

Abstract
The microbial colonization of the intestine during the first months of life constitutes the most important process for the microbiota-induced host-homeostasis. Alterations in this process may entail a high-risk for disease in later life. However, the potential factors affecting this process in the infant are not well known. Moreover, the potential impact of orally administered vaccines upon the establishing microbiome remains unknown. Here we assessed the intestinal microbiome establishment process and evaluated the impact of rotavirus vaccination upon this process. Metagenomic, PCR-DGGE and faecal short chain fatty acids analyses were performed on faecal samples obtained from three infants before and after the administration of each dose of vaccine. We found a high inter-individual variability in the early life gut microbiota at microbial composition level, but a large similarity between the infants' microbiomes at functional level. Rotavirus vaccination did not show any major effects upon the infant gut microbiota. Thus, the individual microbiome establishment and development process seems to occur in a defined manner during the first stages of life and rotavirus vaccination appears to be inconsequential for this process.
The basis of a healthy intestinal microbiota lies in the early neonatal period with the initial steps of establishment of this complex microbial ecosystem1. Microbial colonization of the gut in human newborns is started by facultative anaerobes which contribute to the establishment and development of strict anaerobic bacterial populations by reducing oxygen content2. Different factors including mode of delivery, feeding habits, gestational age or use of medication have been reported to affect this process3,4. The initial microbial colonization has been shown to constitute the most important moment for the microbiota-induced host-homeostasis. This microbe-host interaction in early life is necessary for a proper maturation of the immune system5,6, and results essential for a normal host development and physiology7,8. Therefore, during this relatively unstable and sensitive initial period any alteration in the microbiota development process may increase the risk of disease in later life1,9. After weaning the complexity and diversity of the microbiota increases rapidly and at the age of 2–3 years the infant microbiota reaches an adult-like composition10.
The delivery mode11,12, gestational age13,14,15,16 or antibiotics administration14,17,18,19 are known to affect the microbiota composition. Nevertheless, the impact of other factors, such as other medical interventions in early life, on the process of establishment of the intestinal microbiota in newborns still remains poorly understood as most of the currently available studies have focused on the adult population20. Moreover, most of the studies carried out using modern next-generation-sequencing techniques have applied 16S rRNA gene-sequencing for microbiota analyses whilst few works have assessed the total infant metagenome composition10,20,21,22. To this regard, metagenomic analyses have the advantage of providing not only data at microbial composition level, such as 16S rRNA gene data, but also data on the functions present in the metagenome. Furthermore, most reports on the infant microbiome have evaluated the effect of delivery mode, feeding habits, gestational age or disease, whereas the impact of some common early life medical interventions on the establishing intestinal microbiota remains largely unknown.
Nowadays, vaccination is a very common practice in developed countries. Among the different vaccines some are orally administered and contain attenuated microorganisms or viruses, such as the one for rotavirus, a double stranded RNA virus from the family Reoviridae which produces gastroenteritis and diarrhoea. Oral vaccines are expected to interact with intestinal immune cells eliciting an immune response at mucosal level. Therefore, such vaccination may have the potential for modifying the intestinal environment, thus altering the establishing infant gut microbiome.
The aim of the present study was to assess the process of establishment of the intestinal microbiome in infants and to evaluate the potential impact of oral rotavirus vaccination upon this process. To this end, total metagenomic analyses, in combination with PCR-DGGE and faecal short chain fatty acids (SCFA) determinations, were performed.
Results
PCR-DGGE
In spite of the changes observed over-time, the PCR-DGGE results showed clearly different profiles for the three infants (Figure 1). In addition, no major changes in the DGGE patterns were evidenced when the samples taken before administration of each rotavirus vaccine dose were compared with those obtained after administration of the vaccine dose.
Figure 1. PCR-DGGE faecal microbiota profiles obtained before and after (b or a) each rotavirus vaccination dose (D1, D2 and D3) from each infant.

