

Abstract
The transient receptor potential TRPV1 or vanilloid receptor is a nonselective ligand-gated channel highly expressed in primary sensory neurons where it mediates nociception. TRPV1 is also expressed in the brain where its activation depresses excitatory synaptic transmission. Whether TRPV1 also regulates inhibitory synapses in the brain is unclear. Here, using a combination of pharmacology, electrophysiology, and an in vivo knockdown strategy, we report that TRPV1 activation by capsaicin or by the endocannabinoid anandamide depresses somatic, but not dendritic inhibitory transmission in both rat and mouse dentate gyrus. The effect on somatic inhibition was absent in TRPV1 knock-out mice and was also eliminated by two different TRPV1 shRNAs expressed in dentate granule cells, strongly supporting a functional role for TRPV1 in modulating GABAergic synaptic function. Moreover, TRPV1-mediated depression occurs independently of GABA release, requires postsynaptic Ca2+ rise and activation of calcineurin, and is likely due to clathrin-dependent internalization of GABA receptors. Altogether, these findings reveal a novel form of compartment-specific regulation whereby TRPV1 channels can modify synaptic function in the brain.
Keywords: cannabinoid, endocytosis, feedforward inhibition, GABA, hippocampus, interneuron
Introduction
TRPV1, also known as VR-1 for vanilloid receptor type-1 (Caterina et al., 1997), is a nonselective, ligand-gated cation channel that can be activated by a variety of noxious, physical, and chemical stimuli, including endogenous lipid ligands such as the endocannabinoid (eCB) anandamide (AEA; Ross, 2003). These channels have been well characterized at afferent peripheral sensory neurons, where their activation regulates synaptic transmission associated with pain sensation (Caterina and Julius, 2001). Increasing anatomical, functional, and behavioral evidence supports the presence of functional TRPV1 channels also in the CNS (Sasamura et al., 1998; Mezey et al., 2000; Huang et al., 2002, 2014; Marinelli et al., 2003; D.P. Li et al., 2004; Roberts et al., 2004; Tóth et al., 2005; Cristino et al., 2006; Marsch et al., 2007; Gibson et al., 2008; H.B. Li et al., 2008; Maccarrone et al., 2008; Aguiar et al., 2009; Maione et al., 2009; Musella et al., 2009, 2010; Chávez et al., 2010; Grueter et al., 2010; Bennion et al., 2011; Puente et al., 2011, 2014; P. Han et al., 2013; Terzian et al., 2014). Of note, a recent report suggests that TRPV1 channels are restricted to a few brain areas (Cavanaugh et al., 2011). Thus far, most work in the brain has focused on the role of TRPV1 channels at excitatory synapses where their activation can modify synaptic transmission via both presynaptic (Marinelli et al., 2002, 2003; Gibson et al., 2008; Musella et al., 2009; Köles et al., 2013) and postsynaptic mechanisms (Chávez et al., 2010; Grueter et al., 2010; Puente et al., 2011; Kim et al., 2012). However, the role of TRPV1 channels at inhibitory synapses remains poorly understood.
TRPV1 channels can indirectly modulate inhibition by regulating excitatory afferents onto GABAergic interneurons (Derbenev et al., 2006; Ferrini et al., 2007; Zhou et al., 2007; Gibson et al., 2008; Liao et al., 2011), or by mobilizing eCBs (Maccarrone et al., 2008) that suppress GABA release via type-1 cannabinoid receptors (CB1Rs). In the dentate gyrus (DG), a key relay station that controls information transfer from the entorhinal cortex into the hippocampus, inhibitory interneurons make somatic and dendritic synapses onto dentate granule cells (DGCs; Z.S. Han et al., 1993; Hefft and Jonas, 2005; Liu et al., 2014). GABA release from somatic inhibitory inputs onto DGCs can be modulated by presynaptic CB1Rs (Isokawa and Alger, 2005; Liu et al., 2014), and anatomical evidence suggests CB1R and TRPV1 colocalize in the DG (Cristino et al., 2006). It remains unknown whether TRPV1 directly regulates inhibitory transmission and whether CB1Rs act cooperatively to regulate GABAergic synapses in the DG.
Here we sought to determine the role of TRPV1 in GABAergic transmission. Using rat and mouse hippocampal slices, selective pharmacology, TRPV1 knock-out mice, and in vivo knockdown strategies, we report that TRPV1 activation selectively reduces somatic, but not dendritic inputs in a transmitter release- and CB1R-independent manner. This TRPV1-mediated depression of inhibitory synaptic transmission requires postsynaptic Ca2+ rise and calcineurin activation, and it is likely mediated by the internalization of type-A GABA receptors (GABAAR). These results reveal an unexpected form of eCB-mediated regulation of GABAergic transmission along the somatodendritic axis of DGCs.
Materials and Methods
All procedures outlined in this study were approved by the Animal Care and Use Committee of Albert Einstein College of Medicine, in accordance with National Institutes of Health guidelines.
Hippocampal electrophysiology.
Acute transverse hippocampal slices (400 μm thick), except for AAV-shRNA slices (290 μm thick), were prepared from Wistar or Sprague Dawley rats, postnatal P15–P32, and C57BL/6 mice (Trpv1+/+/Trpv1−/−: P24–P33; cnr1 (CB1R)+/+/cnr1 (CB1R)−/−: P27–P32), either sex, as previously described (Chávez et al., 2010; Rodenas-Ruano et al., 2012). Briefly, the hippocampi were isolated and cut in a solution containing the following (in mm): 215 sucrose, 2.5 KCl, 26 NaHCO3, 1.6 NaH2PO4, 1 CaCl2, 4 MgCl2, 4 MgSO4, and 20 glucose. Thirty minutes post sectioning, slices were incubated in extracellular ACSF recording solution containing the following (in mm): 124 NaCl, 2.5 KCl, 26 NaHCO3, 1 NaH2PO4, 2.5 CaCl2, 1.3 MgSO4, and 10 glucose. All solutions were equilibrated with 95% O2 and 5% CO2, pH 7.4.
Hippocampal slices were visualized using infrared DIC and nontargeting GFP-control and Trpv1-shRNA DGCs were visualized using fluorescence video microscopy on a Nikon eclipse E600FN microscope (Fig. 2A). All experiments, except where indicated, were performed at 28 ± 1°C in a submersion-type recording chamber perfused at ∼1–2 ml/min with ACSF supplemented with 10 μm NBQX and 25 μm d-APV to block AMPA and NMDA receptors, respectively. Whole-cell patch-clamp recordings using a Multiclamp 700A amplifier (Molecular Devices) were made from DGCs voltage-clamped at +5 mV, unless otherwise stated, using patch-type pipette electrodes (∼3–4 MΩ) containing the following (in mm): 131 Cs-gluconate, 8 NaCl, 1 CaCl2, 10 EGTA, 10 glucose, and 10 HEPES; pH 7.4, 285–292 mmol/kg. All recordings were performed from DGCs whose somas were <30 μm from the molecular layer. For BAPTA experiments (Fig. 5A), 20 mm cesium gluconate, 10 mm EGTA, and 1 mm CaCl2 were replaced with 20 mm BAPTA (osmolarity and pH adjusted with CsOH). To isolate excitatory responses (Fig. 4C), NBQX and d-APV were replaced with 100 μm picrotoxin (PTX) to block GABAARs and DGCs were voltage clamped at −60 mV. Series resistance (∼10–25 MΩ) was monitored throughout all experiments with a −5 mV, 80 ms voltage step, and cells that exhibited significant change in series resistance (>20%) were excluded from analysis. To differentially stimulate inhibitory synaptic inputs, two monopolar-stimulating patch-type pipettes were filled with ACSF and placed in the dentate granule layer (DGL; <20 μm from the border with the molecular layer) to activate somatic inputs, and in the middle third of the molecular layer (MML) to stimulate dendritic inputs. Somatic extracellular field inhibitory synaptic potentials (fIPSPs) were recorded using pipettes filled with 1 m NaCl while the stimulating pipette was placed in the DGL. Stimulation intensity was adjusted to produce 0.2–0.4 mV fIPSP amplitude. Exogenous GABA responses (Fig. 3C) were evoked by directly puffing GABA (1 mm, 25 ms, 2 psi) using a Picospritzer III (Parker) connected to a patch pipette (resistance, 3–4 mΩ). The tip of the puffer pipette was positioned close to the recorded cell in the DGL or in the MML ∼30 μm deep from the surface of the slice. All experiments and analyses using Trpv1−/− and CB1R−/− mice were performed in a blind fashion.
