

Abstract
Purified arrestin proteins are necessary for biochemical, biophysical, and crystallographic studies of these versatile regulators of cell signaling. Here we describe a basic protocol for expression in E. coli and purification of tag-free wild type and mutant arrestins. The method includes ammonium sulfate precipitation of arrestins from cell lysates, followed by heparin-Sepharose chromatography. Depending on the arrestin type and/or mutations, this step is followed by Q-Sepharose or SP-Sepharose chromatography. In many cases the non-binding column is used as a pre-filter to bind contaminants without retaining arrestin. In some cases both chromatographic steps need to be performed sequentially to achieve high purity. Purified arrestins can be concentrated up to 10 mg/ml, remain fully functional, and can withstand several cycles of freezing and thawing, provided that overall salt concentration is kept at or above physiological levels.
Keywords: arrestin, recombinant, expression, purification, chromatography
INTRODUCTION
Arrestins are multi-functional adaptor proteins that form complexes with hundreds of G protein-coupled receptors (GPCRs) and dozens of other partners (DeWire et al., 2007; Gurevich and Gurevich, 2006b; Gurevich and Gurevich, 2014). The mammalian arrestin family contains four members, with two visual subtypes, arrestin-1 (a.k.a. S-antigen, 48 kDa protein, visual or rod arrestin) and arrestin-4 (cone or X-arrestin; for unclear reasons its gene is called “arrestin 3” in the HUGO database) in retinal photoreceptors and two ubiquitously expressed non-visual subtypes, arrestin-2 (a.k.a. β-arrestin or β-arrestin1) and arrestin-3 (a.k.a. β-arrestin2 or hTHY-ARRX) (Gurevich and Gurevich, 2006a) present in virtually every cell. Here we use systematic names of arrestin proteins, with the order of cloning indicated after dash.
Originally discovered as GPCR-desensitizing proteins (Attramadal et al., 1992; Lohse et al., 1990), non-visual arrestins have been shown to also serve as versatile regulators of GPCR signaling, sequestration, and trafficking (DeWire et al., 2007; Goodman et al., 1996; Gurevich and Gurevich, 2006a; Laporte et al., 1999; Luttrell et al., 1999). Their broad functional importance has attracted enormous attention and sparked studies that investigate the structural and molecular basis of function. The study of arrestin proteins has been greatly advanced in recent years by the development of methods to purify fully functional arrestins (Gurevich et al., 1997; Lohse et al., 1992; Söhlemann et al., 1995). Purified arrestins were used to obtain the crystal structures of the basal (Han et al., 2001; Hirsch et al., 1999; Sutton et al., 2005; Zhan et al., 2011a) and active-like (Kim et al., 2013; Shukla et al., 2013) conformations, for biophysical studies of receptor-induced conformational changes in arrestins (Kim et al., 2012; Zhuang et al., 2013), the regulation of arrestin-mediated signaling (Kook et al., 2014; Zhan et al., 2011b; Zhan et al., 2013; Zhan et al., 2014), the oligomerization of arrestins (Chen et al., 2014; Hanson et al., 2007b; Hanson et al., 2008; Kim et al., 2011; Song et al., 2013), and the binding surface of arrestins engaged by GPCRs (Hanson et al., 2006b) and other partners (Hanson et al., 2007a; Hanson et al., 2006a; Wu et al., 2006).
The sequential chromatographic protocol of arrestin purification after ammonium sulfate precipitation described below is based on earlier protocols (Lohse et al., 1992; Söhlemann et al., 1995) that were further developed to purify different arrestin isoforms of bovine origin (Gurevich and Benovic, 2000; Gurevich et al., 1999). We have employed this protocol to purify all four bovine wild type (WT) arrestins, arrestins from other species, and numerous mutants. This protocol has worked well for some arrestin isoforms and their variants, for example arrestin-2, while certain modifications in the chromatographic steps were necessary to obtain pure fully functional arrestin proteins from different species and their mutant forms (Basic Protocol 1).
Strategic planning
Before undertaking large-scale expression for purification, it is necessary to ascertain that the arrestin of interest, be it a WT form from another species or a particular mutant of one of the bovine arrestins, expresses at sufficiently high levels. Support Protocol 1 describes test expression in E. coli, performed along with an arrestin protein known to express well as a positive control, to determine whether large-scale expression and purification of that particular arrestin is feasible. In our experience, nine out of ten WT or mutant arrestins express well enough to make large-scale purification possible. Usually the higher the expression level, the higher the overall yield and purity of the final sample. Our rule of thumb is that the expression of soluble arrestin must be greater than 1 mg per liter of bacterial culture to make large-scale purification feasible.
BASIC PROTOCOL 1: BASIC PROTOCOL TITLE: Large-scale expression and purification of arrestins
This protocol describes large-scale expression in E. coli and purification of arrestin proteins. The procedure includes several obligatory steps: cell growth and induction, cell lysis, ammonium sulfate precipitation, and heparin-Sepharose chromatography. Depending on the expression level and overall charge of the protein, pooled heparin-Sepharose fractions contain 20–80% pure arrestins. These are further purified using Q-Sepharose or SP-Sepharose chromatography. It is often advisable to use non-binding column as pre-filter that binds contaminants, but not arrestin itself. In rare cases, sequential Q-Sepharose and SP-Sepharose chromatography are necessary to achieve >95% purity.
Materials
Solutions and Reagents
Luria Broth (LB) (Sigma; prepare according to the manufacturer’s instructions)
100 mg/ml ampicillin stock solution in distilled water (store at 4°C)
100 mM Isopropyl-β-D-thiogalactopyranoside stock solution (IPTG) in distilled water (store at −80°C)
Glycerol (Sigma)
1.0 M Tris-HCl buffer solution, pH 8.0 (Amresco)
0.5 M Ethylene glycol tetraacetic acid solution (EGTA, BioWorld)
1.0 M Glucose stock solution in distilled water (store at −20°C)
Phenylmethylsulfonyl fluoride (PMSF)
Dimethyl sulfoxide (DMSO)
1.0 M Phenylmethanesulfonyl fluoride (PMSF) (prepare a fresh solution in water-free DMSO)
1.0 M Benzamidine (BA) stock solution in distilled water (store at −80°C)
1.0 M Magnesium chloride solution (Sigma)
5.0 M Sodium Chloride solution (Sigma)
4.0 M Dithiothreitol (DTT) stock solution in distilled water (store at −20°C)
0.5 M Ethylenediaminetetraacetic acid (EDTA, Amresco)
2X SDS Sample buffer (Sigma)
Lysozyme from chicken egg white (Sigma)
Protease inhibitor cocktail for E. coli (P-8465, Sigma, prepared according to manufacturer’s instructions)
15,000 U Deoxyribonuclease I from bovine pancreas (DNAse I, Sigma) (stock solution: dissolve in 1 ml of Lysis buffer)
20,000 U Deoxyribonuclease II from porcine spleen (DNAse II, Sigma) (stock solution: dissolve in 1 ml of Lysis buffer)
100 mg/ml Ribonuclease A from bovine pancreas (RNAse A, Sigma) (stock solution: dissolve in 1 ml of Lysis buffer)
Heparin Sepharose 6 Fast Flow (GE Healthcare)
Q Sepharose Fast Flow (GE Healthcare)
SP Sepharose Fast Flow (GE Healthcare)
Special equipment
RC-5B Plus centrifuge (Sorvall)
SLA 3000 rotor (Sorvall)
TL-100 tabletop ultracentrifuge (Beckman)
TLA120.1 rotor (Beckman);
Econo-Column Chromatography Columns, 1.5 × 20 cm (Bio-Rad, cat. #737-1522)
GradiFrac Chromatography system (Pharmacia Biotech, Sweden)
Additional reagents and equipment for performing SDS-PAGE electrophoresis and Western Blotting - described in detail in (Gurevich and Benovic, 2000; Gurevich et al., 1999)).
