

Abstract
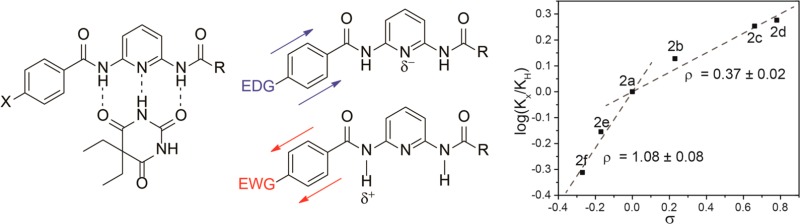
Hydrogen bond strength in host–guest systems is modulated by many factors including preorganization, steric effects, and electronic effects. To investigate how electronic effects impact barbiturate binding in bifurcated Hamilton receptors, a library of receptors with differing electronic substituents was synthesized and 1H NMR titrations were performed with diethyl barbital. The Hammett plot revealed a clear break between the different electronic substituents suggesting a change in binding conformation. The titration data were complimented with computational studies confirming the change in structure.
Host–guest binding plays a key role in many types of chemistry, ranging from molecular recognition to catalysis.1−6 Understanding how structural changes influence such interactions enables control over guest binding and facilitates the molecular design of synthetic supramolecular complexes. Various factors, including steric and electronic effects as well as host–guest preorganization, can all affect host–guest complex stability and guest exchange rates.5,7,8 To better understand the interplay of these forces in hydrogen-bonded systems, we recently investigated barbiturate binding to 2,6-diamidopyridines, a bifurcated form of macrocyclic Hamilton receptors to determine the differential effects of steric interactions and host preorganization on guest binding affinities.9 Toward understanding the impacts of electronic substitution on guest binding in hydrogen-bonded systems, we report here binding studies on 2,6-diaminopyridines with barbital that reveal changes in host–guest structure as a function of electronic substitution.
Because 2,6-diamidopyridines bind barbiturates through complementary hydrogen bonding, we chose to use a system in which one of the amides contained a phenyl substituent with an electron withdrawing or donating group in the para position. This design allowed for electronic changes in the phenyl substituent to modulate the acidity of the amide N–H as well as the basicity of the pyridine nitrogen (Figure 1).10,11 Inclusion of electron withdrawing groups should acidify the amide, making it a better hydrogen bond donor, whereas electron donating groups should not only decrease the amide acidity but also increase the electron density of the pyridine nitrogen, making it a better hydrogen bond acceptor. Although these two effects are opposing, the acidification of the amide NH is expected to be a larger effect due to the closer proximity to the substituted phenyl group.
Figure 1.
Effects of electron donating and withdrawing on the electron density of the 2,6-diamidopyridine receptors.
To prepare the desired compounds, precursor 1a was prepared by coupling 3,3-dimethylbutyryl chloride with 2,6-diaminopyridine using excess diaminopyridine as the base in THF. Compounds 2a–f were prepared by treatment of the corresponding acid chlorides with 1a (Scheme 1). To prepare the p-dimethylamino substituted compound, the pyridine nitrogen of 1a was first oxidized with mCPBA to afford 1b, which was then coupled to p-dimethylamino benzoic acid using EDC and HOBT.12 The resultant N-oxide product (1c) was then reduced with (Bpin)2 to afford 2g.13
Scheme 1. Synthesis of 2,6-Diamidopyridines 2a–g.

To measure the binding affinity of the differently substituted 2a–g with diethyl barbital, 1H NMR titrations were performed for each host–guest system in CDCl3. Because the binding involves hydrogen bonding between the receptor and barbital, the N-H1H NMR resonances of the amides on both the host and the guest change during the course of the titration (Figure 2). Using the tabulated chemical shift data of the barbital N-H, the resultant binding isotherms were fit to a 1:1 model, based on previous studies investigating the binding stoichiometry of compounds such as 2a–g with diethyl barbital.9 All measurements were repeated at least in triplicate to ensure reproducibility of the binding affinities.
Figure 2.
