

Abstract
During tuberculosis (TB), some Mycobacterium tuberculosis bacilli persist in the presence of an active immunity and antibiotics that are used to treat the disease. Herein, by using the Cornell model of TB persistence, we further explored our recent finding that suggested that M. tuberculosis can escape therapy by residing in the bone marrow (BM) mesenchymal stem cells. We initially showed that M. tuberculosis rapidly disseminates to the mouse BM after aerosol exposure and maintained a stable burden for at least 220 days. In contrast, in the lungs, the M. tuberculosis burden peaked at 28 days and subsequently declined approximately 10-fold. More important, treatment of the mice with the antibiotics rifampicin and isoniazid, as expected, resulted in effective clearance of M. tuberculosis from the lungs and spleen. In contrast, M. tuberculosis persisted, albeit at low numbers, in the BM of antibiotic-treated mice. Moreover, most viable M. tuberculosis was recovered from the bone marrow CD271+CD45−-enriched cell fraction, and only few viable bacteria could be isolated from the CD271−CD45+ cell fraction. These results clearly show that BM mesenchymal stem cells provide an antibiotic-protective niche for M. tuberculosis and suggest that unraveling the mechanisms underlying this phenomenon will enhance our understanding of M. tuberculosis persistence in treated TB patients.
Mycobacterium tuberculosis, the causative agent of tuberculosis (TB), is an intracellular pathogen known to infect primarily macrophages and dendritic cells.1–3 However, the viability of M. tuberculosis in these intracellular niches is poor,4 and no evidence exists indicating that these cells can maintain live nonreplicating M. tuberculosis for long periods of time. Therefore, it is unlikely that these cell populations can harbor viable M. tuberculosis during the chronic phase of the disease, which lasts for months or years, as well as during the latent TB infection, which can last for decades.
We have recently shown that M. tuberculosis can reside in bone marrow (BM) within the CD271+CD45− mesenchymal stem cells (BM-MSCs) of individuals treated for pulmonary TB and in mice experimentally infected with M. tuberculosis.5 MSCs can provide an ideal protective niche for M. tuberculosis because these cells have several properties that may promote the pathogen's long-term persistence and survival: i) MSCs are present in TB granulomas of infected mouse and human lung tissue6; ii) stem cells possess the capacity for self-renewal7; iii) stem cells express drug efflux pumps, such as ABCG2, that could contribute to drug evasion by M. tuberculosis5; iv) stem cells have low production of reactive oxygen species,8 which may favor the viability of nonreplicating M. tuberculosis; v) although MSCs have the capacity of self-renewal, they are relatively quiescent,9 and reside in the immune-privileged niche of the BM10,11; and vi) MSCs do not normally express major histocompatibility complex (MHC) class II on their cell surface and their MHC class I molecules are not functionally active (ie, these molecules do not trigger effector functions of cytotoxic T lymphocytes).12 Therefore, it is logical that BM-MSCs constitute a host cell capable of supporting long-term persistence of viable nonreplicating M. tuberculosis. However, many fundamental questions regarding the survival of virulent M. tuberculosis in BM-MSCs remain unanswered. Herein, we confirmed in vivo in the mouse model of TB that virulent M. tuberculosis disseminates rapidly to the BM within 2 weeks after infection with aerosolized organisms and preferentially resides within BM-MSCs. In addition, and more important, we show that antibiotics appeared to efficiently clear the infectious process within the lungs and spleens but fail to do so in the BM.