Numbers indicate the results of the species identification of the corresponding DGGE band.
The DGGE pattern obtained for infant 2 showed less bands than those obtained for the other two infants, suggesting a less complex microbiota (Figure 1). Moreover, the profile of the infant 2 remained largely unchanged during the study with a predominant band that could not be un-ambiguously identified but belonging to the phylum Proteobacteria (the highest homology scores were all obtained with different proteobacteria). Faeces from infant 1 showed initially (first sample, 2 months) a strong band corresponding to Bacteroides, with a band identified as Streptococcus becoming the strongest one at the second sampling point. The samples of the third infant presented a clear band along the sampling period, likely corresponding to proteobacteria, with bands from anaerobes such as Bacteroides and Bifidobacterium becoming apparent in the second and third sampling points (4 and 6 months).
Short chain fatty acids
The SCFA profiles were found to be very stable and not affected by the vaccination, with molar proportions of the three major SCFA (acetic, propionic and butyric) showing little variation along time and among individuals (Supplementary Figure S1). The acetic acid was the most abundant SCFA in the three infants along the study, followed by propionate and then butyrate.
Metagenomic analyses
Metagenomic data showed clear differences in microbiota composition among infants, with all the samples from the same infant clustering together and independently of those of the other infants (Figure 2, Supplementary Figure S2). These differences point to the individual as the main factor determining the microbiota composition; clear differences were found, at genus level, among the intestinal microbiota of the three babies, although their respective microbiotas showed limited variability over time and an almost negligible vaccination-effect.
Figure 2. PCoA analysis of the microbiota composition at genus level obtained from faecal samples before and after (b or a) each rotavirus vaccination dose (D1, D2 and D3) from each infant.
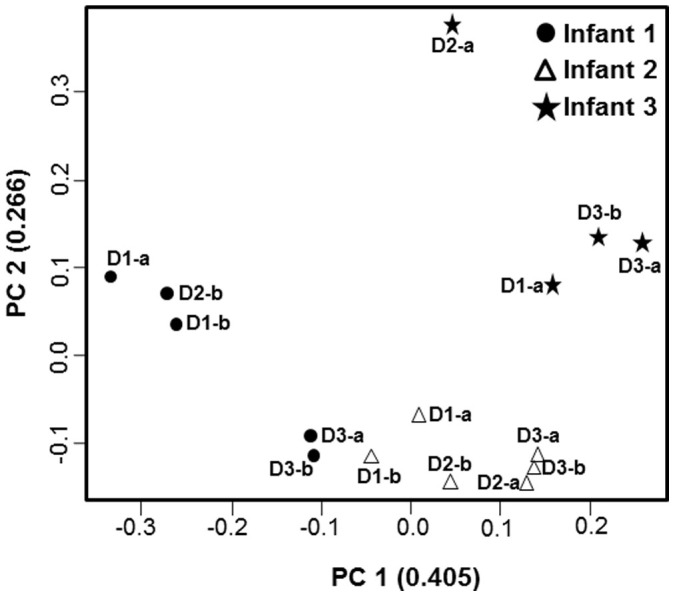
PCoA was performed with Spearman correlation distance.
These results indicate that the vaccine had minimal or no effect on the individual microbiota profile. Thus, in order to assess the microbiome evolution during the first months of life in these three babies we calculated, in each infant and sampling point (vaccine dose), the mean of the relative proportion for each bacterial group obtained before and after vaccine. Microbial composition at different taxonomic levels showed clear differences among infants (Figure 3 and Supplementary Figure S3). At genus level Bacteroides dominated in the first sample of infant 1 (2 months of age) with Bifidobacterium becoming the dominant genus at later sampling points (3 and 4 months of age). The microbiota of infant 2 was dominated by enterobacteria, mainly Escherichia, during the whole sampling period whilst in the case of infant 3 enterobacteria dominated at the first two sampling points (up to 4 months of age), with Bifidobacterium becoming dominant later on.