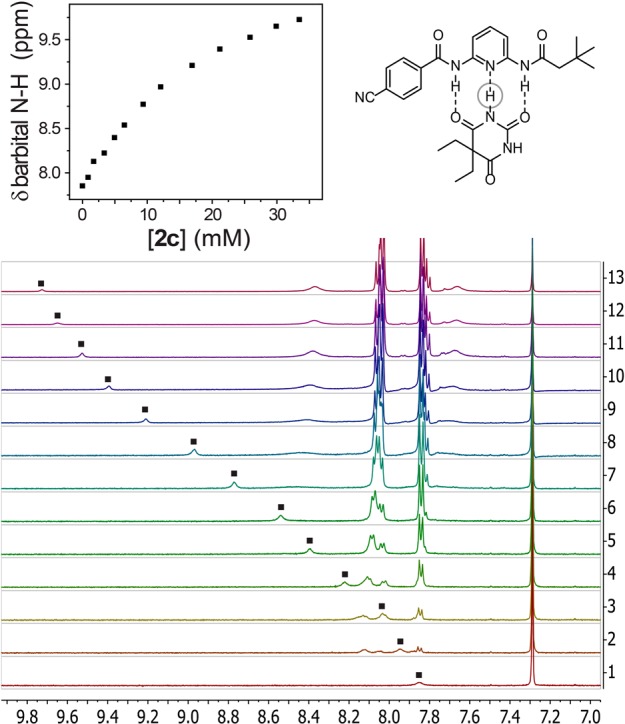
Representative 1H NMR (500 MHz, 25 °C, CDCl3) titration of diethyl barbital with 2c. The stacked 1H NMR titration spectra and tabulated plot of the N-H chemical shift data from the 1H NMR titration are shown.
Measurement of the binding constants of 2a–g with diethyl barbital revealed that electron withdrawing groups result in the highest binding affinity, whereas electron donating groups weakened guest binding. Binding affinities for 2g, which contained the most electron donating group, were too low to measure reproducibly. These results suggest that acidification of the amide N-H plays a larger role in guest binding than increasing the basicity of the pyridine nitrogen, which is consistent with the closer proximity of the amide group to the electronically substituted phenyl group. To quantitatively compare the impacts of electronic effects on barbiturate binding, we used the experimentally determined binding affinities to construct a Hammett plot using the corresponding σp values for each substituent (Figure 3).14,15 The resultant Hammett plot has a positive slope, which confirms that negative charge buildup is stabilized during the guest binding process, an effect that is consistent with both increased amide acidity and increased electron density on the pyridine nitrogen.16,17 The slope of the Hammett plot, however, shows a clear break between the electron donating and withdrawing groups, with ρ values of 1.08 ± 0.08 and 0.37 ± 0.02, respectively.18 This bimodal (or curved) Hammett plot suggests a change in binding conformation between the hosts with electron withdrawing and donating groups, respectively.19−21 Such a change could be due to the shorter hydrogen bonds formed between the host and guest upon acidification of the amides with inclusion of electron withdrawing groups.10,11,22−25
Figure 3.
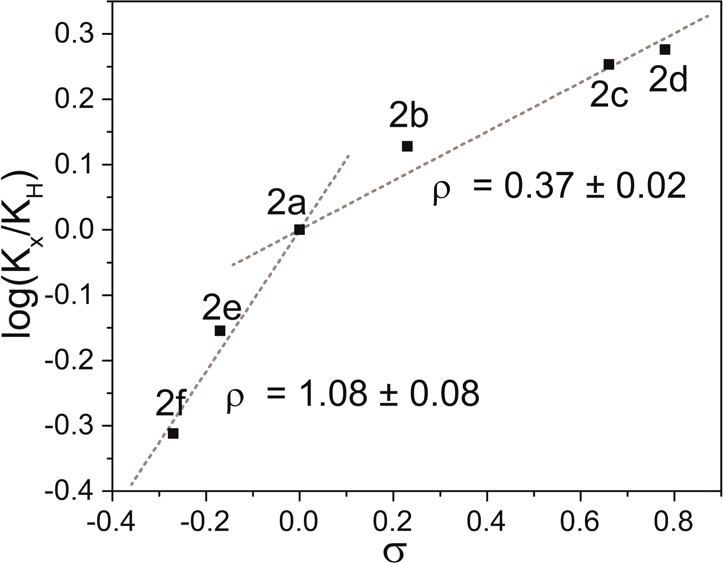
Hammett plot of hosts 2a–f binding diethyl barbital. Binding constants were obtained by following the barbital N–H resonance using 1H NMR spectroscopy (500 MHz, 25 °C, CDCl3).