Buffers
Lysis Buffer: 50 mM Tris-HCl, pH 8.0, 2 mM BA, 1 mM PMSF, 2 mM DTT, 2 mM EGTA, 10 mM glucose, 5 ml of protease inhibitors cocktail for E. coli per 200 ml of lysis buffer.
*Important: Add all protease inhibitors immediately before use, especially PMSF, which is unstable in aqueous solutions.
Multiple Column Buffer (MCB): 10 mM Tris-HCl, pH 7.5; 2 mM BA, 1 mM PMSF, 2 mM EGTA, 2 mM EDTA, 2 mM DTT
Antibodies for Western blot
F4C1 - Mouse monoclonal antibody that detects the epitope DGVVLVD, a sequence that is present in the all known mammalian arrestins (Donoso et al., 1990)
or
F431 - Rabbit polyclonal antibody that detects the same epitope (Song et al., 2011).
Note: While the cells producing monoclonal the F4C1 antibody have been lost, rabbit polyclonal F431 antibody is available from Dr. V.V. Gurevich upon request. Several commercial anti-arrestin antibodies might be suitable for the task, but we have not tested them. We will gladly provide upon request aliquots of purified arrestin proteins to scientists willing to test commercial antibodies and compare them with the F431 that we use.
Step 1. Expression of arrestin proteins in Escherichia coli (BL-21 GOLD)
All arrestin coding sequences can be subcloned into pTrcHisB vector (Life technologies) between Nco I and Hind III sites (this removes the His tag) as described (Gurevich and Benovic, 2000; Gurevich et al., 1999). Expression constructs described in our publications are available upon request from Dr. V.V. Gurevich. The corresponding constructs are transformed into the commercially available BL21 GOLD (Invitrogen) competent cells according to the manufacturer’s instructions. An individual colony is then used to inoculate 3 ml of LB containing 100 μg/ml of ampicillin (LB/Amp) and the cells are grown at 30°C with shaking at 250 rpm for 10–15 h. Glycerol is then added to 10% (v/v) and cell suspensions are aliquoted, frozen, and stored at −80°C until needed. Frozen cells are viable for years. Cells transformed with expression constructs described in our publications are also available upon request from Dr. V.V. Gurevich.
Note that while induction with higher than recommended concentrations of IPTG increase total protein production in E. coli, this results in most of the expressed arrestins in the insoluble fraction, likely because of misfolding. In contrast, induction with 25–35 μM IPTG results in arrestins that remain soluble, correctly folded, and fully functional. The procedure below describes expression in 4.5 L of bacterial culture. It can be scaled up or down, as needed, with proportional adjustment of volumes.
Cell Growth and Arrestin Expression
-
1
Prepare one sterile 500 ml flask with 160 ml of LB medium and four 2 L flasks with 1.1 L LB medium each.
-
2
Add 160 μl of the ampicillin stock solution to 160 ml LB (LB/Amp).
-
3
Inoculate this LB/Amp with selected arrestin/BL21 cells from the glycerol stock (30–60 μl scraped off the cell suspension with a pipette tip without thawing the suspension). Grow the cells at 30°C for 6–8 h (to OD600 ~0.1–0.2) with continuous shaking at 250 rpm.
-
4
Add 40 ml of this culture to each flask with 1.1 liter LB/Amp, grow overnight (to OD600 ~1.0–1.5).
-
5
Add 275 μl or 385 μl (for arrestin-3) of IPTG stock solution to each flask (final concentration 25 μM or 35 μM, respectively) to induce arrestin expression, and keep shaking at 30°C, 250 rpm for additional 4–5 h.
Cell Lysis
-
6
Pellet the cells by centrifugation for 10 min at 6,000 rpm (6,000 ×g) at 4°C (SLA 3000 rotor RC-5B Plus, Sorvall) in six 500 ml centrifuge bottles.
Two rounds of centrifugation are required to pellet cells from ~4.5 L of culture; all bottles should contain roughly equal amounts of cells. -
7
Discard the supernatant carefully, let the tubes stand upside down for 1 min, and wipe all traces of supernatant with Kimwipes.
All further steps are performed on ice or in the cold room. -
8
Add 22 ml of the lysis buffer to each tube and thoroughly resuspend cells by pipetting. It is very important to avoid frothing. Measure the total volume of the cell suspension in each bottle to determine the volume of cells.
-
9
Add 1 ml of fresh 3 mg/ml lysozyme (dissolved in lysis buffer) to each bottle and incubate 30–40 min on ice. Freeze the cells at −80°C.
Cell lysate can be kept frozen at this point for several weeks. -
10
To proceed immediately, freeze cell lysate for 20 min at −80°C, then thaw on ice for 30–60 min.
-
11
Add 80 ml of ice-cold lysis buffer to each bottle (final volume 100 ml per bottle) and carefully resuspend to ensure that frozen cell suspension is fully thawed.
-
12
Sonicate cell lysate three times (15 sec each time, 90% amplitude; Fisher Dismembrator, Model 500). The sonication should be performed in ice-water bath, with 1–2 min intervals between rounds to allow the suspension to cool.
-
13
On ice, add 0.88 ml of 1 M MgCl2 (to final concentration of 7–8 mM, to ensure that free Mg2+ is present), 160 μl of DNAse I (15,000 U/ml), 160 μl of DNAse II (20,000 U/ml), and 160 μl of RNAse A (100 mg/ml) to each bottle, mix thoroughly and incubate on ice for ~40 min. (Optional: take 100 μl aliquot of the homogenate, transfer to the microcentrifuge tube labeled “homogenate” sample, and freeze at −20°C; this sample can be compared by Western Blot with supernatant to determine the fraction of soluble arrestin).
-
14
Pellet the insoluble debris by centrifugation for 90 min at 9,000 rpm (13,000 ×g) at 4°C (SLA 3000 rotor, RC 5B Plus, Sorvall), and carefully collect supernatant into a clean ice-cold 1 L beaker. Determine its volume (usually it equals the total volume of added lysis buffer, i.e., 600 ml). Take 100 μl aliquot of the supernatant, transfer to the microcentrifuge tube labeled “supernatant” sample, and freeze at −20°C).
Ammonium Sulfate Precipitation
-
15
In the cold room (4°C), add 192 g (0.32 g/ml, final concentration 2.4 M) of ammonium sulfate to the 600 ml of the supernatant. Let the stirring continue until ammonium sulfate is completely dissolved.
Do not add the ammonium sulfate all at one time, as it will cause the initial concentration to be much higher than necessary. Add it in four portions, ~25% at a time, while gently stirring in the cold room. Avoid frothing: proteins denature at the water-air interface. -
16
Pellet precipitated protein in two 500 ml bottles by centrifugation for 90 min at 11,000 rpm (20,500 ×g) at 4°C (SLA 3000 rotor, RC-5B Plus centrifuge, Sorvall). Carefully remove supernatant and floating white material.
These pellets can be stored at −80°C until needed in covered bottles. Pellets are stable at least for a month.