Figure 3. Main faecal microbial groups at genus level (A) and gene functional annotations at KEGG level 2 (B) determined from the metagenomic analyses of the samples obtained at different time points from each infant.

Vaccination showed no effects on α-diversity or gene richness (data not shown). In general, infant 1 displayed higher α-diversity than infants 2 and 3 (Table 1). Diversity and gene richness increased over time in infants 1 and 2 but remained almost unchanged in infant 3 (Table 1).
Table 1. α-diversity and gene richness obtained at the different time points analysed for the three infants studied. α-diversity and gene richness were calculated based on genus composition and gene abundance, respectively.
| α-diversity | |||||
|---|---|---|---|---|---|
| Infant | Sample | Shannon | Simpson | Chao1 | Gene Richness |
| 1 | 1 | 0.87 | 0.43 | 89 | 43951 |
| 2 | 1.48 | 0.75 | 120 | 40418 | |
| 3 | 1.44 | 0.64 | 147 | 50516 | |
| 2 | 1 | 0.74 | 0.34 | 103 | 29781 |
| 2 | 0.74 | 0.35 | 98 | 32038 | |
| 3 | 1.14 | 0.52 | 107 | 38032 | |
| 3 | 1 | 0.98 | 0.51 | 130 | 43159 |
| 2 | 0.92 | 0.44 | 99 | 39589 | |
| 3 | 0.88 | 0.42 | 128 | 43023 | |
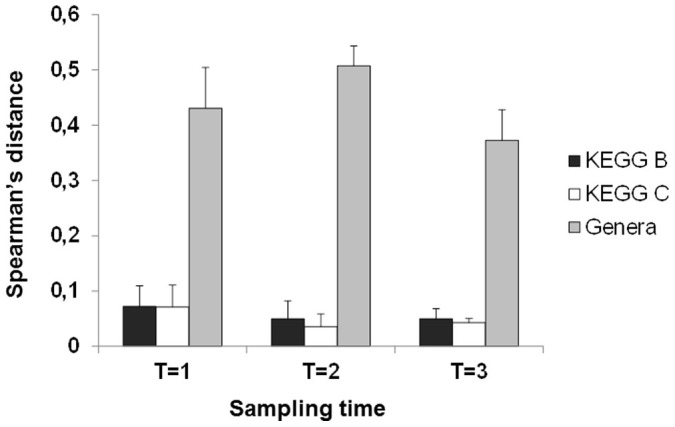
Regarding the functional features of the infants microbiome the KEGG level B categories “enzyme families”, “membrane transport”, “carbohydrate metabolism” and “amino acid metabolism” were the most represented ones in all the infants and samples analysed (Figure 3).The metagenomes showed higher stability and lower variability at functional level than at microbial composition (genus) level (Figure 3 and Supplementary Figure S2). Nevertheless, PCoA analysis of functional data still clustered together the different samples belonging to each infant (Supplementary Figure S4). When spearman correlation distances were calculated using either KEGG (levels B or C) or composition at genus level, it became clear that the distances among samples were lower for KEGG data (Figure 4) than for the genera data. This corroborated the greater similarity of infant metagenomes at functional than at microbial composition level.
Figure 4. Spearman's distances (mean and sd) obtained by comparing data at functional KEGG level B, functional KEGG level C and microbial composition (genus level) from all the samples from the different infants at each time point.
Discussion
The process of intestinal colonization by microorganisms during the postnatal period is very important for later health8,9,23. Therefore, controlling or minimizing the impact of early life medical interventions on the establishing gut microbiome may have a large influence in later health. Unfortunately, our knowledge on the gut microbiome establishment process in the neonatal gut and the effect of perinatal medical interventions upon this process is still limited.