To further investigate this nonlinear Hammett plot, we used computational studies to determine whether the break in the Hammett plot was related to changes in host–guest structure. Structures for 2a–f coordinated to diethyl barbital were optimized at the B3LYP/6-31+G(d,p) level of theory and the IEF-PCM solvation model for CHCl3, which has been shown to correlate well with experimental binding affinities in similar systems.9 Calculations using dispersion-corrected functionals were also performed and provided similar results. For each optimized geometry, the NH–O(barbital) and N–HN(barbital) hydrogen bond lengths were measured and compared to that of 2a ligated to barbital (Figure 4). As expected, the distal N−H of the alkyl amide did not change upon electronic substitution to the phenyl ring because the alkyl N−H is too far away from the electronic modulation to expect a significant contribution. By contrast, the amide N−H proximal to the benzene ring changes linearly with electronic substitution. Similarly, the hydrogen bond to the pyridyl nitrogen changes, although not linearly, with electronic substitution. Taken together, the structural difference upon electronic substation are consistent with a change in equilibrium geometry, which would correspond to a bimodal (or curved) Hammett plot, and is consistent with the observed experimental results.
Figure 4.
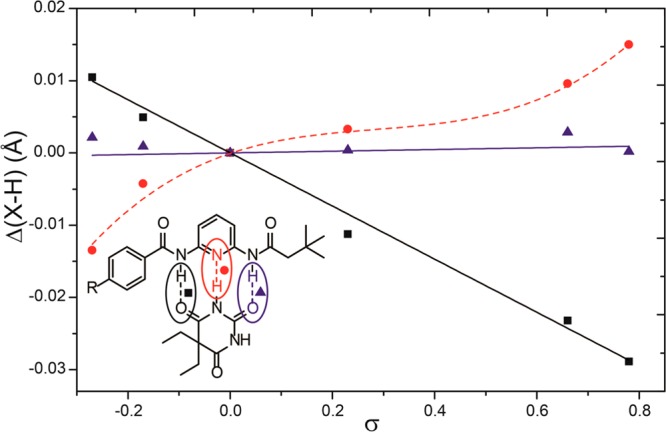
Comparison of the NH–O(barbital) and N–HN(barbital) hydrogen bonding distances for 2a–f hydrogen bonded to diethylbarbital. Calculations were performed in Gaussian using the B3LYP/6-31+G(d,p) level of theory and the IEF-PCM solvation model for CHCl3.
In conclusion, we have shown that binding of barbiturates to 2,6-diamidopyridines with different electronic structures produces a nonlinear Hammett plot, which is characteristic of changes in host–guest structure upon modulation of electronic structure. These results were supported both experimentally, using determined binding affinities, and computationally, using structural comparisons of optimized host–guest geometries. Taken together, these studies highlight how changes in electronic structure in hydrogen bonding assemblies can result not only in changes in binding affinities but also in changes in the assembled structure, thus providing insight into the factors controlling structural changes in the hydrogen-bonded complexes between the receptors and barbiturate guest.
Experimental Section
Materials and Methods
All commercially available reagents were used as received. Deuterated solvents were used as received. Anhydrous solvents used for syntheses were collected from a solvent purification system. Reactions were monitored by TLC on Silicagel 60 F254 plates, and the products were purified on an automated chromatography instrument using SiliaFlash F60 SiO2. NMR spectra were recorded at the indicated frequencies on either a 300 or 500 MHz spectrometer. Chemical shifts are reported in parts per million (δ) and are referenced to residual protic solvent resonances. The following abbreviations are used in describing NMR couplings: (s) singlet, (d) doublet, (t) triplet, (m) multiplet, and (b) broad; coupling constants are reported in hertz (Hz).
General Procedure Binding Constant Determination
Binding studies were performed in CDCl3 for host molecules 2a–g and were monitored by 1H NMR spectroscopy. In a typical CDCl3 titration, 2.00 mL of 1.0 mM barbital was prepared. The guest solution was then divided such that 1.00 mL was placed into an NMR tube and the other 1.00 mL was used to create a second solution containing 50–75 mM host. An initial spectrum of the guest was recorded, after which aliquots (5–100 μL) of the host solution were added until the N–H resonance of barbital no longer shifted. The resultant curves were fit using a 1:1 model, and the Kassoc was obtained.26
Computational Details
Calculations were performed using the Gaussian 0927 software package using the GaussView28 5.0 graphical user interface. Geometry optimizations and unscaled frequency calculations were performed at the B3LYP/6-31+G(d,p) level of theory using the IEF-PCM solvation model for chloroform. Frequency calculations were performed on all converged structures confirming that they corresponded to local minima. In all cases, the lowest energy conformer was used to compare the relative energetics of the calculated species.