Materials and Methods
Six- to eight-week-old C57BL/6 female mice (Charles River Laboratories, Kingston, NY) were exposed (nasally only) to virulent M. tuberculosis Erdman strain using a CH Technologies (Westwood, NJ) aerosol-generating machine contained within a class III biosafety cabinet in the New England Regional Biosafety Laboratory (Grafton, MA). The low-dose exposure deposited approximately 80 bacilli in the lungs. After 28 days of infection, half of the mice received 100 mg/L rifampicin (RIF) with 250 mg/L isoniazid (INH) in the drinking water, and the other half received sterile acidified water. At the time points indicated, mice were euthanized by CO2, and lungs, spleens, and BM were harvested, homogenized in sterile phosphate-buffered saline (PBS), and plated onto oleate-albumin-dextrose-catalase–supplemented 7H11 agar. BM was harvested from both femurs and tibia from each mouse by flushing with sterile PBS, homogenized by pressing through 70-μm nylon mesh screens to yield single-cell suspensions, and resuspended in 2 mL of PBS. In initial experiments, the entire 2 mL of BM from each mouse was plated onto agar for colony-forming unit (CFU) recovery. In subsequent experiments, 500 μL of the 2-mL BM from each mouse was plated onto agar. The remaining 1.5 mL from four to six mice was pooled, and red blood cells were lysed, counted (yielding a calculated average of approximately 3.5 × 107cells per mouse), and enriched for BM-MSCs. The enriched/depleted/unfractioned cells were plated onto agar in duplicate or triplicate at the maximum number possible for BM-MSCs or 1 to 1.25 × 107 cells for the depleted and unfractioned cells. In some experiments, single-cell suspensions were obtained from the BM, and enriched for CD271+CD45− BM-MSCs using Stemcell Technologies (Vancouver, BC, Canada) EasySep Mouse Mesenchymal Stem/Progenitor Cell Enrichment Kit (catalog number 19771) for mouse MSCs, and fractions plated onto agar. Lung and spleen homogenates were serially diluted in sterile PBS before plating. M. tuberculosis CFUs were counted after at least 3 weeks at 37°C. To confirm the quality control of the kit before using it inside the biosafety level 3 facility, BM fractions were obtained from noninfected mice (outside the biosafety level 3 facility) by flushing the marrow out of femur and tibia, followed by softening of bone chips by incubation in solution containing 0.25% collagenase type I in PBS + 20% fetal bovine serum for 45 minutes at 37°C. The mononuclear cells were then filtered through a 70-μm cell strainer and washed twice in PBS containing 2% fetal bovine serum. The CD45− fractions were obtained by performing magnetic sorting, and the isolated cell population was stained for anti-CD45 (17-0451-83, activated protein C labeled; eBiosciences, San Diego, CA), anti-CD271 (ab62122, fluorescein isothiocyanate labeled; Abcam, Cambridge, MA), or isotype-matched controls. Flow cytometry analysis was performed on an AccuriC6 cytometer (BD Biosciences, San Jose, CA). We were able to collect a calculated average of 2.8 × 106 of CD45− cells from 3 × 107 BM cells (n = 5); 33.5% of CD45− cells showed expression of CD271. This analysis confirmed CD271+ cell enrichment using the Stemcell Technologies EasySep Mouse Mesenchymal Stem/Progenitor Cell Enrichment Kit (catalog number 19771) for mouse MSCs. All experiments were approved by Tufts University (Grafton, MA) Institutional Biosafety Committee (GRIA04) and Institutional Animal Care and Use Committee (G2012-151).
Results
Virulent M. tuberculosis Disseminates to the BM within 2 Weeks of Infection
Our initial observation that showed that M. tuberculosis could infect and reside in vivo in the mouse BM-MSCs was done using the i.v. route of infection and an avirulent strain of M. tuberculosis.5 To confirm those findings after a natural route of M. tuberculosis transmission, mice were initially infected with a low dose (approximately 80 CFUs) of aerosolized virulent M. tuberculosis (Erdman strain). Mice were then euthanized at different time points after the infection, and the BM target cells of the infectious process were evaluated. After infection, typical bacillary growth for C57BL/6 mice was observed, peaking at 3 to 4 weeks and then declining by approximately 10-fold to a stable CFU burden (Figure 1).13,14 The sharp slowing of the bacterial growth rate and decline in bacterial burden between days 28 and 42 in the lungs correspond to the timing for the generation and manifestation of acquired anti-M. tuberculosis type 1 helper T-cell immunity.1,14 Interestingly, between days 7 and 14 after aerosol exposure to M. tuberculosis, the bacteria disseminated from the lungs to the BM (Figure 1). This dissemination is similar to that observed in spleen (not shown) and liver.1,15 Thus, it seems that M. tuberculosis enters the BM before the initiation of acquired immunity. Moreover, and in contrast to the lungs, spleen, and liver (not shown in this study), the kinetics of the M. tuberculosis burden in the BM do not follow the pattern of a striking peak and decline in these organs, but instead, in the BM, the bacterial burden increases early and remains stable for at least 120 days (Figure 1). These results are consistent with our former observation pointing to BM as a secondary target organ of the infectious process that follows the lung colonization of virulent aerosolized M. tuberculosis.