Some vaccines such as the one for rotavirus are administered orally and will interact with the intestinal mucosa which may modify the intestinal environment and, therefore, may affect the intestinal microbiome establishment process. To this regard, norovirus infection in adults did not show major effects on the intestinal microbiota in the majority of patients, although in a minority of cases microbiota alterations were observed24. Nevertheless, the impact of other viruses causing enteric infections or that of orally administered vaccines against them remains largely unknown. García-López and co-workers25 used 16S rRNA gene-based analysis to compare the microbiotas of rotavirus vaccinated and unvaccinated children after one year of age (12–15 months) without observing any long-term microbiota differences. In the present study we have used total metagenome analyses to test whether vaccination against rotavirus affected the process of establishment of the intestinal microbiota in infants at the time of vaccination.
Although the limited number of infants does not allow establishing firm conclusions, our PCR-DGGE and metagenomic analyses of the intestinal microbiota of the three infants participating in the study suggest that rotavirus vaccination has no significant effect upon the establishing gut microbiome. In general, our results on microbial composition are in the range of previously reported data for healthy neonates26,27,28,29,30. A significant presence of bifidobacteria (Actinobacteria) was evidenced in the faeces of the two breast-fed infants participating in the study whilst the levels of this microbial group were lower in the formula-fed infant, who presented higher levels of Proteobacteria than the breast-fed babies during the first months of life. This is in contrast to the dominance of Bacteroidetes and Firmicutes occurring in adults31,32,33,34. In spite of the microbiota changes over time we found clearly differentiated individual profiles, with the samples obtained at different times for each infant clustering together. This suggests a predominant role of the individual over the age on determining the gut microbiome during the first months of life and supports data previously obtained by 16S rDNA sequencing showing high inter-individual microbiota variability in neonates29.
In this area, most of the studies performed so far have used 16S rRNA gene-based analyses of the intestinal microbiota of full-term newborns but only a few works have assessed the total metagenome composition in these infants10,20,21. To this regard, our results shed some light on the still limited knowledge of the gut microbiome establishment process, indicating that the inter-individual microbiome shows higher similarity at functional than at microbial composition level. This is also supported by the limited inter-individual variability in faecal metabolites such as SCFA.
To sum up, although the limited sample size precludes establishing definitive conclusions, the present work underlines a high inter-individual variability in the gut microbiota composition and evolution at early life. However, this relatively large microbial community diversity renders few differences between the infants' microbiomes at functional level. Moreover, the individual microbiome establishment and development process seems to occur in a defined manner during the first stages of life and it is not affected by oral rotavirus vaccination.
Methods
Volunteers
The study was approved by the Regional Ethical Committee of Asturias Public Health Service (SESPA) and carried out in accordance with the approved guidelines of the Ethics Committee. Written informed consent was obtained from the parents. The study included three Caucasian male infants. Infant 2 was delivered by caesarean section and received mixed feeding (breast-milk and infant formula) whilst the other two infants (1 and 3) were vaginally delivered and exclusively breast-fed. The three infants received the rotavirus vaccine “RotaTeq®” (Sanofi Pasteur MSD, Lyon, France) which contains attenuated viruses and it is orally administered in three doses during the first six months of life. Infants 1 and 2 received the three doses at 7–8, 11–12 and 15–16 weeks of life, whereas infant 3 received them at 8, 16 and 24 weeks of life.
Faecal Sample Collection and DNA extraction
Faecal samples were collected the day before and the day after (between 24 and 48 hours) the administration of each dose of vaccine. Fresh faecal samples were immediately frozen. For DNA extraction faecal samples were weighed, diluted 1/10 in sterile PBS solution, and homogenized in a LabBlender 400 stomacher (Seward Medical, London, UK) at full-speed for 4 min. The homogenate (1 mL) was then centrifuged and the supernatant obtained was filtered and frozen at −20°C for SCFA analyses. The DNA was extracted from the faecal pellet using the QIAamp DNA stool kit (Qiagen GmbH, Hilden, Germany) following the manufacturer's specifications as previously described14. The extracted DNA was kept frozen (−70°C) until analysis.
