

Abstract
Proteolysis is a critical modification leading to alteration of protein function with important outcomes in many biological processes. However, for the majority of proteases, we have an incomplete understanding of both cellular substrates and downstream effects. Here, we describe detailed protocols and applications for using the rationally engineered peptide ligase, subtiligase, to specifically label and capture protein N-termini generated by proteases either induced or added to complex biological samples. This method allows identification of the protein targets as well as their precise cleavage locations. This approach has revealed >8000 proteolytic sites in healthy and apoptotic cells including >1700 caspase cleavages. One can further determine substrate preferences through rate analysis with quantitative mass spectrometry, physiological substrate specificities, and even infer the identity of proteases operating in the cell. In this chapter, we also describe how this experimental method can be generalized to investigate proteolysis in any biological sample.
1. INTRODUCTION
1.1. Importance of proteolysis
Proteolysis, the hydrolysis of peptide bonds by proteases, is an essential activity in a wide range of cellular functions. Proteases exist in virtually all forms of life, and are classified into five mechanistic categories: serine, threonine, cysteine, acid, and metallo (Lopez-Otin & Matrisian, 2007). In humans alone there are over 550 identified proteases, but their precise roles and substrates are generally poorly understood. The number of substrates for a given protease ranges widely, from a single sites on a few proteins to cleaving a broad swath of the proteome. Proteases function in digesting and recycling proteins, irreversible posttranslational modification via N-terminal methionine processing, signal or transit peptide removal, cleavage of polypeptide chains into their multiple components, and removal of precursor domains. In addition to their role in protein maturation and function, proteolysis is critical for physiological processes including apoptosis. Endoproteolysis can lead to activation, inhibition, or a change of substrate function, allowing proteases to play important roles in signaling. Dysregulation of proteolysis contributes to many pathological states such as arthritis, inflammation, and cancer. Additionally, proteases are used as tools in the laboratory, industrial manufacturing, and commercial products.
An important step to understanding a protease’s role is identification and validation of substrates and cleavage locations. Such information leads naturally to examining the specific functional consequences for individual targets. Thus, there has been a surge in the development of technologies for global and unbiased characterization of proteolysis in complex biological samples (for reviews see Agard & Wells, 2009; Impens et al., 2010; Klingler & Hardt, 2012; Rogers & Overall, 2013). We briefly cover the state-of-the-art in this field and then focus on the detailed implementation and applications of the N-terminomics technology developed in our lab using subtiligase.
1.2. Approaches to substrate cleavages and identification
Historically, the identification of proteases responsible for specific cleavage events has often been driven by knowledge of important substrate proteins that were found cleaved in a biological process. For example, insulin was known to be produced from the precursor pro-insulin leading to the discovery of the protease furin (Smeekens et al., 1992). The processing of pro-IL-1β to IL-1β led to the discovery of the protease caspase-1 (Black, Kronheim, & Sleath, 1989). Until recently, most substrates have been found in a labor intensive, candidate-based approach using a range of focused biochemical approaches.
New proteomic methods have allowed unbiased searches of proteolytic substrates in complex samples (Table 13.1). These global studies often have two goals: to identify all cleavage events in a cell during a particular process and to identify all possible substrates of a given protease. These aims have been greatly aided by the advancements of analytical instruments, specifically liquid chromatography coupled to mass spectrometry (LC–MS). Most methods enrich for proteolytically cleaved peptides by taking advantage of the newly created α-carboxy- or α-amino-termini on either side of the cleavage site. This allows for capture and purification of substrates through specific chemical or enzymatic modification. A single global experiment can generate over a thousand peptide identifications that can be scored and mapped to a specific protein and/or cleavage site. The advantage to capturing the N-terminal side of the cleavage site (“N-terminomics”) is that most proteins are acetylated naturally as they are translated from ribosomes, such that virtually all unblocked α-amines are produced by a post-translational proteolytic event.
Table 13.1.
A summary of current methods for proteolytic cleavage site and substrate identification
| Method | Description | Proteolytic substrates reported | References |
|---|---|---|---|
| Subtiligase | Positive selection of free N-termini α-amines through subtiligase enzymatic labeling with an ester peptide tag. | 8090 peptides (1706 caspase) from untreated and apoptotic human cells | Crawford et al. (2013) and Mahrus et al. (2008) |
| COFRADIC | COmbined FRActional DIagonal Chromatography uses negative selection with a chemical modification at free N-termini (or other modification of interest) to enable separation of modified from unlabeled peptides during chromatography. | 68 caspase substrates from recombinant caspases-2, -3, -7; 9729 carboxypeptidase substrates from in vitro peptide library | Staes et al. (2008), Tanco et al. (2013), and Wejda et al. (2012) |
| TAILS | Terminal amine isotopic labeling uses chemical modifications of protein amines and thiols, sample trypsinization, and negative selection to enrich for neo N- or C-termini. | 288 MMP-2 cleavage sites; >100 GluC cleavage sites | Kleifeld et al. (2010) and Schilling, Huesgen, Barre, and Overall (2011) |
| N-CLAP | N-terminalomics by chemical labeling of the α-amines of proteins. Uses Edman degradation chemistry to block lysine amines to label N-terminal amines with a biotinylated tag for positive selection. | 278 peptides (23 caspase) in apoptotic Jurkat cells | Xu and Jaffrey (2010) and Xu, Shin, and Jaffrey (2009) |
| PROTOMAP | PROtein TOpography and Migration Analysis Platform creates visual peptographs from 1D SDS gel migration patterns and sequence coverage from MS of in-gel digestions to identify cleavages from mass shifts. | 744 proteins with cleavages in apoptotic Jurkat cells | Dix, Simon, and Cravatt (2008) and Dix et al. (2012) |
| GASSP+C- terminal immuno-pull down | Global analyzer of SILAC-derived substrates of proteolysis (GASSP) using differential gel analysis combined with pull down of Asp at C-termini using a specific antibody. | 360 proteolytic sites in Jurkat cells; 160 known caspase sites | Pham et al. (2012) |
| 2D DiGE +MS | Two-dimensional differential gel electrophoresis (2D-DiGE) separates complex mixtures using orthogonal electrophoresis methods and comparison of induced proteolysis and control sample gels reveal shifted spots due to proteolysis. | 21 caspase substrates in Jurkat cells | Tonge et al. (2001) |
| 1D gel+MS | Lysates harvested from in vivo induced proteolysis are run on a large gel, separated into 100 slices and prepared for mass spectrometry with in-gel trypsinization. Substrates are identified as those with less mass than expected values indicating cleavage events. | 37 peptides in apoptotic Jurkat cells | Thiede, Treumann, Kretschmer, Sohlke, and Rudel (2005) |
| 2D SDS PAGE | 2D SDS PAGE gel electrophoresis with protease addition as an intermediate step to look for spots that differentially migrate compared to control indicating proteolysis. | 41 caspase-1 substrates in THP cells | Shao, Yeretssian, Doiron, Hussain, and Saleh (2007) |
| ProC-TEL | Positive selection through carboxy termini tagging using transpeptidation enzymatic reaction. | 76 peptides from Escherichia coli lysates | Xu, Shin, and Jaffrey (2011) |
2. BASIC FEATURES OF SUBTILIGASE METHOD
2.1. Introduction
Here, we describe a global N-terminomics positive enrichment method using the engineered enzyme subtiligase. This method allows one to specifically tag and identify with LC–MS new N-termini generated by endogenous or exogenous proteases (Fig. 13.1A). With this approach, we have identified over 8000 unique α-amines in healthy and apoptotic cell lysate (publicly available at http://wellslab.ucsf.edu/degrabase) as well as quantitatively monitored kinetics of individual cleavage events (Agard et al., 2012). The subtiligase method is easily applied to many different complex biological samples to identify substrates and sites of proteolysis.
Figure 13.1.

An overview of the subtiligase N-terminal labeling method. (A) Proteins with free N-termini in a mixture are selectively tagged using the engineered enzyme, subtiligase. Whole protein samples are incubated with subtiligase and the peptide ester containing a biotin tag. After enzymatic labeling, free N-termini are captured on avidin beads. Proteins are digested by trypsin. The final N-terminal peptide is released from beads via TEV protease cleavage and identified by mass spectrometry. (B) The current peptide ester contains an ester subtiligase acylation site, Abu-tag for positive mass spectrometry identification, a TEV protease site and a biotin label. The peptide ester can be further modified for specific experimental needs.