Figure 2.
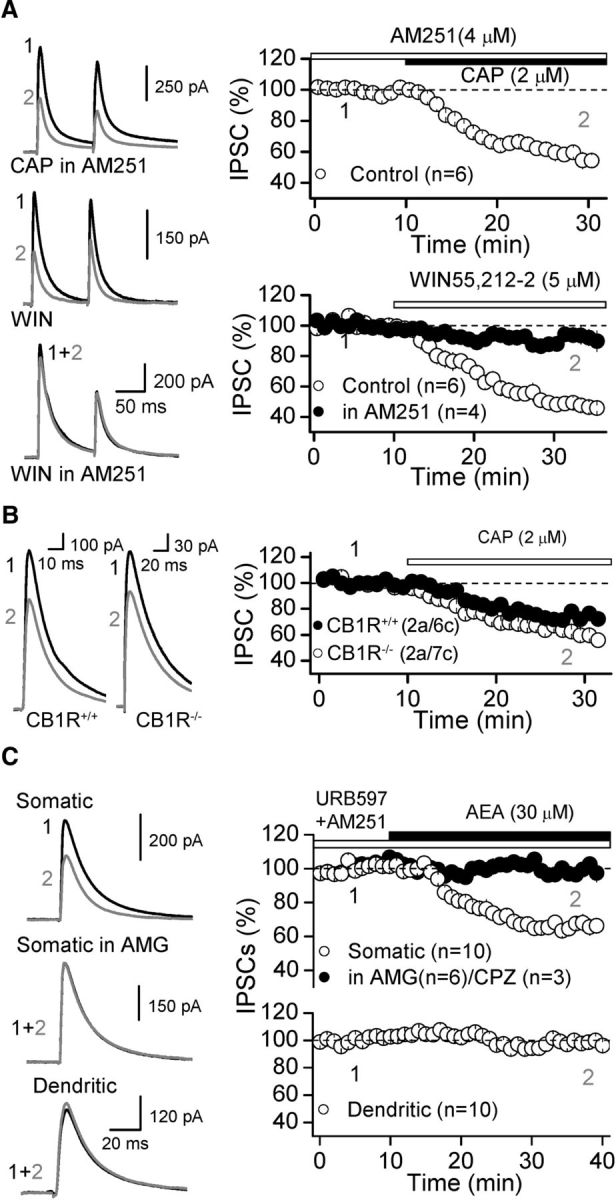
TRPV1 activation depresses inhibitory transmission in a CB1 receptor-independent manner. A, Representative traces (left) and summary data (right) showing that bath application of 2 μm CAP, while blocking CB1R with AM251 (4 μm), depresses somatic IPSCs. In interleaved experiments (bottom), AM251 eliminated the depression of somatic IPSCs observed by bath application of the CB1R agonist WIN55, 212–2 (5 μm). B, CAP-mediated depression was similar in both CB1R+/+ and CB1R−/− mouse, strongly suggesting that CAP depressing IPSCs was CB1R independent. C, Representative traces (left) and summary plots (right) showing that bath application of the eCBs AEA (30 μm) selectively reduced somatic (top), but not dendritic IPSCs (bottom). AEA-mediated depression was eliminated by pretreatment of the slices with TRPV1 antagonist, CPZ or AMG. Summary data represent the mean ± SEM and the number of animals (a) and cells (c) are in parenthesis.
Figure 5.
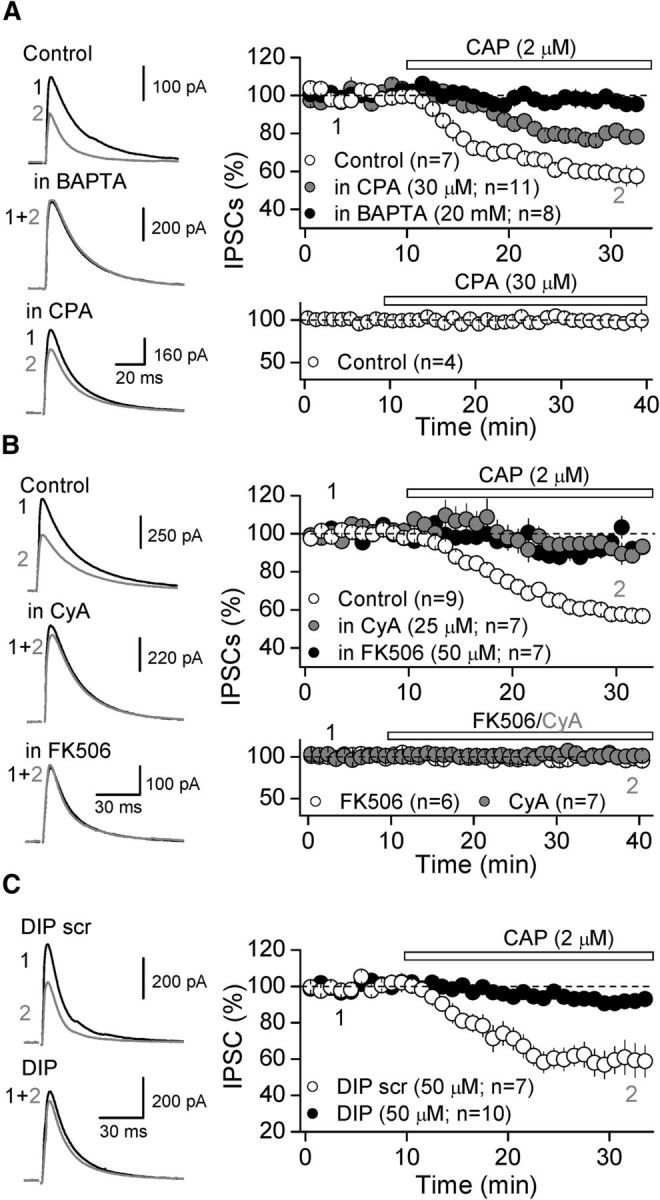
Mechanism underlying TRPV1-mediated depression of inhibitory transmission. A, Representative traces (left) and summary plot (right) showing that loading DGCs with BAPTA (20 mm) completely eliminated, whereas pre-incubation with 30 μm CPA significantly reduces, CAP-mediated depression of GABAergic IPSCs. Note that bath application of CPA for at least 30 min did not affect basal synaptic transmission (bottom). B, Representative traces (left) and summary plot (right) showing that two different calcineurin inhibitors, FK506 (50 μm) and CyA (25 μm), eliminated CAP-mediated depression of inhibition. Bath application of FK506 or CyA for 30 min did not affect GABAergic basal synaptic transmission (bottom). C, Intracellular loading of DIP (50 μm), but not DIP scrambled (DIP scr; 50 μm), eliminated CAP-mediated depression. Averaged sample traces taken at times indicated by numbers are shown next to each summary plot. In all parts, summary data represent the mean ± SEM and the number of cells tested is in parenthesis.
Figure 4.