SCFA Analysis
The analysis of SCFA was carried out in a chromatographic system composed of a 6890N GC (Agilent Technologies Inc., Palo Alto, CA, USA) connected to a FID and a MS 5973N detector as described previously14.
Denaturing Gradient Gel electrophoresis (DGGE)
The profile of the dominant microbial populations in faeces at the different sampling points was determined by PCR-DGGE. PCR-DGGE reaction mixture, conditions and universal primers previously described (357F; TACGGGAGGCAGCAG and 518R; ATTACCGCGGCTGCTGG) were used35. PCR products were separated by DGGE in a DCode system (BioRad Laboratories) in a 30% to 60% gradient of urea-formamide in Tris-Acetate-EDTA (TAE) buffer (pH8). Selected bands were excised from gels and submitted to a new PCR reaction with the same PCR-DGGE primers without the GC clamp. After purification, the amplified PCR products were sequenced in a capillar ABI3730XL DNA Analyzer (Macrogen Europe, Amsterdam, Netherland) and partially identified by comparison (BLAST) with data held in the GenBank database35.
Metagenomic analyses
DNA Sequencing
The extracted faecal DNA (5 μg) was precipitated by standard sodium acetate/ethanol precipitation, submitted to Zhejiang California International Nanosystems Institute (ZCNI, Hangzhou, Zhejiang, China) for library processing and sequenced at BGI (Shezhen, China). Three out of the eighteen samples did not render enough DNA, for the other fifteen samples the libraries were constructed (500 bp insert size, with adapter) and a 2 × 101 Pair-End sequencing strategy was carried out in a Hiseq 2000 sequencing platform (Illumina). CASAVA-1.8.2 was used for base calling with default parameters except –mismatches 1 -mask y100n, I6n, Y10n – adapter-sequence.
Raw Data Processing
The quality control on the pair-end sequenced reads was conducted using the following criteria; i) reads with more than 3 ambiguous bases (N) were removed, ii) reads with adapters' sequences at both ends were discarded, iii) if in one read more than 50 bases presented low quality (Q2) the read was discarded, iv) no more than 15 bases at 3′ end of reads would be trimmed if ambiguous bases or bases with low quality (Q2) occurred and v) when one of the paired-end reads did not passed the control, although its mate may have passed, then the whole pair was discarded.
Assembly, Gene prediction, gene set construct and annotation
We use Soap De novo version 2.04 to perform a genome assembly36 and Glimmer 3.02 to predict genes37. To construct a non-redundant gene set the method introduced by Qin and co-workers32 was used. Non-redundant genes were annotated by KAAS system with SBH mode38.
Abundance profiles of organisms and genes
SOAP aligner version 2.21 was used for aligning the pair-end reads against reference genomes and non-redundant genes (parameters “-r 2 -M 4 -m 100 -x 2000”). Then, the methods described by Arumugan and co-workers33 were applied to generate abundance profiles of microorganisms and genes.
KEGG Function abundance profile
Each gene was assigned into only one KEGG orthologous group, then the abundance of genes was accounted as abundance of their common unique KEGG orthologous. When a KEGG orthologous belonged to different KEGG functional features at B level or C level its abundance was added to all the relative KEGG functional categories to which the orthologous belonged.
α-diversity and gene richness calculations
Chao1, Shannon and Simpson indices were determined with the R program package “vegan” (Oksanen et al. 2013, Vegan: Community Ecology Package 2.0-10. http://cran.r-project.org/web/packages/vegan/index.html). To calculate gene richness we parsed the gene abundance profile and then counted number of observed genes as gene richness.
β-diversity calculations
We used Spearman correlation distance as our β-diversity determination, first we calculated samples's spearman coefficient S, then distance matrix M was calculated by 1 - S. This was determined with the following command “<- 1 – cor (X, method = “spearman”)” in R, where X is profile of KEGG or Genera composition. We used Wilcoxon rank sum to test if two distance sets differs from each other. PCoA analysis was performed by R library “ade4”39.