2.2. Types of samples and peptide tags
The method has been successfully used on purified proteins, cell cultures, peripheral blood plasma, and tissue samples from humans, mice, insects, and worms. In general, proteins in complex biological samples can be solubilized into an appropriate buffer for subtiligase labeling. Additionally, one can design the chemical structure of the peptide ester tag to facilitate downstream purification, identification, and quantification for customization in specific applications (Fig. 13.1B).
2.3. Sample setup introduction
2.3.1 Discovery versus targeted protocols
There are two experimental protocols, we currently use that provide the most complete information for global N-terminomics. The initial “discovery” experiments are designed to identify which proteins and specific sites are cleaved. The discovery experiments are qualitative and focus on high confidence identification of tagged N-terminal peptides. These experiments are important to optimize the labeling procedures, determine background, and establish a list of high confidence peptide identifications. Peptides from discovery experiments are then used in “targeted” mass spectrometry experiments. Targeted experiments allow for the specific monitoring of a subset of peptides in a more sensitive and/or quantitative manner across a wider range of samples, such as selective reaction monitoring (SRM). Examples of both types of experiments will be discussed in more detail below.
2.3.2 Forward versus Reverse experiments
There are two main experimental strategies for subtiligase labeling, which we term “Forward” and “Reverse” (Fig. 13.2). Forward experiments use intact biological systems where endogenous proteolysis is induced such as for apoptosis followed by protein isolation and N-terminal labeling. The panel of substrates is compared to those from an uninduced sample. In contrast, Reverse experiments are performed in vitro where an exogenous pro-tease is added to the total protein from a sample where endogenous proteases have been inactivated. The Forward experiments allow for the identification of biologically relevant protease cleavage events but may not be able to identify the specific protease responsible. The Reverse setup specifically identifies the activity of the added protease, but occurs in lysates where the intracellular structure is disrupted and thus may be less physiologically relevant.
Figure 13.2.
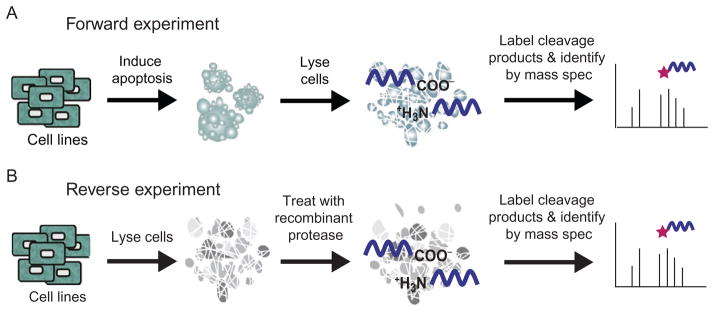
A schematic difference between Forward and Reverse experiments. Forward experiments use samples from intact biological systems, either perturbed or unperturbed that is then harvested, lysed, and labeled. Reverse experiments involve exogenous addition of protease to whole cell or tissue lysate of interest followed by labeling.
3. SUBTILIGASE-BASED LABELING METHOD
3.1. Overview of method
The subtiligase protocol is designed to positively enrich free protein N-termini. Subtiligase itself is a rationally engineered version of the bacterial serine protease subtilisin BPN′. Two-point mutations simultaneously abolish protease activity and allow ligase activity (Abrahmsen et al., 1991). With these modifications, subtiligase can covalently link free peptide α-amines with an ester-containing synthetic peptide. Importantly, the subtiligase enzyme is exquisitely selective for peptide α-amines over the ε-amines of lysine residues (Braisted, Judice, & Wells, 1997). Furthermore, acetylated N-termini present on 80–90% of native eukaryotic proteins (Polevoda & Sherman, 2003) are ignored by the labeling process, greatly reducing background identifications and focusing on N-termini generated by proteolysis.
The protocol follows a catch-and-release strategy (Fig. 13.1A). In combination with a designed synthetic peptide ester (Fig. 13.1B), subtiligase selectively biotinylates free α-amines in the sample. Following avidin bead-mediated immobilization, proteins are then digested with trypsin and nonbiotinylated protein fragments are washed away. The most N-terminal peptide from each substrate is then released from avidin beads through tobacco etch virus (TEV) protease cleavage. TEV is an extremely specific plant viral protease that can be readily purified (Lucast, Batey, & Doudna, 2001) or purchased. TEV recognizes the amino acid sequence ENLYFQ↓S, which importantly is not found in the mammalian proteome. After TEV cleavage, all labeled peptides have a nonnatural amino acid mass tag (α-aminobutyric acid, or Abu-) remaining on the N-terminus. This tag, which is compatible with both subtiligase and TEV, greatly enhances confidence for identifying subtiligase-labeled peptides over nonspecifically bound background. In our experience, >90% of peptides observed by LC–MS incorporate the Abu-mass tag, providing evidence for the specificity of the labeling procedure and recovery method.
3.2. Specialized reagents for subtiligase labeling and enrichment
3.2.1 Subtiligase enzyme
Plasmid vectors and detailed instructions for subtiligase expression are available on request from the Wells laboratory. Subtiligase is expressed in Bacillus subtilis and the enzyme is secreted to the media. The enzyme is purified through ammonium sulfate precipitation, anion exchange, thiopropyl resin capture (for the catalytic cysteine residue in subtiligase), and gel filtration. The enzyme is stored at −80 °C and retains activity for at least 2 years after purification. Activity can be tested and quantified using FRET ester reporters (Shimbo et al., 2012; Yoshihara, Mahrus, & Wells, 2008).
3.2.2 Peptide ester label
The synthetic ester used for labeling is customizable for different experimental goals (Yoshihara et al., 2008). The current version contains four distinct features: (i) an ester linkage for subtiligase acylation and transfer to the free peptide α-amine, (ii) the unique Abu-tag to facilitate MS identification, (iii) the TEV protease cleavage site for elution from avidin beads, and (iv) biotin for initial capture (Fig. 13.1B). The peptide ester is readily synthesized using solid phase fMOC chemistry modified for the more reactive ester bond (Braisted et al., 1997; Jackson et al., 1994). Since each amino acid is added individually, it is possible to change any part of the sequence after the ester bond so long as the first four amino acids can be recognized by subtiligase.
4. EXPERIMENTAL APPLICATIONS OF SUBTILIGASE-BASED N-TERMINOMICS
4.1. Application to cell culture systems undergoing apoptosis (Forward Discovery experiments)
Below, we describe a general protocol using subtiligase-based labeling to identify proteolytic substrates generated during apoptosis in a cell culture system.
4.1.1 Sample size and expected yield
The extent of subtiligase labeling varies but we estimate about 10–15% of the α-amines in a sample are routinely labeled. Hydrolysis of the ester by subtiligase is the biggest impediment to higher labeling efficiency. While the enzyme is very suitable for N-terminomics despite the hydrolysis side-reaction, it requires the use of greater protein sample input. For initial Forward Discovery experiments, we typically use on the scale of 0.5–5×109 cells (~30–300 mg total protein in lysate) to maximize our number of peptides identified by mass spectrometry. The use of more sensitive mass spectrometers or an experimental system which does not require deep coverage allows for smaller amounts of starting sample.
4.1.2 Choice of proteolysis inducer for Forward experiments
The specific proteolysis inducer chosen will depend on the system of interest and research question. Apoptosis can be induced in a cell culture system using a variety of agents but depends on the cell type and organism. For example, we have used both small molecule cytotoxic agents (doxorubicin, bortezomib, staurosporine) to activate the intrinsic pathway of apoptosis and protein-based agents (TRAIL, Fas-ligand) that bind to extracellular surface death receptors and trigger the extrinsic pathway of apoptosis (Agard et al., 2012; Mahrus et al., 2008; Shimbo et al., 2012).