Figure 1.

The M. tuberculosis disseminates, grows, and survives in bone marrow (BM) during early and chronic infection. Mice were infected with 82 ± 52 virulent M. tuberculosis Erdman bacilli by aerosol. At the indicated time points, viable M. tuberculosis CFUs were recovered from the lungs (A) and BM (B). Data are combined from two independent experiments each with four to six mice per time point (n = 8 to 12 mice) and shown as means ± SEM.
Virulent M. tuberculosis that Disseminate from Lung to BM Preferentially Reside within the MSC Subpopulation
To verify the BM cell target of M. tuberculosis, mice were challenged with aerosolized bacteria and their BM cells were subsequently harvested at different time points. These experiments aimed to confirm that viable M. tuberculosis preferentially target BM-MSCs. Harvested BM cells were separated by negative selection (Stemcell Technologies EasySep Mouse Mesenchymal Stem/Progenitor Cell Enrichment Kit) into two subpopulations of cells, one that expresses CD271+CD45− (BM-MSCs) and a second population of cells that are CD271−CD45+. To verify which cell population was preferentially infected with M. tuberculosis, the fractions of BM cells were independently inoculated in oleate-albumin-dextrose-catalase–enriched Middlebrook agar plates and incubated at 37°C for at least 3 weeks, followed by enumeration of the bacterial CFUs. Throughout the infection process, the burden of M. tuberculosis infection (CFU) was ≥10-fold higher in the CD271+CD45−-enriched BM cell fraction (BM-MSCs) than in the CD271-depleted cell population (Figure 2). This preferential BM targeting by M. tuberculosis occurs as early as 14 days after lung challenge and is maintained throughout the chronic phase of the infection. As expected, the CD271+CD45− depleted cell population also contains a relatively high level of infection because this population of cells comprises other cell types, including macrophages, a well-known population of cells that are normally infected by M. tuberculosis. Moreover, the depletion procedure did not non-specifically remove infected CD271− cells because the CFUs recovered from the unfractionated population of cells did not differ from the CD271+-depleted cell population (Figure 2). On the other hand, it is possible that the depleted CD271+ cells still contain CD271+CD45− cells because enrichment procedure does not fully deplete the latter cells. Nonetheless, these results clearly confirm that virulent M. tuberculosis enters the mammalian host through the respiratory tract and subsequently infects MSCs in a remote tissue like the BM.
Figure 2.

The M. tuberculosis disseminates to bone marrow (BM) during early and chronic infection and preferentially infects CD271+ cells. A: Mice were infected with 72 ± 21 virulent M. tuberculosis Erdman bacilli by aerosol. At the indicated time points, BM cell fractions were obtained after negative selection for BM-mesenchymal stem cell. Viable M. tuberculosis colony forming units were recovered from each cell fraction. Data are from four to five mice per time point and shown as means ± SEM. Results were analyzed by one-way analysis of variance with Tukey's post test. B: Representative flow cytometry panel shows the presence of CD271+/CD45− cells in the enriched population for mouse MSCs (blue dots). Isotype controls are indicated in red. ∗∗∗P < 0.001.
