of siRNA-RORγt leads to a ∼50% reduction of RORγt-expressing cells (from 12·1 to 5·8%) (Supporting Information, Fig. S1a). However, IL-17-producing cells were not reduced significantly (Supporting Information, Fig. S1b). As shown in Fig. 4b, the disease onset and severity were similar between the mice receiving siRNA-NS- and siRNA-RORγt-transfected myelin-specific CD4 T cells, while the delayed disease onset and a significant decrease in disease severity was observed only in mice receiving siRNA-Tbet-treated myelin-specific CD4 T cells (Fig. 4b), suggesting that suppressing RORγt expression in myelin-specific CD4 T cells does not reduce T cell encephalitogenicity significantly, especially when compared to myelin-specific CD4 T cells treated with siRNA-Tbet.
As RORγt is the key transcription factor regulating IL-17 production in CD4 T cells and IFN-γ–/– CD4 T cells produce large amounts of IL-17 [20], we determined whether or not RORγt is critical for the encephalitogenicity of IFN-γ–/– myelin-specific CD4 T cells. Splenocytes from naive IFN-γ–/– TCR transgenic mice were transfected with siRNA-NS or siRNA-RORγt, activated with MBP Ac1-11 for 3 days and transferred into naive B10.PL recipient mice. The supernatants were analysed for GM-CSF and IL-3 production. As shown in Fig. 4c, there is no difference in GM-CSF production between siRNA-NS- and siRNA-RORγt-transfected cells. Similarly, the production of IL-3, a gene which is linked closely with the GM-CSF coding gene csf2, was also not altered significantly, indicating that RORγt is not critical to the regulation of the GM-CSF/IL-3 loci. As shown in Fig. 4d, there was no significant difference in disease onset, incidence or severity in siRNA-RORγt-transfected cells. Taken together, our data suggest that RORγt is unlikely to be a more effective therapeutic target for MS therapy.
Discussion
Although there have been important advances in MS treatment in the past several years, most MS therapies target a broad spectrum of cell populations instead of encephalitogenic T cells and are only partially effective. This study was intended to determine whether or not suppressing RORγt could serve as a more effective treatment for MS by targeting encephalitogenic CD4 T cells only. RORγt, a thymus-specific isoform of RORγ, is required for IL-17 production in CD4 T cells. Forced expression of RORγt induces IL-17 expression in naive CD4 T cells [24]. RORγt binds directly to the IL-17 promoter, and this binding is sufficient for activation of the minimum promoter in the HEK 293T cells [46], suggesting that RORγt is one of the transcription factors that directly control IL-17 production. However, at present, no definitive studies support the premise that RORγt is a more effective and specific therapeutic target. RORγt is essential for the generation of fetal lymphoid tissue inducer cells (LTi) [47], and required for the development of lymph nodes and Peyer's patches [25,26]. As a result, RORγt–/– mice lack all peripheral lymph nodes, which make it unclear that the EAE disease resistance observed in RORγt–/– mice is caused by RORγt deficiency in T cells or due to the lack of lymph nodes in those mice. Therefore, to determine the therapeutic potential of RORγt in ameliorating EAE, a siRNA specific for RORγt was used to inhibit RORγt expression in myelin-specific CD4 T cells followed by adoptive transfer into naive recipient mice. Unlike inducing EAE in genetic deficient mice, siRNA-RORγt does not completely silence RORγt gene expression in myelin-specific CD4 T cells, which provides a better evaluation from a therapeutic viewpoint, as no therapy will abolish RORγt expression completely. Our data showed that RORγt inhibition did not reduce disease severity significantly in adoptively transferred EAE. We performed our experiments in mice with two different genetic backgrounds, B10.PL and C57/B6, as our TCR transgenic mice were on a B10.PL background while T-bet and IFN-γ-deficient mice were on a C57/B6 background. Both mouse strains are used commonly in EAE studies. EAE was induced in B10.PL mice by immunization with MBP Ac1-11, while in C57/B6 mice with MOG 35–55. The use of two different mouse strains ensures that the effects we observed are not specific to immunizing peptide or major histocompatibility complex (MHC) of a specific mouse strain. Similarly, we used both antigen stimulation and polyclonal stimulation with anti-CD3/CD28 to activate CD4 T cells, which showed similar effects.