Chromatographic purification
All chromatographic procedures are performed at 4°C. In some cases, an additional SP-Sepharose column is used to improve the purity of arrestin proteins (this step was not described previously (Gurevich and Benovic, 2000; Gurevich et al., 1999)). Arrestin proteins demonstrate different chromatographic profiles on Q- and SP-Sepharose (anion- and cation-exchangers, respectively). Two different strategies of chromatographic purifications are summarized in Fig. 1. Briefly, the first strategy (A) is designed for arrestin proteins that bind weakly to both Q- and SP-Sepharose columns, such as bovine visual arrestin-1. In the process of loading onto Q-Sepharose, the eluate from heparin-Sepharose is diluted to low salt concentration (<10 mM NaCl) via a mixer. SP-sepharose serves as a “filter” to remove some impurities from the eluate from Q-Sepharose column (adjust NaCl to ~100 mM). For arrestin-3 and some truncated visual arrestin mutants, we employ strategy (B). These arrestin proteins bind relatively weakly to Q-Sepharose, while demonstrating a much stronger binding to SP-Sepharose. Dilute and adjust the heparin-Sepharose eluate to ~ 100 mM NaCl, then load onto two sequentially connected Q- and SP-Sepharose columns (Fig. 1). Here, Q-Sepharose serves as the “filter” to remove some impurities while arrestin proteins pass through. The SP-Sepharose binds arrestin proteins, which are eluted (after disconnecting Q-Sepharose column) by a MCB-based NaCl gradient.
Fig. 1.

Two chromatographic strategies for arrestin purification. Heparin-Sepharose is always used as the first step, as it yields the highest purification. Depending on the properties of a particular arrestin, further purification to homogeneity is achieved by chromatography either on a Q-Sepharose or SP-Sepharose column. One of the columns is often used as a “filter”: it binds certain contaminants, but lets arrestin to pass through and then bind to other column, from which it is then eluted with salt gradient (see Table 2).
Heparin-Sepharose chromatography
-
17
To prepare the heparin-Sepharose loading sample, dissolve the protein precipitated by ammonium sulfate in 200 ml of ice-cold MCB. Make sure that the protein pellet is completely dissolved. This typically takes about 40 min of gentle shaking on DS-500 orbital shaker (VWR) or equivalent.
Here and at each step below, add PMSF to the MCB just before use. Starting from this point all further steps are performed on ice or in the cold room
-
18
To remove the insoluble material, centrifuge the protein solution in a 500 ml bottle at 11,000 rpm (20,500 ×g) for 90 min at 4°C (SLA 3000 rotor, RC-5B Plus centrifuge, Sorvall)
-
19
Carefully collect the supernatant while avoiding the pellet, and filter it through a 0.8 μm sterile syringe filter (Corning). Determine the volume of the supernatant to estimate the volume of the initial pellet (usually 15–20 ml). Take a 100 μl aliquot of the supernatant, transfer it to a microcentrifuge tube labeled “heparin input” sample, and freeze at −20°C).
-
20
Dilute the sample to the appropriate salt concentration* before loading it onto the heparin-Sepharose column (Table 1). Generally speaking, the salt concentration of the sample during loading should be ~30–45% of that of the elution peak.
Table 1.
Heparin-Sepharose loading and elution conditions
| Arrestins | Loading conditions | Elution conditions |
|---|---|---|
| Bovine arrestin-1 | 100 mM NaCl | Elution gradient: MCB/100 mM NaCl > MCB/500 mM NaCl Arrestin peak: 190–270 mM NaCl |
| Bovine arrestin-2 | 150 mM NaCl | Elution gradient: MCB/150 mM NaCl > MCB/0.8 M NaCl Arrestin peak: 442–523mM NaCl |
| Bovine arrestin-3 | 200 mM NaCl | Elution gradient: MCB/200 mM NaCl > MCB/1 M NaCl Arrestin peak: 380–500 mM NaCl (full-length) 633–703 mM NaCl (truncated) |
Loading and washing
-
21
Under gravity flow, pack a 25 ml column (1.5×20 cm; Bio-Rad) with heparin-Sepharose 6 (GE Healthcare). Wash this column with 3 M NaCl and then with 200 ml of 200 mM NaCl in MCB (100 mM for arrestin-1 and 150 mM for arrestin-2).
-
22
Load the protein sample onto the column. Collect flow-through for further analysis.
Because the MCB has a very low ionic strength, the addition of MCB to the filtrate sometimes results in precipitation of protein, which might clog the column. Thus, it is advisable to dilute the sample during column loading using a gradient mixer and an appropriate ratio of filtrate and MCB. Because of large volume, loading is usually done overnight at a flow rate that ensures that the column does not dry out by the morning.
-
23
The next morning, wash heparin-Sepharose column with 100 ml MCB/200 mM NaCl. Collect the wash for further analysis.
-
24
Elute proteins from the heparin-Sepharose column with a 400 ml linear gradient (the elution conditions for some arrestin proteins are shown in Table 1) of NaCl and collect 40 10-ml fractions. After the gradient, wash the column with 50 ml of 3 M NaCl in MCB (high salt elution), collecting 10-ml fractions.
Electrophoresis and Western blot
-
25
Take 30 μl aliquots from each 10-ml fraction and transfer to a clean 1.5 ml microcentrifuge tube, along with aliquots of the input, flow-through, and wash (for these samples the aliquots should equal 1:100, 000 of the total volume).
-
26
To precipitate the protein, add 270 μl MeOH (90% final), and centrifuge at 10,000 rpm (16,200 ×g) (Tabletop microcentrifuge, Eppendorf) for 10 min at room temperature
-
27
Carefully decant the supernatant, and add 1 ml of 90% MeOH to the pellets. Vortex thoroughly and pellet protein by centrifugation.
-
28
Discard supernatant and air-dry the samples for 10–15 min (the tubes should not have any MeOH residue).
-
29
Add 15–20 μl of SDS sample buffer to each tube, thoroughly dissolve the pellet by vortexing, and load onto 10% SDS-PAGE gel. The proteins are visualized by Coomassie blue staining (Fig. 2).
When purifying a new arrestin variant, use 90% of the sample for Coomassie gel and 10% for Western blot with anti-arrestin antibodies. This is also important when the overall expression level is not very high, e.g., in case of truncated cys-less enhanced arrestin-1 (Vishnivetskiy et al., 2013)(eVSA-QH-Tr, Fig. 3) because it is challenging to identify the arrestin band on the Coomassie-stained gel of the heparin-Sepharose fractions, and thus it can be difficult to decide which fractions should be pooled for the next step without parallel Western analysis (Fig. 3).
-
30
Carefully pool the fractions according to the gels and estimate the final salt concentration in the combined fractions.
-
31
In contrast to most arrestins that are not subject to in vivo proteolysis in E. coli, the C-tail of arrestin-3 is often cleaved by proteases. Although different kinds of protease inhibitors are added, we observe that up to half of the arrestin-3 is truncated (cleaved at residue 390). The full-length and truncated forms are partially separated on heparin-Sepharose, as shown in Fig. 2. To ensure that only full-length arrestin-3 is purified, carefully pool the fractions containing this form at around 380–500 mM NaCl.
Fig. 2.

Elution of arrestin-3 from heparin-Sepharose (Coomassie gels). Fraction numbers and positions of full-length and in vivo truncated arrestin-3 are shown. Molecular masses of markers are shown on the left of the top gel.
Fig. 3.

Elution of enhanced truncated cys-less arrestin-1 eVSA-QH-Tr (C63V, C128S, C143A, K257Q, E346H; 1-378; this is bovine cys-less arrestin-1 analogue of the most potent in terms of Rh* binding mouse mutant (Vishnivetskiy et al., 2013)) from heparin-Sepharose. Coomassie gels (left panels) do not allow unambiguous identification of fractions to pool, so it is necessary to rely on Western blots (right panels). M, molecular weight markers; S, protein precipitated by ammonium sulfate and then dissolved and loaded onto heparin-Sepharose; Pt, pass-through; W, wash; fraction numbers are shown (numbers greater than 40 indicate high-salt wash).