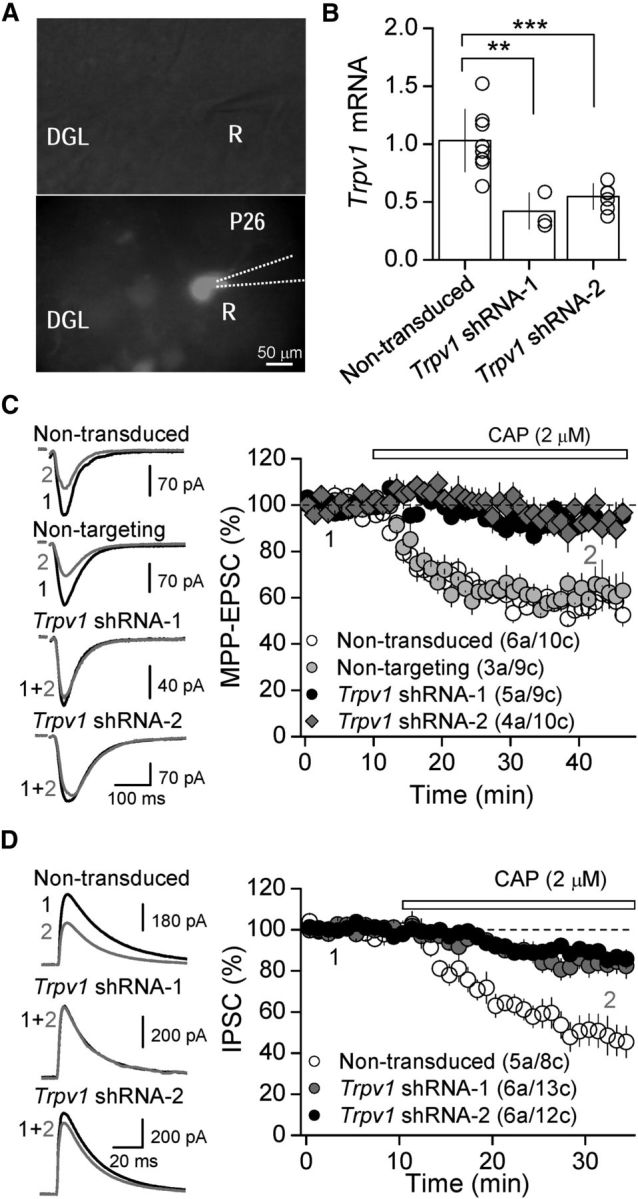
In vivo knockdown of TRPV1 abolishes TRPV1-mediated depression of synaptic transmission in the DG. A, Differential interference contrast (top) and fluorescence (bottom) images showing whole-cell recordings from Trpv1 shRNA-expressing (GFP+) DGCs. R, recording pipette. B, Trpv1 mRNA levels following Trpv1 shRNA-1 and Trpv1 shRNA-2 injection, at P10 and assessed by qRT-PCR at P28–32 in isolated DGCs, were significantly different compared with nontransduced control cells. C, Representative traces (left) and summary data (right) showing that CAP-mediated depression of AMPA-mediated EPSC elicited at the medial perforant path (MPP)-DG synapse was eliminated in both Trpv1 shRNA-1 and Trpv1 shRNA-2-expressing DGCs compared with DGCs expressing nontargeting shRNA or nontransduced cells. D, Both Trpv1 shRNA-1 and Trpv1 shRNA-2 significantly reduce CAP-mediated depression of somatic GABAergic IPSCs. In all parts, averaged sample traces taken at times indicated by numbers are shown next to each summary plot and summary data represent the mean ± SEM. The number of animals (a) and cells (c) are in parenthesis; **p = 0.008; ***p = 0.001.
Figure 3.
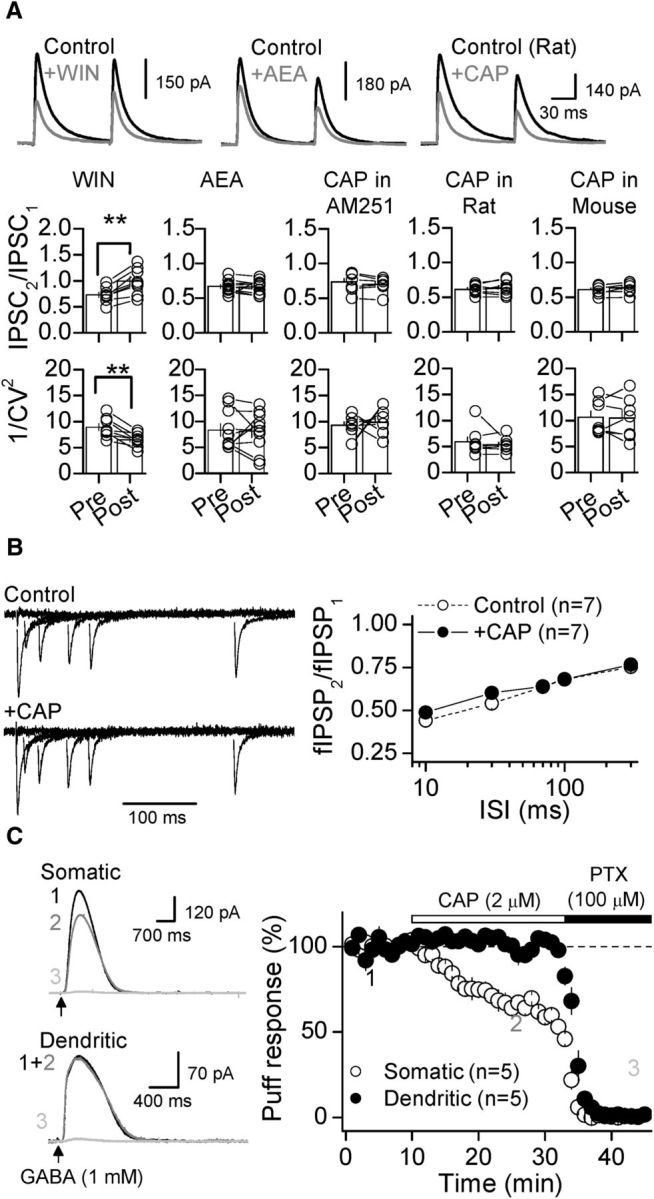
TRPV1-mediated depression of inhibition is postsynaptic. A, Top, Representative traces of two consecutive GABAergic IPSCs (100 ms interstimulus interval) before (black) and after (gray) bath application of WIN, AEA, and CAP. Bottom, Summary plots showing that WIN, but not AEA or CAP, significantly changes the PPR and 1/CV2 of somatic IPSCs. These results were generated from the experiments reported in Figure 1A and D and Figure 2A and C. B, CAP did not alter PPR of fIPSPs recorded as in Figure 1B and measured at different interstimulus intervals (10, 30, 75, 100, and 300 ms). fIPSPs were normalized to the first response. C, Representative traces (left) and summary plot (right) showing that CAP also mediated depression of GABAR-mediated responses evoked by puffing GABA (1 mm, 25 ms) in the DGL (somatic) but not in the middle third of the molecular layer (dendritic). Subsequent application of PTX (100 μm) eliminated the CAP-insensitive component of GABA-evoked responses. In all cases, averaged sample traces taken at times indicated by numbers are shown next to each summary plot. Summary data represent the mean ± SEM. **p < 0.01.
Except where indicated, pharmacological agents were bath applied after a stable baseline was established (∼10–15 min), and their effects were measured after responses reached a new steady state (typically >15 min). Stock solutions were prepared in water or DMSO and added to the ACSF as needed. Total DMSO in the ACSF was maintained at <0.1%. Drugs were obtained from Sigma and Tocris Bioscience, except WIN55, 212-2, d-APV, and NBQX, which were obtained from the NIMH Chemical Synthesis and Drug Supply Program. IPSCs were elicited every 20 s, filtered at 2.4 kHz, and acquired at 5 kHz using a custom-made software written in Igor Pro 4.09A (WaveMetrics). Statistical comparisons were made using paired and unpaired two-tailed Student's t test and ANOVA at the p < 0.05 significance level in OriginPro 7.0 and 8.6 (OriginLab). Unless otherwise indicated, all values are provided as the mean ± SEM and illustrated traces are averages of 31–35 responses.
AAV-shRNA design and cloning.
Trpv1 shRNA-1 (sense: GGCCAGGUAACUCUUACAACAGC) and TrpV1 shRNA-2 (sense: CGCUUGUAUAUGAUCAUCUUCA) with complete complementarity to the coding region of mouse (NM_001001445) and rat (NM_031982) sequences, and a nontargeting control shRNA (GAGGAUCAAAUUGAUAGUAAACC) with no sequence homology to any known gene transcript were designed. Desalted shRNA oligos containing a modified miR155 loop (GTTTTGGCCACTGACTGAC) and overhangs complementary to BamHI and XhoI restriction sites were custom synthesized (Invitrogen), annealed, and cloned into a “CreOff” AAV vector with a floxed cassette that comprises a U6 polymerase III promoter to drive shRNA expression, a CMV promoter to drive eGFP expression (Ventura et al., 2004) for identification of transduced neurons, and a woodchuck post-transcriptional response element to promote transgene expression. AAVs with serotype 9 were subsequently produced by Virovek using a baculovirus expression system (Chen, 2008).
qRT-PCR and stereotaxic injection of Trpv1 AAV-shRNA constructs.