4.1.3 Monitoring proteolysis and sample harvest
Once the desired cell culture system and inducer are chosen, it is recommended to perform validation experiments on a small scale to identify a concentration and time point where proteolysis is most extensive. For apoptosis, we find the maximal number of caspase cleavage events when the extent of apoptosis is >90%. We have primarily used biochemical assays to monitor caspase activity (Caspase-Glo, Promega) and cell viability (Cell-Titer Glo, Promega), though there are a number of other experimental methods also available (Galluzzi et al., 2009). Figure 13.3A demonstrates a typical time course of cell viability and caspase activation with two different doses of apoptotic inducers in two different human malignancy-derived cell lines.
Figure 13.3.

Monitoring apoptosis and proteomic distribution of cleavage substrates. (A) Measuring cell viability and caspase activation. It is important to monitor apoptosis versus time after exposure to drug, as the rate of apoptosis can vary substantially depending on the drug. Caspase activity appears before cell viability decreases. (B) Comparison of caspase substrates identified versus broad range of baseline protein abundance. Protein abundance estimated derived from PaxDB. Extensive distribution overlap indicates that subtiligase-based N-terminomics leads to broad coverage across 6-logs of abundance in the proteome. Figure adapted from Crawford et al. (2013) with permission of the authors.
After cells are grown to scale for Forward Discovery and have undergone apoptosis to the desired extent, cell bodies and debris are pelleted by centrifugation and washed once with ice-cold PBS. The washed cell pellet is then lysed directly or flash-frozen, stored at −80 °C, and thawed prior to lysis. For comparison to background cellular proteolysis, one should include a control sample not exposed to proteolysis inducers.
4.1.4 Protocol for forward discovery labeling
Cell lysis: Prepare 1 mL per sample of 4 × lysis buffer (ratio 4:4:2 of 10% SDS (w/v):1 M bicine pH 8.5:ddH2O). Also prepare stocks of protease inhibitors (Sigma) to quench ongoing endogenous proteolysis: 10 mM z-VAD-fmk (Sigma) caspase inhibitor in DMSO; 10 mM E-64 (Sigma) cysteine protease inhibitor in DMSO; 100 mM 4-(2-aminoethyl)benzenesulfonyl fluoride (AEBSF, Sigma) serine protease inhibitor in DMSO; 0.5 M EDTA pH 8.0 in ddH2O; 100 mM phenylmethylsulfonyl fluoride (PMSF, Sigma) (freshly prepared) in iso-propanol. Add 5 μL of each protease inhibitor stock per 1 mL of 4× lysis buffer. Add 1 mL 4× lysis buffer with inhibitors to cell pellet and lyse completely by probe ultrasonication. Use clarified lysate sample for protein concentration determination.
Cysteine reduction and alkylation: In all proteomic experiments, it is important to first reduce and then irreversibly block-free thiol groups on cysteines to prevent formation of mixed oxidation products that hinder identification by MS. Prepare a fresh stock of 100 mM tris(2-carboxyethyl)phosphine (TCEP, Sigma) in ddH2O. Add TCEP stock to final concentration of 5 mM to each lysed sample and mix. Heat at 95 °C for 15 min to ensure free thiols on cysteines. Allow to cool to room temperature (RT). During cooling, prepare a fresh stock of 200 mM iodoacetamide (IAM). Add IAM stock to final concentration of 10 mM to each sample and mix. Incubate 1 h in the dark at RT to block all cysteines. After incubation, add 1 M dithiothreitol (DTT) stock to final concentration of 25 mM to quench remaining IAM, as any free IAM will block catalytic cysteines of subtiligase at next step. Vortex briefly. Add Triton-X to a final concentration of 2.5% (v/v) to form micelles with SDS (note: subtiligase labeling will not work without removing detergent in this manner).
Subtiligase labeling: Centrifuge each sample for 10 min, ~4000×g to pellet out any insoluble debris and transfer clarified supernatant to new tube. Add ddH2O to final volume of 3.6 mL. Check and adjust pH to 8.5. Add 400 μL of 10 mM peptide ester stock in DMSO to final concentration of 1 mM. Vortex briefly. Add 40 μL of 100 μM subtiligase stock to final concentration of 1 μM. Vortex briefly. Incubate for 1 h at RT (note: labeling for >1 h generally does not improve yields as peptide ester is either ligated to N-termini or hydrolyzed by this time). Labeling can be confirmed through a Western blot against biotin using NeutrAvidin-HRP (Pierce) with comparison to a presubtiligase sample.
Removal of excess peptide ester and exchange into denaturing conditions by protein precipitation: Biotin moieties on excess and hydrolyzed peptide ester will compete for binding sites on avidin beads and render them unavailable for capturing biotinylated proteins. Therefore, precipitate proteins by adding labeled sample dropwise to 35 mL acetonitrile at RT; short peptides, including excess peptide ester, will remain in solution. Vortex gently. Incubate on ice for at least 15 min up to overnight. Centrifuge 8000×g for 30 min at 4 °C. Carefully decant supernatant to waste. Let precipitated protein pellet air dry for ~15 min. Add 1 mL of 8M Guanidine HCl over pellet and let dissolve at RT for 30 min to 1 h. Swirl gently and pipet up and down to dissolve pellet. Add another 1 mL of 8 M Guanidine HCl to fully dissolve; use ultrasonication if necessary to solubilize. Precipitate protein a second time by adding dropwise to 30 mL ice-cold ethanol in a new 50 mL conical vial. Incubate at −80 °C overnight. The next day, centrifuge sample at 30 min, 8000×g, 4 °C to pellet precipitated protein. Decant supernatant and air dry pellet for 15–20 min (note: can also freeze pellet at −80 °C for later use). Add 3 mL, 8 M Guanidine HCl over pellet and let dissolve at RT for 20 min. Add an additional 2 mL Guanidine HCl to complete dissolution. Transfer dissolved protein to new 15 mL conical vial. Rinse prior 50 mL conical vial with 2.5 mL ddH2O, and transfer rinsed solution to same 15 mL conical. Take 8 μL aliquot of total dissolved protein sample for dot blot (below) and store at 4 °C.
Capture on NeutrAvidin resin: We use NeutrAvidin High Capacity resin (Pierce) to maximize capture of biotinylated proteins. For protein from 5×108 cells, we will typically add 1 mL of 50% bead slurry. After adding beads, place overnight at RT with gentle agitation or rotation. A dot blot against NeutrAvidin-HRP (Pierce) is recommended to confirm complete capture of labeled peptides compared to the prebead aliquot, indicated by disappearance of luminescence signal in the postbead sample. If capture is not complete, add additional NeutrAvidin resin and incubate for 2 h up to overnight, dot blot again, and repeat as necessary. Note that incompletely removed peptide ester will increase the amount of beads necessary for complete capture.
On-bead trypsinization: After complete peptide capture, transfer beads to empty polypropylene chromatography column with frit at outlet. Attach the column to vacuum set up and remove the supernatant. Wash beads (add buffer, vortex, flow through) three times with 2 mL biotin wash buffer (10 mM bicine pH 8.0, 1 mM biotin) to occupy unbound avidin sites. Wash beads 5–10× with 5 M Guanidine HCl to remove nonspecifically bound protein from beads. Wash beads 3× in trypsin wash buffer (100 mM bicine pH 8.0, 200 mM NaCl, 20 mM CaCl2, 1 M Guanidine HCl). Add 10–100 μg sequencing grade modified trypsin (trypsin should be added at 1:50 (w/w) to estimated amount of captured protein) in trypsin wash buffer to each sample. Incubate overnight at 37 °C with gentle agitation.
N-terminal peptide elution with TEV protease: Freshly prepare TEV protease buffer (50 mM ammonium bicarbonate pH 8.1, 2 mM DTT, 1 mM EDTA). Remove trypsinization supernatant to waste. Wash beads 5–10× with 5 M Guanidine HCl to remove nonspecifically bound peptides. Wash beads 5× with TEV protease buffer to completely remove guanidine. For each sample, mix 50 μg TEV protease with 1.5 mL TEV protease buffer and add to beads. Incubate overnight at RT with agitation or rotation. The next day, elute supernatant with TEV-cleaved peptides into 1.5 mL tubes. Evaporate to dryness.
Sample cleanup by ZipTip: Resuspend sample in a total of 100 μL 5% TFA to achieve pH≤3. Let stand >10 min at RT. Spin 10 min at 14,000×g at RT to pellet precipitated TEV protease. Transfer supernatant to new tube. For cleanup, we use C18 ZipTips (Millipore) performed with manufacturer protocol with elution into low-protein retention 500 μL tubes. Evaporate to dryness. Peptides may now be stored at −80 °C, resuspended in 0.1% FA to run directly on mass spectrometer, or used for fractionation.