Q-Sepharose and SP-Sepharose
Arrestin-1 and Arrestin-2 (see Table 2)
Table 2.
Purification of WT and mutant arrestins from different species.
| Arrestin | Protein | Elution Peak (NaCl-mM) | Yield (mg/1L of bacterial culture) | |||
|---|---|---|---|---|---|---|
| Organism | Heparin -Sepharose | Q-Sepharose | SP-Sepharose | |||
| Bos taurus | Arrestin-1 | WT | 190–270 | 54–80 | X | 4–5 |
| 1–378 | 350–450 | 12–42 | - | 5 | ||
| Cys-less | 210–290 | 34–100 | X | 8 | ||
| Arrestin-2 | WT | 442–523 | 97–164 | - | 13.5 | |
| 3A | 596–681 | 150–200 | - | 3.9 | ||
| 1–382 | 623–707 | 27–80 | 176–225 | 7.8 | ||
| Cys-less | 426–554 | 120–165 | 92–145 | 10 | ||
| Arrestin-3 | WT | 380–500 | X | 227–327 | 3.4 | |
| 1–382 | 633–703 | 17–43 | - | 0.6 | ||
| Mus musculus | Arrestin-1 | WT | 10–55 | 50–130 | - | 1.35 |
| 3A | 170–300 | 120–226 | - | 9.85 | ||
| Homo sapiens | Arrestin-1 | WT | 183–289 | 18–63 | X | 2.8 |
| Arrestin-4 | WT | 10–55 | 40–130 | - | 0.4 | |
| Ambystoma tigrinum | Arrestin-1 | WT | 114–341 | 105–142 | - | 8.6 |
| 3A | 450–590 | 182–208 | - | 0.44 | ||
| Arrestin-4 | WT | 90–140 | 142–165 | - | 1.4 | |
X, the column is used as a pre-filter to bind impurities and let the arrestin flow through; -, the column is not used.
Q-Sepharose chromatography
For visual arrestin-1 and many of its mutants as well as for arrestin-2, pool the peak arrestin-containing fractions eluted from the heparin-Sepharose.
-
32
Combine these fractions, concentrate in an Amicon filter unit (YM30, Millipore) to approximately 10 ml and filter through a 0.8 μm filter (Sterile Syringe Filter, Corning)
-
33
Take 100 μl aliquot of the supernatant and transfer it to a microcentrifuge tube labeled “heparin” sample, and freeze at −20°C)
-
34
Pack under gravity flow 10 ml Q-Sepharose and SP-Sepharose columns (in 1.5×20 cm columns, Bio-Rad) and wash them with 200 ml of MCB at 4°C.
-
35
Estimate the concentration of NaCl in concentrated pooled fractions (based on the gradient used, taking into account that salt concentration after ~25 ml the column is delayed by 3 fractions; e.g., in case of arrestin-1, the gradient is 100 mM to 500 mM, i.e., the salt in each fraction is higher than in the previous one by ~10 mM, so NaCl in fraction 15 can be estimated as 100 mM + (15-3)×10 mM = 220 mM) and then load the sample onto a 10 ml Q-Sepharose column while diluting the sample (using a gradient mixer) with 0.2x MCB (dilute MCB buffer five-fold) to a salt concentration that is < 20% of elution peak (Table 2).
The protein should be diluted immediately before it is pumped onto the column as arrestins aggregate at low salt concentrations in solution, but are stable in low salt when bound to the resin.
-
36
Wash the column with 50–100 ml of MCB and then elute with a 300 ml linear gradient of NaCl in MCB (0–200 mM for arrestin-1 and 0 – 300 mM for arrestin-2). Collect 30 10 ml fractions and analyze them by 10% SDS-PAGE with Coomassie staining, as above.
Important: for new proteins, run the Western in parallel with a Coomassie-stained gel, using 90% of the sample for the Coomassie and 10% for the Western blot.
-
37
After the gradient, wash the column with 50 ml of 2 M NaCl in MCB (high salt elution), collecting 10-ml fractions.
-
38
Carefully pool the fractions from Q-Sepharose (based on Coomassie gel and Western blot), estimate the final NaCl concentration of the pool, and adjust the final concentration of NaCl to ~100 mM (for arrestin-1).
In cases where overall expression is high, such as arrestin-2 or cys-less arrestin-1 (eNCL, shown in Fig. 4) there is no need to use SP-Sepharose column. Arrestin-2 and eNCL (Fig. 4) come from the Q-Sepharose column very pure (>95% purity). Skip the SP-Sepharose column step.
Fig. 4.

Two-step purification of cys-less arrestin-1 eNCL (C63V, C128S, C143V) (Hanson et al., 2006b; Vishnivetskiy et al., 2010). Coomassie gels for heparin-Sepharose (left panels) and subsequent Q-Sepharose (right panels) are shown. M, molecular weight markers; S, protein precipitated by ammonium sulfate and then dissolved and loaded onto heparin-Sepharose; Pt, pass-through; W, wash; H, pooled fractions from heparin-Sepharose; fraction numbers are shown (numbers greater than 40 after heparin and greater than 30 after Q indicate high-salt wash).
SP-Sepharose chromatography
-
39
Load pooled fractions onto a 10 ml SP-Sepharose column and collect the flow-through. At 100 mM NaCl, visual arrestin-1 directly flows through the SP-Sepharose, whereas some contaminants bind.
-
40
Concentrate the SP-Sepharose flow-through containing visual arrestin-1 (>95% purity) to 3–5 mg/ml in Amicon filter unit (YM30, Millipore). To eliminate aggregates formed during concentration step, filter the concentrated sample through a 0.8 μm filter (Sterile Syringe Filter, Corning). Measure protein concentration.
-
41
Aliquot and freeze arrestin samples at −80°C.
Frozen arrestins do not appreciably reduce their activity for at least 2 years and with 2–3 freeze-thaw cycles.
Alternate Protocol
Arrestin-3 and truncated form of arrestin-1-(1-378) (see Table 2)
After steps 1–31 of Basic Protocol 1, use the following procedure for arrestin-3 and truncated Arrestin-1-(1-378).
Arrestin-3 as well as truncated arrestin-1-(1-378) bind to SP-Sepharose relatively strongly. Therefore, to avoid a time-consuming concentration step, we employ Q-Sepharose as a “filter” column, while binding arrestin proteins to SP-Sepharose.
-
32
Pool heparin-Sepharose fractions (from Step 3), estimate the NaCl concentration. When necessary (Table 2), add 5 M NaCl to the final concentration of 100 mM
-
33
Take 100 μl aliquot of the supernatant, transfer to the microcentrifuge tube labeled “heparin” sample, and freeze at −20°C).
-
34
Pack under gravity flow 10 ml Q-Sepharose and SP-Sepharose columns (in 1.5x20 cm columns, Bio-Rad) and wash them with 200 ml of MCB at 4°C.
-
35
Sequentially connect Q- and SP-Sepharose columns as shown in Fig. 1B. Load samples onto Q-Sepharose column. At ~100 mM NaCl, arrestins flow through Q-Sepharose and only bind to SP-Sepharose.
-
36
Wash Q- and SP-Sepharose columns with 50 ml of 100 mM NaCl in MCB.
-
37
Disconnect SP-Sepharose from Q-Sepharose. Elute arrestin from SP-Sepharose column with a 300 ml linear gradient of NaCl (MCB/100 mM NaCl to MCB/500 mM NaCl). Collect thirty 10 ml fractions. After the gradient, wash the column with 50 ml of 2 M NaCl in MCB (high salt elution), collecting 10-ml fractions. Analyze fractions by SDS-PAGE, as above.