Syntheses
N-(6-Aminopyridin-2-yl)-3,3-dimethylbutanamide (1a)
A round-bottom flask was charged with dry THF (100 mL) and 2,6-diaminopyridine (1.04 g, 9.5 mmol). The flask was then lowered into an ice bath and deoxygenated by sparging with N2. 3,3-Dimethylbutyryl chloride (0.60 mL, 4.3 mmol) was added to an addition funnel containing dry THF (25 mL), and the resultant solution was then added slowly to the diaminopyridine solution over the course of 1 h while stirring at 0 °C under N2. Once the addition of the acid chloride was complete, the ice bath was removed and the reaction was allowed to warm to room temperature overnight while stirring under N2. The precipitate from the reaction was filtered, and the resultant filtrate was concentrated by rotary evaporation. The crude product was purified by column chromatography (SiO2, EtOAc) to afford a solid (0.61 g, 69%). Mp = 113–114 °C. 1H NMR (300 MHz, CDCl3) δ: 7.77 (s, 1H), 7.59 (d, J = 8.0, 1H), 7.48 (t, J = 8.0, 1H), 6.28 (d, J = 7.5, 1H), 4.43 (s, 2H), 2.24 (s, 2H), 1.11 (s, 9H). 13C{1H} NMR (125 MHz, CDCl3) δ: 170.3, 156.7, 149.4, 140.5, 104.2, 103.2, 51.7, 31.3, 29.9. HRMS (ESI-TOF) m/z: [M]+ calcd for [C11H17N3ONa]+, 230.1269; found 230.1275.
2-Amino-6-(3,3-dimethylbutanamido)pyridine 1-Oxide (1b)
1a (0.20 g, 0.97 mmol) was added to a round-bottom flask containing THF (75 mL). mCPBA (0.22, 1.3 mmol) was then added to the flask, and the reaction was allowed to stir overnight at room temperature. The reaction mixture was concentrated, and the residue was taken up in EtOAc and washed 3× with 75 mL of saturated K2CO3. The organic layer was concentrated using a rotary evaporator to yield the product as a yellow solid (0.18 g, 84%). Mp = 119–120 °C. 1H NMR (500 MHz, CDCl3) δ: 9.95 (s, 1H), 7.78 (d, J = 8.5, 1H), 7.19 (t, J = 8.5, 1H), 6.48 (d, J = 8.0, 1H), 5.77 (s, 2H), 2.40 (s, 2H), 1.12 (s, 9H). 13C{1H} NMR (125 MHz, CDCl3) δ: 170.7, 148.6, 142.5, 130.1, 102.7, 102.1, 51.6, 31.3, 29.8. HRMS (ESI-TOF) m/z: [M]+ calcd for [C11H18N3O2]+, 224.1399; found 224.1401.
2-(4-(Dimethylamino)benzamido)-6-(3,3-dimethylbutanamido)pyridine 1-Oxide (1c)
4-Dimethylaminobenzoic acid (80 mg, 0.50 mmol) was added to a vial containing HOBT (70 mg, 0.55 mmol), EDC (0.11 g, 0.55 mmol), NEt3 (80 μL, 0.59 mmol), and CH3CN (15 mL). The reaction mixture was allowed to stir for 1 h at 50 °C before addition of 1b (0.10 g, 0.46 mmol). The reaction mixture was stirred overnight at 50 °C, after which the solvent was removed under vacuum. The crude product was purified by column chromatography (SiO2, 1:1 EtOAc/hexanes) to afford the product as an off-white solid (80 mg, 49%). Mp = 158–159 °C. 1H NMR (500 MHz, CDCl3) δ: 10.63 (s, 1H), 9.85 (s, 1H), 8.31 (d, J = 8.5, 1H), 8.15 (d, J = 8.3, 1H), 7.94 (d, J = 8.5, 2H), 7.42 (t, J = 9.0, 1H), 6.82 (d, J = 8.5, 2H), 3.11 (s, 6H), 2.42 (s, 2H), 1.16 (s, 9H). 13C{1H} NMR (125 MHz, CDCl3) δ: 170.2, 164.9, 162.9, 149.8, 149.4, 140.9, 129.1, 126.3, 114.1, 109.6, 109.4, 55.5, 51.9, 31.4, 29.8. HRMS (ESI-TOF) m/z: [M + H]+ calcd for [C20H27N4O3]+, 371.2079; found 371.2083.
