

Abstract
The Mur E enzyme of Mur pathway of Mycobacterium tuberculosis is an attractive drug target as it is unique to bacteria and is absent in mammalian cells. The virtual screening of large libraries of drug like molecules against a protein target is a common strategy used to identify novel inhibitors. However, the method has a large number of pitfalls, with large variations in accuracy caused in part by inaccurate protocols, use of improper standards and libraries, and system dependencies such as the potential for nonspecific docking from large active-site cavities. The screening of drug-like small molecules from diversity sets can, however, be used to short-list potential fragments as building blocks to generate leads with improved specificity. We describe a protocol to implement this strategy, which involves an analysis of the active site and known inhibitors to identify orthospecific determinants, virtual screening of a drug-like diversity library to identify potential drug primitives, and inspection of the potential docked fragments for both binding potential and toxicity. The protocol is implemented on the M.tb Mur E protein which has a large active site with poor enrichment of known positives and a set of drug-like molecules that meets this criteria is presented for further analysis.
Abbreviations
MTB - Mycobacterium tuberculosis, NCI - National Cancer Institute, PDB - Protein Databank.
Keywords: PDB ID: 2XJA, Mycobacterium tuberculosis, Virtual Screening, Molecular Docking, Mur E inhibitors
Background
Tuberculosis (TB) persists as a major global health issue. An estimated 8.6 million people developed TB and 1.3 million deaths were observed in 2012 which included 320,000 deaths among HIV positive people. About 25 percent of TB cases and deaths occur among men but TB is one among the top three killer of women worldwide. Majority of cases were observed in South-East Asia 29%, African and Western Pacific 27% and 19% regions respectively. India alone accounted for 26 % of the total cases and 12% of the cases were observed in China [1].
Tuberculosis is primarily an airborne disease. Infection of Mycobacterium tuberculosis is established as this bacterium overcomes challenges brought forward by host immune system. Mycobacteria invade and persist silently within host macrophages for many years establishing a chronic infection upon failure of host defense mechanisms.
The duration and administration of the drug regimen for tuberculosis is long and complicated requiring directly observed therapy (DOT) through a health professional. The recent appearance of drug resistant strains threatens to make the present treatment obsolete. Despite the occurrence of drug resistance and the inadequacy of present drug regimen, no new drug has been developed in the past 50 years. Infection with resistant strains of M.tb decreases the probability of cure along with increase in the cost of treatment [2].
Novel drug targets of MTB should be explored to kill drug sensitive as well as drug resistant bacteria. A good drug target is a protein unique to the pathogen, and critical for its survival within the host. One such pathway containing multiple enzymes as potential targets is peptidoglycan biosynthesis, widely conserved in bacteria and involves two stages [3, 4, 5]. The formation of UDP-N-acetylmuramoyl pentapeptide, the monomeric building block is the first stage which occurs in cytoplasm and is catalyzed by the Mur enzymes. Transfer of an enolpyruvate residue from phosphoenolpyruvate (PEP) to UDP N-acetylglucosamine is catalysed by MurA and is followed by reduction of enolpyruvate to D -lactate by catalysis through MurB enzyme yielding UDP N-acetylmuramate and is the first committed step of this pathway. Formation of UDP Nacetylmuramyl pentapeptide is a result of stepwise addition of the pentapeptide side-chain on the newly reduced D -lactyl group by ATP-dependent amino acid ligases (MurC, MurD, MurE and MurF). Mur C adds L-Alanine to the UDP-Mur-NAc followed by addition of D-Glutamine by Mur D enzyme. LLysine is added to this precursor UDP-Mur-NAc-L-Ala-D-Glu by the action of Mur E enzyme. The product of this reaction is UDP-Mur-NAc-Tripeptide which later is used by Mur F enzyme of ligase family that adds D-Ala-D-Ala which completes the synthesis of UDP-Mur-NAc-Pentapeptide. These ligase reactions are essentially ATP dependent. Of these protein, only the crystal structures of the M.tb Mur E are available, making it an appropriate target for prediction of potential inhibitors.