GM-CSF has been shown to be crucial for EAE development [40,42,43]. Myelin-specific CD4 T cells from immunized mice, when cultured with myelin antigens and αIFN-γ/IL-12, are mainly GM-CSF-producing cells and highly encephalitogenic following adoptive transfer into naive recipients [43]. Furthermore, GM-CSF production by Th1 and Th17 cells is required for their encephalitogenicity, as neither GM-CSF–/– Th1 cells nor GM-CSF–/– Th17 cells transfer EAE adoptively. Although IL-23 and IL-1β have been shown to up-regulate GM-CSF production by Th17 cells, the role of RORγt in regulating GM-CSF production in CD4 T cells remains controversial. One study showed that RORγt–/– CD4 T cells produce minimal amounts of GM-CSF, even under GM-CSF-skewing conditions, while CD4 T cells transduced retrovirally with RORγt have increased GM-CSF production, suggesting that RORγt drives GM-CSF production in CD4 T helper cells [43]. However, another study showed that RORγt–/– CD4 T cells activated in vitro produced similar or more GM-CSF compared to WT cells, depending on the cytokine environment, suggesting that RORγt is not required for GM-CSF production [42]. In the current study, CD4 T cells differentiated with TGF-β1 and IL-6 have the highest levels of RORγt expression, but produce almost no GM-CSF, which suggests that RORγt does not drive GM-CSF production. Moreover, siRNA-RORγt transfection did not change the production of GM-CSF or IL-3. Similar to Th1 cells, the encephalitogenic Th17 cells induced by IL-6 in the absence of Th1- and Th2-differentiating signals produce considerable amounts of GM-CSF, suggesting that inhibition of GM-CSF production in CD4 T cells by TGF-β1 and IL-6 may contribute to the lack of encephalitogenicity of TGF-β1/IL-6-induced natural Th17 (nTh17) cells. On a related note, TGF-β3 and IL-6 were shown recently to contribute to the induction of pathogenic Th17 cells, which was associated with enhanced GM-CSF expression [48]. However, it remains unclear how TGF-β1 and TGF-β3 signalling would result in different T cell phenotypes, as they share the same receptor.
During the search of encephalitogenic factor(s) regulating myelin-specific CD4 T cells in the past several decades, T-bet was shown originally to be critical for T cell encephalitogenicity. T-bet–/– mice were resistant to EAE induced by active immunization [49]. In-vitro and in-vivo suppression of T-bet by siRNA inhibited EAE development [18], and therapeutic administration of siRNA to T-bet significantly improved the clinical course of established EAE [19]. However, recent studies have challenged this concept, and suggested that T-bet is not universally required for T cell encephalitogenicity [50–52]. Contrary to previous data, T-bet-deficient mice have been shown recently to be fully susceptible to EAE induction by immunization. Furthermore, T-bet is shown to be absolutely required for the encephalitogenicity of Th1 cells, but not Th17 cells. Together, all these data suggested that pathogenic myelin-reactive T cells are heterogenetic, and therefore there might be different major determinants of T cell encephalitogenicity for different pathogenic myelin-reactive T populations, which makes the search for therapeutic targets even more complicated.
The traditional concept of CD4 T cell differentiation is that each CD4 T lineage has its own master transcription factor and signature cytokines, such as T-bet for IFN-γ-producing Th1, GATA3 for IL-4-producing Th2 and RORγt for IL-17-producing Th17 cells. However, recent studies have challenged this concept [53]. Several lineage specific transcription factors are expressed in more than one lineage. For example, T-bet is the master transcription factor for Th1 differentiation and IFN-γ production in Th1 cells, but it is also expressed in encephalitogenic Th17 cells [20,54] and contributes to the encephalitogenicity of Th17 cells [20]. Similarly, Bcl-6, the key transcription factor for T follicular helper cells (Tfh) [55,56], is also expressed in early-stage Th1 cells [57,58], although the detailed function in Th1 cells have not been well characterized. These data suggest that lineage-defining transcription factors have functions beyond driving lineage-specific cytokine production. Therefore, it is important to determine the possible roles of each key transcription factor in regulating T cell encephalitogenicity. Although genetic deletion of RORγt results in the loss of disease susceptibility, therapeutically targeting RORγt is unlikely to be a more effective strategy for ameliorating immune-mediated demyelinating disease.