Antibiotics Are Not as Effective at Treating Virulent M. tuberculosis in the BM Compared with Lungs and Spleens
Our initial observation in humans indicated that TB patients successfully treated and considered cured still harbored viable M. tuberculosis in their BM-MSCs.5 It is possible that this biological state of resistance to therapy could be a consequence of efflux pump transporters present in these cells.5,16,17
To begin to evaluate a role for BM-MSCs as a protective niche allowing M. tuberculosis to evade therapy by antibiotic drugs, we took advantage of a well-established animal model of dormant TB. In this model, known for >50 years as the Cornell model,18–20 mice are infected with virulent M. tuberculosis and the resultant infection is typically treated for 12 weeks with the anti-mycobacterial drugs, which eliminates cultivable tubercle bacilli from the lungs, spleen, and livers. However, the condition can revert spontaneously or be induced by immune-suppressive drugs, and M. tuberculosis can again be cultured from lungs and spleens of approximately 50% of the animals. This indicates that in some mice, tubercle bacilli were present somewhere and somehow protected from the antibiotics. Because BM is a major niche of MSCs, we hypothesized that viable M. tuberculosis could still be present in BM even after successful sterilization of the lungs and spleen. In M. tuberculosis-infected mice, which received continuous treatment with RIF and INH starting 28 days after infection, viable M. tuberculosis were still present in BM for at least 100 days after no more viable M. tuberculosis could be recovered from the lungs and spleens (Figure 3). Interestingly, after the initiation of the antibiotic therapy, there was a steep and equal reduction of the viable M. tuberculosis that could be recovered from lungs and spleens, which lead to undetectable levels of viable M. tuberculosis (CFU). In contrast, no sterilization occurred in the BM, although there was a reduction in viable M. tuberculosis that reached the maximum at day 91 after infection (7 weeks of continuous antibiotic therapy). From then on, M. tuberculosis bacilli plateaued at low numbers until the end of the experiment at day 220 after infection (after >6 months of continuous antibiotic therapy). The level of detection in our assays was equivalent for the lungs, spleens, and BM.
Figure 3.
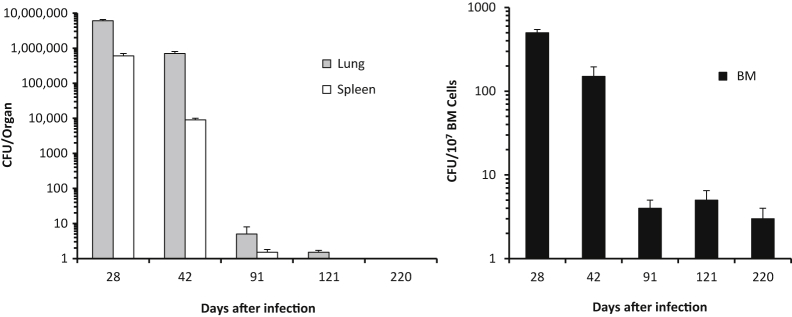
The M. tuberculosis survives in the bone marrow (BM) after anti-tuberculosis therapy. Mice were infected with 82 ± 52 M. tuberculosis Erdman bacilli by aerosol. Starting at day 28 of M. tuberculosis infection, mice received rifampicin and isoniazid continuously in the drinking water. At the indicated time points, viable M. tuberculosis colony forming unit (CFU) were recovered from lungs, spleens, and BM. Data are from two independent experiments each with four to six mice per time point (n = 8 to 12 mice) and shown as the means ± SEM. The last time points that M. tuberculosis could be recovered from the spleens and lungs were after 65 and 93 days, respectively, of continuous antibiotic therapy (days 121 and 220 after infection, respectively). In contrast, viable M. tuberculosis CFUs could be recovered from BM at all of the time points tested. Control CFU yields for mice receiving sterile water (instead of antibiotics) are depicted in Figure 1 (experiments were performed concomitantly).
To identify the cell population in the BM that hosts and protects the M. tuberculosis from RIF/INH, mice were infected for 28 days, followed by treatment with the antibiotics for 4 weeks. BM cells were obtained and separated as described above into CD271+CD45− cells and CD271−CD45+ cells. M. tuberculosis bacilli were 100 times more numerous in the CD271+CD45− cell population than in the CD271−CD45+ cell fraction (Figure 4). These results clearly indicate that BM-MSCs constitute a unique cellular niche, which, in the Cornell model of M. tuberculosis persistence, protects the bacteria from therapeutic properties of antibiotics that are used to treat TB.