Structure based virtual screening is typically employed to dock a large library of small molecules against a known protein target to score their potential as orthosteric or allosteric inhibitors. Pitfalls in its implementation arise from unreal assumptions and expectations - where scoring is typically, and wrongly, assumed to be highly correlated with potency; data design and content - where the lack of knowledge of positive and negative controls, and improper filters in the library creation can lead to erroneous interpretations; and errors that arise from conformational sampling and the software used [6]. In addition, the typical use case requires commercial proprietary software and high-performance computing as the large size of libraries makes the problem computationally intractable.
We revise the aim of virtual screening to provide as many diverse starting points for the hit-to-lead, and lead optimization phases of drug discovery, and not as the endpoint of the selection of highly potent molecules. The protocol is designed to minimize the pitfalls normally associated with virtual screening, and use commonly and freely available docking and diversity libraries which can be performed on normal computational equipment.
The Mur E protein is monomeric in its stoichiometry and is present as a unit of four monomer in biological form with four chains A, B, C, D. All of the chains being identical monomeric units. Each chain/monomeric unit has a natural ligand (Uridine-5'-Diphosphate-N-Acetylmuramoyl-L-Alanine-DGlutamate) bound to the protein along with an ADP molecule and two magnesium ions present as cofactor with presence of a single water molecule. The structure of Mur E PDB ID 2XJA [7] is used in present docking study. The Chain A of protein was selected for docking. The bound ligand was removed from active site of the receptor retaining ADP and the two magnesium ions. Critical amino acids with which the natural substrate is interacting are identified using residue information from PDBsum [8] and this site is selected for virtual screening.
Methodology
Identification of Positive Control:
Positive control dataset consist of molecules identified for their inhibitory effect against Mur E enzyme from a survey of the literature. Phosphinate inhibitor of the meso-diaminopimelic acid-adding enzyme [9], compound SID 103691194 [10], compound 6a [11], 3-methoxynordomesticine [12] and five Nmethyl- 2-alkenyl-4-quinolones [13] are present in positive control library. Structure of these compounds were sketched using Avogadro Software [14] before being converted to 3D SDF format using Open Babel [15].
Generation of Decoys:
DUD Gen Program of DUD-E [16] was used for generation of decoys. This program generates 50 decoys for every active provided, with input and output being in SMILES format. The decoys generated were converted into sdf format using Open Babel [15].
Standard Diversity Set:
For the benefit of the general research community with the goal of identifying novel chemical leads and biological mechanisms a compound screening program is operated by the Developmental Therapeutics Program (DTP) ( http://dtp.nci.nih.gov/docs/misc/common_files/submit_compounds.html). NCI diversity set IV [17] is a dataset containing 1596 molecule which are derived from almost 140,000 compounds available for distribution from the DTP repository and was used as a library for docking against the protein 2XJA.
Molecule Pre-Processing:
Preprocessing of ligand molecules involved conversion of dimension from 2D to 3D and conversion of file format to .pdb using Open Babel software [15]. To bring the receptor and the ligands in the same coordinate space an in-house written python and perl script for receptor and ligand, respectively was used for transformation. The center of active site is used for transformation of the coordinate of the receptor. The docking site was centered at the following residues of the protein (Ser222, Asp247, His248, Thr195, Thr196, Arg230, Glu198, Leu67, Ser84, Ala69,Gln70, Thr86, Thr85). The geometric center of the ligand is used to transform the molecule to the same coordinate space.
Molecular Docking:
Docking study was performed using AutoDock Vina [18] Preparation of required input files for AutoDock Vina are prepared using AutoDock program. Preparation of files through AutoDock involved addition of polar hydrogen atoms and gasteiger charges. The size of grid box in AutoDock Vina was kept as 25,25,25 for X,Y,Z. The energy range was kept as 4 which is default setting. Vina is implemented through shell script provided by AutoDock Vina developers. The binding affinity of ligand is observed in as a negative score with Kcal/mol as unit. AutoDock Vina script generates nine poses of ligand with distinguished binding energy for each ligand input. The pose of ligand with best binding affinity is extracted from the docked complex using in-house perl script. Docking through Vina [18] is done in similar fashion for the three ligand dataset i.e., active, inactives and NCI diversity set IV [17].
Results & Discussion
Identification of residues involved in substrate specificity and binding:
Amino acids which are involved in interaction with the natural substrate were identified using residue information from PDBsum and this site is selected for virtual screening. X,Y,Z coordinates are derived by averaging the coordinates of the residues derived from PDBsum [8].