N-(6-(3,3-Dimethylbutanamido)pyridin-2-yl)benzamide (2a)
A round-bottom flask was charged with dry THF (30 mL), 1a (114.5 mg, 0.55 mmol), and NEt3 (0.12 mL, 0.83 mmol). The flask was then lowered into an ice bath and deoxygenated by sparging with N2. Benzoyl chloride (70 μL, 0.61 mmol) was added to an addition funnel containing dry THF (10 mL), and the resultant acid chloride solution was then slowly added to the diaminopyridine solution while stirring in the ice bath under N2. Once the addition of the acid chloride was complete, the ice bath was removed and the reaction was allowed to warm to room temperature overnight while stirring under N2. The reaction was concentrated by rotary evaporation, and the crude product was taken up in EtOAc and washed 3× with 1 M NaOH. The organic layer was kept and concentrated under vacuum to afford the product as a white solid (0.15 g, 87%). Mp = 140–141 °C. 1H NMR (500 MHz, CDCl3) δ: 8.34 (s, 1H), 8.08 (d, J = 8.0, 1H), 7.99 (d, J = 8.0, 1H), 7.90 (d, J = 7.5, 2H), 7.78–7.73 (m, 2H), 7.58 (t, J = 7.5, 1H), 7.51 (d, J = 8.0, 2H), 2.26 (s, 2H), 1.12 (s, 9H). 13C{1H} NMR (125 MHz, CDCl3) δ: 170.3, 165.4, 149.6, 140.9, 134.2, 132.3, 128.9, 127.1, 109.7, 109.6, 51.8, 31.4, 29.8. HRMS (ESI-TOF) m/z: [M]+ calcd for [C18H21N3O2Na]+, 334.1531; found 334.1532.
4-Chloro-N-(6-(3,3-dimethylbutanamido)pyridin-2-yl)benzamide (2b)
Disubstituted diaminopyridine 2b was prepared according to the general procedure outlined for 2a with the following quantities: 4-chlorobenzoyl chloride (50 μL, 0.37 mmol) in THF (10 mL) was added slowly to 1a (58.2 mg, 0.281 mmol) and triethylamine (0.10 mL, 0.70 mmol) in THF (30 mL). The solvent was removed, and the residue purified by column chromatography (SiO2, 4:1 hexanes/EtOAc) to afford a white crystalline solid (95 mg, 97%). Mp = 149–151 °C. 1H NMR (500 MHz, DMSO) δ: 10.49 (s, 1H), 10.07 (s, 1H), 8.01 (d, J = 8.5, 2H), 7.88 (d, J = 7.0, 1H), 7.81 (t, J = 7.5, 1H), 7.75 (d, J = 7.0, 1H), 7.60 (d, J = 8.5, 2H), 3.35 (s, 2H), 1.03 (s, 9H). 13C{1H} NMR (125 MHz, DMSO) δ: 171.4, 165.3, 151.0, 150.5, 140.4, 137.2, 133.4, 130.3, 128.9, 111.3, 110.3, 49.5, 31.4, 30.1. HRMS (ESI-TOF) m/z: [M]+ calcd for [C18H21N3O2Cl]+, 346.1311; found 346.1322.