Acknowledgments
This study is supported by grants from National Institute of Health (R01 NS067441) and National Multiple Sclerosis Society (RG3812). P. K. N.-G. is supported by NIH Postbacculaureate Research Education Program (PREP), grant R25 GM089571. We thank Todd Shawler for his technical support on flow cytometry and C. Pannell for his support of our mouse studies.
Disclosure
We declare that we have no significant competing financial, professional or personal interests that might have influenced the performance or presentation of the work described in this manuscript.
Supporting information
Additional Supporting information may be found in the online version of this article at the publisher's web-site:
Fig. S1. Splenocytes from naive T cell receptor (TCR) Vα2·3/Vβ8·2 transgenic mice were transfected with siRNA-NS and siRNA-RORγt for 18 h. The cells were then activated with myelin basic protein (MBP) Ac1-11 for 3 days. RORγt expression and cytokine production [interleukin (IL)-17 and IFN-γ] was analysed by flow cytometry. Data are representative of multiple independent experiments.
{kind=link}
References
- 1.Frohman EM, Racke MK, Raine CS. Multiple sclerosis – the plaque and its pathogenesis. N Engl J Med. 2006;354:942–955. doi: 10.1056/NEJMra052130. [DOI] [PubMed] [Google Scholar]
- 2.McFarland HF, Martin R. Multiple sclerosis: a complicated picture of autoimmunity. Nat Immunol. 2007;8:913–919. doi: 10.1038/ni1507. [DOI] [PubMed] [Google Scholar]
- 3.Lovett-Racke AE, Yang Y, Racke MK. Th1 versus Th17: are T cell cytokines relevant in multiple sclerosis? Biochim Biophys Acta. 2011;1812:246–251. doi: 10.1016/j.bbadis.2010.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mosmann TR, Cherwinski H, Bond MW, Giedlin MA, Coffman RL. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J Immunol. 1986;136:2348–2357. [PubMed] [Google Scholar]
- 5.Mosmann TR, Coffman RL. TH1 and TH2 cells: different patterns of lymphokine secretion lead to different functional properties. Annu Rev Immunol. 1989;7:145–173. doi: 10.1146/annurev.iy.07.040189.001045. [DOI] [PubMed] [Google Scholar]
- 6.Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, Glimcher LH. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell. 2000;100:655–669. doi: 10.1016/s0092-8674(00)80702-3. [DOI] [PubMed] [Google Scholar]
- 7.Mowen KA, Glimcher LH. Signaling pathways in Th2 development. Immunol Rev. 2004;202:203–222. doi: 10.1111/j.0105-2896.2004.00209.x. [DOI] [PubMed] [Google Scholar]
- 8.Murphy KM, Reiner SL. The lineage decisions of helper T cells. Nat Rev Immunol. 2002;2:933–944. doi: 10.1038/nri954. [DOI] [PubMed] [Google Scholar]
- 9.Leonard JP, Waldburger KE, Schaub RG, et al. Regulation of the inflammatory response in animal models of multiple sclerosis by interleukin-12. Crit Rev Immunol. 1997;17:545–553. [PubMed] [Google Scholar]
- 10.Pettinelli CB, McFarlin DE. Adoptive transfer of experimental allergic encephalomyelitis in SJL/J mice after in vitro activation of lymph node cells by myelin basic protein: requirement for Lyt 1+ 2– T lymphocytes. J Immunol. 1981;127:1420–1423. [PubMed] [Google Scholar]
- 11.Ando DG, Clayton J, Kono D, Urban JL, Sercarz EE. Encephalitogenic T cells in the B10.PL model of experimental allergic encephalomyelitis (EAE) are of the Th-1 lymphokine subtype. Cell Immunol. 1989;124:132–143. doi: 10.1016/0008-8749(89)90117-2. [DOI] [PubMed] [Google Scholar]
- 12.Waldburger KE, Hastings RC, Schaub RG, Goldman SJ, Leonard JP. Adoptive transfer of experimental allergic encephalomyelitis after in vitro treatment with recombinant murine interleukin-12. Preferential expansion of interferon-gamma-producing cells and increased expression of macrophage-associated inducible nitric oxide synthase as immunomodulatory mechanisms. Am J Pathol. 1996;148:375–382. [PMC free article] [PubMed] [Google Scholar]