-
38
Pool the arrestin-containing fractions and estimate the final NaCl concentration. Concentrate using an Amicon filter unit (YM30, Millipore) to 3–5 ml. To eliminate aggregates formed during concentration step, filter the concentrated sample through a 0.8 μm filter (Sterile Syringe Filter, Corning). Aliquot and freeze arrestin samples at −80°C.
-
39
Measure protein concentration.
Fig. 5 shows typical purity of the protein (arrestin-3) at different steps of purification, whereas Table 3 shows usual protein yields at each step (the last number is the yield of purified arrestin).
Fig. 5.

Purity of arrestin-3 through purification steps. The positions of molecular weight markers (M) and protein content of lysate, supernatant after dissolution of ammonium sulfate-precipitated protein, pooled heparin-Sepharose (Heparin) and SP-Sepharose (SP) fractions are shown.
Table 3.
Typical yields of total protein at sequential steps of purification (these are achieved by a moderately experienced person; yields can vary +/− 50%)
| Purification step | Protein (mg) | ||
|---|---|---|---|
| Bovine arrestin-1 | Bovine arrestin-2 | Bovine arrestin-3 | |
| Homogenate | ~2,200 | ~2,600 | ~2,900 |
| Supernatant | ~1,100 | ~1,500 | ~1,600 |
| Heparin-Sepharose | ~580 | ~100 | ~30 |
| Q-Sepharose (pure) | ~20 | ~50 | - |
| SP-Sepharose (pure) | - | - | ~13 |
SUPPORT PROTOCOL 1: Small-scale test-expression of WT and mutant arrestins in E. coli
Introductory paragraph
The aim of this protocol is to determine the relative expression level of different arrestins in E. coli. Purification of any protein is usually a time- and effort-consuming process. In the case of arrestin it takes 5–6 days to obtain pure high quality protein. Not all arrestins behave identically in the E. coli expression system. For example, bovine arrestin-2 is expressed at levels twice those of bovine arrestin-1, whereas bovine arrestin-3 expresses at lower levels then either arrestin-1 or -2. Sometimes substitution of a single amino acid in bovine arrestin-1 can dramatically change its behavior. In our experience, nine out of ten mutants express well, whereas some simply do not express at a sufficient level to make their purification feasible. Test expression of the desired arrestins using either the well-characterized bovine arrestin-1 or another highly expressing species (as a positive control) in a small volume of E. coli cells provides an estimate of what to expect and whether large-scale purification should be performed. As an example, the results of the test expression of the bovine cys-less arrestin-1 and its full-length and truncated mutants with single or double cysteine substitutions are shown in Fig. 6.
Fig. 6.
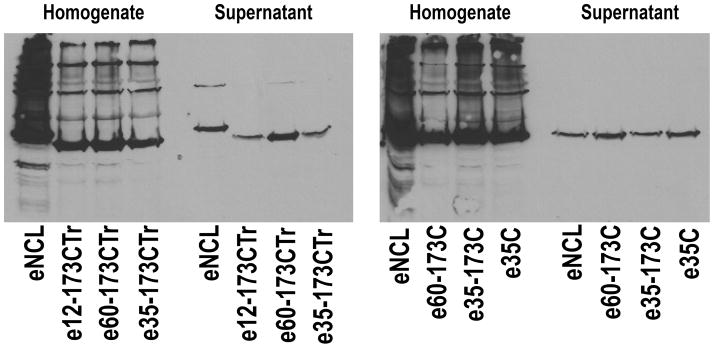
Western blots of test-expression samples of truncated (1-378; left panel) and full-length (right panel) bovine arrestin-1 mutants on cys-less VSA (C63V, C128S, C143A) background with cysteines in the indicated positions, as compared to eNCL (C63V, C128S, C143V). The blots of supernatant are much more informative than those of homogenate. Note several-fold differences in expression levels, although all proteins express well enough to perform a big prep.
Materials (same as in Basic protocol 1)
Step 1. Cell Growth and Arrestin Expression
Prepare one sterile 15 ml tube with 3 ml LB medium for each arrestin protein to be test-expressed (including positive control). Add 3 μl of the ampicillin stock solution into each tube.
Inoculate each tube with frozen arrestin/BL21 cells (20–30 μl that can be scraped with a pipette tip off the glycerol stock without thawing it) and grow overnight (12–16 h) at 30°C with continuous shaking at 250 rpm.
Add 7.5 μl or 10.5 μl (for arrestin-3) of 0.01 M IPTG to each tube (final concentrations 25 μM and 35 μM, respectively) to induce the expression of arrestin and keep shaking at 30°C, 250 rpm for additional 3–4 h.
-
Pellet cells by centrifugation for 10 min at 3,000 rpm (6,000 ×g) at 4°C. Discard the supernatant carefully; let the tubes stand for 1 min upside down to remove all traces of LB medium.
When performing test-expression of several proteins, prepare solutions sufficient for an extra sample. For example, when expressing 9 proteins, prepare solutions for 10 to allow for loss and pipetting errors. -
Add 2 μl of 1.0 M glucose (final concentration 2 mM) and 2 μl PMSF to 200 μl of the lysis buffer.
Starting from this point all remaining steps are performed on ice. Here and at each step below add PMSF to the lysis buffer immediately before use. Add 200 μl of the ice-cold lysis buffer with glucose and PMSF to the bacterial pellet and resuspend the cells by vortexing. Transfer the cell suspension to a 1.5 ml microcentrifuge tube.
Add 200 μl of the ice-cold lysis buffer with 0.1 mg/ml lysozyme to 200 μl of the cell suspension and vortex. Keep on ice for ~40 minutes.
-
Freeze cells at −80°C for approximately 20 minutes or until fully frozen. After that allow the cells to thaw on ice.
The freeze-thaw cycle helps to break the bacterial cell membrane because the ice crystals forming upon freezing cause the bacteria to swell. Add 200 μl of the lysis buffer with PMSF to thawed cell suspension and sonicate for 15 seconds at 12% amplitude (Fisher Dismembrator, Model 500) in ice-water bath.
-
Add 4 μl of 1 M MgCl2, 2 μl RNase A (100 mg/ml) and 2 μl DNase I (15,000 U/ml) to the sample and incubate on ice for ~40 minutes.
Sonication as well as DNAse and RNAse treatment helps to shear DNA and RNA after lysis of bacterial cells. Transfer 200 μl to a new microcentrifuge tube labeled “homogenate” and freeze at −80°C.
Transfer the remaining 400 μl to the 500 μl centrifuge tube (Beckman) and centrifuge at 100,000 rpm (356,000 ×g) for 30 min at 4°C (TLA 120.1 rotor, TL-100 tabletop ultracentrifuge, Beckman).
Transfer the supernatant to new microcentrifuge tube labeled “supernatant” and freeze at −80°C.
To prepare samples for Western analysis, take 5 μl of the “homogenate” and “supernatant” and dissolve them in 15 μL of the 2X sample buffer.
Run 2 μl of each sample on 10% SDS-PAGE gel, transfer to PVDF membrane (Millipore) and develop with F4C1 or F431 antibody (or any other arrestin-specific antibody). Compare the intensity of the arrestin band with that of known protein test-expressed in parallel (e.g., bovine arrestin-1 or arrestin-2).