Nucleotide sequence accession numbers
The raw sequences reported in this article have been deposited in the EMBL European Nucleotide Archive (accession number PRJEB6972).
Author Contributions
G.S., C.G.R.-G. and M.G. designed the study. G.S. and M.S. recruited the volunteers and collected the samples. L.A., S.A., G.L., Y.C. and Q.N. conducted the metagenomic, PCR-DGGE and SCFA determinations. L.A. and M.G. evaluated the results and wrote the manuscript. All authors discussed the results, reviewed and accepted the manuscript.
Supplementary Material
Supplementary information
Acknowledgments
This work was funded by a CSIC intramural project (Ref. 201370E019) and Spanish Ministry of Economy and Competitiveness project AGL2013-43770R. We show our greatest gratitude to all the infants participating in the study and their families.
References
- Sekirov I., Russell S. L., Antunes L. C. & Finlay B. B. Gut microbiota in health and disease. Physiol. Rev. 90, 859–904 (2010). [DOI] [PubMed] [Google Scholar]
- Wopereis H., Oozeer R., Knipping K., Belzer C. & Knol J. The first thousand days- intestinal microbiology of early life: establishing a symbiosis. Pediatr. Allergy Immunol. 25, 428–438 (2014). [DOI] [PubMed] [Google Scholar]
- Penders J. et al. Factors influencing the composition of the intestinal microbiota in early infancy. Pediatrics 118, 511–521 (2006). [DOI] [PubMed] [Google Scholar]
- Johnson C. L. & Versalovic J. The human microbiome and its potential importance to pediatrics. Pediatrics 129, 950–960 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renz H., Brandtzaeg P. & Hornef M. The impact of perinatal immune development on mucosal homeostasis and chronic inflammation. Nat. Rev. Imunol. 12, 9–23 (2012). [DOI] [PubMed] [Google Scholar]
- El Aidy S., Hooiveld G., Tremaroli V., Bäckhed F. & Kleerebezem M. The gut microbiota and mucosal homeostasis: colonized at birth or at adulthood, does it matter? Gut Microbes 4, 118–124 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins S. M., Surette M. & Bercik P. The interplay between the intestinal microbiota and the brain. Nat. Rev. Microbiol. 10, 735–742 (2012). [DOI] [PubMed] [Google Scholar]
- Sommer F. & Bäckhed F. The gut microbiota–masters of host development and physiology. Nat. Rev. Microbiol. 11, 227–238 (2013). [DOI] [PubMed] [Google Scholar]
- Sim K. et al. The neonatal gastrointestinal microbiota: the foundation of future health? Arch. Dis. Child. Fetal Neonatal Ed. 98, F362–364 (2013). [DOI] [PubMed] [Google Scholar]
- Yatsunenko T. et al. Human gut microbiome viewed across age and geography. Nature 486, 222–227 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez-Bello M. G. et al. Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc. Natl. Acad. Sci. USA 107, 11971–11975 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakobsson H. E. et al. Decreased gut microbiota diversity, delayed Bacteroidetes colonization and reduced Th1 responses in infants delivered by caesarean section. Gut 63, 559–566 (2014). [DOI] [PubMed] [Google Scholar]
- Rouge C. et al. Investigation of the intestinal microbiota in preterm infants using different methods. Anaerobe 16, 362–370 (2010). [DOI] [PubMed] [Google Scholar]
- Arboleya S. et al. Establishment and development of intestinal microbiota in preterm neonates. FEMS Microbiol. Ecol. 79, 763–772 (2012). [DOI] [PubMed] [Google Scholar]
- Arboleya S. et al. Deep 16S rRNA metagenomics and quantitative PCR analyses of the premature infant fecal microbiota. Anaerobe 18, 378–380 (2012). [DOI] [PubMed] [Google Scholar]
- Barrett E. et al. The individual-specific and diverse nature of the preterm infant microbiota. Arch. Dis. Child. Fetal Neonatal. Ed. 98, F334–340 (2013). [DOI] [PubMed] [Google Scholar]
- Tanaka S. et al. Influence of antibiotic exposure in the early postnatal period on the development of intestinal microbiota. FEMS Immunol. Med. Microbiol. 56, 80–87 (2009). [DOI] [PubMed] [Google Scholar]