For qRT-PCR, GFP+ DGCs were suctioned with a patch pipette from acute slices and RNA was extracted by means of the Dynabeads mRNA direct micro kit (Invitrogen). Real-time quantitative qPCR was performed with TaqMan probes (Applied Biosystems) for Trpv1 (Rn00583117) and hypoxanthine phosphoribosyltransferase; RN01527838_g1 (Hprt). Reactions were run in triplicate in a StepOnePlus real-time PCR system (Applied Biosystems). mRNA abundance was calculated by means of the comparative cycle threshold method at a threshold of 0.02. Data were analyzed using the software tool REST (http://www.REST.de.com/), on the basis of the group mean for the target transcript, versus reference Hprt transcript or Gapdh transcript.
For in vivo experiments Trpv1-shRNA was delivered by stereotaxic injection into the DG of live rats at P10 (Rodenas-Ruano et al., 2012) and all recorded neurons were assessed at P28–P35. Rats were placed in a stereotaxic frame and anesthetized with isoflurane and concentrated viral solution (2 μl) was injected into the right DG (3.4 mm posterior to bregma, 3.2 mm lateral to bregma, and 3.1 mm ventral from dura) at a flow rate of 0.2 μl/min.
Results
TRPV1 activation selectively depresses somatic inhibition onto DGCs
To evaluate the role of TRPV1 on GABAergic inputs onto DGCs, putative somatic and dendritic IPSCs were monitored from the same cell (see Materials and Methods), and TRPV1 channels were activated with the specific agonist capsaicin (CAP; 2 μm). We found that CAP bath application selectively depressed somatic, but not dendritic IPSCs in rat (Somatic: 62.6 ± 4.07% of baseline, n = 8, p < 0.001 paired t-test; Dendritic: 94.7 ± 2.7% of baseline, n = 8, p > 0.05 paired t-test; Fig. 1A). Subsequent application of the GABAARs antagonist PTX (100 μm) eliminated the CAP-insensitive component (Fig. 1A). CAP also depressed less invasive, PTX-sensitive, extracellular fIPSPs recorded in DGL (see Materials and Methods; fIPSP: 74.1 ± 2.9% of baseline, n = 8, p < 0.001 paired t-test; Fig. 1B). Pretreating slices (∼10–20 min) with two different TRPV1 antagonists, capsazepine (CPZ, 10 μm) or AMG9810 (AMG, 3 μm), eliminated CAP-mediated effects on somatic IPSCs compared with interleaved control slices (Control: 66.7 ± 3.7% of baseline, n = 6 vs CPZ: 94.5 ± 1.4% of baseline, n = 7, p < 0.001 ANOVA and AMG: 95.2 ± 1.6% of baseline, n = 9, p < 0.001 ANOVA; Fig. 1C). Moreover, this CAP-mediated effect on somatic inhibition was absent in slices from TRPV1 knock-out mice (Trpv1+/+: 56.9 ± 2.4% of baseline, 3 animals, 8 cells; Trpv1−/−: 90.1 ± 2.0% of baseline, 3 animals, 7 cells, p < 0.001 ANOVA; Fig. 1D). Finally, transient TRPV1 activation with CAP (for 10 min), chased with CPZ, induced a long-lasting suppression of somatic inhibitory transmission (74.7 ± 4.5% of baseline; n = 6, p < 0.005, paired t-test; Fig. 1E). These results indicate that in DG, TRPV1 channels can suppress GABAergic synaptic transmission in a compartment-specific and long-lasting manner.
Figure 1.
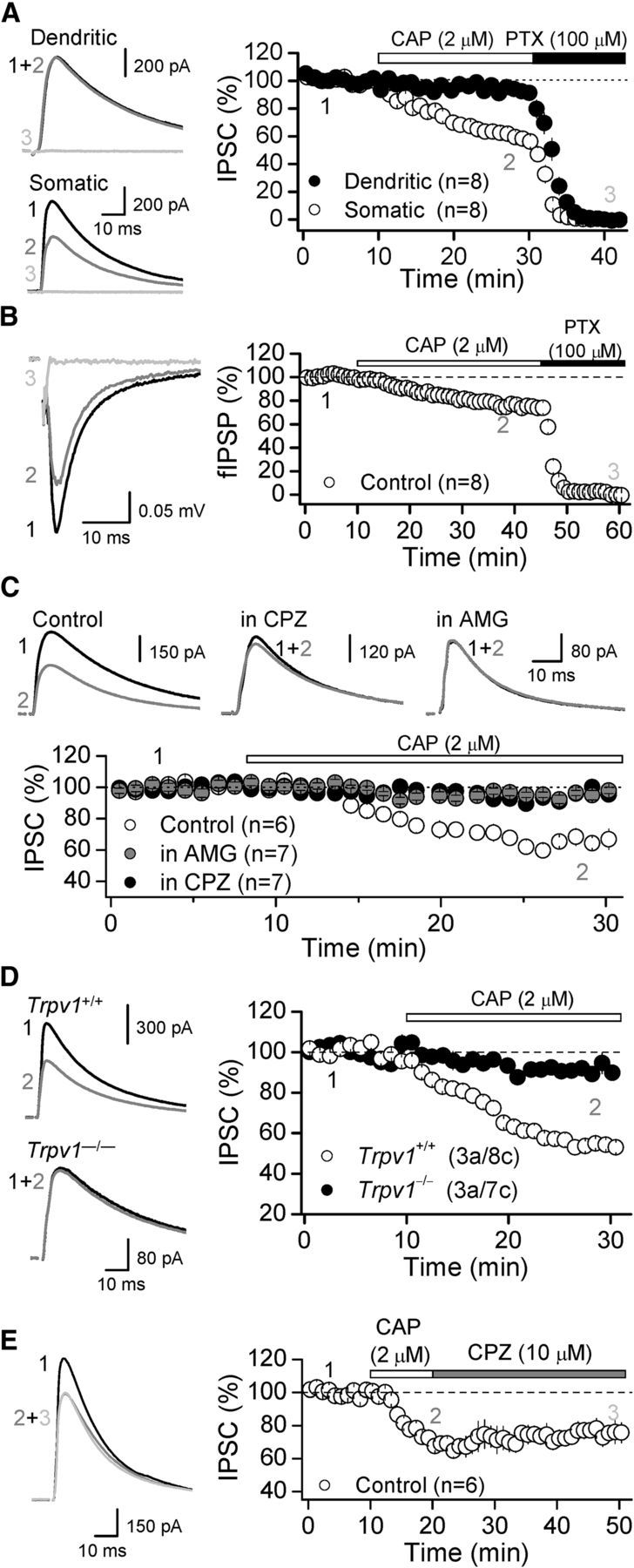
Functional expression of TRPV1 channels at inhibitory synapses in the dentate gyrus. A, Pharmacological activation of TRPV1 receptors with the agonist CAP (2 μm) depressed GABAergic IPSCs in a compartment-specific manner. Left, Representative traces of GABAergic IPSCs before (black) and after (gray) bath application of CAP at both dendritic and somatic inputs (see Materials and Methods). Right, Summary plot showing the effect of CAP on IPSCs. Subsequent application of the GABAARs antagonist PTX (100 μm) eliminated the CAP-insensitive component. B, Somatic fIPSP was also depressed by bath application of CAP and eliminated by subsequent application of PTX. C, Pretreatment with the TRPV1 antagonist CPZ (10 μm) or AMG (3 μm) eliminated CAP-mediated depression of somatic IPSCs. D, CAP suppression was observed in wild-type (Trpv1+/+), but not in TRPV1 knockdown mice (Trpv1−/−). E, Transient application of CAP (2 μm, 10 min) followed by the TRPV1 antagonist CPZ (10 μm) triggers a long-lasting suppression of inhibitory transmission in rat. Averaged sample traces taken at times indicated by numbers are shown next to each summary plot. In all parts, summary data represent the mean ± SEM and the number of animals (a) and cells (c) are in parenthesis.