Commentary
a. Background Information
The protocols described here for arrestin expression in E. coli are designed to yield soluble, properly folded protein that is fully functional. After lysis, insoluble material (which includes considerable amounts of some arrestins) is pelleted along with cell debris. Partial purification, as well as elimination of DNA and RNA fragments that would preclude heparin-Sepharose binding, is achieved by ammonium sulfate precipitation (Lohse et al., 1992). The first chromatographic step, heparin-Sepharose, is based on the finding that arrestins bind heparin with relatively high affinity (Gurevich et al., 1994; Palczewski and Hargrave, 1991) and that considerable purification can be achieved by heparin-Sepharose chromatography (Söhlemann et al., 1995). An anion exchange step using Q-Sepharose was also used earlier, although without preceding heparin-Sepahrose step (Lohse et al., 1992).
Based on many years of experience, we combined previously tested steps and cation exchange on SP-Sepharose into a unified procedure. The method described here is general; it works for the purification of all four mammalian arrestins along with most mutants, such as the phosphorylation-independent truncated and 3A forms (Gurevich, 1998; Song et al., 2009), and the cysteine-free forms (Hanson et al., 2006b; Hanson et al., 2007b; Kim et al., 2012; Vishnivetskiy et al., 2011; Zhuo et al., 2014). The two chromatographic steps described originally (Gurevich and Benovic, 2000; Gurevich et al., 1999) are supplemented with an additional SP-Sepharose step, where either Q- or SP-Sepharose column serves as a “filter” removing certain impurities, while arrestin binds the other column and is then eluted with a NaCl gradient. These modifications eliminate the time-consuming and often yield-lowering concentration steps. The higher arrestin-binding capacity of SP-Sepharose than of Q-Sepharose also dramatically increases the yields of purified arrestin proteins, especially arrestin-3, which does not express as readily as other subtypes and is subject to in vivo proteolysis in E. coli.
b. Critical Parameters
Note that the described induction by low concentrations of IPTG at 30°C is specifically optimized for maximum yields of properly folded functional arrestin in bacterial cytoplasm. While higher overall expression can be achieved at higher IPTG concentrations, in this case most arrestins end up in the insoluble fraction (likely inclusion bodies). Since multi-milligram quantities of most arrestins pure that are enough for either crystallization (Han et al., 2001; Hirsch et al., 1999; Sutton et al., 2005; Zhan et al., 2011a) or demanding biophysical studies (Hanson et al., 2007a; Hanson et al., 2006b; Kim et al., 2011; Kim et al., 2012; Vishnivetskiy et al., 2011; Zhuang et al., 2013; Zhuo et al., 2014) can be produced in a soluble form, there has not been an attempt to refold arrestins from inclusion bodies. Accordingly, the most critical parameter for the success of purification of arrestins by this protocol is the expression level of correctly-folded protein, which is estimated by small-scale test-expressions (Support protocol 1). We found that it is virtually impossible to purify any WT or mutant arrestin that expresses at < 1 mg/L of bacterial culture.
c. Troubleshooting
The most common problem is low expression. We found that ~10% of mutants do not express at levels necessary for subsequent purification. It is very important to compare total arrestin expression (in cell homogenates) with the soluble fraction (see Support protocol 1). In some cases the protein appears to express, but remains insoluble (i.e., it is high in homogenate, but low in the supernatant). While counterintutitive, the yield of soluble arrestin can often be increased by reducing the IPTG concentration (e.g., to 15–20 μM). This may be combined with the more classical method or reducing the temperature to 18–25°C following induction, with correspondingly longer cell growth times. If neither of these approaches works, the best strategy is to use a different mutant or select WT arrestin from a different species. For example, we were unable to express rat arrestin-2 and -3, whereas the closely related bovine, mouse, and human homologs expressed well.
The tendency of all arrestins to aggregate at salt concentrations below 100 mM NaCl can also result in losses during dilution. If dilution below 100 mM NaCl is necessary for column binding, the arrestin sample should be diluted with low-salt buffer using gradient mixer immediately before it goes onto the column since arrestins bound to the column matrix are significantly more resistant to low ionic strength. Similarly, when arrestins elute at low salt concentration from any column (Table 2), the ionic strength of the fractions should be adjusted to >100 mM NaCl before ultrafiltration. In addition, concentrating most arrestins above 10 mg/ml should be avoided, as it often leads to aggregation. In our experience, arrestins do not have significant aggregation upon freezing and therefore can survive several freeze-thaw cycles. However, when even a small fraction of aggregated protein is detrimental for the experiment, thawed arrestins should be centrifuged for 1 h at 4°C at > 100,000xg, and the top four fifths of the supernatant (to exclude any traces of the pellet) should be used. Modification of single-cysteine arrestins with spin (Hanson et al., 2007a; Hanson et al., 2006a; Hanson et al., 2006b; Hanson et al., 2007b; Vishnivetskiy et al., 2010; Vishnivetskiy et al., 2011; Zhuo et al., 2014) or fluorescent labels (Ahmed et al., 2011) for 1–2 h at room temperature often results in aggregation. These procedures can be safely performed overnight at 4°C.
d. Anticipated Results
Table 3 shows the expected yields of bovine WT arrestins, all of which were obtained at >95% purity. The yields of certain mutants and/or arrestins from different species can be much lower. A good guide is an estimate of the amount of arrestin of interest in soluble form in low-scale test-expression (Support protocol 1), with expected ~30% overall yield after purification. Thus, if the arrestin of interest yields ~1 mg/L of soluble protein, i.e., ~4–5 mg per 4.5 L preparation as described here, about 1.5–2 mg of pure protein can be obtained in the end, which is the minimum that makes purification feasible. Lower expression levels result in disproportionally lower yields due to losses in the process.
Sample data and data manipulation instructions
When purifying a new protein, or any arrestin with lower yield, it is important to analyze all chromatography fractions both using a Coomassie-stained SDS-PAGE gel and using a Western blot (Fig. 3). It should be noted that all arrestins run anomalously slowly on SDS-PAGE: arrestin-1 is 44 kDa, but runs at ~50 kDa, arrestin-2 is 46 kDa, but runs above 50 kDa. Western blots are certainly not quantitative, but indicate where the majority of the protein of interest elutes, which allows ready identification of the Coomassie band that corresponds to the expressed protein. In contrast, Coomassie staining is more quantitative. As a result, we recommend that the fractions are pooled on the basis of the Coomassie-stained gels. In the end, it is useful to run samples from different stages of purification side-by-side (Fig. 5), loading the same amount of total protein (~ 1–2 μg). To evaluate purity of the final product, it is advisable to run different amounts of it on the same gel, from ~5 μg to visualize even minor impurities to ~ 0.2 μg to see possible closely running proteolytic products.
e. Time Considerations
There are several points in the expression and purification procedure where it can be paused and then resumed days or even weeks later. Cell growth and induction takes ~ 1.5 days. It is important to resuspend the cells in the first portion of lysis buffer and incubate them with lysozyme right after pelleting the cells. After that, the suspension must be frozen and can be stored at −80°C for up to a few weeks, possibly longer (we never tried more than 2 months). Thus, it is feasible to express several proteins staggered by one day and accumulate numerous cell suspensions for future processing. The continuation of lysis, pelleting the debris, and protein precipitation by ammonium sulfate should be done in one day, but the resultant pellet can again be stored at −80°C for up to a few weeks. Subsequent steps, starting with dissolving pellet in MCB and all necessary chromatographic steps should be performed in close succession. Usually it is convenient to load the heparin-Sepharose column overnight, then elute with a gradient, analyze the fractions, pool the protein, concentrate, and start loading the next column in a single day. For some proteins, it might be feasible to load the next column in 2–3 h, and then elute with a gradient overnight. For others, loading overnight is necessary, with elution and fraction analysis the next day. If the protein requires a third chromatographic step, it takes another night (loading or gradient elution) and next day (fraction analysis, concentration, aliquoting, etc). Thus, purification of proteins that require two chromatographic steps takes three consecutive days: loading of heparin-Sepharose should start on Monday, loading the next column on Tuesday, and final fraction analysis, concentration, and aliquoting is on Wednesday. Those that require three steps take one extra day. The first day involves limited hands-on time, whereas the days when fractions are analyzed are labor intensive.