Fractionation using reverse-phase high-pH chromatography (optional but recommended): To obtain the greatest depth of peptide coverage and greatest number of substrate identifications in Discovery experiments, it is advisable to perform a separation step prior to MS analysis. We use reverse-phase high-pH chromatography as it has been shown to offer similar separation capabilities to strong cation exchange and does not require additional ZipTip clean up of fractions (Yang, Shen, Camp, & Smith, 2012). If desired, after separation fractions can be pooled for analysis. Evaporate to dryness. Store at −80 °C or resuspend each fraction in 0.1% FA for evaluation by MS.
4.1.5 Mass spectrometry analysis and bioinformatics
Mass spectrometry analysis of samples is carried out essentially like any other proteomic-based method, incorporating low-pH reverse-phase chromatography in-line with the mass spectrometer, as described in detail by others (for review, see Aebersold & Mann, 2003). Samples are analyzed in data-dependent acquisition mode, with exact parameters dependent on instrument used.
-
General protein database search to identify substrates: To identify N-termini, MS data must be searched against a database of known proteins specific for the organism of interest. Such searches can be performed with a variety of resources (Kapp & Schutz, 2007); we typically use Protein Prospector (http://prospector.ucsf.edu). This search algorithm is able to search a semitryptic peptide space: while the C-terminus has trypsin cleavage (at Arg/Lys), the N-terminus is allowed to be any amino acid in order to capture all potential proteolytic cleavages. In addition to this feature, Protein Prospector also allows a search with a constant N-terminal Abu-modification, which has a mass orthogonal from any naturally occurring amino acid. The completed database search across all analyzed fractions (typically at a false discovery rate of <1% based on a decoy database set) results in a list of N-terminal peptides identified in the sample. Further analysis of the sequence prior to the identified N-terminus (derived from database protein sequence) can reveal proteolytic specificity. In our prior studies, we have found that after apoptosis, there is a large increase in caspase-related Asp residues at the P1 position immediately N-terminal to the identified cleavage site. In nonapoptotic samples, we find a preponderance of Arg or Lys at P1 (Crawford et al., 2013).
We have found it best for downstream analysis to collect and store all experimental data in a central database. For each experiment, we upload the relevant details (date, cell line, Forward or Reverse, inducer), links to the raw MS files, MS run parameters, protein database search parameters, and the protein database output file. We designed a FileMakerPro database (available at wellslab.ucsf.edu/degrabase) that also contains lookups to UniProtKb (www.uniprot.org) to add further details about the protein. This central database creates an easy and consistent workflow for importing and storing datasets and allows for easy export of data for analysis in another program or publication.
Comparison to other substrate databases: The list of potential proteolytic substrates can be compared to existing databases that focus on protease specificity or already identified substrates. Examples include MEROPS (Rawlings, Barrett, & Bateman, 2012), TopFind (Lange, Huesgen, & Overall, 2012), and the DegraBase (Crawford et al., 2013).
Analysis of biological function: Additionally, biological function of substrates can be analyzed using tools such as GoMiner (Zeeberg et al., 2005) or Ingenuity Pathway Analysis (www.ingenuity.com). Comparison to a general database of human cellular protein abundance, PaxDB (Wang et al., 2012), demonstrates that subtiligase offers substantial coverage of proteolytic substrates across the concentration range of the cellular proteome (Fig. 13.3B).
4.2. Identifying specific substrates of recombinant proteases in cell lysates (Reverse Discovery experiments)
In a Reverse Discovery experiment, the subtiligase-based method is modified to examine cleavage events catalyzed by the addition of a specific recombinant protease. The Reverse protocol is effective for the study of individual proteases of biological interest and for systems that may not be amenable to cell culture-based experiments.
4.2.1 Choice of sample and preparation
4.2.1.1 Preparation of cell lysate
For Reverse experiments, we typically start with 0.5–1×109 cells (30–60 mg of total protein in lysate). Our protocol has been optimized for caspase experiments but can be customized for the requirements of other enzymes.
4.2.1.2 Inhibition of endogenous proteases
In order to reduce background protease activity as thoroughly as possible, we include protease inhibitors in our lysis buffer. However, as the lysis buffer is not typically exchanged before protease addition, it is important to not include any inhibitors that would target the protease of interest. Therefore, for Reverse experiments with caspases, we include EDTA, AEBSF, and PMSF but omit the cysteine protease inhibitors z-VAD-fmk and E-64. One also needs to be aware that if the added protease activates an endogenous one of the same class that is not inhibited, then one would get a sum of the protease substrates. This is a distinct possibility when testing individual caspases and will not be eliminated even when comparing with a control sample with no protease added.
4.2.2 Preparation of proteases and characterization in lysate system
The purity and activity of the protease of interest is the most important part of a Reverse experiment. For example, for caspase-3, we express and purify our own enzymes and then perform kinetic activity assays using z-DEVD-AFC fluorescent substrate (CalBioChem) in either a small volume (50 μL−1 mL) of cellular lysate or optimal enzyme buffer. We use a range of caspase-3 concentrations from 1 to 1000 nM, near the expected physiological caspase concentration of ~50–200 nM. Ideally, activity in both lysate and buffer will be similar. Experiments at this small scale allows for lysis buffer modifications as necessary to enhance activity. These experiments can also determine appropriate enzyme concentration and time points where proteolysis has reached completion for full-scale studies.
4.2.3 Reverse experimental protocol
Cell lysis and caspase inactivation: For 1×109 cells in a caspase experiment, lyse using 10 mL of cold 100 mM HEPES pH 7.4, 0.1% Triton-X-100, 10 mM IAM, and 5 mM EDTA, 1 mM AEBSF, and 1 mM PMSF. Use 50 μL of lysed sample for protein concentration determination. The lysate is kept in the dark for 15 min at 4 °C for irreversible alkylation of endogenous caspase catalytic cysteines by IAM. Excess IAM is quenched by the addition of 20 mM DTT to prevent inhibition of recombinant caspase added in the next step. The lysate is cleared 2× by centrifugation at 4000×g for 5 min.
Protease addition and quenching: Prepare a 1 mL volume of active protease buffer (for caspases: 10 mM DTT, 0.1% CHAPS, 20 mM Pipes pH 7.2, 100 mM NaCl, 1 mM EDTA, and 10% sucrose). Add active protease to the 1 mL buffer, adjusted for the desired final concentration in ~15 mL after addition of enzyme in buffer to lysate. After a desired amount of time (determined in validation experiments), appropriate protease inhibitors are added to quench proteolysis. For caspases, we use 10–1000 nM for 10 min to 3 h and quench with 100 μM z-VAD-fmk.
Subtiligase labeling: To the lysate, add 10 mM ester peptide stock in DMSO to final concentration of 1 mM. Vortex briefly. Check and adjust pH to 8.5. Add 100 μM subtiligase stock to final concentration of 1 μM. Vortex briefly. Incubate for 1 h at RT. Labeling can be confirmed through a Western blot against biotin.
The remaining steps are similar to those in the Forward protocol (Section 4.1.4). Samples are desalted by acetonitrile precipitation. After resuspension of the pellet in 8 M Guanidine HCl, thiols are reduced by TCEP and alkylated by IAM. Protein is then precipitated again in EtOH overnight. After resuspension of protein in 8 M Guanidine HCl, all steps from NeutrAvidin capture to MS analysis (Section 4.1.4, Steps 5–9) are identical.
4.3. Quantification and kinetics
Thus far, we have described methods by which to identify proteolytic substrates, either in intact cells or as substrates of a specific recombinant enzyme. Next, we describe methods of quantitative proteomics combined with subtiligase-based labeling to determine the rates that substrates are cleaved.