4-Cyano-N-(6-(3,3-Dimethylbutanamido)pyridin-2-yl)benzamide (2c)
Disubstituted diaminopyridine 2c was prepared according to the general procedure outlined for 2a with the following quantities: 4-cyanobenzoyl chloride (228 mg, 1.38 mmol) in THF (10 mL) was added slowly to 1a (238 mg, 1.15 mmol) and triethylamine (0.32 mL, 2.29 mmol) in THF (40 mL). The solvent was removed, and the residue was purified by column chromatography (SiO2, 1:1 EtOAc/hexanes) to afford a white crystalline solid (0.31 g, 80%). Mp = 202–203 °C. 1H NMR (500 MHz, CDCl3) δ: 8.27 (s, 1H), 8.07–8.02 (m, 4H), 7.85–7.79 (m, 3H), 7.55 (s, 1H), 2.28 (s, 2H), 1.13 (s, 9H). 13C{1H} NMR (125 MHz, CDCl3) δ: 170.3, 163.6, 149.6, 148.9, 141.2, 138.1, 132.7, 127.8, 117.8, 115.9, 110.3, 109.7, 51.8, 31.4, 29.8. HRMS (ESI-TOF) m/z: [M]+ calcd for [C19H20N4O2Na]+, 359.1484; found 359.1499.
N-(6-(3,3-Dimethylbutanamido)pyridin-2-yl)-4-nitrobenzamide (2d)
The disubstituted diaminopyridine 2d was prepared according to the general procedure outlined for 2a with the following quantities: 4-nitrobenzoic acid was stirred in thionyl chloride (3 mL) overnight at 65 °C. The thionyl chloride was removed, and the residue was taken up in THF (10 mL) and was then added slowly to 1a (126.6 mg, 0.61 mmol) and NEt3 (0.17 mL, 1.22 mmol) in THF (30 mL). The solvent was removed, and the residue was purified by column chromatography (SiO2, 1:1 EtOAc/hexanes) to afford a white crystalline solid (0.20 g, 84%). Mp = 162–163 °C. 1H NMR (500 MHz, CDCl3) δ: 8.38 (d, J = 9.0, 2H), 8.10 (d, J = 8.5, 2H), 8.07–8.02 (m, 2H), 7.81 (t, J = 8.0, 1H), 7.61 (s, 2H), 2.29 (s, 2H), 1.14 (s, 9H). 13C{1H} NMR (125 MHz, CDCl3) δ: 170.4, 163.5, 150.0, 149.6, 148.9, 141.2, 139.6, 128.4, 124.1, 110.3, 109.7, 51.8, 31.4, 29.8. HRMS (ESI-TOF) m/z: [M]+ calcd for [C20H27N4O2]+, 355.2134; found 355.2123.
N-(6-(3,3-Dimethylbutanamido)pyridin-2-yl)-4-methylbenzamide (2e)
Disubstituted diaminopyridine 2e was prepared according to the general procedure outlined for 2a with the following quantities: p-tolulic acid was stirred in thionyl chloride (3 mL) overnight at 65 °C. The thionyl chloride was then removed under vacuum, and the residue taken up in THF (10 mL) and was then added slowly to 1a (160.1 mg, 0.77 mmol) and triethylamine (0.22 mL, 1.54 mmol) in THF (30 mL). The solvent was removed, and the residue was purified by column chromatography (SiO2, 1:1 EtOAc/hexanes) to afford a white crystalline solid (0.22 g, 81%). Mp = 159–161 °C. 1H NMR (500 MHz, CDCl3) δ: 8.28 (s, 1H), 8.09 (d, J = 8.5, 1H), 7.98 (d, J = 7.5, 1H), 7.83 (d, J = 8.0, 2H), 7.78 (t, J = 8.0, 1H), 7.60 (s, 1H), 7.32 (d, J = 8.0, 2H), 2.46 (s, 3H), 2.28 (s, 2H), 1.14 (s, 9H). 13C{1H} NMR (125 MHz, CDCl3) δ: 170.3, 165.5, 149.6, 149.3, 143.0, 141.1, 131.3, 129.6, 127.1, 109.6, 109.5, 51.8, 31.4, 29.8, 21.6. HRMS (ESI-TOF) m/z: [M]+ calcd for [C19H24N3O2]+, 326.1869; found 326.1877.