Acknowledgments
This work was supported in part by NIH grants EY011500, GM077561, and GM081756 (to VVG) and GM095633 (to TMI).
Footnotes
As an example on how to calculate sample dilution we use arrestin-3. The arrestin-3 is eluted from heparin-Sepharose at ~380–500 mM NaCl, therefore, the salt concentration of loading sample was set at 200 mM NaCl. The pellet contains 2.4 M ammonium sulfate, which is equivalent to 7.2 M NaCl solution in terms of ionic strength. Therefore, for a 20 ml pellet, final volume of the loading sample should be 720 ml (36-fold dilution relative to pellet).
References
- Ahmed MR, Zhan X, Song X, Kook S, Gurevich VV, Gurevich EV. Ubiquitin ligase parkin promotes Mdm2-arrestin interaction but inhibits arrestin ubiquitination. Biochemistry. 2011;50:3749–3763. doi: 10.1021/bi200175q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attramadal H, Arriza JL, Aoki C, Dawson TM, Codina J, Kwatra MM, Snyder SH, Caron MG, Lefkowitz RJ. Beta-arrestin2, a novel member of the arrestin/beta-arrestin gene family. J Biol Chem. 1992;267:17882–17890. [PubMed] [Google Scholar]
- Chen Q, Zhuo Y, Kim M, Hanson SM, Francis DJ, Vishnivetskiy SA, Altenbach C, Klug CS, Hubbell WL, Gurevich VV. Self-association of arrestin family members. Handb Exp Pharmacol. 2014;219:205–223. doi: 10.1007/978-3-642-41199-1_11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeWire SM, Ahn S, Lefkowitz RJ, Shenoy SK. Beta-arrestins and cell signaling. Annu Rev Physiol. 2007;69:483–510. doi: 10.1146/annurev.physiol.69.022405.154749. [DOI] [PubMed] [Google Scholar]
- Donoso LA, Gregerson DS, Smith L, Robertson S, Knospe V, Vrabec T, Kalsow CM. S-antigen: preparation and characterization of site-specific monoclonal antibodies. Curr Eye Res. 1990;9:343–355. doi: 10.3109/02713689008999622. [DOI] [PubMed] [Google Scholar]
- Goodman OB, Jr, Krupnick JG, Santini F, Gurevich VV, Penn RB, Gagnon AW, Keen JH, Benovic JL. Beta-arrestin acts as a clathrin adaptor in endocytosis of the beta2-adrenergic receptor. Nature. 1996;383:447–450. doi: 10.1038/383447a0. [DOI] [PubMed] [Google Scholar]
- Gurevich EV, Gurevich VV. Arrestins are ubiquitous regulators of cellular signaling pathways. Genome Biology. 2006a;7:236. doi: 10.1186/gb-2006-7-9-236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurevich VV. The selectivity of visual arrestin for light-activated phosphorhodopsin is controlled by multiple nonredundant mechanisms. J Biol Chem. 1998;273:15501–15506. doi: 10.1074/jbc.273.25.15501. [DOI] [PubMed] [Google Scholar]
- Gurevich VV, Benovic JL. Arrestin: mutagenesis, expression, purification, and functional characterization. Methods Enzymol. 2000;315:422–437. doi: 10.1016/s0076-6879(00)15859-8. [DOI] [PubMed] [Google Scholar]
- Gurevich VV, Chen CY, Kim CM, Benovic JL. Visual arrestin binding to rhodopsin: Intramolecular interaction between the basic N-terminus and acidic C-terminus of arrestin may regulate binding selectivity. J Biol Chem. 1994;269:8721–8727. [PubMed] [Google Scholar]
- Gurevich VV, Gurevich EV. The structural basis of arrestin-mediated regulation of G protein-coupled receptors. Pharm Ther. 2006b;110:465–502. doi: 10.1016/j.pharmthera.2005.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurevich VV, Gurevich EV. Overview of Different Mechanisms of Arrestin-Mediated Signaling. Curr Prot Pharmacol. 2014 doi: 10.1002/0471141755.ph0210s67. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurevich VV, Orsini MJ, Benovic JL. Characterization of arrestin expression and function. Receptor Biochemistry and Methodology. 1999;IV:157–178. [Google Scholar]
- Gurevich VV, Pals-Rylaarsdam R, Benovic JL, Hosey MM, Onorato JJ. Agonist-receptor-arrestin, an alternative ternary complex with high agonist affinity. J Biol Chem. 1997;272:28849–28852. doi: 10.1074/jbc.272.46.28849. [DOI] [PubMed] [Google Scholar]
- Han M, Gurevich VV, Vishnivetskiy SA, Sigler PB, Schubert C. Crystal structure of beta-arrestin at 1.9 A: possible mechanism of receptor binding and membrane translocation. Structure. 2001;9:869–880. doi: 10.1016/s0969-2126(01)00644-x. [DOI] [PubMed] [Google Scholar]
- Hanson SM, Cleghorn WM, Francis DJ, Vishnivetskiy SA, Raman D, Song X, Nair KS, Slepak VZ, Klug CS, Gurevich VV. Arrestin mobilizes signaling proteins to the cytoskeleton and redirects their activity. J Mol Biol. 2007a;368:375–387. doi: 10.1016/j.jmb.2007.02.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson SM, Francis DJ, Vishnivetskiy SA, Klug CS, Gurevich VV. Visual arrestin binding to microtubules involves a distinct conformational change. J Biol Chem. 2006a;281:9765–9772. doi: 10.1074/jbc.M510738200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson SM, Francis DJ, Vishnivetskiy SA, Kolobova EA, Hubbell WL, Klug CS, Gurevich VV. Differential interaction of spin-labeled arrestin with inactive and active phosphorhodopsin. Proc Natl Acad Sci U S A. 2006b;103:4900–4905. doi: 10.1073/pnas.0600733103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson SM, Van Eps N, Francis DJ, Altenbach C, Vishnivetskiy SA, Arshavsky VY, Klug CS, Hubbell WL, Gurevich VV. Structure and function of the visual arrestin oligomer. EMBO J. 2007b;26:1726–1736. doi: 10.1038/sj.emboj.7601614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson SM, Vishnivetskiy SA, Hubbell WL, Gurevich VV. Opposing effects of inositol hexakisphosphate on rod arrestin and arrestin2 self-association. Biochemistry. 2008;47:1070–1075. doi: 10.1021/bi7021359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch JA, Schubert C, Gurevich VV, Sigler PB. The 2.8 A crystal structure of visual arrestin: a model for arrestin’s regulation. Cell. 1999;97:257–269. doi: 10.1016/s0092-8674(00)80735-7. [DOI] [PubMed] [Google Scholar]
- Kim M, Hanson SM, Vishnivetskiy SA, Song X, Cleghorn WM, Hubbell WL, Gurevich VV. Robust self-association is a common feature of mammalian visual arrestin-1. Biochemistry. 2011;50:2235–2242. doi: 10.1021/bi1018607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M, Vishnivetskiy SA, Van Eps N, Alexander NS, Cleghorn WM, Zhan X, Hanson SM, Morizumi T, Ernst OP, Meiler J, Gurevich VV, Hubbell WL. Conformation of receptor-bound visual arrestin. Proc Nat Acad Sci USA. 2012;109:18407–18412. doi: 10.1073/pnas.1216304109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YJ, Hofmann KP, Ernst OP, Scheerer P, Choe HW, Sommer ME. Crystal structure of pre-activated arrestin p44. Nature. 2013;497:142–146. doi: 10.1038/nature12133. [DOI] [PubMed] [Google Scholar]