4.3.1 MS quantification methods
There are a number of methods to quantify peptides by mass spectrometry. The relative advantages and disadvantages have been reviewed in detail by others (Bantscheff, Lemeer, Savitski, & Kuster, 2012; Bantscheff, Schirle, Sweetman, Rick, & Kuster, 2007; Liebler & Zimmerman, 2013; Nikolov, Schmidt, & Urlaub, 2012). These quantitative methods can be classified as either labeling or label-free, as well as either untargeted or targeted mode. Notably, essentially all can be implemented in combination with subtiligase-based N-terminomics. Untargeted labeling approaches include metabolic stable isotope labeling (SILAC) (Bushell et al., 2006) or isobaric mass tags coupled to peptides after tryptic digestion (Bantscheff et al., 2012). However, these methods can limit the number of samples that can be compared simultaneously.
Various untargeted, label-free approaches have been implemented too, including the method of “spectral counting” as well as others that rely on the peak area in the MS extracted ion chromatogram (Nahnsen, Bielow, Reinert, & Kohlbacher, 2013). Like all untargeted methods, a significant limitation is that these methods frequently do not identify all peptides of interest across all tested conditions. Targeted, label-free quantification, while limited to a few hundred proteins per run, offers highly sensitive and reproducible quantification as the MS instrument focuses only detecting peptides of interest in a complex sample. The most common method in this category is selected reaction monitoring (SRM; also known as multiple reaction monitoring) (Picotti & Aebersold, 2012). SRM methods can typically be run on unfractionated samples, leading to greatly reduced MS instrument time compared to other methods described. In addition, SRM assays can easily be applied to an unlimited number of samples (Huttenhain et al., 2012; Li et al., 2013). Below, we outline our approach to development of SRM assays to monitor proteolysis during apoptosis.
4.3.2 Design of a kinetic time course
We have primarily used SRM to quantify kinetics of proteolysis across a time course (Agard et al., 2012; Shimbo et al., 2012; Wiita et al., 2013). Similar to the Forward and Reverse Discovery experiments, SRM kinetic analysis can also be performed in intact cells as well as with recombinant pro-teases. Intact cells may present more variable kinetic data as they will initiate the apoptotic process at different times after inducer addition. Nonetheless, we have found that the kinetics of caspase cleavage in Reverse experiments with recombinant caspase matches quite well with cleavage kinetics in intact cells (Agard et al., 2012). In terms of experimental design, it is important to identify time points which will provide the most information on the proteolytic process of interest. A small scale experiment and Western blotting for cleavage of known substrates or a fluorescent reporter assay can fill this role.
4.3.3 Development and use of SRM assays
The SRM method relies on the use of a triple quadrupole mass spectrometer. In this system, a total peptide sample is injected onto an LC directly in-line with the MS instrument (Fig. 13.4). As peptides are eluted, the first quadrupole is used as a mass filter to only isolate peptides with a targeted m/z. The second quadrupole serves as a collision cell to break the peptide into fragments. The third quadrupole functions as a second mass filter for specified m/z fragments from the initial parent peptide. Each of these parent-fragment ion pairs is termed a “transition,” and transition intensity is recorded by the detector. The coelution of multiple fragments from a single parent peptide indicates the specific identification of the peptide of interest. The total peak area reflects the relative abundance of the peptide across conditions (Fig. 13.4B).
Figure 13.4.

Selected reaction monitoring (SRM) for proteolytic substrate quantification. (A) Schematic diagram of triple quadrupole (Q1–Q2–Q3) mass spectrometer used for SRM. ESI, electrospray ionization, representing ionized peptides eluted from liquid chromatography column into the mass spectrometer. (B) Example data from SRM monitoring caspase-cleaved peptide from ATF4 protein during bortezomib-induced apoptosis. Each individual trace represents a parent-fragment ion pair (transition). Coelution of multiple transitions from the same peptide confirms peptide identity. Peak area can be used for quantification, with kinetic parameters derived based on change across the time course.
Development of a spectral library: As SRM is a targeted method, Forward or Reverse Discovery experimental MS identification is required to develop an SRM assay. From this Discovery dataset, a “spectral library” of identified peptides is generated, including the parent peptide mass, fragment ions, and MS signal intensity information. From this spectral library, peptides of further interest are specified for quantitative study. As an alternative source of a peptide data, our laboratory has made mass spectra available for peptides identified in Forward Discovery experiments in apoptotic and nonapoptotic cells (wellslab.ucsf.edu/degrabase).
SRM method development: Once a spectral library is obtained and peptides of interest are selected, the next step of method development is selection of the optimal transitions. One of the biggest recent advances in SRM is the implementation of the open-source, freely available Skyline software (MacLean et al., 2010). Skyline quickly generates transition lists from the imported spectral library. For targeted peptides, we first run unscheduled SRM validation runs (no LC retention time information) monitoring up to seven transitions per protein and ~200 transitions per run, ideally using the same Discovery samples. We typically require a minimum of five of seven coeluting transitions and a retention time consistent with Discovery experiments to proceed with development. Synthetic peptide standards can also aid in confirmation. We then apply collision energy optimization and then create a scheduled method incorporating LC retention time information. Scheduling allows for monitoring significantly more peptides and transitions per run. We have found using the AB SCIEX QTRAP 5500 instrument up to ~250 peptides (~1000 transitions) can be included in a single SRM run. The method development process typically can be completed in less than a week.
SRM sample preparation and analysis: Samples are prepared according to the same protocols as in Sections 4.1 or 4.2. The main difference is we have had success with smaller sample input, on the scale of 2×108 cells (~10 mg of total protein). Samples are not fractionated and the entire population of N-terminal labeled peptides is analyzed directly in a single SRM run. Skyline software is then used to determine peak area for each identified peptide in each sample. Overall sample intensity must be normalized to account for differences in labeling efficiency and MS conditions across runs. Prior to subtiligase labeling, we typically spike in purified proteins not endogenously present in the sample as internal normalization standards. Using SRM to monitor recombinant caspase cleavage of substrates in cell lysates, we were also able to derive plots of substrate appearance versus time, analogous to more traditional enzymology experiments but capable of tracking hundreds of substrates simultaneously (Agard et al., 2012; Fig. 13.5).
Figure 13.5.

Monitoring kinetics of recombinant caspase cleavage. (A) SRM transitions show increase in intensity across time course after caspase addition. (B) Peptide intensities are fit to pseudo-first-order kinetic equations to determine kinetic efficiency (kcat/Km) for each substrate. (C and D) Rank order of catalytic efficiencies for substrates of caspases-3 and -7 span at least two orders of magnitude. Caspase-3 plot indicates substrates with rapid, medium, and slow cleavage. Figure adapted from Agard et al. (2012) with permission of the authors.
4.4. Applications to human plasma and serum
N-terminal labeling by subtiligase can be modified for human plasma and serum samples to reveal insights into the complex proteolysis in the circulation (Wildes & Wells, 2010). Blood collection tubes should be centrifuged as soon as possible after sampling to separate plasma/serum from cellular components. Plasma/serum can be stored at −80 °C or used immediately for labeling. The labeling process is somewhat simplified compared to cell culture samples. To plasma/serum volumes of 0.5–2 mL, add 1 M bicine pH 8.5 to a final concentration of 100 mM. Then, add AEBSF to a final concentration of 1 mM to inhibit plasma serine proteases. Incubate at RT for 10 min. Add DTT to a final concentration of 2 mM and ester peptide to a final concentration of 1 mM. Vortex briefly. Add subtiligase enzyme to a final concentration of 1 μM and incubate at RT for 1 h. For plasma samples, we have had success removing excess ester with NAP-25 chromatography columns (GE Healthcare) per manufacturer protocol with equilibration buffer of 50 mM bicine pH 8.0. After elution in 2.5 mL of 50 mM bicine pH 8.0, add 7.5 mL of 8 M Guanidine HCl to desalted sample. Reduce and alkylate thiols as in the protocols above. Then, add NeutrAvidin beads, typically at a slurry volume similar to the initial plasma volume. Sample preparation now proceeds identically as from Step 5 in Section 4.1.4. Enriched N-terminal peptides can be used for either Discovery or Targeted MS. Of note, general plasma proteomic studies are often confounded by the extremely high abundance of serum albumin, as signal intensity from albumin precludes detection of biologically interesting changes in low-abundance proteins (Anderson & Anderson, 2002). Fortunately, subtiligase labeling leads to extremely limited pull down of albumin, enabling detection of even very low-abundance plasma proteins (Fig. 13.6).