- 13.Langrish CL, Chen Y, Blumenschein WM, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201:233–240. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ferber IA, Brocke S, Taylor-Edwards C, et al. Mice with a disrupted IFN-gamma gene are susceptible to the induction of experimental autoimmune encephalomyelitis (EAE) J Immunol. 1996;156:5–7. [PubMed] [Google Scholar]
- 15.Willenborg DO, Fordham S, Bernard CC, Cowden WB, Ramshaw IA. IFN-gamma plays a critical down-regulatory role in the induction and effector phase of myelin oligodendrocyte glycoprotein-induced autoimmune encephalomyelitis. J Immunol. 1996;157:3223–3227. [PubMed] [Google Scholar]
- 16.Lublin FD, Knobler RL, Kalman B, et al. Monoclonal anti-gamma interferon antibodies enhance experimental allergic encephalomyelitis. Autoimmunity. 1993;16:267–274. doi: 10.3109/08916939309014645. [DOI] [PubMed] [Google Scholar]
- 17.Heremans H, Dillen C, Groenen M, Martens E, Billiau A. Chronic relapsing experimental autoimmune encephalomyelitis (CREAE) in mice: enhancement by monoclonal antibodies against interferon-gamma. Eur J Immunol. 1996;26:2393–2398. doi: 10.1002/eji.1830261019. [DOI] [PubMed] [Google Scholar]
- 18.Lovett-Racke AE, Rocchini AE, Choy J, et al. Silencing T-bet defines a critical role in the differentiation of autoreactive T lymphocytes. Immunity. 2004;21:719–731. doi: 10.1016/j.immuni.2004.09.010. [DOI] [PubMed] [Google Scholar]
- 19.Gocke AR, Cravens PD, Ben L-H, et al. T-bet regulates the fate of Th1 and Th17 lymphocytes in autoimmunity. J Immunol. 2007;178:1341–1348. doi: 10.4049/jimmunol.178.3.1341. [DOI] [PubMed] [Google Scholar]
- 20.Yang Y, Weiner J, Liu Y, et al. T-bet is essential for encephalitogenicity of both Th1 and Th17 cells. J Exp Med. 2009;206:1549–1564. doi: 10.1084/jem.20082584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nath N, Prasad R, Giri S, Singh AK, Singh I. T-bet is essential for the progression of experimental autoimmune encephalomyelitis. Immunology. 2006;118:384–391. doi: 10.1111/j.1365-2567.2006.02385.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Komiyama Y, Nakae S, Matsuki T, et al. IL-17 plays an important role in the development of experimental autoimmune encephalomyelitis. J Immunol. 2006;177:566–573. doi: 10.4049/jimmunol.177.1.566. [DOI] [PubMed] [Google Scholar]
- 23.Haak S, Croxford AL, Kreymborg K, et al. IL-17A and IL-17F do not contribute vitally to autoimmune neuro-inflammation in mice. J Clin Invest. 2009;119:61–69. doi: 10.1172/JCI35997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ivanov II, McKenzie BS, Zhou L, et al. The orphan nuclear receptor RORgamma t directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 2006;126:1121–1133. doi: 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]
- 25.Eberl G, Littman DR. The role of the nuclear hormone receptor RORgammat in the development of lymph nodes and Peyer's patches. Immunol Rev. 2003;195:81–90. doi: 10.1034/j.1600-065x.2003.00074.x. [DOI] [PubMed] [Google Scholar]
- 26.Sun Z, Unutmaz D, Zou YR, et al. Requirement for RORgamma in thymocyte survival and lymphoid organ development. Science. 2000;288:2369–2373. doi: 10.1126/science.288.5475.2369. [DOI] [PubMed] [Google Scholar]