- Kook S, Zhan X, Cleghorn WM, Benovic JL, Gurevich VV, Gurevich EV. Caspase-cleaved arrestin-2 and BID cooperatively facilitate cytochrome C release and cell death. Cell Death Differ. 2014;21:172–184. doi: 10.1038/cdd.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laporte SA, Oakley RH, Zhang J, Holt JA, Ferguson sSG, Caron MG, Barak LS. The 2-adrenergic receptor/arrestin complex recruits the clathrin adaptor AP-2 during endocytosis. Proc Nat Acad Sci USA. 1999;96:3712–3717. doi: 10.1073/pnas.96.7.3712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohse MJ, Andexinger S, Pitcher J, Trukawinski S, Codina J, Faure JP, Caron MG, Lefkowitz RJ. Receptor-specific desensitization with purified proteins. Kinase dependence and receptor specificity of beta-arrestin and arrestin in the beta 2-adrenergic receptor and rhodopsin systems. J Biol Chem. 1992;267:8558–8564. [PubMed] [Google Scholar]
- Lohse MJ, Benovic JL, Codina J, Caron MG, Lefkowitz RJ. beta-Arrestin: a protein that regulates beta-adrenergic receptor function. Science. 1990;248:1547–1550. doi: 10.1126/science.2163110. [DOI] [PubMed] [Google Scholar]
- Luttrell LM, Ferguson SS, Daaka Y, Miller WE, Maudsley S, Della Rocca GJ, Lin F, Kawakatsu H, Owada K, Luttrell DK, Caron MG, Lefkowitz RJ. Beta-arrestin-dependent formation of beta2 adrenergic receptor-Src protein kinase complexes. Science. 1999;283:655–661. doi: 10.1126/science.283.5402.655. [DOI] [PubMed] [Google Scholar]
- Palczewski K, Hargrave PA. Studies of ligand binding to arrestin. J Biol Chem. 1991;266:4201–4206. [PubMed] [Google Scholar]
- Shukla AK, Manglik A, Kruse AC, Xiao K, Reis RI, Tseng WC, Staus DP, Hilger D, Uysal S, Huang LY, Paduch M, Tripathi-Shukla P, Koide A, Koide S, Weis WI, Kossiakoff AA, Kobilka BK, Lefkowitz RJ. Structure of active β-arrestin-1 bound to a G-protein-coupled receptor phosphopeptide. Nature. 2013;497:137–141. doi: 10.1038/nature12120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Söhlemann P, Hekman M, Puzicha M, Buchen C, Lohse MJ. Binding of purified recombinant beta-arrestin to guanine-nucleotide-binding-protein-coupled receptors. Eur J Biochem. 1995;232:464–472. [PubMed] [Google Scholar]
- Song X, Seo J, Baameur F, Vishnivetskiy SA, Chen Q, Kook S, Kim M, Brooks EK, Altenbach C, Hong Y, Hanson SM, Palazzo MC, Chen J, Hubbell WL, Gurevich EV, Gurevich VV. Rapid degeneration of rod photoreceptors expressing self-association-deficient arrestin-1 mutant. Cell Signal. 2013;25:2613–2624. doi: 10.1016/j.cellsig.2013.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song X, Vishnivetskiy SA, Gross OP, Emelianoff K, Mendez A, Chen J, Gurevich EV, Burns ME, Gurevich VV. Enhanced Arrestin Facilitates Recovery and Protects Rod Photoreceptors Deficient in Rhodopsin Phosphorylation. Curr Biol. 2009;19:700–705. doi: 10.1016/j.cub.2009.02.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song X, Vishnivetskiy SA, Seo J, Chen J, Gurevich EV, Gurevich VV. Arrestin-1 expression in rods: balancing functional performance and photoreceptor health. Neuroscience. 2011;174:37–49. doi: 10.1016/j.neuroscience.2010.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton RB, Vishnivetskiy SA, Robert J, Hanson SM, Raman D, Knox BE, Kono M, Navarro J, Gurevich VV. Crystal Structure of Cone Arrestin at 2.3Å: Evolution of Receptor Specificity. J Mol Biol. 2005;354:1069–1080. doi: 10.1016/j.jmb.2005.10.023. [DOI] [PubMed] [Google Scholar]
- Vishnivetskiy SA, Chen Q, Palazzo MC, Brooks EK, Altenbach C, Iverson TM, Hubbell WL, Gurevich VV. Engineering visual arrestin-1 with special functional characteristics. J Biol Chem. 2013;288:11741–11750. doi: 10.1074/jbc.M112.445437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vishnivetskiy SA, Francis DJ, Van Eps N, Kim M, Hanson SM, Klug CS, Hubbell WL, Gurevich VV. The role of arrestin alpha-helix I in receptor binding. J Mol Biol. 2010;395:42–54. doi: 10.1016/j.jmb.2009.10.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vishnivetskiy SA, Gimenez LE, Francis DJ, Hanson SM, Hubbell WL, Klug CS, Gurevich VV. Few residues within an extensive binding interface drive receptor interaction and determine the specificity of arrestin proteins. J Biol Chem. 2011;286:24288–24299. doi: 10.1074/jbc.M110.213835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu N, Hanson SM, Francis DJ, Vishnivetskiy SA, Thibonnier M, Klug CS, Shoham M, Gurevich VV. Arrestin binding to calmodulin: a direct interaction between two ubiquitous signaling proteins. J Mol Biol. 2006;364:955–963. doi: 10.1016/j.jmb.2006.09.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan X, Gimenez LE, Gurevich VV, Spiller BW. Crystal structure of arrestin-3 reveals the basis of the difference in receptor binding between two non-visual arrestins. J Mol Biol. 2011a;406:467–478. doi: 10.1016/j.jmb.2010.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan X, Kaoud TS, Dalby KN, Gurevich VV. Non-visual arrestins function as simple scaffolds assembling MKK4- JNK3α2 signaling complex. Biochemistry. 2011b;50:10520–10529. doi: 10.1021/bi201506g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan X, Kaoud TS, Kook S, Dalby KN, Gurevich VV. JNK3 binding to arrestin-3 differentially affects the recruitment of upstream MAP kinase kinases. J Biol Chem. 2013;288:28535–28547. doi: 10.1074/jbc.M113.508085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan X, Perez A, Gimenez LE, Vishnivetskiy SA, Gurevich VV. Arrestin-3 binds the MAP kinase JNK3α2 via multiple sites on both domains. Cell Signal. 2014;26:766–776. doi: 10.1016/j.cellsig.2014.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang T, Chen Q, Cho MK, Vishnivetskiy SA, Iverson TI, Gurevich VV, Sanders CR. Involvement of Distinct Arrestin-1 Elements in Binding to Different Functional Forms of Rhodopsin. Proc Nat Acad Sci USA. 2013;110:942–947. doi: 10.1073/pnas.1215176110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuo Y, Vishnivetskiy SA, Zhan X, Gurevich VV, Klug CS. Identification of Receptor Binding-induced Conformational Changes in Non-visual Arrestins. J Biol Chem. 2014;289:20991–21002. doi: 10.1074/jbc.M114.560680. [DOI] [PMC free article] [PubMed] [Google Scholar]