The active site of MurE is a large cavity, and from a structural point of view can be divided into three sections. Two sections impart specificity to the natural ligand, which maybe divided into the UDP moiety - consisting of the uracil and pyrophosphate rings, and the linear di-peptide L-Ala-D-Glu. The interactions between the uracil ring of UDP moiety of UAG is through residues in the loop connecting β2 and β3 while the pyrophosphate moiety makes hydrogen bonds with the main chain of the loop connecting β3 and α3, all three secondary structures being part of the same domain. The O6׳ of Nacetylmuramic acid ring forms a hydrogen bond with carboxylate of Glu198 a conserved residue in the MurE family, present in a second domain. For the linear di-peptide, O19 of the carbonyl group of the peptide bond of UAG interacts with NH1 and NH2 of Arg 230 that is again highly conserved. The α- carboxylate of d-Glu forms a hydrogen bond with main chain nitrogen of Thr195 and the side chain of Ser222. The other carboxylate is coordinated with a Magnesium ion, involved with the catalytic site with the binding of ATP. The third section of the active site contains other conserved residues from the MurE family associated with ATP binding - which is a generic function present in a number of catalytic reactions. Potential inhibitors of this section of the cavity would have a problem of cross-reactivity with ATP binding, and have a high chance of being toxic. In order to exclude this section of the active-site cavity from the screen, the active site is prepared with the ADP and Mg2+ ligands retrieved from the crystal structure. The residues responsible for substrate specificity identified from the active site are Ser222, Asp247, His248, Thr195, Thr196, Arg 230, Glu198, Leu67, Ser84, Ala69, Gln70, Thr8 and Thr85 (Figure 1b).
Figure 1.
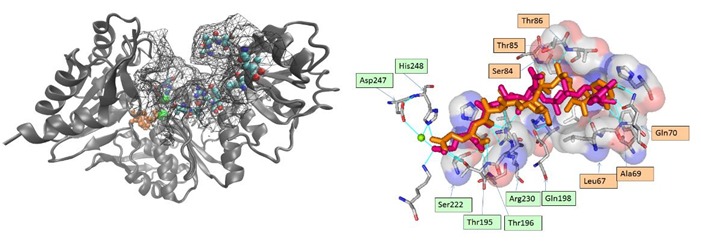
A) The M. tb Mur E enzyme (2xja) with the active site grid used for docking visible as a mesh. Critical residues of importance for natural ligand are shown as spheres. The associated ADP and magnesium in orange and green color respectively; B) Critical residues responsible for binding can be defined from the X-RAY and docked ligand: Ser222, Asp247, His248, Thr195, Thr196, Arg 230, Glu198, Leu67, Ser84, Ala69,Gln70, Thr8 and Thr85 residues interact with the natural substrate Uridine-5'- Diphosphate-N-Acetylmuramoyl-L-Alanine-D-Glutamate. LIGPLOT superimposition shows overlap at eighteen places which supports the docking algorithm being followed. The post docking ligand showed proximity with 10 out of the 13 earlier identified specificity determining residues that interact with the natural substrate. The superimposed pre and post docked conformations of the of natural substrate shows that the linear tail corresponding to the di-peptide (C21 to C28) is correctly docked in the groove of the active site (average RMSD of C21-C28 = 0.9671), while the UDP moiety has a higher RMSD of 3.753. The orange and green labels correspond to section I & II, respectively.
Virtual screening with Mur E does not have high specificity:
The enzyme MurE does have a range of known inhibitors, though not all have been tested against M.tb. Enrichment curves from virtual screening of inhibitors and their decoys can evaluate the discriminating ability of the docking program as they rank the known positives relative to decoys. The enrichment curve of the known inhibitors against MurE show that docking with AutoDock Vina cannot separate six of the seven known ligands from their respective decoys (Figure 2a).
Figure 2.
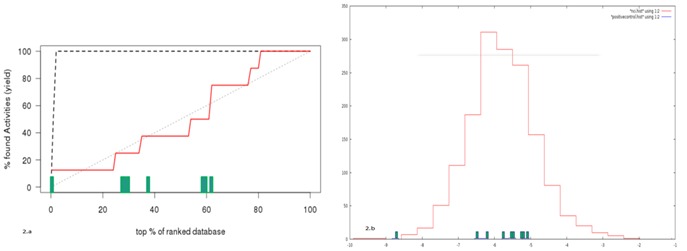
A) Rank plot of known positives after docking. SID 103691194 is separated adequately from the decoys; B) Prediction of potential inhibitors from libraries: A histogram plot of the docking energies of the NCI diversity set. The energies of known positives are shown as bars on the X-axis.