N-(6-(3,3-Dimethylbutanamido)pyridin-2-yl)-4-methoxybenzamide (2f)
Disubstituted diaminopyridine 2f was prepared according to the general procedure outlined for 2a with the following quantities: p-anisic acid (73.9 mg, 0.489 mmol) was stirred in thionyl chloride (3 mL) at 65 °C overnight. The thionyl chloride was removed under vacuum, and the resulting residue taken up in THF (10 mL) was added slowly to 1a (91.5 mg, 0.442 mmol) and triethylamine (0.12 mL, 0.882 mmol) in THF (30 mL). After solvent removal, the residue was purified by column chromatography (SiO2, 1:1 EtOAc/hexanes) to afford a white crystalline solid (0.18 g, 95%). Mp = 171–172 °C. 1H NMR (500 MHz, CDCl3) δ: 8.36 (s, 1H), 8.11 (d, J = 8.5, 1H), 7.98 (d, J = 8.0, 1H), 7.98 (d, J = 8.5, 2H), 7.82 (t, J = 7.5, 1H), 7.64 (s, 1H), 7.02 (d, J = 9.0, 2H), 3.92 (s, 3H), 2.28 (s, 2H), 1.15 (s, 9H). 13C{1H} NMR (125 MHz, CDCl3) δ: 170.4, 165.0, 162.9, 149.7, 149.4, 141.1, 129.1, 126.2, 114.1, 109.6, 109.4, 55.5, 51.8, 31.4, 29.8. HRMS (ESI-TOF) m/z: [M]+ calcd for [C19H23N3O3Na]+, 364.1637; found 364.1635.
4-(Dimethylamino)-N-(6-(3,3-dimethylbutanamido)pyridin-2-yl)benzamide (2g)
1c (48 mg, 0.13 mmol) was placed in a vial in a glovebox. A solution of bis(pinacolato)diboron (36 mg, 0.14 mmol) in CH3CN (10 mL) was then added to 1c and allowed to stir for 24 h. The solvent was then removed, and the residue purified using column chromatography (SiO2, 3:1 EtOAc/hexanes) to afford a white, chalky solid (40 mg, 87%). Mp = 207–209 °C. 1H NMR (500 MHz, CDCl3) δ: 8.23 (s, 1H), 8.09 (d, J = 8.5, 1H), 7.94 (d, J = 7.5, 1H), 7.82 (d, J = 8.5, 2H), 7.75 (t, J = 8.0, 1H), 7.66 (s, 1H), 6.72 (d, J = 9.0, 2H), 3.07 (s, 6H), 2.27 (s, 2H), 1.13 (s, 9H). 13C{1H} NMR (125 MHz, CDCl3) δ: 170.3, 165.3, 152.9, 150.2, 149.4, 140.8, 128.9, 120.4, 111.1, 109.6, 109.0, 51.8, 40.1, 31.4, 29.8. HRMS (ESI-TOF) m/z: [M]+ calcd for [C20H26N4O2Na]+, 377.1953; found 377.1966.
Acknowledgments
This work was supported by funding from the University of Oregon. The NMR facilities at the UO are supported by the NSF/ARRA (CHE-0923589), and the computational infrastructure is supported by the OCI (OCI-096054). The Biomolecular Mass Spectrometry Core of the Environmental Health Sciences Core Center at Oregon State University is supported, in part, by the NIEHS (P30ES000210) and the NIH.
Supporting Information Available
NMR spectra of new compounds, titration data, optimized geometries. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Izatt R. M.; Pawlak K.; Bradshaw J. S.; Bruening R. L. Chem. Rev. 1995, 95, 2529–2586. [Google Scholar]
- Evans N. H.; Beer P. D. Angew. Chem., Int. Ed. 2014, 44, 11716–11754. [DOI] [PubMed] [Google Scholar]
- Weisman G. A.; Camden J. M.; Peterson T. S.; Ajit D.; Woods L. T.; Erb L. Mol. Neurobiol. 2012, 46, 96–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cetina M.; Rissanen K. Croat. Chem. Acta. 2012, 85, 319–325. [Google Scholar]
- Schneider H. J.; Yatsimirsky A. K. Chem. Soc. Rev. 2008, 37, 263–277. [DOI] [PubMed] [Google Scholar]
- Therrien B.; Vieille-Petit L.; Tschan M.; Romakh V. B.; Suss-Fink G. Chimia 2003, 57, 593–596. [Google Scholar]
- Schneider H. J. Angew. Chem., Int. Ed. 2009, 48, 3924–3977. [DOI] [PubMed] [Google Scholar]
- Reek J. N. H.; Priem A. H.; Engelkamp H.; Rowan A. E.; Elemans J. A. A. W.; Nolte R. J. M. J. Am. Chem. Soc. 1997, 119, 9956–9964. [Google Scholar]