TRPV1-mediated effects are CB1 receptor independent and likely postsynaptic in nature
We next investigated the mechanism by which TRPV1 activation depresses somatic GABAergic transmission. One possibility is that TRPV1 could indirectly depress somatic GABA release by mobilizing eCBs that activate presynaptic CB1Rs (Maccarrone et al., 2008). However, CB1R blockade with AM251 (4 μm) had no effect on the CAP-mediated depression of somatic IPSCs (to 60.1 ± 3.0% of baseline, n = 6, p < 0.001 paired t-test; Fig. 2A), whereas in interleaved experiments, AM251 abolished the depression of GABAergic transmission induced by the CB1R agonist WIN55, 212-2 (5 μm; WIN: 50.2 ± 4.0 of baseline, n = 8 vs WIN + AM251: 90.6 ± 3.4 of baseline, n = 4, p < 0.001 ANOVA; Fig. 2A). In addition, CAP-mediated depression was observed in both wild-type and CB1R knock-out mice (CB1R+/+: 75.5 ± 3.8% of baseline, n = 6; CB1−/−: 64.4 ± 2.1% of baseline, n = 7; Fig. 2B). Moreover, activation of TRPV1 channels with the eCB AEA (30 μm), in the presence of the fatty acid amide hydrolase (FAAH) inhibitor URB597 (1 μm) to block AEA degradation, and the CB1R antagonist AM251 (4 μm), selectively depressed somatic, but not dendritic IPSCs (Somatic: 65.9 ± 3.2% of baseline, n = 10, p < 0.001 paired t-test; Dendritic: 97.8 ± 2.5% of baseline, n = 10, p = 0.356 paired t-test; Fig. 2C). This AEA-mediated depression was eliminated by two TRPV1 antagonists AMG (100.0 ± 3.8% of baseline, n = 6, p = 0.799 paired t-test) or CPZ (95.1 ± 2.7% of baseline, n = 3, p = 0.220 paired t-test; Fig. 2C). These results strongly argue that TRPV1-mediated depression of GABAergic transmission is CB1R independent.
To determine whether TRPV1 channels could directly depress GABA release, we examined the effects of CAP on both paired-pulse ratio (PPR; Fig. 3A,B) and coefficient of variation (1/CV2; Fig. 3A), two different measures that estimate changes in probability of release. Unlike WIN-mediated depression of inhibition (Fig. 3A), AEA- and CAP-mediated depression were not accompanied by significant changes in either PPR (Fig. 3A,B) or 1/CV2 (Fig. 3A) in both rat and mouse DG. It is therefore unlikely that a reduction in GABA release underlies AEA- and CAP-mediated effects on inhibitory transmission. To directly test whether TRPV1 channels could act postsynaptically, we monitored GABAergic responses elicited by brief puffs of GABA (1 mm), a manipulation that shortcuts transmitter release. GABA-evoked responses (monitored at 0 mV) elicited by puffing in DGL (i.e., DGC soma), but not in MML (i.e., DGC dendrites), were significantly depressed by 2 μm CAP (soma: 64.07 ± 2.6% of baseline, n = 5, p < 0.001 paired t-test; dendrites: 100.47 ± 3.3% of baseline, n = 5, p = 0.890 paired t-test; Fig. 3C). At the end of each experiment, we confirmed that these responses were blocked by 100 μm PTX (Fig. 3C). These results indicate that TRPV1 channels depress inhibitory synaptic transmission postsynaptically rather than by reducing GABA release.
In vivo knockdown of TRPV1 eliminates TRPV1-mediated effects on synaptic transmission
As a complementary approach to TRPV1 pharmacology, we used two different RNA interferences targeted to different Trpv1 regions to knockdown TRPV1 in DGCs. We stereotaxically injected synthetic shRNA targeted to TRPV1 (Trpv1 shRNA-1 and Trpv1 shRNA-2) or nontargeting shRNA directly into the rat DG (Fig. 4A; see Materials and Methods). Two to three weeks post injections, we validated the efficacy of the TRPV1 knockdown by suctioning DGCs from hippocampal slices using a patch-type pipette and obtaining samples enriched with GFP+-expressing neurons. Despite the presence of GFP− cells in our sample, qRT-PCR analysis revealed that the levels of Trpv1 mRNA in DGCs were significantly reduced by both Trpv1 shRNA-1 and Trpv1 shRNA-2 relative to nontransduced control cells (Trpv1 shRNA-1: 40 ± 9.0% of control, three animals, p = 0.008, Trpv1 shRNA-2: 55.8 ± 6.5% of control, 6 animals, p = 0.001; see Materials and Methods; Fig. 4B). To assess the functional impact of knocking down TRPV1 in DGCs, we first examined excitatory synaptic inputs (i.e., MPP synapses) where we previously reported that TRPV1 activation suppresses transmission (Chávez et al., 2010). We found that CAP-mediated depression of AMPARs-EPSCs was similar between nontargeting shRNA and nontransduced neurons and thus, these neurons were pooled for further analysis (9 nontargeting vs 10 nontransduced cells; p = 0.98 ANOVA; Fig. 4C). In contrast, the CAP-mediated depression was significantly reduced in neurons expressing Trpv1 shRNA-1 (94.6 ± 3.0% of baseline, 5 animals/9 cells; Fig. 4C) or Trpv1 shRNA-2 (93.7 ± 4.9% of baseline, 4 animals/10 cells; Fig. 4C) when compared with control neurons (60.2 ± 3.2% of baseline, 9 animals/19 cells; Control vs Trpv1 shRNA-1 or Trpv2 shRNA-2, p < 0.001, ANOVA; Fig. 4C). Importantly, we also found that CAP-mediated depression of GABAergic transmission was reduced in neurons expressing Trpv1 shRNA-1 (83.4 ± 4.6% of baseline, 6 animals/13 cells) and Trpv1 shRNA-2 (88.7 ± 3.0% of baseline, 6 animals/12 cells) when compared with nontransduced control neurons recorded in interleaved experiments (control: 51.2 ± 6.8% of baseline, 5 animals/6 cells; shRNA-1 vs Control p = 0.001 ANOVA; shRNA-2 vs Control p < 0.001, ANOVA; Fig. 4D). Thus, our findings using complementary TRPV1 knockdown (Fig. 4) and selective pharmacology (Figs. 1–3) demonstrate that activation of TRPV1 channels in DGCs depresses both excitatory and inhibitory transmission by regulating postsynaptic AMPA and GABA receptors, respectively.
Mechanisms underlying TRPV1-mediated suppression of inhibition
At excitatory synapses, TRPV1 reduces transmission by promoting postsynaptic Ca2+ rise and clathrin/dynamin-dependent endocytosis of AMPARs (Chávez et al., 2010; Grueter et al., 2010). Because GABAARs can also be endocytosed in a clathrin/dynamin-dependent manner (Luscher et al., 2011), we next tested whether internalization of GABAARs might underlie TRPV1-mediated depression of somatic inhibitory transmission. First, we evaluated the requirement of postsynaptic Ca2+ rise by loading DGCs with the Ca2+ chelator BAPTA (20 mm). Under this condition, CAP failed to depress GABAergic transmission compared with interleaved control neurons (BAPTA: 98.1 ± 2.7% of baseline, n = 8, p = 0.419 paired t-test; Control: 62.1 ± 3.3% of baseline, n = 7, p < 0.001 paired t-test; BAPTA vs Control: p < 0.001 ANOVA; Fig. 5A). In addition, pretreatment of slices with 30 μm cyclopiazonic acid (CPA), a manipulation aimed to deplete intracellular Ca2+ stores, significantly reduced CAP-mediated depression of somatic IPSCs compared with interleaved control neurons (CPA: 80.1 ± 1.7%, n = 11, CPA vs Control: p < 0.001 ANOVA; Fig. 5A). In interleaved experiments, CPA did not affect basal synaptic transmission (98.7 ± 8.1% of control, n = 4, p = 0.898 paired t test; Fig. 5A, bottom). Next, we tested whether the Ca2+/calmodulin-dependent protein phosphatase calcineurin (CaN) could also participate in the TRPV1-mediated suppression of GABAergic transmission, as we previously reported at excitatory synapses in the DG (Chávez et al., 2010). Pretreatment of slices for 30 min with CaN inhibitors, FK506 (50 μm) and Cyclosporin A (CyA; 25 μm) eliminated CAP-mediated depression of GABAergic transmission compared with control slices (Control: 62.6 ± 2.5% of baseline, n = 9 vs FK506: 92.0 ± 3.5% of baseline, n = 7, p < 0.001 ANOVA; CyA: 94.0 ± 3.0% of baseline, n = 7, p < 0.001 ANOVA; Fig. 5B). Application of FK506 and CyA for 30 min did not affect basal synaptic transmission (FK506: 98.6 ± 3.7% of baseline, n = 6, p = 0.538, paired t test; CyA: to 100.5 ± 6.1% of baseline, n = 7, p = 0.998, paired t test; Fig. 5B, bottom). Finally, we investigated whether dynamin-dependent endocytosis of GABAARs could be involved in the TRPV1-mediated depression of inhibitory transmission. To test this possibility, we loaded DGCs with the dynamin inhibitory peptide (DIP; 50 μm), and found that this peptide significantly reduced CAP-mediated depression of inhibition compared with a scrambled DIP (DIP: 92.7 ± 4.9% of baseline, n = 10 vs scrambled DIP: 59.3 ± 6.5% of baseline, n = 7, p < 0.001, ANOVA; Fig. 5D). Thus, our results strongly suggest that activation of TRPV1 channels leads to an increase in postsynaptic Ca2+ rise which, by activating the CaN-dynamin complex, likely promotes GABAARs endocytosis.
Discussion
The results presented in this study demonstrate that TRPV1 channels, in addition to modulating excitatory synaptic inputs (Chávez et al., 2010; Grueter et al., 2010; Puente et al., 2011), can also target inhibitory synapses in rat and mouse DG. We report that activation of TRPV1 channels, presumably by promoting internalization of GABAARs, selectively depresses somatic but not dendritic GABAergic synaptic inputs onto DGCs. We also show that the eCB AEA depresses inhibitory synaptic transmission by targeting TRPV1 channels, and this effect can occur while CB1Rs are pharmacologically blocked. To our knowledge, we provide the first evidence that TRPV1 channels can directly regulate inhibitory transmission at central synapses.
In the CNS, there is good evidence that activation of TRPV1 channels regulates excitatory transmission either presynaptically by modulating glutamate release (for review, see Kauer and Gibson, 2009), or postsynaptically by promoting AMPAR removal from the synapse (Chávez et al., 2010; Grueter et al., 2010; Kim et al., 2012). In contrast, previous attempts have not identified any effect of TRPV1 activation on synaptic inhibition (Yang et al., 1998; Marinelli et al., 2003; D.P. Li et al., 2004; Xing and Li, 2007). Our finding that TRPV1 channels suppress somatic but not dendritic inhibitory inputs (Figs. 1, 2) is reminiscent of previous observations showing that the TRPV1-mediated depression of excitatory transmission occurs at some synaptic inputs but not others (Chávez et al., 2010; Grueter et al., 2010; Puente et al., 2011). Thus, input specificity could be a general property of TRPV1-mediated effects throughout the brain.
The precise mechanism underlying input specificity of TRPV1-mediated suppression of synaptic transmission is unclear. For example, it could reflect the selective expression of TRPV1 channels at some synapses, although spatially constrained signaling downstream of TRPV1 channels may also contribute. In support of TRPV1 synaptic segregation, a recent anatomical immunoelectron microscopy study in DG showed that TRPV1 is highly concentrated in postsynaptic dendritic spines receiving perforant path inputs, whereas expression is weak at more proximal excitatory hilar mossy cell synapses (Puente et al., 2014). This subcellular anatomical distribution is consistent with the selective TRPV1-mediated suppression of excitatory transmission at medial perforant path but not hilar mossy cell synapses (Chávez et al., 2010). GABAergic interneurons in the DG comprise a diverse group of cell types (Houser, 2007) with domain-specific innervation and distinct functional properties (Z.S. Han et al., 1993; Hefft and Jonas, 2005; Liu et al., 2014), raising the possibility that TRPV1 channels also could be expressed at subtypes of inhibitory synapses. Regardless of the precise synapse type, by selectively suppressing somatic inhibition, TRPV1 channels may control DGC spike initiation (Miles et al., 1996). The fact that TRPV1 channels modulate both excitatory (Chávez et al., 2010) and inhibitory transmission in a compartment-specific manner indicates that TRPV1 might play a subtle role in regulating synaptic integration and DGC output. While exogenous activation of TRPV1 channels is expected to suppress both excitatory and inhibitory inputs onto DGCs, it remains to be seen whether both actions can occur simultaneously by endogenous TRPV1 activation.
TRPV1 can be activated by lipid messengers such as the eCB AEA (Ross, 2003). Typically, eCBs are mobilized from the postsynaptic compartment and act retrogradely to suppress transmitter release by activating presynaptic CB1Rs (Kano et al., 2009; Castillo et al., 2012), and in most synapses, this form of retrograde signaling is likely mediated by the eCB 2-AG (Ohno-Shosaku and Kano, 2014). Growing evidence, however, indicates that eCBs can also regulate synaptic function in an unconventional manner by activating postsynaptic TRPV1 channels (Castillo et al., 2012). Our findings that AEA regulates GABAergic transmission in a postsynaptic and CB1R-independent manner provide further support for this way of regulating synaptic function via eCBs. Previous work in the DG showed that retrograde eCB signaling suppresses GABA release (Isokawa and Alger, 2005), suggesting eCB-mediated modulation of synaptic inhibition is complex. In addition, AEA acting on TRPV1 channels could facilitate 2-AG production and by this means suppress synaptic inhibition via CB1Rs, as previously reported in dorsal striatum (Maccarrone et al., 2008). However, we show that AEA in the DG suppresses GABAergic transmission in a CB1R-independent manner by activating TRPV1 channels. It has recently been reported that specific patterns of activity mobilizes AEA or 2-AG from the same neuron and, by activating postsynaptic TRPV1 channels or presynaptic CB1Rs, respectively, these eCBs trigger mechanistically different forms of long-term depression (LTD) at excitatory synapses (Grueter et al., 2010; Puente et al., 2011). Whether endogenous activation of TRPV1 and CB1Rs can trigger similar forms of long-term plasticity at inhibitory synapses remains to be determined.
Recent studies have shown that activation of TRPV1 channels promotes AMPAR internalization at excitatory synapses in the DG (Chávez et al., 2010), nucleus accumbens (Grueter et al., 2010), and spinal cord (Kim et al., 2012). In the DG, we have reported that TRPV1-mediated endocytosis of AMPARs occurs in a CaN/dynamin-dependent manner (Chávez et al., 2010), and our new findings indicate that this mechanism also operates for GABAARs (Fig. 5). Notably, CaN has been involved in TRPV1-mediated downregulation of voltage-gated Ca2+ channels in primary sensory neurons (Wu et al., 2005) and TRPV1-induced LTD in hippocampal interneurons (Jensen and Edwards, 2012). Together, these observations substantiate the notion that CaN is a common downstream target of TRPV1-mediated effects.
In summary, our data provide further evidence in support of functional TRPV1 channels as regulators of synaptic transmission in the CNS. Such regulation, at least in DG, is input specific, includes excitatory and inhibitory synapses, and could play a role in controlling the relative weight of excitatory and inhibitory inputs. Although our understanding of brain TRPV1 in vivo is currently rather limited, studies using TRPV1 knock-out mice suggest that this channel might be involved in drug addiction (Grueter et al., 2010), anxiety, and fear condition (Marsch et al., 2007). In addition, TRPV1 protein levels are reportedly increased in the DG of an animal model of epilepsy (Bhaskaran and Smith, 2010), suggesting that TRPV1 could be regulated by aberrant neural activity. Brain TRPV1-mediated actions deserve attention given that TRPV1 antagonism has emerged as an alternative strategy for pain management. Additional work is necessary to fully understand under what conditions TRPV1 modulates excitatory and inhibitory inputs and ultimately, to determine how exactly modulation of these inputs affects brain function.
Footnotes
This work was supported by National Institutes of Health grants to P.E.C. (DA017392 and MH081935) and C.S.C. (NS069777 and NS069777-S1). A.E.C. was partially supported by a Ruth L. Kirschstein Award (F32 NS071821) and a National Alliance for Research on Schizophrenia and Depression Young Investigator Grant from the Brain & Behavior Research Foundation. We thank Thomas Younts for critical reading of this manuscript.
The authors declare no competing financial interests.
References
- Aguiar DC, Terzian AL, Guimarães FS, Moreira FA. Anxiolytic-like effects induced by blockade of transient receptor potential vanilloid type 1 (TRPV1) channels in the medial prefrontal cortex of rats. Psychopharmacology. 2009;205:217–225. doi: 10.1007/s00213-009-1532-5. [DOI] [PubMed] [Google Scholar]
- Bennion D, Jensen T, Walther C, Hamblin J, Wallmann A, Couch J, Blickenstaff J, Castle M, Dean L, Beckstead S, Merrill C, Muir C, St Pierre T, Williams B, Daniel S, Edwards JG. Transient receptor potential vanilloid 1 agonists modulate hippocampal CA1 LTP via the GABAergic system. Neuropharmacology. 2011;61:730–738. doi: 10.1016/j.neuropharm.2011.05.018. [DOI] [PubMed] [Google Scholar]
- Bhaskaran MD, Smith BN. Effects of TRPV1 activation on synaptic excitation in the dentate gyrus of a mouse model of temporal lobe epilepsy. Exp Neurol. 2010;223:529–536. doi: 10.1016/j.expneurol.2010.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillo PE, Younts TJ, Chávez AE, Hashimotodani Y. Endocannabinoid signaling and synaptic function. Neuron. 2012;76:70–81. doi: 10.1016/j.neuron.2012.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caterina MJ, Julius D. The vanilloid receptor: a molecular gateway to the pain pathway. Annu Rev Neurosci. 2001;24:487–517. doi: 10.1146/annurev.neuro.24.1.487. [DOI] [PubMed] [Google Scholar]
- Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D. The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature. 1997;389:816–824. doi: 10.1038/39807. [DOI] [PubMed] [Google Scholar]
- Cavanaugh DJ, Chesler AT, Jackson AC, Sigal YM, Yamanaka H, Grant R, O'Donnell D, Nicoll RA, Shah NM, Julius D, Basbaum AI. Trpv1 reporter mice reveal highly restricted brain distribution and functional expression in arteriolar smooth muscle cells. J Neurosci. 2011;31:5067–5077. doi: 10.1523/JNEUROSCI.6451-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chávez AE, Chiu CQ, Castillo PE. TRPV1 activation by endogenous anandamide triggers postsynaptic long-term depression in dentate gyrus. Nat Neurosci. 2010;13:1511–1518. doi: 10.1038/nn.2684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H. Intron splicing-mediated expression of AAV Rep and Cap genes and production of AAV vectors in insect cells. Mol Ther. 2008;16:924–930. doi: 10.1038/mt.2008.35. [DOI] [PubMed] [Google Scholar]
- Cristino L, de Petrocellis L, Pryce G, Baker D, Guglielmotti V, Di Marzo V. Immunohistochemical localization of cannabinoid type 1 and vanilloid transient receptor potential vanilloid type 1 receptors in the mouse brain. Neuroscience. 2006;139:1405–1415. doi: 10.1016/j.neuroscience.2006.02.074. [DOI] [PubMed] [Google Scholar]
- Derbenev AV, Monroe MJ, Glatzer NR, Smith BN. Vanilloid-mediated heterosynaptic facilitation of inhibitory synaptic input to neurons of the rat dorsal motor nucleus of the vagus. J Neurosci. 2006;26:9666–9672. doi: 10.1523/JNEUROSCI.1591-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrini F, Salio C, Vergnano AM, Merighi A. Vanilloid receptor-1 (TRPV1)-dependent activation of inhibitory neurotransmission in spinal substantia gelatinosa neurons of mouse. Pain. 2007;129:195–209. doi: 10.1016/j.pain.2007.01.009. [DOI] [PubMed] [Google Scholar]
- Gibson HE, Edwards JG, Page RS, Van Hook MJ, Kauer JA. TRPV1 channels mediate long-term depression at synapses on hippocampal interneurons. Neuron. 2008;57:746–759. doi: 10.1016/j.neuron.2007.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grueter BA, Brasnjo G, Malenka RC. Postsynaptic TRPV1 triggers cell type-specific long-term depression in the nucleus accumbens. Nat Neurosci. 2010;13:1519–1525. doi: 10.1038/nn.2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han P, Korepanova AV, Vos MH, Moreland RB, Chiu ML, Faltynek CR. Quantification of TRPV1 protein levels in rat tissues to understand its physiological roles. J Mol Neurosci. 2013;50:23–32. doi: 10.1007/s12031-012-9849-7. [DOI] [PubMed] [Google Scholar]
- Han ZS, Buhl EH, Lörinczi Z, Somogyi P. A high degree of spatial selectivity in the axonal and dendritic domains of physiologically identified local-circuit neurons in the dentate gyrus of the rat hippocampus. Eur J Neurosci. 1993;5:395–410. doi: 10.1111/j.1460-9568.1993.tb00507.x. [DOI] [PubMed] [Google Scholar]
- Hefft S, Jonas P. Asynchronous GABA release generates long-lasting inhibition at a hippocampal interneuron-principal neuron synapse. Nat Neurosci. 2005;8:1319–1328. doi: 10.1038/nn1542. [DOI] [PubMed] [Google Scholar]
- Houser CR. Interneurons of the dentate gyrus: an overview of cell types, terminal fields and neurochemical identity. Prog Brain Res. 2007;163:217–232. doi: 10.1016/S0079-6123(07)63013-1. [DOI] [PubMed] [Google Scholar]
- Huang SM, Bisogno T, Trevisani M, Al-Hayani A, De Petrocellis L, Fezza F, Tognetto M, Petros TJ, Krey JF, Chu CJ, Miller JD, Davies SN, Geppetti P, Walker JM, Di Marzo V. An endogenous capsaicin-like substance with high potency at recombinant and native vanilloid VR1 receptors. Proc Natl Acad Sci U S A. 2002;99:8400–8405. doi: 10.1073/pnas.122196999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang WX, Min JW, Liu YQ, He XH, Peng BW. Expression of TRPV1 in the C57BL/6 mice brain hippocampus and cortex during development. Neuroreport. 2014;25:379–385. doi: 10.1097/WNR.0000000000000105. [DOI] [PubMed] [Google Scholar]
- Isokawa M, Alger BE. Retrograde endocannabinoid regulation of GABAergic inhibition in the rat dentate gyrus granule cell. J Physiol. 2005;567:1001–1010. doi: 10.1113/jphysiol.2005.094219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen T, Edwards JG. Calcineurin is required for TRPV1-induced long-term depression of hippocampal interneurons. Neurosci Lett. 2012;510:82–87. doi: 10.1016/j.neulet.2012.01.006. [DOI] [PubMed] [Google Scholar]
- Kano M, Ohno-Shosaku T, Hashimotodani Y, Uchigashima M, Watanabe M. Endocannabinoid-mediated control of synaptic transmission. Physiol Rev. 2009;89:309–380. doi: 10.1152/physrev.00019.2008. [DOI] [PubMed] [Google Scholar]
- Kauer JA, Gibson HE. Hot flash: TRPV channels in the brain. Trends Neurosci. 2009;32:215–224. doi: 10.1016/j.tins.2008.12.006. [DOI] [PubMed] [Google Scholar]
- Kim YH, Back SK, Davies AJ, Jeong H, Jo HJ, Chung G, Na HS, Bae YC, Kim SJ, Kim JS, Jung SJ, Oh SB. TRPV1 in GABAergic interneurons mediates neuropathic mechanical allodynia and disinhibition of the nociceptive circuitry in the spinal cord. Neuron. 2012;74:640–647. doi: 10.1016/j.neuron.2012.02.039. [DOI] [PubMed] [Google Scholar]
- Köles L, Garção P, Zádori ZS, Ferreira SG, Pinheiro BS, Da Silva-Santos CS, Ledent C, Köfalvi A. presyntaptic TRPV1 vanilloid receptor function is age- but not CB1 cannabinoid receptor-dependent in the rodent forebrain. Brain Res Bull. 2013;97:126–135. doi: 10.1016/j.brainresbull.2013.06.007. [DOI] [PubMed] [Google Scholar]
- Li DP, Chen SR, Pan HL. VR1 receptor activation induces glutamate release and postsynaptic firing in the paraventricular nucleus. J Neurophysiol. 2004;92:1807–1816. doi: 10.1152/jn.00171.2004. [DOI] [PubMed] [Google Scholar]
- Li HB, Mao RR, Zhang JC, Yang Y, Cao J, Xu L. Antistress effect of TRPV1 channel on synaptic plasticity and spatial memory. Biol Psychiatry. 2008;64:286–292. doi: 10.1016/j.biopsych.2008.02.020. [DOI] [PubMed] [Google Scholar]
- Liao HT, Lee HJ, Ho YC, Chiou LC. Capsaicin in the periaqueductal gray induces analgesia via metabotropic glutamate receptor-mediated endocannabinoid retrograde disinhibition. Br J Pharmacol. 2011;163:330–345. doi: 10.1111/j.1476-5381.2011.01214.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YC, Cheng JK, Lien CC. Rapid dynamic changes of dendritic inhibition in the dentate gyrus by presynaptic activity patterns. J Neurosci. 2014;34:1344–1357. doi: 10.1523/JNEUROSCI.2566-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luscher B, Fuchs T, Kilpatrick CL. GABAA receptor trafficking-mediated plasticity of inhibitory synapses. Neuron. 2011;70:385–409. doi: 10.1016/j.neuron.2011.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maccarrone M, Rossi S, Bari M, De Chiara V, Fezza F, Musella A, Gasperi V, Prosperetti C, Bernardi G, Finazzi-Agrò A, Cravatt BF, Centonze D. Anandamide inhibits metabolism and physiological actions of 2-arachidonoylglycerol in the striatum. Nat Neurosci. 2008;11:152–159. doi: 10.1038/nn2042. [DOI] [PubMed] [Google Scholar]
- Maione S, Cristino L, Migliozzi AL, Georgiou AL, Starowicz K, Salt TE, Di Marzo V. TRPV1 channels control synaptic plasticity in the developing superior colliculus. J Physiol. 2009;587:2521–2535. doi: 10.1113/jphysiol.2009.171900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinelli S, Vaughan CW, Christie MJ, Connor M. Capsaicin activation of glutamatergic synaptic transmission in the rat locus coeruleus in vitro. J Physiol. 2002;543:531–540. doi: 10.1113/jphysiol.2002.022863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinelli S, Di Marzo V, Berretta N, Matias I, Maccarrone M, Bernardi G, Mercuri NB. Presynaptic facilitation of glutamatergic synapses to dopaminergic neurons of the rat substantia nigra by endogenous stimulation of vanilloid receptors. J Neurosci. 2003;23:3136–3144. doi: 10.1523/JNEUROSCI.23-08-03136.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsch R, Foeller E, Rammes G, Bunck M, Kössl M, Holsboer F, Zieglgänsberger W, Landgraf R, Lutz B, Wotjak CT. Reduced anxiety, conditioned fear, and hippocampal long-term potentiation in transient receptor potential vanilloid type 1 receptor-deficient mice. J Neurosci. 2007;27:832–839. doi: 10.1523/JNEUROSCI.3303-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mezey E, Tóth ZE, Cortright DN, Arzubi MK, Krause JE, Elde R, Guo A, Blumberg PM, Szallasi A. Distribution of mRNA for vanilloid receptor subtype 1 (VR1), and VR1-like immunoreactivity, in the central nervous system of the rat and human. Proc Natl Acad Sci U S A. 2000;97:3655–3660. doi: 10.1073/pnas.97.7.3655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miles R, Tóth K, Gulyás AI, Hájos N, Freund TF. Differences between somatic and dendritic inhibition in the hippocampus. Neuron. 1996;16:815–823. doi: 10.1016/S0896-6273(00)80101-4. [DOI] [PubMed] [Google Scholar]
- Musella A, De Chiara V, Rossi S, Prosperetti C, Bernardi G, Maccarrone M, Centonze D. TRPV1 channels facilitate glutamate transmission in the striatum. Mol Cell Neurosci. 2009;40:89–97. doi: 10.1016/j.mcn.2008.09.001. [DOI] [PubMed] [Google Scholar]
- Musella A, De Chiara V, Rossi S, Cavasinni F, Castelli M, Cantarella C, Mataluni G, Bernardi G, Centonze D. Transient receptor potential vanilloid 1 channels control acetylcholine/2-arachidonoylglicerol coupling in the striatum. Neuroscience. 2010;167:864–871. doi: 10.1016/j.neuroscience.2010.02.058. [DOI] [PubMed] [Google Scholar]
- Ohno-Shosaku T, Kano M. Endocannabinoid-mediated retrograde modulation of synaptic transmission. Curr Opin Neurobiol. 2014;29C:1–8. doi: 10.1016/j.conb.2014.03.017. [DOI] [PubMed] [Google Scholar]
- Puente N, Cui Y, Lassalle O, Lafourcade M, Georges F, Venance L, Grandes P, Manzoni OJ. Polymodal activation of the endocannabinoid system in the extended amygdala. Nat Neurosci. 2011;14:1542–1547. doi: 10.1038/nn.2974. [DOI] [PubMed] [Google Scholar]
- Puente N, Reguero L, Elezgarai I, Canduela MJ, Mendizabal-Zubiaga J, Ramos-Uriarte A, Fernandez-Espejo E, Grandes P. The transient receptor potential vanilloid-1 is localized at excitatory synapses in the mouse dentate gyrus. Brain Struct Funct. 2014 doi: 10.1007/s00429-014-0711-2. doi: 10.1007/s00429-014-0711-2. Advance online publication. [DOI] [PubMed] [Google Scholar]
- Roberts JC, Davis JB, Benham CD. [3H]Resiniferatoxin autoradiography in the CNS of wild-type and TRPV1 null mice defines TRPV1 (VR-1) protein distribution. Brain Res. 2004;995:176–183. doi: 10.1016/j.brainres.2003.10.001. [DOI] [PubMed] [Google Scholar]
- Rodenas-Ruano A, Chávez AE, Cossio MJ, Castillo PE, Zukin RS. REST-dependent epigenetic remodeling promotes the developmental switch in synaptic NMDA receptors. Nat Neurosci. 2012;15:1382–1390. doi: 10.1038/nn.3214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross RA. Anandamide and vanilloid TRPV1 receptors. Br J Pharmacol. 2003;140:790–801. doi: 10.1038/sj.bjp.0705467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasamura T, Sasaki M, Tohda C, Kuraishi Y. Existence of capsaicin-sensitive glutamatergic terminals in rat hypothalamus. Neuroreport. 1998;9:2045–2048. doi: 10.1097/00001756-199806220-00025. [DOI] [PubMed] [Google Scholar]
- Terzian AL, dos Reis DG, Guimarães FS, Corrêa FM, Resstel LB. Medial prefrontal cortex transient receptor potential vanilloid type 1 (TRPV1) in the expression of contextual fear conditioning in Wistar rats. Psychopharmacology. 2014;231:149–157. doi: 10.1007/s00213-013-3211-9. [DOI] [PubMed] [Google Scholar]
- Tóth A, Boczán J, Kedei N, Lizanecz E, Bagi Z, Papp Z, Edes I, Csiba L, Blumberg PM. Expression and distribution of vanilloid receptor 1 (TRPV1) in the adult rat brain. Brain Res Mol Brain Res. 2005;135:162–168. doi: 10.1016/j.molbrainres.2004.12.003. [DOI] [PubMed] [Google Scholar]
- Ventura A, Meissner A, Dillon CP, McManus M, Sharp PA, Van Parijs L, Jaenisch R, Jacks T. Cre-lox-regulated conditional RNA interference from transgenes. Proc Natl Acad Sci U S A. 2004;101:10380–10385. doi: 10.1073/pnas.0403954101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu ZZ, Chen SR, Pan HL. Transient receptor potential vanilloid type 1 activation down-regulates voltage-gated calcium channels through calcium-dependent calcineurin in sensory neurons. J Biol Chem. 2005;280:18142–18151. doi: 10.1074/jbc.M501229200. [DOI] [PubMed] [Google Scholar]
- Xing J, Li J. TRPV1 receptor mediates glutamatergic synaptic input to dorsolateral periaqueductal gray (dl-PAG) neurons. J Neurophysiol. 2007;97:503–511. doi: 10.1152/jn.01023.2006. [DOI] [PubMed] [Google Scholar]
- Yang K, Kumamoto E, Furue H, Yoshimura M. Capsaicin facilitates excitatory but not inhibitory synaptic transmission in substantia gelatinosa of the rat spinal cord. Neurosci Lett. 1998;255:135–138. doi: 10.1016/S0304-3940(98)00730-7. [DOI] [PubMed] [Google Scholar]
- Zhou HY, Zhang HM, Chen SR, Pan HL. Increased nociceptive input rapidly modulates spinal GABAergic transmission through endogenously released glutamate. J Neurophysiol. 2007;97:871–882. doi: 10.1152/jn.00964.2006. [DOI] [PubMed] [Google Scholar]