However, the large inhibitors Phosphoinate, SID 103691194 and the natural ligand could be separated from the other decoys. There is a known correlation between large molecular size and its corresponding high binding energy [19]. This is found to be the case with MurE: only the large molecular inhibitors rank well against the set of decoys. Virtual screening would not be able to select for smaller inhibitors like the quininoles, given the large size of the active site. However, the actual mechanism of quininoles in MurE inhibition is thought to be through the interference of ATP, a site which has been modified in our screening process, as we wish to select only for molecules specific to Section I and II of the active site defined in the earlier section of this manuscript. Given the range of inhibitors and low enrichment, virtual screening for the MurE protein system would be expected to result in a low sensitivity.
Prediction of potential fragments from a diversity library:
However, as three out of the known inhibitors still score well with docking, virtual screening can still be successfully used to select similar molecules, and maintain a high specificity if appropriate thresholds and filters were used to select potential inhibitors from the library. We have chosen to use a cutoff of 1 % of the highest scoring library compounds for further analysis (Figure 2b). The top 16 ligands obtained after virtual screening through AutoDdock Vina were further studied for their potential as inhibitor against Mur E enzyme of Mycobacterium tuberculosis. Docked complexes were analyzed using LIGPLOT [20], to identify interactions with residues critical for binding to the natural substrate, shown in (Figure 3, Figure 4). Interestingly, seven of the sixteen molecules dock preferentially to section I (Figure 1b) of the active site, while three dock only with section II, allowing for the potential pairing of the two sets of molecules to create a more active inhibitor. A visual examination of decoys with high energies, shows that this selective docking is not present - residue interactions being present across both sections of the active site.
Figure 3.
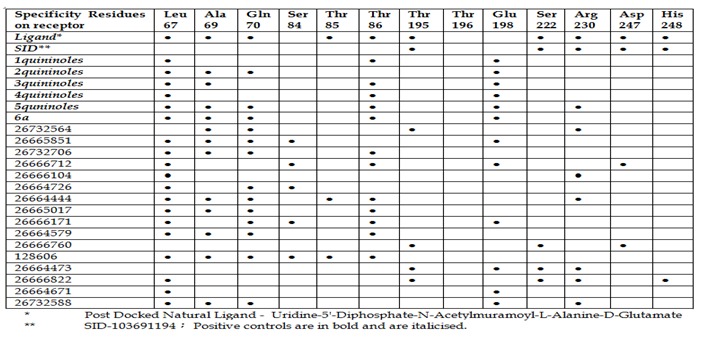
Comparison of positive controls and predicted inhibitors: interacting residues after docking
Figure 4.
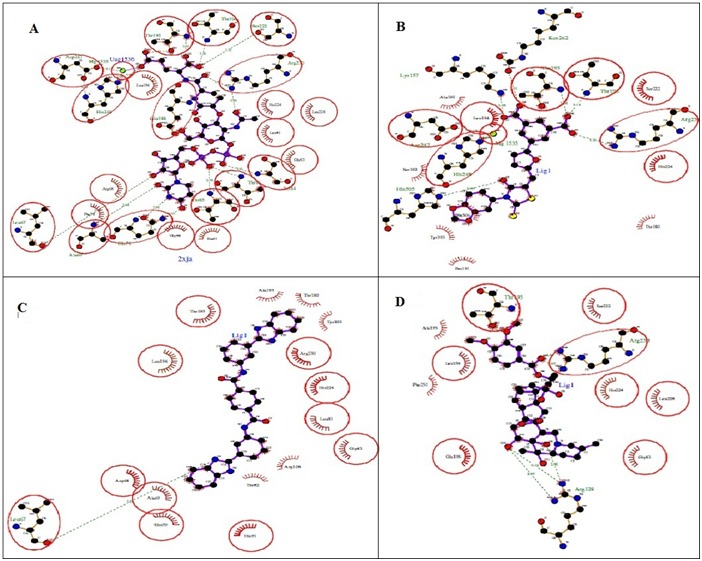
Ligplot results for post-docking poses of selected ligands A) Uridine-5'-Diphosphate-N-Acetylmuramoyl-L-Alanine-DGlutamate- (Natural Ligand.); B) SID 103691194 (Known inhibitor for E.Coli); C) SID 26732564 and; D) SID 26664473.