Figure 4.
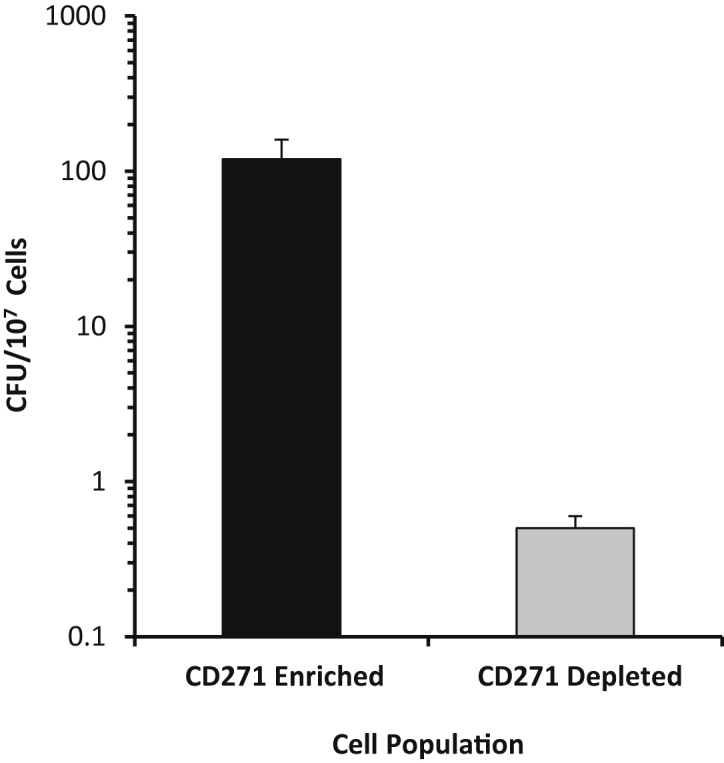
The M. tuberculosis bacilli are preferentially recovered from CD271+CD45− bone marrow cells after anti-tuberculosis therapy. Mice were infected with 72 ± 21 M. tuberculosis Erdman bacilli by aerosol. Starting at day 28 of M. tuberculosis infection, mice received rifampicin and isoniazid in the drinking water. After 4 weeks of antibiotic therapy, BM was harvested, followed by separation of CD271+CD45− and CD271−CD45+ cell fractions by negative selection. Cells were dispersed for M. tuberculosis culture, and CFU was determined after at least 3 weeks of incubation at 37°C. Data are from four to six mice per group and shown as the means ± SEM. Control colony forming unit (CFU) yields for mice receiving sterile water (instead of antibiotics) are depicted in Figure 2 (experiments were performed concomitantly).
Discussion
Our previous work established that BM cells, in particular, the CD271+CD45− BM-MSCs, harbor viable M. tuberculosis bacilli.5 However, these studies did not address the kinetics of dissemination from the lungs and persistence of virulent M. tuberculosis in the BM or explore temporal relationships between growth of the M. tuberculosis in the BM, compared with the primary site of infection and disease (lungs) or other secondary sites (spleen). Understanding these fundamental processes in vivo is important because the results have mechanistic implications for M. tuberculosis latent TB infection, which affects billions of people across the globe.21 Because these experiments cannot be performed in humans, we used the most common inbred mouse strain in M. tuberculosis research: C57BL/6 mice. C57BL/6 mice are well-known to be relatively resistant to low-dose aerosol infection of virulent M. tuberculosis (as we used herein) with survival >1 year,22 attributed to strong antimycobacterial immunity due to numerous and potent antigen-specific T cells.13,23 Thus, we used C57BL/6 mice as a model for studying M. tuberculosis in the BM of hosts capable of mounting strong type 1 helper T-cell–polarized anti-M. tuberculosis immunity.