- 27.Solt LA, Kumar N, Nuhant P, et al. Suppression of TH17 differentiation and autoimmunity by a synthetic ROR ligand. Nature. 2011;472:491–494. doi: 10.1038/nature10075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xiao S, Yosef N, Yang J, et al. Small-molecule RORγt antagonists inhibit T helper 17 cell transcriptional network by divergent mechanisms. Immunity. 2014;40:477–489. doi: 10.1016/j.immuni.2014.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Goverman J, Woods A, Larson L, Weiner LP, Hood L, Zaller DM. Transgenic mice that express a myelin basic protein-specific T cell receptor develop spontaneous autoimmunity. Cell. 1993;72:551–560. doi: 10.1016/0092-8674(93)90074-z. [DOI] [PubMed] [Google Scholar]
- 30.Manel N, Unutmaz D, Littman DR. The differentiation of human T(H)-17 cells requires transforming growth factor-beta and induction of the nuclear receptor RORgamma t. Nat Immunol. 2008;9:641–649. doi: 10.1038/ni.1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang XO, Pappu BP, Nurieva R, et al. T helper 17 lineage differentiation is programmed by orphan nuclear receptors ROR alpha and ROR gamma. Immunity. 2008;28:29–39. doi: 10.1016/j.immuni.2007.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, TG SB. Fbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24:179–189. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 33.Bettelli E, Carrier Y, Gao W, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 34.Mangan PR, Harrington LE, O'Quinn DB, et al. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature. 2006;441:231–234. doi: 10.1038/nature04754. [DOI] [PubMed] [Google Scholar]
- 35.Ghoreschi K, Laurence A, Yang X-P, et al. Generation of pathogenic T(H)17 cells in the absence of TGF-β signalling. Nature. 2010;467:967–971. doi: 10.1038/nature09447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Das J, Ren G, Zhang L, et al. Transforming growth factor beta is dispensable for the molecular orchestration of Th17 cell differentiation. J Exp Med. 2009;206:2407–2416. doi: 10.1084/jem.20082286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McGeachy MJ, Bak-Jensen KS, Chen Y, et al. TGF-beta and IL-6 drive the production of IL-17 and IL-10 by T cells and restrain T(H)-17 cell-mediated pathology. Nat Immunol. 2007;8:1390–1397. doi: 10.1038/ni1539. [DOI] [PubMed] [Google Scholar]
- 38.Cua DJ, Sherlock J, Chen Y, et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature. 2003;421:744–748. doi: 10.1038/nature01355. [DOI] [PubMed] [Google Scholar]
- 39.Langrish CL, McKenzie BS, Wilson NJ, Malefyt R, Kastelein RA, Cua DJ. IL-12 and IL-23: master regulators of innate and adaptive immunity. Immunol Rev. 2004;202:96–105. doi: 10.1111/j.0105-2896.2004.00214.x. [DOI] [PubMed] [Google Scholar]
- 40.Ponomarev ED, Shriver LP, Maresz K, Pedras-Vasconcelos J, Verthelyi D, Dittel BN. GM-CSF production by autoreactive T cells is required for the activation of microglial cells and the onset of experimental autoimmune encephalomyelitis. J Immunol. 2007;178:39–48. doi: 10.4049/jimmunol.178.1.39. [DOI] [PubMed] [Google Scholar]
- 41.McQualter JL, Darwiche R, Ewing C, et al. Granulocyte macrophage colony-stimulating factor: a new putative therapeutic target in multiple sclerosis. J Exp Med. 2001;194:873–882. doi: 10.1084/jem.194.7.873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.El-Behi M, Ciric B, Dai H, et al. The encephalitogenicity of T(H)17 cells is dependent on IL-1- and IL-23-induced production of the cytokine GM-CSF. Nat Immunol. 2011;12:568–575. doi: 10.1038/ni.2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Codarri L, Gyülvészi G, Tosevski V, et al. RORγt drives production of the cytokine GM-CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nat Immunol. 2011;12:560–567. doi: 10.1038/ni.2027. [DOI] [PubMed] [Google Scholar]
- 44.Burnett JC, Rossi JJ, Tiemann K. Current progress of siRNA/shRNA therapeutics in clinical trials. Biotechnol J. 2011;6:1130–1146. doi: 10.1002/biot.201100054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Davidson BL, McCray PB. Current prospects for RNA interference-based therapies. Nat Rev Genet. 2011;12:329–340. doi: 10.1038/nrg2968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ichiyama K, Yoshida H, Wakabayashi Y, et al. Foxp3 inhibits RORgamma t-mediated IL-17A mRNA transcription through direct interaction with RORgamma t. J Biol Chem. 2008;283:17003–17008. doi: 10.1074/jbc.M801286200. [DOI] [PubMed] [Google Scholar]
- 47.Eberl G, Marmon S, Sunshine M-J, Rennert PD, Choi Y, Littman DR. An essential function for the nuclear receptor RORgamma(t) in the generation of fetal lymphoid tissue inducer cells. Nat Immunol. 2004;5:64–73. doi: 10.1038/ni1022. [DOI] [PubMed] [Google Scholar]
- 48.Lee Y, Awasthi A, Yosef N, et al. Induction and molecular signature of pathogenic TH17 cells. Nat Immunol. 2012;13:991–999. doi: 10.1038/ni.2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bettelli E, Sullivan B, Szabo SJ, Sobel RA, Glimcher LH, Kuchroo VK. Loss of T-bet, but not STAT1, prevents the development of experimental autoimmune encephalomyelitis. J Exp Med. 2004;200:79–87. doi: 10.1084/jem.20031819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.O'Connor RA, Cambrook H, Huettner K, Anderton SM. T-bet is essential for Th1-mediated, but not Th17-mediated, CNS autoimmune disease. Eur J Immunol. 2013;43:2818–2823. doi: 10.1002/eji.201343689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Grifka-Walk HM, Lalor SJ, Segal BM. Highly polarized Th17 cells induce EAE via a T-bet independent mechanism. Eur J Immunol. 2013;43:2824–2831. doi: 10.1002/eji.201343723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Duhen R, Glatigny S, Arbelaez CA, Blair TC, Oukka M, Bettelli E. Cutting edge: the pathogenicity of IFN-γ-producing Th17 cells is independent of T-bet. J Immunol. 2013;190:4478–4482. doi: 10.4049/jimmunol.1203172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Oestreich KJ, Weinmann AS. Master regulators or lineage-specifying? Changing views on CD4+ T cell transcription factors. Nat Rev Immunol. 2012;12:799–804. doi: 10.1038/nri3321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen Y, Langrish CL, McKenzie B, et al. Anti-IL-23 therapy inhibits multiple inflammatory pathways and ameliorates autoimmune encephalomyelitis. J Clin Invest. 2006;116:1317–1326. doi: 10.1172/JCI25308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Johnston RJ, Poholek AC, DiToro D, et al. Bcl6 and Blimp-1 are reciprocal and antagonistic regulators of T follicular helper cell differentiation. Science. 2009;325:1006–1010. doi: 10.1126/science.1175870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yu D, Rao S, Tsai LM, et al. The transcriptional repressor Bcl-6 directs T follicular helper cell lineage commitment. Immunity. 2009;31:457–468. doi: 10.1016/j.immuni.2009.07.002. [DOI] [PubMed] [Google Scholar]
- 57.Oestreich KJ, Huang AC, Weinmann AS. The lineage-defining factors T-bet and Bcl-6 collaborate to regulate Th1 gene expression patterns. J Exp Med. 2011;208:1001–1013. doi: 10.1084/jem.20102144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nakayamada S, Kanno Y, Takahashi H, et al. Early Th1 cell differentiation is marked by a Tfh cell-like transition. Immunity. 2011;35:919–931. doi: 10.1016/j.immuni.2011.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Splenocytes from naive T cell receptor (TCR) Vα2·3/Vβ8·2 transgenic mice were transfected with siRNA-NS and siRNA-RORγt for 18 h. The cells were then activated with myelin basic protein (MBP) Ac1-11 for 3 days. RORγt expression and cytokine production [interleukin (IL)-17 and IFN-γ] was analysed by flow cytometry. Data are representative of multiple independent experiments.