- McGrath J. M.; Pluth M. D. J. Org. Chem. 2014, 79, 711–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang S. Y.; Kim H. S.; Chang K. J.; Jeong K. S. Org. Lett. 2004, 6, 181–184. [DOI] [PubMed] [Google Scholar]
- Legrand Y. M.; Gray M.; Cooke G.; Rotello V. M. J. Am. Chem. Soc. 2003, 125, 15789–15795. [DOI] [PubMed] [Google Scholar]
- Londregan A. T.; Storer G.; Wooten C.; Yang X. J.; Warmus J. Tetrahedron Lett. 2009, 50, 1986–1988. [Google Scholar]
- Kokatla H. P.; Thomson P. F.; Bae S.; Doddi V. R.; Lakshman M. K. J. Org. Chem. 2011, 76, 7842–7848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordwell F. G.; Mccallum R. J.; Olmstead W. N. J. Org. Chem. 1984, 49, 1424–1427. [Google Scholar]
- Hansch C.; Leo A.; Taft R. W. Chem. Rev. 1991, 91, 165–195. [Google Scholar]
- Pellizzaro M. L.; Fisher J.; Wilson A. J. Rsc. Adv. 2013, 3, 3103–3108. [Google Scholar]
- Gooch A.; McGhee A. M.; Pellizzaro M. L.; Lindsay C. I.; Wilson A. J. Org. Lett. 2011, 13, 240–243. [DOI] [PubMed] [Google Scholar]
- The binding constants do not trend with the σ+ or σ– Hammett parameters indicating that the effect of the electronic group is due to inductive rather than resonance effects. Additionally, the 1H NMR chemical shifts of the phenyl hydrogens do not shift during the titrations, suggesting that there are no significant changes in resonance structures upon barbiturate binding.
- Ul Hoque M. E.; Guha A. K.; Kim C. K.; Lee B. S.; Lee H. W. Org. Biomol. Chem. 2009, 7, 2919–2925. [DOI] [PubMed] [Google Scholar]
- Creary X.; O’Donnell B. D.; Vervaeke M. J. Org. Chem. 2007, 72, 3360–3368. [DOI] [PubMed] [Google Scholar]
- Um I. H.; Chung E. K.; Kwon D. S. Tetrahedron Lett. 1997, 38, 4787–4790. [Google Scholar]
- Chae M. K.; Cha G. Y.; Jeong K. S. Tetrahedron Lett. 2006, 47, 8217–8220. [Google Scholar]
- Leventis N.; Rawaswdeh A. M. M.; Zhang G. H.; Elder I. A.; Sotiriou-Leventis C. J. Org. Chem. 2002, 67, 7501–7510. [DOI] [PubMed] [Google Scholar]
- Li Y. L.; Flood A. H. J. Am. Chem. Soc. 2008, 130, 12111–12122. [DOI] [PubMed] [Google Scholar]
- Wilcox C. S.; Kim E.; Romano D.; Kuo L. H.; Burt A. L.; Curran D. P. Tetrahedron 1995, 51, 621–634. [Google Scholar]
- Thordarson P. Chem. Soc. Rev. 2011, 40, 1305–1323. [DOI] [PubMed] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Mennucci B.; Petersson G. A.; Nakatsuji H.; Caricato M.; Li X.; Hratchian H. P.; Izmaylov A. F.; Bloino J.; Zheng G.; Sonnenberg J. L.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Montgomery J. A. Jr.; Peralta J. E.; Ogliaro F.; Bearpark M.; Heyd J. J.; Brothers E.; Kudin K. N.; Staroverov V. N.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Rega N.; Millam J. M.; Klene M.; Knox J. E.; Cross J. B.; Bakken V.; Adamo C.; Jaramillo J.; Gomperts R.; Stratmann R. E.; Yazyev O.; Austin A. J.; Cammi R.; Pomelli C.; Ochterski J. W.; Martin R. L.; Morokuma K.; Zakrzewski V. G.; Voth G. A.; Salvador P.; Dannenberg J. J.; Dapprich S.; Daniels A. D.; Farkas Ö.; Foresman J. B.; Ortiz J. V.; Cioslowski J.; Fox D. J.. Gaussian 09; Gaussian, Inc.; Wallingford, CT, 2009. [Google Scholar]
- Dennington R.; Keith T.; Millam J.. GaussView Version 5; Semichem Inc.: Shawnee Mission, KS, 2009. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.