Herein, we define that, after lung infection of mice with virulent M. tuberculosis, the infectious process disseminates not only to organs like spleen and liver but to BM as well. Interestingly, the pattern of growth and control of the M. tuberculosis in the lungs, spleen, and liver1,13–15 is not the same to that occurring with M. tuberculosis in the BM. In contrast to the lung, spleen, and liver, where growth is limited and burden declines because of robust immunity, the growth of the bacteria in the BM does not decline during the chronic phase of infection. Although we have not yet formally investigated the biological and molecular mechanisms that contribute to this phenomenon, it is possible that M. tuberculosis within the BM is protected from the host immune response because this tissue is generally considered an immune-privileged niche,10,11 and this is supported by our observation that no granulomas formed within the BM (not shown).
More important, the present studies clearly demonstrate that M. tuberculosis, in addition to the CD45+ cells, also resides in the BM within the unique MSC population (CD271+CD45− cells). The results that supported this conclusion (Figure 2) indicated that proportionally the CD271+CD45− cells were more infected than the CD45+ population of cells. However, it is important to keep in mind that during the in vivo infectious process, the CD45+ cell population constitutes a major target of the M. tuberculosis infection because these cells are disparately more numerous than the CD271+CD45− cell population. Nonetheless, because of the unique properties of the MSCs, the results strongly support the proposed hypothesis that infection of these cells constitutes an important M. tuberculosis escape mechanism of survival within the host. At this point, nothing is known about the mechanism of internalization of M. tuberculosis into the BM-MSCs. It is possible that the final localization of the bacteria within these cells may be a stepwise process (ie, the M. tuberculosis may be initially internalized into conventional host cells, like macrophages, followed by subsequent dedifferentiation into stem cells). Indeed, this set of events has recently and elegantly been demonstrated to be the case for Mycobacterium leprae–infected Schwann cells.24 This report shows that the leprosy bacterium induces the macrophage-like adult Schwann cells, the preferred host cells for M. leprae, to reprogram their differentiation to a stage of progenitor/stem-like cells by down-regulating cell lineage/differentiation-associated genes and up-regulating genes mostly of mesoderm development. Although hypothetical, this is an attractive mechanism to explain our observations that M. tuberculosis can be found internalized and likely persist within BM-MSCs.
The persistence of M. tuberculosis may represent an important escape mechanism from both the host immune response and may mask them from the reach of anti-TB drugs. The evasion of the immune response is likely to occur within the BM-MSCs because these cells do not normally express MHC class II on their cell surface and their MHC class I molecules are not functionally active and do not trigger effector functions of cytotoxic T lymphocytes.12 Protection from drugs is likely to be achieved because BM-MSCs have potent efflux pump transporters of ABCG2,5 an ATP-binding cassette family member that can pump a diversity of organic molecules, including the anti-TB antibiotic RIF, out of the cells.16,17 Indeed, our data lend support to this hypothesis. By using the well-established Cornell mouse model to study M. tuberculosis persistence after drug therapy, for the first time, our results clearly showed that even with prolonged and continuous treatment for >6 months with INH and RIF, the bacterium could still be recovered from the BM. In contrast and as expected from the Cornell model, no M. tuberculosis could be isolated from the lungs or spleen. These results identify a hidden location of the residual and viable M. tuberculosis present in mice in the Cornell model, which has been elusive for >50 years. More important, our results clearly identified that within the BM cells of antibiotic-treated mice, the pathogen was isolated primarily from the CD271+CD45− (BM-MSCs). In contrast, only few M. tuberculosis could be isolated from the CD271−CD45+ cells (monocytes, macrophages, and other phagocytic cells). However, because the CD271+CD45− enriched cell population was obtained by negative selection, other cells besides BM-MSCs may be present in this BM cell fraction. Therefore, other possible targets of infection by M. tuberculosis that could also help the pathogen escape antibiotic treatment cannot be excluded. However, the fact that the depleted fraction contained few organisms strongly supports the hypothesis that the BM-MSCs can protect M. tuberculosis from long-term administration of RIF/INH. These observations are highly relevant, because they may explain why it can be exceedingly difficult to achieve complete microbial sterilization of TB patients with antibiotics.