Figure 13.6.

Plasma N-terminomics. Proteins identified with free N-termini in plasma demonstrate over six-order of magnitude range of abundance, demonstrating ability of subtiligase labeling to track low-abundance plasma proteins. Figure adapted from Wildes and Wells (2010) with permission of the authors.
4.5. Application of N-terminal labeling by subtiligase to any biological sample
While we have focused on intact cells, cellular lysates, and human plasma, subtiligase-based enrichment can easily be applied to study proteolysis in virtually any biological sample. The general protocol would be highly similar to those shown above. The key step is obtaining the total protein sample in a buffer compatible with subtiligase labeling: nondenaturing conditions (i.e., no free detergent or denaturant) and pH ~8.5. Similar to plasma labeling, labeling of biological fluids (CSF, urine, etc.) can likely be achieved without any significant sample manipulation. Tissue samples can be processed similarly to cell culture samples. Alternatively, we have had success using trichloroacetic acid precipitation of total protein following by resuspension in guanidine-containing buffer. Buffer is then exchanged with desalting columns into subtiligase-compatible conditions. From as little as 40 μg of starting protein, we can identify ~50–200 proteolytically cleaved peptides released into the culture media after cellular apoptosis (Wiita, Hsu, Lu, Esensten, & Wells, 2014).
5. LIMITATIONS TO THE SUBTILIGASE LABELING METHOD
Subtiligase labeling has many advantages in proteolysis research: (i) it is an unbiased, enzymatically driven method without chemical protein modification, (ii) it can identify thousands of peptides over six orders of magnitude in abundance from complex biological samples, (iii) it can be combined with highly reproducible, label-free quantification, and (iv) single labeling with positive enrichment allows not only identification of the target, but reveals the precise site of proteolysis. However, there are limitations to this approach.
As described earlier, subtiligase labeling efficiency is the biggest current limitation of application to systems of interest. To counter this inefficiency, we use large amounts of starting material. This is relatively easy for cell culture-based experiments, but can be more complicated for animal or human samples. Additionally, there are a few N-terminal amino acids, notably proline, valine, and isoleucine, that subtiligase is relatively slow to ligate in vitro (Chang, Jackson, Burnier, & Wells, 1994). Nonetheless, we have found that subtiligase can indeed label these N-terminal amino acids in our cell-based experiments. Our N-terminomics data show that N-terminal labeling is only a small bias and does not significantly affect the utility of the method.
For Discovery experiments, these methods suffer the same limitation as all MS experiments: there can be incomplete overlap between peptides identified in one run to another, even under the same conditions. This is due to the stochastic nature of sampling of low-abundance peptides by the MS instrument. This limitation may be circumvented through the use of biological and technical replicates. A particular limitation for our strategy is that short peptides labeled by subtiligase will not be precipitated with other proteins (in Step 4 of protocol in Section 4.1.4) and will be discarded. However, semi-tryptic peptides can rescue many of these. Furthermore, the tryptic digestion of biotinylated proteins may lead to N-terminal peptides either too long or too short to be identified by MS.
Of note, we have provided the subtiligase plasmid and expression strains under a standard materials transfer agreement to more than 30 laboratories.
6. SUMMARY OF FINDINGS FROM SUBTILIGASE-BASED N-TERMINOMICS
As discussed in Section 1, there are multiple methods for enrichment and identification of proteolytically cleaved peptides in biological samples (Rogers & Overall, 2013). The subtiligase method has been extensively applied to the study of caspases. These studies showed that caspases can cleave more than 1000 cellular substrates during apoptosis, greatly expanding the scope of caspase biology. Initial studies revealed that caspases have a general preference for disordered structural elements in substrates (Mahrus et al., 2008). Further experiments across multiple cell lines allowed us to compile over 1700 caspase cleavage sites in nearly 1300 substrates as well as over 6000 noncaspase proteolytic sites (Crawford et al., 2013). With this large database, publicly available at http://wellslab.ucsf.edu/degrabase, we identified conserved motifs of caspase cleavage (Fig. 13.7) as well as sequence features of noncaspase endoproteases in nonapoptotic cells (Crawford et al., 2013). Furthermore, by combining recombinant enzyme purification with subtiligase labeling, we characterized specific substrates of the inflammatory caspases-1, -4, and -5 as well as the apoptotic caspases-3, -7, -8, and -9 (Agard et al., 2012; Agard, Maltby, & Wells, 2010). A combination of Forward and Reverse experiments allowed us to find evolutionarily conserved relationships between caspase cut site, protein substrates, and pathway-level relationships across organisms (Crawford et al., 2012). With a similar experimental combination, we probed proteolytic cleavage in human plasma which may relate to disease signatures (Wildes & Wells, 2010). The discovery of a broad range of caspase catalytic efficiencies across hundreds of substrates in parallel (Agard et al., 2012), was facilitated by the combination of label-free quantification with N-terminomics to determine catalytic efficiencies of natural substrates in complex mixtures. Furthermore, we showed that quantitative signatures of caspase cleavage can be used to monitor chemotherapeutic effects in cancer cells (Shimbo et al., 2012; Wiita et al., 2013, 2014). In summary, this method has revealed extensive information about proteolytic cleavage during apoptosis.
Figure 13.7.

Sequence features of identified N-termini. Aggregate plots across all N-termini shown using IceLogo, where amino acids favored at a given site are above the baseline while those disfavored are below (Colaert, Helsens, Martens, Vandekerckhove, & Gevaert, 2009). Cleavage occurs between position P1 (left of lightning bolt) and P1′. (A) Across all peptides identified in Forward Discovery experiments in apoptotic cells, Asp at P1 is highly enriched. (B) Focusing on only peptides shown in (A) with Asp at P1, a signature of caspase cleavage, we identify the canonical D-E-V-D cleavage motif for caspases from P4 to P1 site. (C and D) This motif is also conserved in N-termini identified in cell lysate incubated with recombinant caspase-3 during Reverse Discovery experiments.
7. FUTURE DIRECTIONS
To broaden the range of applications of our methods, we are using protein engineering to improve subtiligase labeling efficiency and decrease the need for large amounts of starting material. In tandem, new highly sensitive mass spectrometers will allow for both more comprehensive substrate identification and potentially label-free quantification with less protein input (Gallien et al., 2012; Hebert et al., 2014; Peterson, Russell, Bailey, Westphall, & Coon, 2012). Both of these advances will allow for in-depth analysis of many more recombinant proteases and biological systems. In intact cells, caspases have only been fully profiled for substrate generation during apoptosis; their substrate profiles in processes such as differentiation or nonapoptotic cell stress (Kuranaga, 2012) have yet to be elucidated. Others have recently shown that there is significant cross-talk between protein phosphorylation and caspase cleavage (Dix et al., 2012). N-terminomics can further be combined with new enrichment methods to investigate the relation between caspase cleavage and other posttranslational modifications, such as ubiquitination and lysine acetylation (Mertins et al., 2013). Alternatively, isolating intracellular organelles or secreted domains from cell membrane proteins will allow one to monitor proteolysis specific to different cellular perturbations in different cellular compartments. This knowledge may lead to information relevant to therapeutic and diagnostic development. This subtiligase technique is poised for wide use in proteolysis research.
Acknowledgments
This project was supported by Grants from National Institutes of Health R01 GM081051, R01 GM097316, and RO1 CA154802 (J. A. W.); The Rogers Family Foundation (J. A. W.), an award from the Sandler Program in Biological Sciences (J. A. W.); and the UCSF Stephen and Nancy Grand Multiple Myeloma Translational Initiative (J. A. W.). A. P. W. is a Damon Runyon Fellow supported by the Damon Runyon Cancer Research Foundation (DRG 111-12). J. E. S is supported by National Institutes of Health Training Grant T32 GM007175 and the Global Healthy Living Foundation. Mass spectrometry was performed at the Bio-Organic Biomedical Mass Spectrometry Resource at UCSF, which is supported by grants from the National Center for Research Resources (5P41RR001614) and the National Institute of General Medical Sciences (8P41GM103481 and 1S10RR015804) from the National Institutes of Health. We thank our long time collaborator Professor Alma Burlingame and his group for providing expert advice and open access to the UCSF Mass Spectrometry Center.
References
- Abrahmsen L, Tom J, Burnier J, Butcher KA, Kossiakoff A, Wells JA. Engineering subtilisin and its substrates for efficient ligation of peptide bonds in aqueous solution. Biochemistry. 1991;30:4151–4159. doi: 10.1021/bi00231a007. [DOI] [PubMed] [Google Scholar]
- Aebersold R, Mann M. Mass spectrometry-based proteomics. Nature. 2003;422:198–207. doi: 10.1038/nature01511. [DOI] [PubMed] [Google Scholar]
- Agard NJ, Mahrus S, Trinidad JC, Lynn A, Burlingame AL, Wells JA. Global kinetic analysis of proteolysis via quantitative targeted proteomics. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:1913–1918. doi: 10.1073/pnas.1117158109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agard NJ, Maltby D, Wells JA. Inflammatory stimuli regulate caspase substrate profiles. Molecular and Cellular Proteomics. 2010;9:880–893. doi: 10.1074/mcp.M900528-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agard NJ, Wells JA. Methods for the proteomic identification of protease substrates. Current Opinion in Chemical Biology. 2009;13:503–509. doi: 10.1016/j.cbpa.2009.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson NL, Anderson NG. The human plasma proteome: History, character, and diagnostic prospects. Molecular and Cellular Proteomics. 2002;1:845–867. doi: 10.1074/mcp.r200007-mcp200. [DOI] [PubMed] [Google Scholar]
- Bantscheff M, Lemeer S, Savitski MM, Kuster B. Quantitative mass spectrometry in proteomics: Critical review update from 2007 to the present. Analytical and Bioanalytical Chemistry. 2012;404:939–965. doi: 10.1007/s00216-012-6203-4. [DOI] [PubMed] [Google Scholar]
- Bantscheff M, Schirle M, Sweetman G, Rick J, Kuster B. Quantitative mass spectrometry in proteomics: A critical review. Analytical and Bioanalytical Chemistry. 2007;389:1017–1031. doi: 10.1007/s00216-007-1486-6. [DOI] [PubMed] [Google Scholar]
- Black RA, Kronheim SR, Sleath PR. Activation of interleukin-1 beta by a co-induced protease. FEBS Letters. 1989;247:386–390. doi: 10.1016/0014-5793(89)81376-6. [DOI] [PubMed] [Google Scholar]
- Braisted AC, Judice JK, Wells JA. Synthesis of proteins by subtiligase. Methods in Enzymology. 1997;289:298–313. doi: 10.1016/s0076-6879(97)89053-2. [DOI] [PubMed] [Google Scholar]
- Bushell M, Stoneley M, Kong YW, Hamilton TL, Spriggs KA, Dobbyn HC, et al. Polypyrimidine tract binding protein regulates IRES-mediated gene expression during apoptosis. Molecular Cell. 2006;23:401–412. doi: 10.1016/j.molcel.2006.06.012. [DOI] [PubMed] [Google Scholar]
- Chang TK, Jackson DY, Burnier JP, Wells JA. Subtiligase: A tool for semisynthesis of proteins. Proceedings of the National Academy of Sciences of the United States of America. 1994;91:12544–12548. doi: 10.1073/pnas.91.26.12544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colaert N, Helsens K, Martens L, Vandekerckhove J, Gevaert K. Improved visualization of protein consensus sequences by iceLogo. Nature Methods. 2009;6:786–787. doi: 10.1038/nmeth1109-786. [DOI] [PubMed] [Google Scholar]
- Crawford ED, Seaman JE, Agard N, Hsu GW, Julien O, Mahrus S, et al. The DegraBase: A database of proteolysis in healthy and apoptotic human cells. Molecular and Cellular Proteomics. 2013;12:813–824. doi: 10.1074/mcp.O112.024372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford ED, Seaman JE, Barber AE, 2nd, David DC, Babbitt PC, Burlingame AL, et al. Conservation of caspase substrates across metazoans suggests hierarchical importance of signaling pathways over specific targets and cleavage site motifs in apoptosis. Cell Death and Differentiation. 2012;19:2040–2048. doi: 10.1038/cdd.2012.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dix MM, Simon GM, Cravatt BF. Global mapping of the topography and magnitude of proteolytic events in apoptosis. Cell. 2008;134:679–691. doi: 10.1016/j.cell.2008.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dix MM, Simon GM, Wang C, Okerberg E, Patricelli MP, Cravatt BF. Functional interplay between caspase cleavage and phosphorylation sculpts the apoptotic proteome. Cell. 2012;150:426–440. doi: 10.1016/j.cell.2012.05.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallien S, Duriez E, Crone C, Kellmann M, Moehring T, Domon B. Targeted proteomic quantification on quadrupole-orbitrap mass spectrometer. Molecular and Cellular Proteomics. 2012;11:1709–1723. doi: 10.1074/mcp.O112.019802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L, Aaronson SA, Abrams J, Alnemri ES, Andrews DW, Baehrecke EH, et al. Guidelines for the use and interpretation of assays for monitoring cell death in higher eukaryotes. Cell Death and Differentiation. 2009;16:1093–1107. doi: 10.1038/cdd.2009.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert AS, Richards AL, Bailey DJ, Ulbrich A, Coughlin EE, Westphall MS, et al. The one hour yeast proteome. Molecular and Cellular Proteomics. 2014;13:339–347. doi: 10.1074/mcp.M113.034769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttenhain R, Soste M, Selevsek N, Rost H, Sethi A, Carapito C, et al. Reproducible quantification of cancer-associated proteins in body fluids using targeted proteomics. Science Translational Medicine. 2012;4:142ra194. doi: 10.1126/scitranslmed.3003989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Impens F, Colaert N, Helsens K, Plasman K, Van Damme P, Vandekerckhove J, et al. MS-driven protease substrate degradomics. Proteomics. 2010;10:1284–1296. doi: 10.1002/pmic.200900418. [DOI] [PubMed] [Google Scholar]
- Jackson DY, Burnier J, Quan C, Stanley M, Tom J, Wells JA. A designed peptide ligase for total synthesis of ribonuclease A with unnatural catalytic residues. Science. 1994;266:243–247. doi: 10.1126/science.7939659. [DOI] [PubMed] [Google Scholar]
- Kapp E, Schutz F. Overview of tandem mass spectrometry (MS/MS) database search algorithms. Current Protocols in Protein Science. 2007;Chapter 25(Unit 25.2) doi: 10.1002/0471140864.ps2502s49. [DOI] [PubMed] [Google Scholar]
- Kleifeld O, Doucet A, auf dem Keller U, Prudova A, Schilling O, Kainthan RK, et al. Isotopic labeling of terminal amines in complex samples identifies protein N-termini and protease cleavage products. Nature Biotechnology. 2010;28:281–288. doi: 10.1038/nbt.1611. [DOI] [PubMed] [Google Scholar]
- Klingler D, Hardt M. Profiling protease activities by dynamic proteomics workflows. Proteomics. 2012;12:587–596. doi: 10.1002/pmic.201100399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuranaga E. Beyond apoptosis: Caspase regulatory mechanisms and functions in vivo. Genes to Cells. 2012;17:83–97. doi: 10.1111/j.1365-2443.2011.01579.x. [DOI] [PubMed] [Google Scholar]
- Lange PF, Huesgen PF, Overall CM. TopFIND 2.0—Linking protein termini with proteolytic processing and modifications altering protein function. Nucleic Acids Research. 2012;40:D351–D361. doi: 10.1093/nar/gkr1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li XJ, Hayward C, Fong PY, Dominguez M, Hunsucker SW, Lee LW, et al. A blood-based proteomic classifier for the molecular characterization of pulmonary nodules. Science Translational Medicine. 2013;5:207ra142. doi: 10.1126/scitranslmed.3007013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebler DC, Zimmerman LJ. Targeted quantification of proteins by mass spectrometry. Biochemistry. 2013;52:3797–3806. doi: 10.1021/bi400110b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Otin C, Matrisian LM. Emerging roles of proteases in tumour suppression. Nature Reviews Cancer. 2007;7:800–808. doi: 10.1038/nrc2228. [DOI] [PubMed] [Google Scholar]
- Lucast LJ, Batey RT, Doudna JA. Large-scale purification of a stable form of recombinant tobacco etch virus protease. Biotechniques. 2001;30:544. doi: 10.2144/01303st06. [DOI] [PubMed] [Google Scholar]
- MacLean B, Tomazela DM, Shulman N, Chambers M, Finney GL, Frewen B, et al. Skyline: An open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics. 2010;26:966–968. doi: 10.1093/bioinformatics/btq054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahrus S, Trinidad JC, Barkan DT, Sali A, Burlingame AL, Wells JA. Global sequencing of proteolytic cleavage sites in apoptosis by specific labeling of protein N termini. Cell. 2008;134:866–876. doi: 10.1016/j.cell.2008.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertins P, Qiao JW, Patel J, Udeshi ND, Clauser KR, Mani DR, et al. Integrated proteomic analysis of post-translational modifications by serial enrichment. Nature Methods. 2013;10:634–637. doi: 10.1038/nmeth.2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nahnsen S, Bielow C, Reinert K, Kohlbacher O. Tools for label-free peptide quantification. Molecular and Cellular Proteomics. 2013;12:549–556. doi: 10.1074/mcp.R112.025163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolov M, Schmidt C, Urlaub H. Quantitative mass spectrometry-based proteomics: An overview. Methods in Molecular Biology. 2012;893:85–100. doi: 10.1007/978-1-61779-885-6_7. [DOI] [PubMed] [Google Scholar]
- Peterson AC, Russell JD, Bailey DJ, Westphall MS, Coon JJ. Parallel reaction monitoring for high resolution and high mass accuracy quantitative, targeted proteomics. Molecular and Cellular Proteomics. 2012;11:1475–1488. doi: 10.1074/mcp.O112.020131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham VC, Pitti R, Anania VG, Bakalarski CE, Bustos D, Jhunjhunwala S, et al. Complementary proteomic tools for the dissection of apoptotic proteolysis events. Journal of Proteome Research. 2012;11:2947–2954. doi: 10.1021/pr300035k. [DOI] [PubMed] [Google Scholar]
- Picotti P, Aebersold R. Selected reaction monitoring-based proteomics: Workflows, potential, pitfalls and future directions. Nature Methods. 2012;9:555–566. doi: 10.1038/nmeth.2015. [DOI] [PubMed] [Google Scholar]
- Polevoda B, Sherman F. N-terminal acetyltransferases and sequence requirements for N-terminal acetylation of eukaryotic proteins. Journal of Molecular Biology. 2003;325:595–622. doi: 10.1016/s0022-2836(02)01269-x. [DOI] [PubMed] [Google Scholar]
- Rawlings ND, Barrett AJ, Bateman A. MEROPS: The database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Research. 2012;40:D343–D350. doi: 10.1093/nar/gkr987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers L, Overall CM. Proteolytic post translational modification of proteins: Proteomic tools and methodology. Molecular and Cellular Proteomics. 2013;12:3532–3542. doi: 10.1074/mcp.M113.031310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schilling O, Huesgen PF, Barre O, Overall CM. Identification and relative quantification of native and proteolytically generated protein C-termini from complex proteomes: C-terminome analysis. Methods in Molecular Biology. 2011;781:59–69. doi: 10.1007/978-1-61779-276-2_4. [DOI] [PubMed] [Google Scholar]
- Shao W, Yeretssian G, Doiron K, Hussain SN, Saleh M. The caspase-1 digestome identifies the glycolysis pathway as a target during infection and septic shock. Journal of Biological Chemistry. 2007;282:36321–36329. doi: 10.1074/jbc.M708182200. [DOI] [PubMed] [Google Scholar]
- Shimbo K, Hsu GW, Nguyen H, Mahrus S, Trinidad JC, Burlingame AL, et al. Quantitative profiling of caspase-cleaved substrates reveals different drug-induced and cell-type patterns in apoptosis. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:12432–12437. doi: 10.1073/pnas.1208616109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smeekens SP, Montag AG, Thomas G, Albigesrizo C, Carroll R, Benig M, et al. Proinsulin processing by the subtilisin-related proprotein convertases furin, PC2, and PC3. Proceedings of the National Academy of Sciences of the United States of America. 1992;89:8822–8826. doi: 10.1073/pnas.89.18.8822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staes A, Van Damme P, Helsens K, Demol H, Vandekerckhove J, Gevaert K. Improved recovery of proteome-informative, protein N-terminal peptides by combined fractional diagonal chromatography (COFRADIC) Proteomics. 2008;8:1362–1370. doi: 10.1002/pmic.200700950. [DOI] [PubMed] [Google Scholar]
- Tanco S, Lorenzo J, Garcia-Pardo J, Degroeve S, Martens L, Aviles FX, et al. Proteome-derived peptide libraries to study the substrate specificity profiles of carboxypeptidases. Molecular and Cellular Proteomics. 2013;12:2096–2110. doi: 10.1074/mcp.M112.023234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiede B, Treumann A, Kretschmer A, Sohlke J, Rudel T. Shotgun pro-teome analysis of protein cleavage in apoptotic cells. Proteomics. 2005;5:2123–2130. doi: 10.1002/pmic.200401110. [DOI] [PubMed] [Google Scholar]
- Tonge R, Shaw J, Middleton B, Rowlinson R, Rayner S, Young J, et al. Validation and development of fluorescence two-dimensional differential gel electrophoresis proteomics technology. Proteomics. 2001;1:377–396. doi: 10.1002/1615-9861(200103)1:3<377::AID-PROT377>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Wang M, Weiss M, Simonovic M, Haertinger G, Schrimpf SP, Hengartner MO, et al. PaxDb, a database of protein abundance averages across all three domains of life. Molecular and Cellular Proteomics. 2012;11:492–500. doi: 10.1074/mcp.O111.014704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wejda M, Impens F, Takahashi N, Van Damme P, Gevaert K, Vandenabeele P. Degradomics reveals that cleavage specificity profiles of caspase-2 and effector caspases are alike. Journal of Biological Chemistry. 2012;287:33983–33995. doi: 10.1074/jbc.M112.384552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiita AP, Ziv E, Wiita PJ, Urisman A, Julien O, Burlingame AL, et al. Global cellular response to chemotherapy-induced apoptosis. eLife. 2013;2:e01236. doi: 10.7554/eLife.01236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiita AP, Hsu GW, Lu CM, Esensten JH, Wells JA. Circulating proteolytic signatures of cell death in humans identified by N-terminal labeling. Proceedings of the National Academy of Sciences of the United States of America. 2014 doi: 10.1073/pnas.1405987111. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wildes D, Wells JA. Sampling the N-terminal proteome of human blood. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:4561–4566. doi: 10.1073/pnas.0914495107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu G, Jaffrey SR. N-CLAP: Global profiling of N-termini by chemoselective labeling of the alpha-amine of proteins. Cold Spring Harbor Protocols. 2010;2010 doi: 10.1101/pdb.prot5528. pdb.prot5528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu G, Shin SB, Jaffrey SR. Global profiling of protease cleavage sites by chemoselective labeling of protein N-termini. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:19310–19315. doi: 10.1073/pnas.0908958106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu G, Shin SB, Jaffrey SR. Chemoenzymatic labeling of protein C-termini for positive selection of C-terminal peptides. ACS Chemical Biology. 2011;6:1015–1020. doi: 10.1021/cb200164h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F, Shen Y, Camp DG, 2nd, Smith RD. High-pH reversed-phase chromatography with fraction concatenation for 2D proteomic analysis. Expert Review of Proteomics. 2012;9:129–134. doi: 10.1586/epr.12.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshihara HA, Mahrus S, Wells JA. Tags for labeling protein N-termini with subtiligase for proteomics. Bioorganic and Medicinal Chemistry Letters. 2008;18:6000–6003. doi: 10.1016/j.bmcl.2008.08.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeeberg BR, Qin H, Narasimhan S, Sunshine M, Cao H, Kane DW, et al. High-throughput GoMiner, an ‘industrial-strength’ integrative gene ontology tool for interpretation of multiple-microarray experiments, with application to studies of common variable immune deficiency (CVID) BMC Bioinformatics. 2005;6:168. doi: 10.1186/1471-2105-6-168. [DOI] [PMC free article] [PubMed] [Google Scholar]