Conclusion
Virtual screening of small molecule libraries, which is a common strategy to shortlist potential inhibitors for screening, does not have high accuracy. Algorithms for exploring conformational space in the ligand are well-developed, but the lack of accurate scoring functions, and other pitfalls including improper protocols contribute to their inherently low sensitivity.
We propose the use of virtual screening from a diversity set of small molecules to be used as fragments to build larger, and more specific inhibitors. Compounds that had higher or equal scores than the natural ligand and SID 103691194, corresponding to 1% (16 in no.) of the library, were selected as potential building blocks. These were further evaluated by visual inspection of their docked poses. Interestingly, the two sections of the active site defined in this manuscript, serve as separate sites for the eventual docking of the drug-like molecules, allowing for the identification of two sets of molecules, which can be combined to create more specific leads. This property in high-scoring drug-like molecules is not found in a sampling of high-scoring decoys, and maybe used as a filter to further improve the sensitivity of docking.
Acknowledgments
Shilpi Singh acknowledges the award of a Tata-CSIR-OSDD Scholarship (TCOS).
Footnotes
Citation:Singh et al, Bioinformation 10(11): 697-702 (2014)
References
- 1.World Health Organization. Global tuberculosis report. 2013 [Google Scholar]
- 2.Ormerod LP, et al. Br Med Bull. 2005;73:17. doi: 10.1093/bmb/ldh047. [DOI] [PubMed] [Google Scholar]
- 3.El Zoeiby A, et al. Mol Microbol. 2003;47:1. doi: 10.1046/j.1365-2958.2003.03289.x. [DOI] [PubMed] [Google Scholar]
- 4.Smith CA, et al. J Mol Biol. 2006;362:640. doi: 10.1016/j.jmb.2006.07.066. [DOI] [PubMed] [Google Scholar]
- 5.Barreteau H, et al. FEMS Microbiol Rev. 2008;32:168. doi: 10.1111/j.1574-6976.2008.00104.x. [DOI] [PubMed] [Google Scholar]
- 6.Scior T, et al. J Chem Inf Model. 2012;52:867. doi: 10.1021/ci200528d. [DOI] [PubMed] [Google Scholar]
- 7.Basavannacharya C, et al. Protein cell. 2010;1:1011. doi: 10.1007/s13238-010-0132-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Laskowski RA. Nucleic Acids Res. 2001;29:221. doi: 10.1093/nar/29.1.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zeng, et al. The Journal of Organic Chemistry. 1998;63:10081. [Google Scholar]
- 10.Bugg TD, et al. Trends in biotechnol. 2011;29:167. doi: 10.1016/j.tibtech.2010.12.006. [DOI] [PubMed] [Google Scholar]
- 11.TomaŠić T, et al. ChemMedChem. 2010;5:286. doi: 10.1002/cmdc.200900449. [DOI] [PubMed] [Google Scholar]
- 12.Guzman JD, et al. J Antimicrob Chemother. 2010;65:2101. doi: 10.1093/jac/dkq313. [DOI] [PubMed] [Google Scholar]
- 13.Guzman JD, et al. J Antimicrob Chemother. 2011;66:1766. doi: 10.1093/jac/dkr203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hanwell MD, et al. J Cheminform. 2012;4:17. doi: 10.1186/1758-2946-4-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Boyle OL, et al. J Cheminf. 2011;3:33. [Google Scholar]
- 16.Mysinger MM, et al. J Med Chem. 2012;55:6582. doi: 10.1021/jm300687e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. http://dtp.nci.nih.gov/branches/dscb/div2_explanation. html.
- 18.Trott O, Olson AJ. J Comput Chem. 2010;31:455. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Verdonk ML, et al. J Chem inf Comput Sci. 2004;44:793. doi: 10.1021/ci034289q. [DOI] [PubMed] [Google Scholar]
- 20.Wallace AC, et al. Protein Eng. 1995;8:127. doi: 10.1093/protein/8.2.127. [DOI] [PubMed] [Google Scholar]