In conclusion, by using a stringent in vivo model of TB, these results strongly support the proposal that BM-MSCs are important target cells of the infection process that provides a robust antibiotic-protective niche for M. tuberculosis to survive therapy within its host. In addition, these studies suggest that unraveling the mechanisms underlying the M. tuberculosis and BM-MSC interactions will enhance our understanding of M. tuberculosis persistence under immune and antibiotic pressures in TB patients.
Acknowledgments
We thank Melanie Harwood, Curtis Rich, Donald Girouard, and Donna Akiyoshi (New England Regional Biosafety Laboratory, Tufts University Cummings School of Veterinary Medicine, Grafton, MA) for providing their expertise.
Footnotes
Supported by NIH grants R01AI076425 (A.C.-N.) and R56 AI111823 (A.C.-N. and G.B.), The Cummings School of Veterinary Medicine (Tufts University), and by the KaviKrishna Foundation (Sualkuchi, Assam, India).
Disclosures: None declared.
Contributor Information
Gillian Beamer, Email: gillian.beamer@tufts.edu.
Antonio Campos-Neto, Email: acampos@forsyth.org.
References
- 1.Cooper A.M. Cell-mediated immune responses in tuberculosis. Annu Rev Immunol. 2009;27:393–422. doi: 10.1146/annurev.immunol.021908.132703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Flynn J.L., Chan J., Lin P.L. Macrophages and control of granulomatous inflammation in tuberculosis. Mucosal Immunol. 2011;4:271–278. doi: 10.1038/mi.2011.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bodnar K.A., Serbina N.V., Flynn J.L. Fate of Mycobacterium tuberculosis within murine dendritic cells. Infect Immun. 2001;69:800–809. doi: 10.1128/IAI.69.2.800-809.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Keane J., Balcewicz-Sablinska M.K., Remold H.G., Chupp G.L., Meek B.B., Fenton M.J., Kornfeld H. Infection by Mycobacterium tuberculosis promotes human alveolar macrophage apoptosis. Infect Immun. 1997;65:298–304. doi: 10.1128/iai.65.1.298-304.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Das B., Kashino S.S., Pulu I., Kalita D., Swami V., Yeger H., Felsher D.W., Campos-Neto A. CD271(+) BM mesenchymal stem cells may provide a niche for dormant Mycobacterium tuberculosis. Sci Transl Med. 2013;5:170ra13. doi: 10.1126/scitranslmed.3004912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Raghuvanshi S., Sharma P., Singh S., Van Kaer L., Das G. Mycobacterium tuberculosis evades host immunity by recruiting mesenchymal stem cells. Proc Natl Acad Sci U S A. 2010;107:21653–21658. doi: 10.1073/pnas.1007967107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sacchetti B., Funari A., Michienzi S., Di Cesare S., Piersanti S., Saggio I., Tagliafico E., Ferrari S., Robey P.G., Riminucci M., Bianco P. Self-renewing osteoprogenitors in BM sinusoids can organize a hematopoietic microenvironment. Cell. 2007;131:324–336. doi: 10.1016/j.cell.2007.08.025. [DOI] [PubMed] [Google Scholar]
- 8.Das B., Antoon R., Tsuchida R., Lotfi S., Morozova O., Farhat W., Malkin D., Koren G., Yeger H., Baruchel S. Squalene selectively protects mouse BM progenitors against cisplatin and carboplatin-induced cytotoxicity in vivo without protecting tumor growth. Neoplasia. 2008;10:1105–1119. doi: 10.1593/neo.08466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aria F., Suda T. Harvard Stem Cell Institute; Cambridge, MA: 2008. Quiscent stem cells in the niche. StemBook; pp. 1–11. [PubMed] [Google Scholar]
- 10.Fujisaki J., Wu J., Carlson A.L., Silberstein L., Putheti P., Larocca R., Gao W., Saito T.I., Lo Celso C., Tsuyuzaki H., Sato T., Cote D., Sykes M., Strom T.B., Scadden D.T., Lin C.P. In vivo imaging of Treg cells providing immune privilege to the haematopoietic stem-cell niche. Nature. 2011;474:216–219. doi: 10.1038/nature10160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tormin A., Li O., Brune J.C., Walsh S., Schutz B., Ehinger M., Ditzel N., Kassem M., Scheding S. CD146 expression on primary nonhematopoietic BM stem cells is correlated with in situ localization. Blood. 2011;117:5067–5077. doi: 10.1182/blood-2010-08-304287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jacobs S.A., Roobrouck V.D., Verfaillie C.M., Van Gool S.W. Immunological characteristics of human mesenchymal stem cells and multipotent adult progenitor cells. Immunol Cell Biol. 2013;91:32–39. doi: 10.1038/icb.2012.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Turner J., Gonzalez-Juarrero M., Saunders B.M., Brooks J.V., Marietta P., Ellis D.L., Frank A.A., Cooper A.M., Orme I.M. Immunological basis for reactivation of tuberculosis in mice. Infect Immun. 2001;69:3264–3270. doi: 10.1128/IAI.69.5.3264-3270.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Beamer G.L., Flaherty D.K., Vesosky B., Turner J. Peripheral blood gamma interferon release assays predict lung responses and Mycobacterium tuberculosis disease outcome in mice. Clin Vaccine Immunol. 2008;15:474–483. doi: 10.1128/CVI.00408-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Flynn J.L., Chan J. Immunology of tuberculosis. Annu Rev Immunol. 2001;19:93–129. doi: 10.1146/annurev.immunol.19.1.93. [DOI] [PubMed] [Google Scholar]
- 16.Robey R.W., Ierano C., Zhan Z., Bates S.E. The challenge of exploiting ABCG2 in the clinic. Curr Pharm Biotechnol. 2011;12:595–608. doi: 10.2174/138920111795163913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Janvilisri T., Shahi S., Venter H., Balakrishnan L., van Veen H.W. Arginine-482 is not essential for transport of antibiotics, primary bile acids and unconjugated sterols by the human breast cancer resistance protein (ABCG2) Biochem J. 2005;385:419–426. doi: 10.1042/BJ20040791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Scanga C.A., Mohan V.P., Joseph H., Yu K., Chan J., Flynn J.L. Reactivation of latent tuberculosis: variations on the Cornell murine model. Infect Immun. 1999;67:4531–4538. doi: 10.1128/iai.67.9.4531-4538.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McCune R.M., Jr., Tompsett R. Fate of Mycobacterium tuberculosis in mouse tissues as determined by the microbial enumeration technique, I: the persistence of drug-susceptible tubercle bacilli in the tissues despite prolonged antimicrobial therapy. J Exp Med. 1956;104:737–762. doi: 10.1084/jem.104.5.737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McCune R.M., Jr., McDermott W., Tompsett R. The fate of Mycobacterium tuberculosis in mouse tissues as determined by the microbial enumeration technique, II: the conversion of tuberculous infection to the latent state by the administration of pyrazinamide and a companion drug. J Exp Med. 1956;104:763–802. doi: 10.1084/jem.104.5.763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lin P.L., Flynn J.L. Understanding latent tuberculosis: a moving target. J Immunol. 2010;185:15–22. doi: 10.4049/jimmunol.0903856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Medina E., North R.J. Resistance ranking of some common inbred mouse strains to Mycobacterium tuberculosis and relationship to major histocompatibility complex haplotype and Nramp1 genotype. Immunology. 1998;93:270–274. doi: 10.1046/j.1365-2567.1998.00419.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Woodworth J.S., Wu Y., Behar S.M. Mycobacterium tuberculosis-specific CD8+ T cells require perforin to kill target cells and provide protection in vivo. J Immunol. 2008;181:8595–8603. doi: 10.4049/jimmunol.181.12.8595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Masaki T., Qu J., Cholewa-Waclaw J., Burr K., Raaum R., Rambukkana A. Reprogramming adult Schwann cells to stem cell-like cells by leprosy bacilli promotes dissemination of infection. Cell. 2013;152:51–67. doi: 10.1016/j.cell.2012.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]