- Fouhy F. et al. High-Throughput sequencing reveals the incomplete, short-term recovery of infant gut microbiota following parenteral antibiotic treatment with ampicilin and gentamicin. Antimicrob. Agents Chemother. 56, 5811–5820 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faa G. et al. Factors influencing the development of a personal tailored microbiota in the neonate, with particular emphasis on antibiotic therapy. J. Matern. Fetal Neonatal Med. 26, 35–43 (2013). [DOI] [PubMed] [Google Scholar]
- Valles Y. et al. Microbial succession in the gut: directional trends of taxonomic and functional change in a birth cohort of Spanish infants. PLoS Genet. 10, e1004406 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenig J. E. et al. Succession of microbial consortia in the developing infant gut microbiome. Proc. Natl. Acad. Sci. USA 108 (Suppl 1), 4578–4585 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claud E. C. et al. Bacterial community structure and functional contributions to emergence of health or necrotizing enterocolitis in preterm infants. Microbiome 1, 20 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matamoros S., Gras-Leguen C., Le Vacon F., Potel G. & de La Cochetiere M. F. Development of intestinal microbiota in infants and its impact on health. Trends Microbiol. 21, 167–173 (2013). [DOI] [PubMed] [Google Scholar]
- Nelson A. M. et al. Disruption of the human gut microbiota following Norovirus infection. PLoS One 7, e48224 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-López R., Pérez-Brocal V., Diez-Domingo J. & Moya A. Gut microbiota in children vaccinated with rotavirus vaccine. Pediatr. Infect. Dis. J. 31, 1300–1302 (2012). [DOI] [PubMed] [Google Scholar]
- Jost T., Lacroix C., Braegger C. P. & Chassard C. New insights in gut microbiota establishment in healthy breast fed neonates. PLoS One 7, e44595 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azad M. B. et al. Gut microbiota of healthy Canadian infants: profiles by mode of delivery and infant diet at 4 months. Can. Med. Assoc. J. 185, 385–394 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milani C. et al. Assessing the fecal microbiota: an optimized Ion Torrent 16S rRNA gene-based analysis protocol. PLoS One 8, e68739 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avershina E. et al. Major fecal microbiota shifts in composition and diversity with age in a geographically restricted cohort of mothers and their children. FEMS Microbiol. Ecol. 87, 280–290 (2014). [DOI] [PubMed] [Google Scholar]
- Bergström A. et al. Establishment of intestinal microbiota during early life: a longitudinal, explorative study of a large cohort of Danish infants. Appl. Environ. Microbiol. 80, 2889–2900 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckburg P. B. et al. Diversity of the human intestinal microbial flora. Science 308, 1635–1638 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin J. et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464, 59–65 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arumugan M. et al. Enterotypes of the human gut microbiome. Nature 473, 174–180 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature 486, 207–214 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salazar N., Gueimonde M., Hernández-Barranco A. M., Ruas-Madiedo P. & de los Reyes-Gavilán C. G. Exopolysaccharides produced by intestinal Bifidobacterium strains act as fermentable substrates for human intestinal bacteria. Appl. Environ. Microbiol. 74, 4737–4745 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo R. et al. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. GigaScience 1, 18 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delcher A. L., Bratke K. A., Powers E. C. & Salzberg S. L. Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics 23, 673–679 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriya Y., Itoh M., Okuda S., Yoshizawa A. & Kanehisa M. KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 35, W182–W185 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dray S. & Dufour A. B. The ade4 package: implementing the duality diagram for ecologists. J. Stat. Soft. 22, 1–20 (2007). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary information