

Abstract
Biodegradable, injectable depot formulations for long-term controlled drug release have improved therapy for a number of drug molecules and led to over a dozen highly successful pharmaceutical products. Until now, success has been limited to several small molecules and peptides, although remarkable improvements have been accomplished in some of these cases. For example, twice-a-year depot injections with leuprolide are available compared to the once-a-day injection of the solution dosage form. Injectable depots are typically prepared by encapsulation of the drug in poly(lactic-co-glycolic acid) (PLGA), a polymer that is used in children every day as a resorbable suture material, and therefore, highly biocompatible. PLGAs remain today as one of the few “real world” biodegradable synthetic biomaterials used in US FDA-approved parenteral long-acting-release (LAR) products. Despite their success, there remain critical barriers to the more widespread use of PLGA LAR products, particularly for delivery of more peptides and other large molecular drugs, namely proteins. In this review, we describe key concepts in the development of injectable PLGA controlled-release depots for peptides and proteins, and then use this information to identify key issues impeding greater widespread use of PLGA depots for this class of drugs. Finally, we examine important approaches, particularly those developed in our research laboratory, toward overcoming these barriers to advance commercial LAR development.
Keywords: Controlled release, Depot, PLGA, Protein, Peptide, Biodegradable
1. Introduction
When bioactive agents are microencapsulated in biodegradable polymers, such as poly(lactic-co-glycolic acid)s (PLGAs) and related polymers, in a manner suitable for injection, it is possible to extend the duration of action of peptides from 1 day (e.g., for a solution dosage form) to 6 months [1]. Such a remarkable improvement for delivery of the luteinizing hormone-releasing hormone (LHRH) agonist, leuprolide, has been obtained with the Lupron Depot® microspheres and in situ forming gels of Eligard® [2]. Also used clinically is the Zoladex® implant, which extends duration to as long as 3 months with another LHRH agonist, goserelin, utilizing PLGA cylinders on the millimeter scale (which we have referred to as “millicylinders”) [3,4], similar to a mechanical pencil lead. For patients with prostate cancer and other LHRH-indications, such long-acting-release products (LARs) not only improve lifestyle by minimizing exposure to the needle, but also generally improve patient outcomes by improving patient compliance and reducing peak-and-valley blood levels [5].
The “large molecule” class of drugs is special — usually requiring injections — owing to the difficulty to deliver these molecules by noninvasive routes. A second salient feature of these molecules is the commonly short half-lives when administered in blood, meaning that injections are not only required, but patients experience the needle frequently. Improving the delivery of large molecules is one of the top issues in the drug delivery field and controlled release is not the only approach to this problem. For example, the ability to extend peptide and protein half-lives in blood, usually by modification of the peptide/ protein molecule, has been accomplished by PEGylation [6], protein fusion (e.g., to albumin or Fc region of the antibody) [7,8], and lipidation [9]. These methods also have been extremely successful, and the first two methods have been used in extending duration of protein pharmaceuticals [10,11].
Second, a plethora of noninvasive approaches have been on the horizon for quite some time, including oral, nasal, pulmonary, and transdermal delivery, just to name a few. There are some limited examples where approvals have come for administration of peptides by mucosal routes. If metabolic, transport, residence time, and safety barriers are traversed, there are significant opportunities, particularly as the oral route is the most commonly used route for drug administration. Third, the technology for injecting solutions, namely the pen injectors, have improved patient acceptance and raised the bar to compete with standard injections [12]. Patient acceptance can clearly be a significant obstacle for the delivery systems for large molecules, as evidenced by difficulties of Bydureon® (exenatide LAR) and Exubera® (pulmonary insulin) to attain a dominant market share. For example, Bydureon® has just recived FDA approval of a dual chamber pen containing diluent and drug in a single syringe/needle presentation to simplify the reconstitution and injection of the medication. The pen replaced a presentation in which drug and diluent were in separate containers. It is expected this new format will improve patient acceptance and sales of this drug, which faces stiff competition from the lipidated glucagon-like peptide-1 (GLP-1), liraglutide, and in the face of incoming GLP-1s (e.g., an albuminated GLP-1, abluglutide, was just approved by the FDA earlier this year) [7]. Thus, the battle of the GLP-1s between controlled release and peptide-modification is expected to continue as the controlled release depots also move to extend release to once-a-month or even longer.
So what is unique about PLGA depots? The prospect of sustained drug levels in the blood or target tissue for 1–3 months following a single injection is the holy grail for numerous drug companies for delivery of their polypeptide drugs. This long duration is not so readily attainable by non-PLGA systems, which do not have commercial precedence and lack the so-called “real world biomaterial” status. The noninvasive and peptide/protein modification strategies have typically been limited to weekly dosing. However, in order to accomplish 1–3 month controlled release, typically high total doses have to be initially encapsulated in the polymer. Once administered to the body, the polymer reacts with water to initiate and sustain erosion of the polymer, and the physical–chemical events that take place during this time are more complex than previously believed. Trying to reduce the complexity is one of the objectives for this review, as outlined below.
Numerous protein and peptide drugs could provide greater therapeutic benefit if the plasma concentration could be maintained for an extended period of time. The initial clinical application of PLGA implants focused on hormonal peptide therapeutics (leuprolide, octreotide, buserelin, and others), diabetes GLP-1 peptide, and growth hormone. However, a number of other products will benefit from sustained delivery of systemic dose such as blood coagulation factors, metabolic peptides, monoclonal antibodies and antibody fragments, enzymes, and cytokines. In addition, by using a polymer implant to maintain systemic levels of therapeutic drug, it is also of a great advantage to deliver drugs for local/site-specific activity. For example, protein delivery to the eye or brain is an excellent application of this polymer, and multiple products are currently in development for these applications. Delivery of growth factors held great promise for regenerative medicine for treatment of peripheral vascular and ischemic heart diseases. However, systemic administration of growth factors has led to mixed clinical outcomes. Increasing growth factor levels locally in ischemic tissue for extended time periods alone or in combination with cell-based regenerative therapy could lead to improved benefits. The local delivery of protein and peptides is an advantage of polymer implants that will not be furnished by other technologies, i.e., PEGylation, and the others mentioned above that are normally used for half-life extensions.
The purpose of this review is to (a) discuss key concepts in the development of injectable PLGA controlled release depots for peptides and proteins, (b) use this information to identify key issues impeding more widespread use of PLGA depots for this class of drugs, and (c) describe important approaches, particularly those developed in our laboratory, toward overcoming these obstacles and achieving more commercial depot formulations. Note that our goal with this review is to organize and describe the necessary concepts efficiently with supporting examples. The total body of literature in this area is immense and therefore, there are certain to be many important publications omitted.
2. Evaluation of key PLGA concepts to identify issues in depot development
One common misconception is that the release kinetics from PLGA are undesirable and do not provide continuous zero-order type release. In fact, it has been known for more than 30 years that, by incorporating a low molecular weight PLGA into the polymer matrix at elevated drug loading, continuous release of polypeptides is commonly observed without an induction time before polymer mass loss (should the polypeptide remain soluble) [13]. There are also PLGA products that release highly water-soluble peptides without a significant burst release [14]. Another perception about PLGA is that proteins are unstable when encapsulated in PLGAs. This issue has been more persistent, and complicated by the analytical difficulties associated with analysis of proteins in the release media and in the polymer. We see many papers about microencapsulation, but we see far fewer about issues that take place in real manufacturing plants. Are we microencapsulating the correct way? What about needle size? Clearly, smaller is generally better, and this is another topic that has received less attention in publications. Lastly, are there other polymers for accomplishing long-term release of peptides and proteins? There certainly are, yet the vast majority of these have not made it to clinical trials and for the few that have, there are still issues. These important concepts are examined, as their analysis is essential to identifying key issues limiting biodegradable polymer depot development.
2.1. Release kinetics
Drug release from PLGAs can have multiple phases, which typically include (a) an initial burst phase lasting over one to several days or even a week, (b) a lag (or induction) phase where low levels of polymer erode and sometimes little or no drug release is observed, and (c) an active erosion phase, where polymer mass is continuously lost from the polymer and drug release becomes continuous. The middle phase generally occurs when there is little or no pore diffusion of peptides and proteins throughout the polymer matrix and the molecular weight (MW) of the polymer is too high. The high polymer MW is accompanied by the lack of availability of water-soluble polymer monomers and oligomers that are necessary for polymer mass loss to occur. Overcoming this lag phase is one of the keys to generating continuous release. A discussion of these aspects is given in Sections 2.1.1 and 2.1.2. In addition, the interaction of the peptide and protein with itself (e.g., protein aggregation) and with the polymer (e.g., peptide acylation) is less well-recognized as a rate-controlling pathway to release. One final important point is that once the polymer is hydrated, there are typically three separate domains in which the drug may be located (or at the interfaces thereof), including (a) dissolved in the polymer phase, (b) dispersed in a non-polymer containing drug solid state, and (c) dissolved or bound within the aqueous pores in the polymer. The extent to which the drug distributes in each of these domains will often strongly affect both drug release and stability.
2.1.1. Concepts regarding initial burst
The initial burst has recently been reviewed [15,16]. If significant drug is not encapsulated, and either physically adhered to the surface of the polymer or present as a free flowing solid among the PLGA polymer, this drug fraction will obviously contribute to the initial burst. Attention is usually paid to the drug fraction, which is either well-encapsulated (i.e. having at least a polymer membrane all around the drug) or located within the polymer in a manner where the drug particle has a porous escape route to the polymer surface (i.e., located within a pore percolating to the surface of the polymer). Let us assume for the moment the very common case when the peptide or protein is highly water-soluble and located primarily within aqueous pores. There is a general misconception that these drug domains deep within the polymer cannot significantly contribute to the initial burst. In fact, drug that is initially encapsulated can certainly leave the polymer during this early release phase (see below).
For this discussion it is worth while to discuss competing rates. The first rate is how fast the water is taken up by the polymer matrix, which can be subdivided into the rates of water penetration into non-polymer domains to create aqueous pores and water uptake into polymer domains, causing plasticization and increased polymer chain mobility. Virtually nothing can diffuse out of the polymer matrix until it is hydrated. A second competing rate is that of opening of the pore-network (as well as surface pores and/or cracks), which creates the diffusion pathways for the drug. A third rate is the self-diffusion of the drug once the diffusion path is created. A final common competing rate is the rate of pore closing, or what is commonly referred to in material science as “passive polymer healing” [17]. This last rate process is considered responsible for shutting off the rapid diffusive escape route of the drug [18,19]. Naturally, if the drug has significant solubility in, and/or interactions with, the polymer phase, there are additional rates to consider, and depending on unique circumstances additional competing rates may arise (e.g., loss of protein solubility due to protein aggregation).
The pore healing step, which we described while breaking down these aforementioned aspects [18], remarkably has been absent from consideration in the early scientific literature of polymer controlled release processes despite its understanding much earlier in material science. We showed that the burst release of octreotide acetate stopped in an acetate buffered release media corresponding to self-healing of the surface pores of the polymer [18]. Reduced uptake kinetics of a fluorescent pore marker, dextran tetramethyl rhodamine (3000 Da), during the initial burst confirmed the closure of the pore diffusion pathway [18]. We also quantified the influence of the healing on the simultaneous initial burst release of both dextran and bovine serum albumin (BSA) from microspheres of PLGA-glu, a glucose star polymer of PLGA 50/50, by evaluating the role of temperature on the release of the two macromolecules [19]. As seen in Table 1, at the two lowest temperatures (4 and 25 °C), expected to be < Tg of PLGA-glu, much more of the macromolecules were released (45–48%) than at the higher temperatures of 37 °C (15–20%) and 45 °C (8–11%). Scanning electron micrographs of the same polymer formulations showed the steady disappearance of surface pores at 37 °C but not at 4 °C after 2 days [19].
Table 1.
Fraction releasable a during initial burst release of biomacromolecules from PLGA-glumicrospheres [19].
| Temperature (°C) | ||||
|---|---|---|---|---|
| Encapsulated agent | 4 | 25 | 37 | 45 |
| BSA | 0.45 | 0.48 | 0.20 | 0.11 |
| Dextran (70 kD) | 0.45 | 0.45 | 0.15 | 0.08 |
Determined by fitting release to integrated Fick's second law of diffusion equation.
From initial studies, it appears that pore creation, which is expected to result from polymer matrix swelling and uneven osmotic pressures, can typically occur much faster than pore closure (see, for example, the initial burst time-sequence of scanning electron micrographs in [18]). However, it is important to note that pore closure will depend on pore size [20], as it takes longer for polymer chains to fill in larger pore volumes than smaller ones. This may be one factor to potentially explain the observation that reduced pore size in microspheres tends to decrease the initial burst release of peptides [21]. Therefore, clearly for large pores on the micron scale, pore closing at 37 °C for PLGA 50/50 is slow. However, as pore size is reduced to 30–100 nm or so, it is possible that in fact the pore closing rates might approach rates of pore opening, a topic which has not been significantly addressed in the literature to our knowledge.
2.1.2. Five common ways to accomplish continuous drug release
Continuous release of drugs from PLGAs has been accomplished for many years. Some of these methods are useful to large molecules and some are not. It is worthwhile to describe methods for small molecules too, as small molecule movement in and out of PLGAs is also important. Small molecule release events can indirectly affect large molecule release and/or stability. For example, release of either low molecular weight acids, produced by the degradation of the polymer, or low molecular weight excipients often added to the formulation, can affect degradation rates and microclimate pH development [3]. We describe below five methods that have been used to control the release of drugs continuously from PLGA.
2.1.2.1. Method 1 — use low polymer molecular weight fraction
Hutchinson showed that continuous peptide and protein release could be accomplished by including a significant or whole fraction of PLGA having reduced molecular weight [13]. Increased drug loading also was found to be beneficial. Low MW PLGA typically contains oligomers of the polymer that have limited water solubility to trigger polymer erosion, and upon hydrolysis generates a continuous supply of polymer chains in the ∼1 kD range or less [22]. The continuous in vitro release of peptide from low MW PLGA films by erosion control was also found to be mostly independent of film thickness [13].
2.1.2.2. Method 2 — co-encapsulate a poorly soluble base
Bernstein et al. [23] showed that when poorly soluble bases (e.g., MgCO3, Mg(OH)2, ZnCO3) are added to PLGAs of moderate MW, instead of observing a lag phase, continuous release is observed. These bases are the same type of excipients that our group has used extensively to attenuate the microclimate pH in the polymer [3,24]. The behavior can be rationalized by the following arguments: (a) the base reacts with acids (likely low MW acids) produced upon hydrolysis to form salts, which in turn, generates osmotic pressure and new pores for release of large molecules; (b) as the base reacts with the acids produced by PLGA hydrolysis, acids are removed from the polymer phase, which in turn reduces acid-catalyzed hydrolysis slowing the PLGA degradation rate [24]; and (c) the inclusion of these acids induces significantly more water uptake into the polymer matrix [3]. Positive effects have been found typically at base loading of 3–10% w/w [23,24].
2.1.2.3. Method 3 — blend in a water-soluble polymer
Polyethylene glycol has partial miscibility with PLGA [25]. When blended in a very slow-degrading PLGA, like 100% PLA, it can cause continuous release of protein for a month with little pH-induced damage to BSA. Formaldehyde-treated BSA, a model formalinized protein antigen (e.g., for tetanus and diphtheria toxoids) was also essentially completely released when additional stabilizing amino acids were included to inhibit the formaldehyde-aggregation pathway [26].
2.1.2.4. Method 4 — utilize classic diffusion pathways described for nondegradable polymer matrix systems
Four cases (I–IV) for pore and polymer diffusion pathways have been described for drug release from nondegradable polymers for controlled release (e.g., poly(ethylene-co-vinyl acetate) (EVA) and silicone rubber) [27]. If the drug has solubility in the PLGA, it can be released via case I (loading < drug solubility in polymer) or case II (loading > solubility in the polymer). We monitored BODIPY diffusion into, and out of, PLGA microspheres by confocal microscopy and successfully fit the solution to Fick's second law of diffusion to the resulting concentration profiles in the polymer [19]. If the drug is loaded at a level above the lower percolation threshold (typically occurs at >20–30% w/w), then the drug can be released via pore diffusion case III (drug water solubility < drug loading) and case IV (drug water solubility > drug loading). Zhang [28] observed pore diffusion of gentamycin through PLGA coated PLGA/gentamycin rods when the drug loading was 30–50%.
2.1.2.5. Method 5 — utilize osmotic-mediated drug release
When significant levels (typically at or below the lower percolation threshold) of low molecular weight water-soluble excipients, or the drug itself, are incorporated into the polymer, significant osmotic pressures can be generated. In the aforementioned study by Zhang et al. [28], when the loading was fixed at 30% and the length of the cylinder was increased, steadily more osmotically-mediated release of gentamycin was observed. The osmotic effect was counteracted by increasing the osmotic pressure of the release media, which reduced the drug release when the water mechanism was operative.
2.2. Stability of PLGA-encapsulated peptides and proteins
PLGA and other polymers for controlled release are reaction vessels in a sense. Unlike nondegradable polymers, PLGA is reacting with water all the time and contains acid end-groups and ester bonds that can interact physically and/or chemically with the peptide or protein of interest. The microclimate pH is often acidic and not well controlled, which can be deleterious for many peptides and proteins. Therefore, stability of peptides and proteins when encapsulated in PLGA has been reviewed extensively [29–37], as it is commonly considered the most significant issue impeding development of PLGA depots for proteins [38].
In order to address this issue on a mechanistic level, the general approach that has been taken is based on analyzing the deleterious physical–chemical events occurring from the time of encapsulation of the drug until the point when the drug is released in vivo [29]. This analysis and ensuing experimental evidence has identified several concepts [29–33]. For example, during encapsulation, protein damage is common during the micronization step, which generally involves a significant input of energy to break up cohesive forces in the protein phase to create a large surface area and tiny liquid or solid protein particles [29,31]. Likewise, the exposure to the organic solvent, particularly if the protein has mobility, either before, during or after micronization, can cause protein unfolding and associated instability mechanisms, particularly protein aggregation [29–31]. Drying is also an important step whether it is at elevated temperature under vacuum or by freeze-drying.
During release, four common stresses on the protein appear to be dominant. (1) Exposure to moisture — hydration and long-term exposure to moisture of the protein can place the protein at a deleterious water level [39]. For example, recent estimates of a formulation of simple BSA-loaded PLGA microspheres without additional stabilizers placed the protein concentration in the polymer pores at ∼500 mg/mL [40]. (2) Uncontrolled and often acidic microclimate pH — the polymer produces acid over a range of rates with PLGA 50/50 being the fastest to PLA being the slowest. In addition, the low MW acids, which have been shown to control the pH in the polymer pores (i.e., the microclimate pH, μpH), may have differing levels of diffusion rates depending on the polymer porosity, polymer molecular weight, plasticization, and others [19]. Reduced levels of pH < 5 are frequently problematic for proteins because of acid unfolding. (3) Polymer interactions (physical) — physical interactions with the polymer are known to occur on different levels [41]. Simple single-layer adsorption is expected to be reversible or irreversible. For proteins, particularly susceptible to surface-induced unfolding, this mechanism may prove to be important. Whether the protein can enter the polymer phase when the polymer chains get particularly short (i.e. later in release or when using oligomeric PLGA) has not been carefully investigated to our knowledge. However, in such cases the protein would be expected to unfold owing to the decreased water activity, which is necessary to remain high to maintain the folded structure of the protein. (4) Polymer interactions (chemical) — direct chemical reactions of the polymer with peptides do occur and have been documented for numerous therapeutic peptides [42–44]. Recent evidence suggests that, in certain cases, peptides can actually penetrate into the polymer phase (e.g., with cationic octreotide and leuprolide in low MW PLGAs with acid endcapping [45]). This fact helps to clarify the significant extent to which certain peptides form amide bonds in the acylation reaction as the peptide within the polymer phase is in intimate contact with polymer chains in the presence of reduced levels of water. For proteins, acylation is less clear and warrants further investigation.
From these stresses, several physical and chemical mechanisms of instability act on the peptides and proteins. Among the most common are unfolding, soluble and insoluble protein aggregation, hydrolysis, deamidation (and related racemization), and oxidation [33]. A lack of analytical equipment and training available in certain academic settings to rigorously characterize peptide and protein instability pathways continues to hinder efforts. Analytical columns are quite costly, and the entirety of analytical equipment (e.g., CD, fluorescence, FTIR, LC/MS, and NMR, just to name a few) is not always available. Moreover, separating the peptide or protein from the polymer at various times of release incubation for analysis is not always trivial and requires proper validation. Common protein assays and bioassays also can have interference with components of the release media. The total polypeptide loading in PLGA can be determined definitively by amino acid analysis after acid hydrolysis [45–47]. Knowing this value irrespective of the drug's stability is always important when beginning formulation work.
2.3. Microencapsulation and manufacturing
Most peptides and proteins cannot be terminally sterilized by gamma or e-beam radiation without damage. This requires that, for LAR products, these molecules must be microencapsulated aseptically on a commercial scale in the presence of organic solvents. These common processes — namely solvent evaporation, coacervation, and spray-drying — have generally reached large scale and produced numerous peptide-PLGA LARs that have passed the test of time on the market. However, drug makers have also suffered significant unanticipated costs in product scale-up and need to manage the presence of one or more residual organic solvents. Impurities in PLGA delivery systems have long been known to cause issues with depots. For example, residual osmotically-active water-soluble acids in the polymer have been shown to be capable of increasing the initial burst release of leuprolide (as well as causing shelf-life and encapsulation issues) [48]. Most residual organic solvents have significant solubility in water, and therefore are also osmotically active. When dissolved in water, organic solvents are surface active and typically reduce surface tension [49], and can also affect polymer aging [50]. Keeping a uniform particle size distribution and maintaining high product yield are further challenges.
2.4. Who uses the smallest needle and does it matter?
The type of needle used for parenteral injections depends on such factors as the drug formulation, route of administration and the intended patient population [51]. Two important aspects are the needle length and the needle gauge (diameter); the higher the gauge number, the smaller the diameter. For example, standard intramuscular (IM) injections require longer needles with a thicker diameter (typically 23G to 18G and 2.5 to 7.5 cm long) [52]. Smaller needles are generally used for standard subcutaneous (SC) injections (25G to 23G with a length of 1.5 to 2 cm) [52], although newer technologies have significantly decreased this size range (e.g., BD Ultra-Fine pen needles).
The needle geometry is also determined by the formulation for injection. The Lupron Depot®, the LAR suspension for leuprolide acetate, is supplied as lyophilized PLA microspheres and a diluent in a prefilled, dual-chamber syringe with a 23G needle. The microspheres are mixed with the diluent to form a suspension prior to IM injection [1]. Bydureon®, the microsphere formulation for the extended release of exenatide, is available as both a prefilled syringe plus vial and as an injection pen. The pen contains PLGA microspheres and diluent, which are mixed together and then injected subcutaneously through a 23G needle. The injection pen is a simpler alternative to the syringe and vial preparation of Bydureon® [53,54].
Eligard® and the Zoladex® implant are two polymer matrix-based sustained release formulations. Eligard®, an in situ forming injectable depot implant for the delivery of leuprolide acetate, is packaged as two separate syringes whose contents are combined directly prior to SC injection. One syringe contains leuprolide acetate and the other contains the ATRIGEL® delivery system, a matrix composed of PLGA dissolved in a biocompatible solvent system as N-methyl pyrrolidone. The two components are combined immediately before injection with a short 20G needle and form a solid implant at the injection site [2,55]. The Zoladex® implant is supplied as a prefilled syringe that contains goserelin acetate dispersed in a cylindrical PLGA matrix. It is administered by SC injectionin the abdomen with a 16G or a 14G needle depending on the dose [56].
To develop an effective and pharmaceutically acceptable PLGA injectable suspension, a number of characteristics must be evaluated. As for any injectable suspension, critical characteristics defining pharmaceutical acceptability include syringeability, injectability, clogging, resuspendability, and viscosity [57]. Syringeability refers to the ability of an injectable suspension to pass through a hypodermic needle from the vial to the syringe. It includes an ease of withdrawal, clogging and foaming tendencies, and accuracy of dose measurements. Increases in the viscosity, density, particle size, and concentration of solids in the suspension, all hinder the syringeability of suspensions [57]. Injectability refers to suspension performance during injection, i.e., pressure required for injection, evenness of flow, aspiration qualities, and freedom from clogging. Clogging may occur because of a single large particle or particle aggregate. Clogging involves a number of factors, such as the injection vehicle, wetting of particles, particle size and distribution, particle shape, viscosity, and flow characteristics of the suspension. Resuspendability describes the ability of the suspension to uniformly disperse with minimal shaking after it has been at rest. “Caking” upon rest, or settling, and fusion of the deflocculated particles present a problem to resuspendability.
A key issue associated with the use of needles is pain and discomfort from injection, which can cause burden on the patient and reduce treatment compliance. A survey conducted by Rubin et al. interviewed patients being treated with insulin. Almost half of the patients indicated that they would like to ease the pain related to injections, and the majority of patients wished to reduce the number of daily injections [58]. Needle diameter has been directly associated with injection pain and incidence of bleeding, with patients feeling less pain and experiencing a lower frequency of bleeding with thinner needles [59].
Local reactions from the injection of SC PLGA formulations are common. SC administration of PLGA microspheres may form nodules at the injection site, however these nodules are temporary and usually resolve without medical aid [54]. Patients treated with Eligard® reported injection site reactions consistent with reactions from other SC-administered drugs; overall, these reactions were mild and transient [60]. In the case of Zoladex®, icing the injection site has been found to reduce pain associated with the larger needle size [61]. One study demonstrated that there was no statistical difference in pain experienced by patients treated with Zoladex® (16G needle) or Prostap® (leuprorelin acetate, 23G needle) when blind to the needle size [62]. However, even discomfort from seeing a larger needle can influence pain and should be given due consideration [61].
2.5. Is PLGA the only game in town?
A very large number of natural and synthetic polymers have been developed over the years for the delivery of pharmaceutically active compounds [27,63–68]. As newer molecules have been formulated for a wide variety of diseases and therapies, they demand different properties for optimal delivery. A range of biodegradable biopolymers has been developed to meet these challenges [42,66,67,69–71]. It is beyond the scope of this review to discuss each of these, but rather to discuss a few interesting examples in the biomacromolecular controlled release context. Of the many biodegradable polymers, only a few reach the clinic and beyond in large part because of the high costs and risk associated with placing unproven biomaterials in pharmaceutical LAR products. In addition, others fail because of poor drug-polymer compatibility, inherent polymer toxicity, immunogenicity, low drug loading and poor preclinical performance [36].
In spite of these obstacles, some promising clinical results have been reported. For example, Locetron®, a poly(ether-ester) microsphere formulation for the delivery of interferon α2b, had promising phase II trials [72,73]. PhaseBio pharmaceuticals has reported promising clinical results for Lucemera® and Insumera®, which are based on its propriety elastin-like polypeptide delivery system [74]. The extensively studied polycaprolactone polymer has been a part of FDA-approved therapies in the past, making it an ideal candidate for developing controlled release products [75]. Poly(d,l-lactide-co-hydroxymethyl glycolide) (PLHMGA) microspheres have been shown to develop lower acidic conditions compared to equivalent PLGAs [40] and have been reported to release stable octreotide over 60 days [76]. Copolymerization of hydrophobic polymers (e.g. PLGA and PLA) with hydrophilic polymers also has been accomplished to overcome the issue of acidic degradation [36]. A polyoxalate based polymer system has been shown to be biodegradable, biocompatible, and provide better cell viability as compared to PLGA [77]. Polyketal copolymers have been shown to deliver imatinib effectively, but the inflammatory response warrants further investigation [78].
As is evident, new polymers promising better therapeutic outcomes are many but the challenges remain the same, namely safety and efficacy. Hence, a number of new biodegradable polymers have been developed over the years but they face regulatory and clinical hurdles. Judicious and early use of cellular and animal studies need to be adopted to ensure that biocompatibility issues are overcome for the timely development of safe and effective drug delivery systems. Another plausible factor limiting the alternatives to PLGA is the gap between the research work carried out developing new polymers and the developmental work in the pharmaceutical industry. Most new polymer systems are tested exclusively in preclinical studies, and rarely undergo further clinical research and development. These materials fall into the “valley of death”, which arguably impedes development of new discoveries into therapies [79,80]. Thus, in the absence of robust characterization and developmental work and a more straightforward regulatory path to close this gap, the industry usually prefers to work with very well characterized and polymeric biomaterials like PLGA, already used in FDA approved parenteral and implantable products, to avoid delays, risks and high costs associated with regulatory approval.
3. Approaches to overcome issues impeding depot development
There are numerous specific design criteria in a commercial PLGA formulation, and accomplishing these at a reasonable cost of goods is certainly a significant challenge. For example, adding stabilizing excipients for the drug can in turn affect the release kinetics, as observed with the poorly soluble bases (e.g., MgCO3) described above. Living with some undesirable product attributes such as residual solvents and broad particle size distributions may be required to achieve acceptable yields. We have grouped these important issues into four categories: (a) release kinetics, (b) drug stability, (c) microencapsulation, manufacturing, and aspects associated with organic solvent use, and (d) needle size. Efforts to overcome these are described below.
3.1. Improving release kinetics
Besides making more uniform microspheres, most of the research in this area recently has been focused on improving release kinetics and has involved attempting to reduce the initial burst release. The strategies to reduce burst can be broadly divided into three categories. The first has to do with controlling microsphere surface properties by alteration of process variables, including special steps for surface modifications and use of excipients to alter surface morphology. This strategy is used in numerous commercial PLGA commercial products, and modifications of this approach have been patented [15,81,82]. Generally it involves effective plasticizing of the surface of the microparticle to allow surface pore closure and to remove residual stresses. This is accomplished by addition of surfactants or plasticizers [5,83], sealing the surface by organic solvent vapors [84], and washing or incubating microparticles in alcohol solutions at temperatures slightly above the effective polymer Tg [84]. This concept may also help to explain why the relative amount of residual solvent significantly affects the initial burst, since in addition to affecting osmotic pressure, residual solvents influence effective Tg and surface plasticity. Another surface modification approach involves annealing the polymer above the Tg to facilitate pore closure and removal of bulk and surface stresses [82]. These considerations may also explain why the freeze-dried microspheres typically have higher bursts than those vacuum-dried at room temperature as the latter likely anneal during drying [85]. Other surface focused approaches include minimizing surface drug crystals by controlling solvent evaporation/solvent extraction rate [86] or by utilizing non-aqueous emulsion systems (oil-in-oil) by emulsifying polymer solution in silicone or cottonseed oils [87]. Yet another surface modification involves coating the surface of formed microspheres with a new layer of PLGA [88].
A second category includes increasing physical attraction of drug and polymer by either modifying the peptide to increase organic solvent partitioning, modifying the polymer to increase drug binding or using an excipient to bind the polypeptide and reduce its net charge or solubility. For example, peptides were modified with PEG or lipid tails to increase organic solvent partitioning to co-dissolve the peptide with the polymer [89]. Alternatively, the polymer composition was altered by introduction of polyethylene oxide, dextran or chitosan in the peptide chains [90]. Use of ion-pairing excipient to increase peptide hydrophobicity was found to be an effective strategy to reduce initial burst [91, 92]. Complexing therapeutic protein with other proteins, polysaccharide or cyclodextrin, was also utilized to reduce bursts and improve stability of encapsulated agents [15,93]. The third category involves optimizing the polymer microstructure by optimizing process variables. Examples include, producing denser/non-porous microspheres by using more concentrated polymer solutions, micronizing drug to obtain fine powder for suspension in the polymer phase and minimizing inner water phase emulsion droplet size [15,94].
3.2. Devising ways to improve peptide and protein stability
Significant progress has been made in the last several years on methods to improve stability of PLGA-encapsulated proteins and peptides. As proposed early on, an important way to manage the protein with higher order structure is to keep the biomacromolecule entirely dry or fully hydrated during processing and release incubation [29]. For example, in the 1990s encapsulation methodologies were developed to encapsulate the protein in the solid state in the absence of water. Examples included (a) the ProLease® spray-congealing strategy to spray micronized and precipitated human growth hormone suspended in methylene chloride/PLGA into a bath of liquid nitrogen before cold ethanol extraction of the organic solvent [95,96], and (b) suspension of solid and ground tissue plasminogen activator, BSA, and/or basic fibroblast growth factor in PLGA/acetone before extruding into silicone rubber molds to remove the solvent under vacuum [3,4]. Although Alkermes and Genentech were able to obtain FDA approval for the rhGH/PLGA Nutropin depot, which utilized the ProLease process, sustained commercialization of this and other methodologies involving the encapsulation of solid-sate proteins in PLGA microspheres overall has been quite limited. Similarly, to our knowledge no attempts have been made to commercially develop cylindrical implants from anhydrous extrusion of proteins in PLGA akin to the highly successful Zoladex® implant releasing the LHRH peptide agonist. Therefore, water-based encapsulation (when protein is fully hydrated but without organic solvent) appeared to us as a logical alternative, which is described below. During release, efforts are also described to overcome variations in microclimate pH and interactions with the polymer, in order to minimize protein unfolding and aggregation, chemical changes, and acylation adducts. Finally, efforts to shield polymer–polypeptide interactions have also been developed either to prevent direct covalent reactions (i.e., peptide acylation) or to help retain the protein's native conformation.
3.2.1. Aqueous microencapsulation
Common microencapsulation methods (solvent evaporation, coacervation and spray-drying) were not designed with process-sensitive molecules such as proteins in mind. The alternative to anhydrous encapsulation, which does not require micronization and organic solvents, is to remotely load the protein in preformed microspheres in water. This process has shown improvement in stability of lysozyme and tetanus toxoid [46,97,98] and is described below in Section 3.3.
3.2.2. Monitoring, manipulating, and predicting microclimate pH
There have been several steps in our evolution of understanding of the microclimate pH (μpH) in PLGA systems, which all produce acidic byproducts upon hydrolysis of the PLGA polyester. After many studies pointed to an acidification within the polymer, we demonstrated definitively that BSA encapsulated in PLGA 50/50 cylindrical rods forms acid-induced noncovalent aggregates and is hydrolyzed [3]. The structural features of BSA aggregates formed under acidic and moist conditions indeed matched BSA aggregates extracted from the polymer during release [99]. Raising the pH within both PLGA cylindrical implants and microspheres with a variety of poorly soluble bases showed a strong reduction in the acid-induced instability of PLGA-encapsulated BSA and therapeutic proteins [24,100]. These and other studies strongly motivated the development of research to understand and measure the μpH in an effort to obviate its deleterious effects.
For example, when bFGF was added to a similar BSA/PLGA formulation as described above, but with heparin added to stabilize the native state of the growth factor [101], bFGF was also released continuously for over 1 month with high retention of immunoreactivity and bioactivity [3]. These formulations were tested in SCID mice, whose primary arteries and veins feeding their right hindlimbs had been ligated and cut, such that, without treatment the limbs did not heal. In the presence of the stabilized formulations containing bFGF not only did the limbs survive (>90% animals retaining limbs after 6 weeks) (see Table 2) but Doppler imaging showed that blood flow was largely restored relative to the healthy left limb (Figs. 1–2), with animals remaining ambulatory. By contrast, in the controls where the growth factor was not properly stabilized or not present, by 6 weeks most of the ischemic limbs did not survive (Table 2) and any that remained had severely impaired blood flow (Fig. 2). Hence, the improved stability of the released growth factor conferred by the incorporation of the basic additive, Mg(OH)2 or MgCO3, demonstrated dramatic therapeutic effects. Additional success with the basic additives was observed with t-PA in cylindrical implants [100] and with tetanus toxoid in microspheres [102].
Table 2.
Rescue of ischemic hindlimbs in the bFGF/PLGA treatment (S) group versus controls (PS, US, B) as a function of time after induction of limb ischemia and treatment.
| Surviving treated limbs/total treated limbs | |||
|---|---|---|---|
|
|
|||
| Formulation | 2 weeks | 4 weeks | 6 weeks |
| S | 14/15 | 9/10 | 5/5 |
| PS | 11/14 | 5/9 | 1/4 |
| US | 12/15 | 4/10 | 0/4 |
| B | 11/12 | 7/8 | 1/4 |
Data from [103].
Fig. 1.
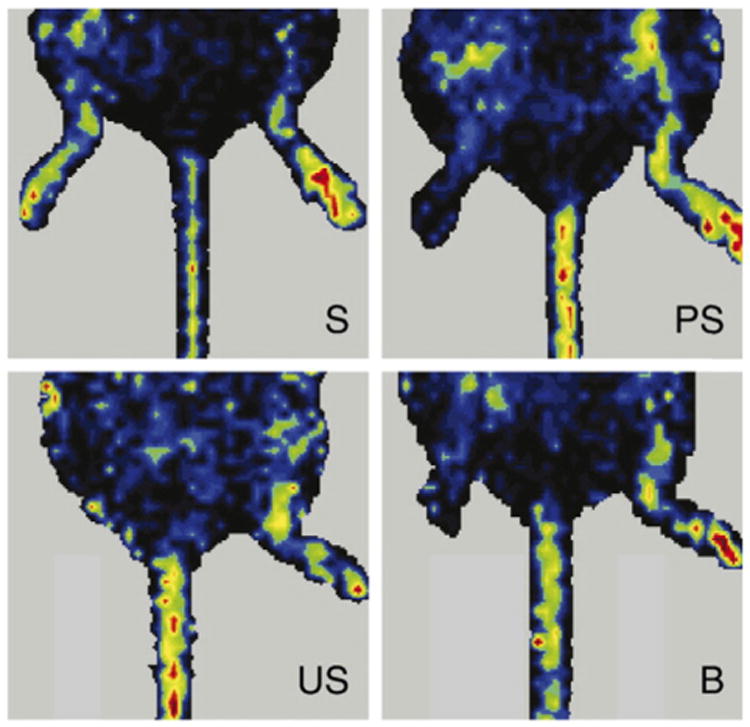
Representative LDPI images of mouse hindlimbs at 6 weeks post-surgery; S — stabilized (bFGF + standard stabilizers + bulk excipient + microclimate control), PS — partially stabilized (bFGF + standard stabilizers + bulk excipient microclimate control), US — unstabilized (bFGF only), and B — blank (no drug + standard stabilizers + bulk excipient + microclimate control). The right hindlimbs (left in the images) were subjected to the surgery to develop ischemia at the beginning. The left hindlimbs (right in the images) were kept intact and represent healthy controls. Standard stabilizers: 0.01% heparin, 0.01% EDTA, and 2.3% sucrose; bulk excipient was 15.7% gum arabic for PS and 12.7% BSA for S and B; microclimate control: 3% Mg(OH)2.
Reproduced with permission from [103].
Fig. 2.
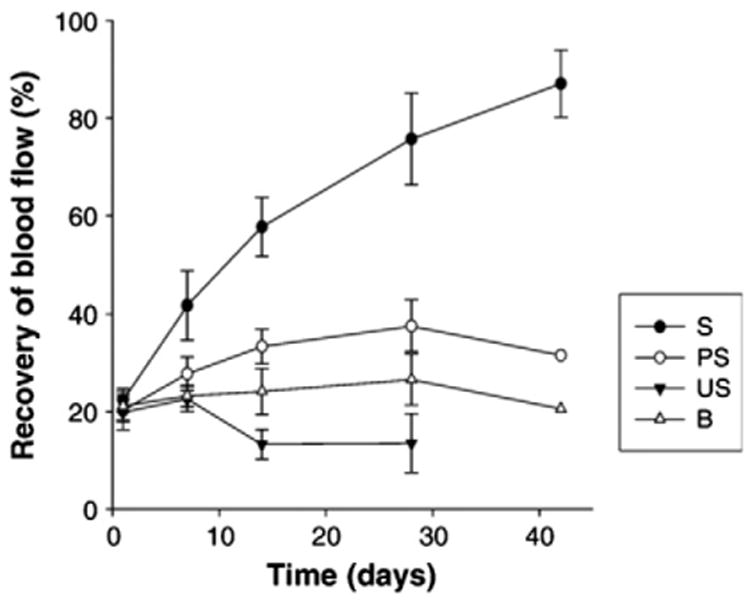
Recovery of hindlimb blood flow over 6 weeks post surgery. The intensity ratios of the right (ligated) to left (healthy) limbs from LDPI images were calculated only for mice with remaining limbs; the values were expressed as mean ± SEM.
Reproduced with permission from [103].
A key step toward defining the role of acid build-up in the polymer was to develop an assay to measure the different acids within the polymer. We accomplished this by derivatizing the acids with the chromophore, bromophenacyl bromide, to form bromophenacyl esters, which then could be distinguished according to their hydrophobicity via their adjusted retention times during reverse phase HPLC analysis [104]. This amino acid analysis like assay was validated, and it clearly showed the kinetics of the distribution of the water-soluble acids, and the significant initial presence of lactoyllactic acid owing from residual lactide in the polymer.
A second important step was measurement of the μpH itself, for which a number of interesting methods have been developed [105–108]. We initially developed a simple method that was based coating thin films of PLGA on working glass electrodes [109]. The method was validated by encapsulating buffering salts that fixed the pH at various values, which were similarly determined by the calibrated glass electrode. This measurement showed that the μpH in PLGA 50/50 could in fact become remarkably low (<3) at the outset, due to the acidic impurities in the polymer. It also showed the tendency for the pH to rise just after incubation, consistent with a burst release of water-soluble acids (before the hydrolytic rate overcame the acid diffusion rate to once again lower the pH value) [109]. The return to acidic levels was not observed with higher lactic-content PLGAs during the one-month time scale of the experiment.
We combined the two earlier studies on assay development in order to test our ability to predict μpH in the thin PLGA films coating glass electrodes [110]. We hypothesized that the water-soluble acids that are present as impurities and then by hydrolysis of the PLGA rapidly partition between the aqueous pore and the polymer. Once in the pore these acids dissociate and provide protons that lower μpH. In the first mathematical model, shown below, we assumed that the water-soluble acids are at equilibrium throughout the polymer, which is a reasonable assumption for a large size polymer film. By making several additional rudimentary assumptions, we could estimate the pH in the pores of the PLGA films by the following transcendental equation (see [110] for details):
| (1) |
where μpHR is the μpH that satisfies the root of the above equation (i.e., when F = 0); Pi is the polymer/water partition coefficient for each water-soluble acid in the polymer; ρp and ρw are the density of the pure polymer and water phases in the polymer matrix; φw is the water uptake; Kai and pKai are the acid distribution coefficient of each acid and is the total molar content of water-soluble acids in the polymer matrix; and XXAi is the mole fraction of each water soluble acid in the polymer matrix. Therefore, the pre-derivatization assay for the water-soluble acids could be used to determine and XXAi as a function of time during PLGA film erosion and simple gravimetry could be used for φw. However, the values of Pi were still needed.
Therefore, Pi was determined directly for the primary μpH-determining acids, GA (glycolic acid), LA (lactic acid) and L2A (lactoyllactic acid), and indirectly for larger acids by extrapolating the linear free energy theory curve between the directly measured Pi's and the pre-derivatization HPLC adjusted retention times [104]. We used the fact that (a) the composition of water soluble acids in films was found to remain fairly constant in the first week of degradation of medium MW PLGA 50/50 (i.v. = ∼0.6 dL/g) [104] and (b) if the films were made to be very thin, the water soluble acids should equilibrate with the polymer as diffusion should be very fast relative to degradation (see for example, speed of BODIPY diffusion [100]).
As seen in Fig. 3, there was a linear relationship between the concentration of GA, LA, and L2A in the polymer (corrected for distribution of acids in the water pores) and the concentration in the aqueous solution. The data indicated that for a medium MW end-capped PLGA, the monomers and oligomers slightly preferred the polymer phase from ∼6 for glycolic acid to ∼100 for one of the oligomers that had significant concentrations in the later part of the release [104,110]. From the linear free energy theory (log(tR(m)′) – log(tR(n)′) = b(m – n)) [111], the data in Table 3 for LA and L2A predict the LA trimer at 1.54 min (oligomer 2) and LA tetramer at 2.09 min (oligomer 4). The absence of any GA dimer in our studies, which would be expected to elute between LA and L2A strongly suggests that this dimer is fairly unstable in water. These studies provide a potential framework to generally evaluate the partitioning behavior of monomers and oligomers in biodegradable polymers.
Fig. 3.

Determination of Pi for primary acids responsible for the lowering of microclimate pH(L2A, LA and GA) by equilibration in very thin PLGA films at 37 °C before significant hydrolysis could occur.
Data from [110].
Table 3.
Determination of Pi of water-soluble acidic monomers and oligomersin PLGA 50/50 films.
By using Eq. (1), and directly measuring via pre-derivatization HPLC, water-soluble acid content in the PLGA films as a function of time (i.e., to give and XXAi as a function of time) the μpH in the glass electrode PLGA film coatings was predicted over a variety of conditions and found to correspond extremely well with the potentiometric determined value. For example, the experimental and predicted values for 3 different polymers are compared in Table 4. The μpH begins low, originating from acidic impurities and begins to rise, particularly for the PLA, which does not produce the acids fast enough to compete with their release from the polymer matrix. We have disclosed initial models that describe the same type of prediction for microspheres taking into account the production and release kinetics of the low MW acids [112], and these results will be published in the near future.
Table 4.
Predicted vs. experimental μpH in PLGA films as a function of composition during incubation in PBST at 37 °C.
The next step to make more practical use of this analysis directly in microsphere products were to monitor the μpH distribution in the polymer. Fu et al. devised a method based on ratiometric imaging of a pH-sensitive dye [113], which we later perfected for two different ranges of measurements, namely, 2.8 < μpH < 5.8 (Lysosensor yellow/blue-dextran) [114] and 5.8 < μpH < 8.0 (SNARF1-dextran) [115] by utilizing a single dye and careful signal processing to create a Gaussian distribution of the standard pH images [114]. These data confirmed that PLGA microspheres of different molecular weights and lactic/glycolic ratios could reach very low values (μpH < 3). These studies were however conducted in the absence of buffering agents of polypeptides loaded within the polymer. Later we conducted a similar study using the acidic dye but in the presence of proteins, BSA and lysozyme [40]. Interestingly, the presence of high concentrations of protein in the polymer pores interfered with the measurement, requiring that standard curves be used in the presence of the relevant concentration of the interfering co-encapsulated drug [40]. From this analysis, the pH in 50/50 microspheres with elevated loading of BSA was commonly in the pH 4–5 range.
3.2.3. Minimize interactions with the polymer
A couple of interesting approaches have been used to decrease the interaction of cationic peptides with the carboxylic acids of acid-end group PLGAs [42,44], which leads to peptide acylation of octreotide [43,44]. As Na and DeLuca postulated [43] that upon peptide adsorption to the polymer, the ε-amino group of octreotide first forms a salt with the PLGA carboxylic acid end group before the α-amino group attacks the polymer backbone to form an amide bond. To block this reaction, the authors PEGylated at either the α- or ε-amino group, or both, and found significant inhibition of the polymer-peptide interaction in a strongly phosphate buffered solution. More significantly, the acylation was completely absent in the presence of either the N-terminal PEGylated or di-PEGylated peptide, but still occurred slightly with the PEGylated peptide at the ε-amino group. The N-terminal PEGylated peptides (2 K and 5 K PEG) were also shown to be biologically active with similar pharmacokinetics in rats with the native peptide [43].
Similarly, we sought to minimize the peptide–polymer interaction by outcompeting the peptide for the polymer carboxylates [44,116, 117]. By adding various water-soluble salts of divalent cations, such as Mn2+ and Ca2+, to suspensions of acid-end group PLGA and the peptide, the octreotide–polymer interaction was strongly inhibited, resulting in a modest decrease in peptide acylation [44]. This strategy also translated into sharp decreases in octreotide acylation when encapsulated in microspheres when formulation conditions were optimized [116]. The approach was found to be augmented in certain cases when carboxymethyl chitosan was added to the formulation to help retain the divalent cationic salts [117].
A second interesting approach to minimize protein polymer interactions, apart from the well-known protein insolubilization strategy with Zn2+ (e.g., Zn-precipitation of human growth hormone in the Nutropin Depot [95]) is to immobilize the protein on a solid or gel support. This was accomplished by binding protein antigens with the aluminum adjuvant, Al(OH)3 ionomeric gel [46,97,98]. We have also disclosed binding of lysozyme to dextran sulfate [118]. When tetanus toxoid (TT) was encapsulated in PLGA microspheres by binding with pre-encapsulated Al(OH)3, the antigen was released with full immunoreactivity over 1 month (see data below). This is in contrast to the extraordinary instability of this antigen recorded by numerous groups with the common issue of protein aggregation [26,29] and loss of immunoreactivity [102,119]. A third possibility to minimize protein/polymer interactions is to introduce PEG blocks in the PLGA or add PEG to the protein [120–122].
3.3. New opportunities for microencapsulation
Interesting microencapsulation approaches have focused on ways to increase control of the polymer size, and to shift the paradigm to remote loading in water. The ability to form unimodal emulsion size distributions has provided numerous examples of successful microsphere formulations of controlled size. Secondly, employing spontaneous self-assembly of polymer chains via healing and absorption processes has allowed our group to create an aqueous remote loading paradigm for large molecules in PLGA.
3.3.1. Exploring the concept of remote loading in PLGA
The Doxil® liposomal formulation for doxorubicin provides precedence for aqueous remote loading of a drug in a parenteral colloidal drug formulation that has reached the clinic. In this example, an empty liposome with a pH gradient across the bi-layer is placed in contact with doxorubicin, which in its freely basic form diffuses across the bi-layer and precipitates as a sulfate salt to achieve high drug loading and encapsulation efficiency [123].
Below two different scientific concepts are described, both of which our group unexpectedly discovered while carrying out mechanistic studies. The initial burst release of peptides and peptide–polymer interactions and processes rely on spontaneous polymer chain self-assembly at temperatures above the hydrated polymer Tg. If we presume that such a remote loading approach could be applied to PLGA-encapsulation of large molecules and commercialized, it is interesting to consider the potential advantages. These advantages can be subdivided into the following general improvements (see also [46]): (a) increased stability of proteins, owing to the absence of organic solvents, interfaces, and high sheer stresses associated with protein micronization; (b) reduced cost of goods associated with manufacture, owing to the ability to (i) sterilize the drug-free polymer before encapsulation allowing the bulk of the manufacturing to be conducted under non-sterile conditions, and (ii) test the drug-free polymer on a small scale before large-scale use encapsulation, (c) the ability to work at lower operating yields with drug-free polymer microspheres (as the API is not yet present) in order to enhance product attributes, such as particle size distribution (useful to reduce needle size); (d) the ability to test controlled release in earlier phases of drug discovery because much lower levels of API are needed for encapsulation; (e) high loading can be achieved from much lower API concentrations than with the solvent evaporation method; and (f) the potential to formulate drug-free microparticles under conditions not normally used because of the presence of an API.
3.3.1.1. Self-healing encapsulation
After discovering the remarkable ability of micron-size or small pores in the surface of PLGA to close spontaneously in water and shut down the initial burst release [18], we have devised a paradigm to microencapsulate large molecules in water by self-assembly of polymer chains to heal defects [46,124]. This paradigm involves first preparing drug-free microspheres (or other polymer geometry/configuration), in which a percolating pore network is created. Percolation can be accomplished by a variety of methods such as encapsulating high levels of osmotic agents, e.g., sugars such as trehalose or sucrose by the solvent evaporation methods [46]. Second, the porous self-encapsulating (SE) microspheres are placed in an aqueous solution containing the drug for encapsulation under mild agitation at a temperature below the hydrated polymer (Tg) to allow entry of the drug deep within the polymer matrix. Third, the polymer is healed by raising the temperature >Tg (typically ∼37–43 °C depending on the polymer MW, end-capping, and presence or absence of an additional plasticizer), which causes both closing of the pores at the surface of the polymer but also a separation of pores within the polymer matrix. The surface pore closure thus encapsulates the drug in the polymer for later controlled release (Fig. 4).
Fig. 4.
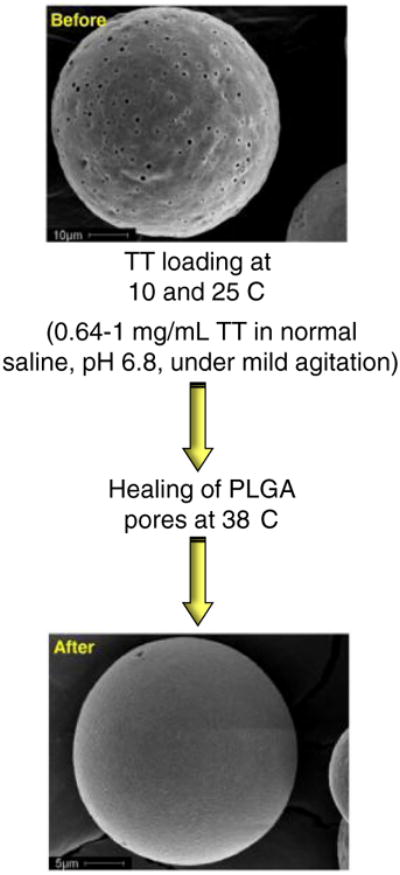
Active self-healing microencapsulation of tetanus toxoid (TT) in PLGA microspheres. The surface morphology of 3.2 wt.% Al(OH)3-PLGA-3.5 wt.% trehalose-5 wt.% diethyl phthalate microspheres before and after encapsulation of the antigen.
Reproduced from [97] with permission.
Note that the above approach can be done either passively, without imposing an additional incentive for the drug to enter the polymer pores, or actively, e.g., by placing an excipient in the SE polymer matrix that traps the drug as it enters the polymer. We refer to this excipient as a “trapping agent.” The first example we used to accomplish active self-encapsulation (ASE) was PLGA with lyoprotected Al(OH)3 adjuvant, which reversibly binds and stabilizes protein antigens. The passive SE approach is limited in terms of the encapsulation efficiency. However, the ASE strategy has achieved >97% efficiency of loading and (with 1–2% w/w antigen load) full immunoreactivity of the tetanus toxoid, an antigen notoriously unstable to PLGA-encapsulation and release (Fig. 5). Moreover, the ASE microspheres could be sterilized by gamma-irradiation before successful encapsulation and release with little change in microsphere performance [98].
Fig. 5.

Controlled release of immunoreactive TT from self-healed PLGA Al(OH)3/PLGA microspheres. The release kinetics of TT is shown for the initial burst (A) and long-term release (B) into PBST + 0.2% BSA at 37 °C Formulations were unencapsulated Al(OH)3 (●), TT/PLGA microspheres prepared by w/o/w emulsion solvent evaporation (▲), and TT/adjuvant/plasticizer/PLGA microspheres prepared by self-healing encapsulation 3.2 wt.% Al(OH)3-PLGA-3.5 wt.% trehalose-5 wt.% diethyl phthalate (DEP) (□), 3.2 wt.% Al(OH)3-PLGA-3 wt.% trehalose-5 wt.% tributyl acetylcitrate (TBAC) (◆), and 3.2 wt.% Al(OH)3-PLGA-1.5 wt.%trehalose-1.5% MgCO3-5 wt.% TBAC (○).
Reproduced from [97] with permission.
3.3.1.2. Peptide absorption encapsulation
Another exciting approach to compliment existing microencapsulation techniques for peptides is based on absorption of peptides in acid end-group PLGA (PLGA-COOH). It has been known for years that small molecules could partition in PLGA (e.g., BODIPY or monomer/oligomer partitioning described above). However, recently while studying the interaction of peptides susceptible to the acylation reaction, we observed a predicted maximal molar sorption of small cationic peptides >600 Da, leuprolide and octreotide, in the vicinity of the molar capacity of end groups of the PLGA (Table 5). Moreover, this sorption was rapid, requiring less than 1 day of exposure to PLGA particles at 37 °C This high level of sorption suggested to us that the peptide was entering the polymer phase. We conducted several studies including (a) peptide sorption at constant surface area but varying polymer thickness, (b) microtoming peptide-sorbed films, (c) stimulated Raman scattering of peptide-sorbed films, and (d) confocal microscopy of uptake of fluorescently labeled peptide [45]. In each case, when temperature was raised above a critical temperature (likely the hydrated Tg of the polymer), we observed evidence to support peptide absorption as follows: (a) steadily more peptide was extracted as film thickness was increased at constant surface area, (b) microtomed films showed steadily less peptide recovery after steadily removing the surface layers of peptide-sorbed films, (c) SRS imaging of peptide-sorbed films bound peptide throughout a ∼20-μm film, and (d) fluorescent confocal imaging showed uniform uptake of dye-conjugated octreotide [45].
Table 5.
Langmuir model fitted parameters a and estimated fraction of acids occupied at maximal peptide absorption after 1 day at 37 °C. Data from [45].
| Peptide | Polymer | K (μM−1) | Γ0 (μmol/g PLGA) | Γmax (μmol/g PLGA) | Total acids (μmol/g PLGA) | Fraction acids occupied b |
|---|---|---|---|---|---|---|
| Leuprolide | RG 502H | 0.77 | 1.5 | 229 | 185 | 1.24 |
| Octreotide | RG 502H | 1.6 | 8.5 | 163 | 185 | 0.88 |
| Octreotide | RG 503H | 1.2 | 2.9 | 81 | 94 | 0.86 |
Sorption values determined by fitting to 1 day absorption data using where Γmax = Γ1 + Γ0.
Fraction acids occupied estimated from ratio of Γmax to total acids determined in polymer.
To demonstrate encapsulation, aqueous leuprolide solution was incubated with low molecular weight PLGA with acid end groups, which had been ground and sieved suitable for injection, at 37 °C for a day. After concentration adjustment during incubation, the peptide had absorbed into the polymer at 17% by wt. (∼70% of acid end groups bound). The polymer controlled the release over several weeks in vitro and demonstrated effective testosterone suppression in rats for 2 months, when injected biweekly as good as that achieved by 2 doses of the 1-month Lupron Depot® [45].
3.3.2. Exploring the concept of particle size and microstructure control
Recent advances have been made for controlling microsphere size during production. Traditional microsphere preparation methods tend to suffer from batch-to-batch variation with large size distribution and issues with microsphere uniformity. Reproducibility and narrow size distributions are important for quality control and syringeability [125]. Microfluidic technology, particularly flow-focusing devices, has been employed to produce uniform, monodisperse PLGA microspheres [126]. In flow-focusing devices, one phase (e.g., dispersed phase) is injected into a stream of another phase (e.g., continuous phase) to form droplets; the fluid streams determine the microsphere size [126, 127]. Other fabrication techniques that allow for fine control of size and uniformity include microsieve emulsification (Nanomi) and Envisia's PRINT® technology [125,126]. In microsieve emulsification, one phase is dispersed into a second phase through a silicon-based microsieve with very precise pore size [125]. PRINT® technology involves rolling a pre-particle liquid into a mold that possesses micro-/ nano-sized features. Once the liquid has hardened (e.g. by solvent evaporation) in the mold cavities, the particle array can be removed and collected. Removal is carried out using an adhesive layer to pull the particles out of the mold; the adhesive is then dissolved, thereby releasing the particles for collection [126].
3.4. What can be done about needle size?
An important aspect that must be considered when designing PLGA microsphere formulations for parenteral delivery is microsphere size. The size affects the syringeability of the formulation and the needle gauge to be used for injection. Generally, smaller microspheres have better syringeability and require a smaller needle diameter. However, below a certain size, microspheres are more prone to phagocytosis. Therefore, it is important to find a size range that balances depot formation, syringeability, and needle gauge [125]. With the advent of microsphere size control, as described above by direct control of emulsion size or with lower yields (without the API) by tightly controlling size between screen sizes according to the aqueous encapsulation paradigm, the prospect of improving needles for patients is highly intriguing. Keep in mind that this can usually be done without large changes to long-term polypeptide release if the Hutchinson [13] approach to continuous release is used, which is not expected to be strongly influenced by microsphere size.
Viscosity enhancers are also typically added to microsphere injection vehicles in order to retard settling of the particles. However, viscosity is typically kept not too high, in order to facilitate mixing, re-suspension of the particles with the vehicle, and to use less force during injection. The Lupron Depot® (mean particle size of ∼8 μm) utilizes an injection vehicle with a viscosity of approximately 5.4 cp at room temprature. The fluid phase of a suspension of Decapeptyl® (mean particle size of ∼40 μm) has a viscosity of approximately 19.7 cp [128]. A systematic study of the effects of microsphere particle size, concentration, diluent density and viscosity on PLGA microsphere solution syringeability and injectability was performed by Alkermes [128]. As expected the use of sieves to eliminate large particles (>180 μm, >150 μm and >125 μm) improved syringeability and injectability of microsphere suspensions. Interestingly the viscosity of diluent was a defining parameter for injection failures. Diluents with viscosities >20 cp at 20 °C had statistically significantly less injection failures due to syringe blockage than the same size and concentration of microspheres suspended in diluents of viscosities of 11 or 2 cp. The higher density of diluents with the same viscosity had a positive effect on injectability, whereas an increase of microsphere concentration from 150 to 300 mg/mL did not appear to have much of the effect. Therefore, the composition of diluent is very important for injectability and selection of needle size. Carboxymethyl cellulose is usually added to increase viscosity at ∼1.5–3% (w/w), sorbitol (up to 30% v/v) is added to increase solution density, polysorbates are added to improve wetting of the microspheres and NaCl, sucrose and buffering salts are used to control pH and osmolality. Strategies are described to improve injectability that involve hydrating microsphere powder with diluent of low viscosity containing wetting agents to assure rapid hydration followed by either adding a second diluent to increase viscosity or by adjusting temperature until the desired viscosity increase is obtained [128].
3.5. Alternative delivery technologies for polypeptides
Several alternative approaches to PLGA delivery of peptides and proteins are currently on the market or in development. Attaching polyethylene glycol chains to protein in order to extend the plasma half-life is the most widely used, as described above. There are a number of approved extended circulation pegylated products such as Cimzia®, Neulasta®, PegIntron® and Somavert® [129]. Other circulation extension approaches include fusing the target protein with human serum albumin (HSA) or the Fc fragment of the antibody as well as altering glycosylation pattern [129]. There are also other technologies including fusion with carboxyl terminal peptide (CTP, Prolor Biotech/Opko Health) [130], hydrophobic amino acid tail fusion using XTEN® technology (Versartis) [131], elastin like polypeptides or ELP technology (PhaseBio). Moreover, several non-invasive protein delivery approaches have reached the market and are currently in development. Exubera®, an inhalable insulin product of Pfizer that uses Nektar's protein inhalation technology was approved by the FDA and EMA in 2003, but pulled from the market [132]. Still, multiple inhalable peptide/protein products are currently being developed, including Afrezza® (MannKind) which has an NDA under review by the FDA as well as a formulation of GLP-1 peptide in the pipeline [133]. A number of technologies for oral absorption of proteins and peptides are in the early stages of preclinical/clinical development that involve the use of permeation enhancers, adhesion patches, nanoparticles and lipid-based nanoemulsions, as reviewed by Park et al. [134].
4. What's next?
There has been significant progress over the last 10–15 years to deepen our understanding and to create new opportunities for controlling the release of large molecules from PLGAs and related polymers. Despite this progress, the most significant advance will be to accelerate commercialization of LAR formulations for large molecules. For proteins, we anticipate that the most significant direct development obstacles are protein stability, manufacturing, and microencapsulation issues. For peptides, the primary issue appears to be the elevated costs of goods associated with manufacturing. Significant advances have been made in PLGAs despite the growing age of this material: for example, understanding of and development of new assays for key phenomena such as polymer healing, peptide absorption, and microclimate pH. Significant strides have also been made in development of new encapsulation approaches that allow fine particle size control and even encapsulation of large molecules in the absence of organic solvent. Both aqueous encapsulation methods (healing and absorption) described here were devised based on mechanistic studies. We believe that with further mechanistic analysis and continued effort in these areas as well as with the aid of PLGA-related materials, it will be possible to develop many more commercial LARs for large molecules. With both peptide and protein LARs, the competition from alternative approaches to controlled release is also significant, particularly with peptide and protein modification and the improved devices and needles to deliver daily soluble injections. This competition makes decreasing needle size and injection convenience of LARs a very significant and under-studied issue to overcome. However, there are still local–regional controlled-release approaches and new therapies such as cell and tissue engineering, which await LARs and reasonably cannot be duplicated by competing technologies with controlled release. Hence, this research area remains a very important and vibrant area to the controlled release and related fields.
Acknowledgments
The authors would like to thank all of the Schwendeman lab members and collaborators who contributed to the data described here. We would also like to thank Dr. Mark Tracy for helpful comments on the manuscript. Support is acknowledged from NIH R01 HL 68345, NIH R21 EB 08873 and FDA U01 FD 005014.
References
- 1.Lupron Depot(R) AbbVie Inc; 2013. package insert. [Google Scholar]
- 2.Eligard(R) TOLMAR Pharmaceuticals; 2014. package insert. [Google Scholar]
- 3.Zhu GZ, Mallery SR, Schwendeman SP. Stabilization of proteins encapsulated ininjectable poly(lactide-co-glycolide) Nat Biotechnol. 2000;18:52–57. doi: 10.1038/71916. [DOI] [PubMed] [Google Scholar]
- 4.Zhou TH, Lewis H, Foster RE, Schwendeman SP. Development of a multiple-drug delivery implant for intraocular management of proliferative vitreoretinopathy. J Control Release. 1998;55:281–295. doi: 10.1016/s0168-3659(98)00061-3. [DOI] [PubMed] [Google Scholar]
- 5.Buse JB, Drucker DJ, Taylor KL, Kim T, Walsh B, Hu H, Wilhelm K, Trautmann M, Shen LZ, Porter LE. DURATION-1: exenatide once weekly produces sustained glycemic control and weight loss over 52 weeks. Diabetes Care. 2010;33:1255–1261. doi: 10.2337/dc09-1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Harris JM, Chess RB. Effect of pegylation on pharmaceuticals. Nat Rev Drug Discov. 2003;2:214–221. doi: 10.1038/nrd1033. [DOI] [PubMed] [Google Scholar]
- 7.GSK; London UK: Apr 15, 2014. Press release: GSK receives US approval for once-weekly type 2 diabetes treatment, TanzeumTM (albiglutide) http://www.gsk.com/media/press-releases/2014/gsk-receives-us-approval-for-once-weekly-type-2-diabetes-treatme.html. [Google Scholar]
- 8.Jiang XR, Song A, Bergelson S, Arroll T, Parekh B, May K, Chung S, Strouse R, Mire-Sluis A, Schenerman M. Advances in the assessment and control of the effector functions of therapeutic antibodies. Nat Rev Drug Discov. 2011;10:101–110. doi: 10.1038/nrd3365. [DOI] [PubMed] [Google Scholar]
- 9.Victoza(R) Novo Nordisk; 2013. package insert. [Google Scholar]
- 10.Alprolix(R) Biogen; 2014. package insert. [Google Scholar]
- 11.Pegasys(R) Hoffmann-La Roche Inc; 2013. package insert. [Google Scholar]
- 12.Fuchs GS, Mikkelsen S, Knudsen TK, Kappelgaard AM. Ease of use and acceptability of a new pen device for the administration of growth hormone therapy in pediatric patients: an open-label, uncontrolled usability test. Clin Ther. 2009;31:2906–2914. doi: 10.1016/j.clinthera.2009.12.014. [DOI] [PubMed] [Google Scholar]
- 13.Hutchinson FG. Continuous release pharmaceutical compositions. U.P. Office; USA: 1982. [Google Scholar]
- 14.Fineman M, Flanagan S, Taylor K, Aisporna M, Shen L, Mace K, Walsh B, Diamant M, Cirincione B, Kothare P, Li WI, MacConell L. Pharmacokinetics and pharmacodynamics of exenatide extended-release after single and multiple dosing. Clin Pharmacokinet. 2011;50:65–74. doi: 10.2165/11585880-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 15.Yeo Y, Park K. Control of encapsulation efficiency and initial burst in polymeric microparticle systems. Arch Pharm Res. 2004;27:1–12. doi: 10.1007/BF02980037. [DOI] [PubMed] [Google Scholar]
- 16.Allison SD. Analysis of initial burst in PLGA microparticles. Expert Opin Drug Deliv. 2008;5:615–628. doi: 10.1517/17425247.5.6.615. [DOI] [PubMed] [Google Scholar]
- 17.Wool RP. Self-healing materials: a review. Soft Matter. 2008;4:400–418. doi: 10.1039/b711716g. [DOI] [PubMed] [Google Scholar]
- 18.Wang J, Wang BM, Schwendeman SP. Characterization of the initial burst release of a model peptide from poly(d, l-lactide-co-glycolide) microspheres. J Control Release. 2002;82:289–307. doi: 10.1016/s0168-3659(02)00137-2. [DOI] [PubMed] [Google Scholar]
- 19.Kang J, Schwendeman SP. Pore closing and opening in biodegradable polymers and their effect on the controlled release of proteins. Mol Pharm. 2006;4:104–118. doi: 10.1021/mp060041n. [DOI] [PubMed] [Google Scholar]
- 20.Mazzara JM, Balagna MA, Thouless MD, Schwendeman SP. Healing kinetics of microneedle-formed pores in PLGA films. J Control Release. 2013;171:172–177. doi: 10.1016/j.jconrel.2013.06.035. [DOI] [PubMed] [Google Scholar]
- 21.Wright SG, Troy C, Yeoh T, Rickey ME, Hotz JM, Kumar R, Costantino HR. Polymer-based Sustained Release Device, US 8461105. 2013 [Google Scholar]
- 22.Husmann M, Schenderlein S, Luck M, Lindner H, Kleinebudde P. Polymer erosion in PLGA microparticles produced by phase separation method. Int J Pharm. 2002;242:277–280. doi: 10.1016/s0378-5173(02)00187-4. [DOI] [PubMed] [Google Scholar]
- 23.Howard Bernstein YZ, Amin Khan M, Tracy Mark A. US 6,749,866. Modulated Release From Bio-compatible Polymers. 2004
- 24.Zhu G, Schwendeman SP. Stabilization of proteins encapsulated in cylindrical poly(lactide-co-glycolide) implants: mechanism of stabilization by basic additives. Pharm Res. 2000;17:351–357. doi: 10.1023/a:1007513425337. [DOI] [PubMed] [Google Scholar]
- 25.Jiang W, Schwendeman SP. Stabilization and controlled release of bovine serum albumin encapsulated in poly(d, l-lactide) and poly(ethylene glycol) microsphere blends. Pharm Res. 2001;18:878–885. doi: 10.1023/a:1011009117586. [DOI] [PubMed] [Google Scholar]
- 26.Jiang W, Schwendeman SP. Stabilization of a model formalinized protein antigen encapsulated in poly(lactide-co-glycolide)-based microspheres. J Pharm Sci. 2001;90:1558–1569. doi: 10.1002/jps.1106. [DOI] [PubMed] [Google Scholar]
- 27.Langer RS, Peppas NA. Present and future applications of biomaterials in controlled drug delivery systems. Biomaterials. 1981;2:201–214. doi: 10.1016/0142-9612(81)90059-4. [DOI] [PubMed] [Google Scholar]
- 28.Xichen Z, Wyss UP, Pichora D, Goosen MFA. A mechanistic study of antibiotic release from biodegradable poly(d,l-lactide) cylinders. J Control Release. 1994;31:129–144. [Google Scholar]
- 29.Schwendeman SP, Cardamone M, Brandon MR, Klibanov A, Langer R. Stability of proteins and their delivery from biodegradable polymer microspheres. In: Bernstein H, Cohen S, editors. Microparticulate Systems for the Delivery of Proteins and Vaccines. Marcel Dekker Inc; New York: 1996. pp. 1–49. [Google Scholar]
- 30.Putney SD, Burke PA. Improving protein therapeutics with sustained-release formulations. Nat Biotechnol. 1998;16:153–157. doi: 10.1038/nbt0298-153. [DOI] [PubMed] [Google Scholar]
- 31.Kissel T, Koneberg R. Injectable biodegradable microspheres for vaccine delivery. In: Bernstein H, Cohen S, editors. Microparticulate Systems for the Delivery of Proteins and Vaccines. Marcel Dekker Inc; New York: 1996. pp. 51–88. [Google Scholar]
- 32.van de Weert M, Hennink WE, Jiskoot W. Protein instability in poly(lactic-co-glycolic acid) microparticles. Pharm Res. 2000;17:1159–1167. doi: 10.1023/a:1026498209874. [DOI] [PubMed] [Google Scholar]
- 33.Schwendeman SP. Recent advances in the stabilization of proteins encapsulated in injectable PLGA delivery systems. Crit Rev Ther Drug Carrier Syst. 2002;19:73–98. doi: 10.1615/critrevtherdrugcarriersyst.v19.i1.20. [DOI] [PubMed] [Google Scholar]
- 34.Pérez C, Castellanos IJ, Costantino HR, Al-Azzam W, Griebenow K. Recent trends in stabilizing protein structure upon encapsulation and release from bioerodible polymers. J Pharm Pharmacol. 2002;54:301–313. doi: 10.1211/0022357021778448. [DOI] [PubMed] [Google Scholar]
- 35.Tamber H, Johansen P, Merkle HP, Gander B. Formulation aspects of biodegradable polymeric microspheres for antigen delivery. Adv Drug Deliv Rev. 2005;57:357–376. doi: 10.1016/j.addr.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 36.Brown LR. Commercial challenges of protein drug delivery. Expert Opin Drug Deliv. 2005;2:29–42. doi: 10.1517/17425247.2.1.29. [DOI] [PubMed] [Google Scholar]
- 37.van der Walle CF, Sharma G, Kumar MR. Current approaches to stabilising and analysing proteins during microencapsulation in PLGA. Expert Opin Drug Deliv. 2009;6:177–186. doi: 10.1517/17425240802680169. [DOI] [PubMed] [Google Scholar]
- 38.Fu K, Klibanov AM, Langer R. Protein stability in controlled-release systems. Nat Biotechnol. 2000;18:24–25. doi: 10.1038/71875. [DOI] [PubMed] [Google Scholar]
- 39.Volkin DB, Klibanov AM. Minimizing protein inactivation. In: Creighton TE, editor. Protein Function: A Practical Approach. Oxford Univ Press; Oxford: 1989. pp. 1–24. [Google Scholar]
- 40.Liu Y, Schwendeman SP. Mapping microclimate pH distribution inside protein-encapsulated PLGA microspheres using confocal laser scanning microscopy. Mol Pharm. 2012;9:1342–1350. doi: 10.1021/mp200608y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Butler SM, Tracy MA, Tilton RD. Adsorption of serum albumin to thin films of poly(lactide-co-glycolide) J Control Release. 1999;58:335–347. doi: 10.1016/s0168-3659(98)00173-4. [DOI] [PubMed] [Google Scholar]
- 42.Okada H, Toguchi H. Biodegradable microspheres in drug delivery. Crit Rev Ther Drug Carrier Syst. 1995;12:1–99. doi: 10.1615/critrevtherdrugcarriersyst.v12.i1.10. [DOI] [PubMed] [Google Scholar]
- 43.Na DH, DeLuca PP. PEGylation of octreotide: I. Separation of positional isomers and stability against acylation by poly(d,l-lactide-co-glycolide) Pharm Res. 2005;22:736–742. doi: 10.1007/s11095-005-2589-4. [DOI] [PubMed] [Google Scholar]
- 44.Sophocleous AM, Zhang Y, Schwendeman SP. A new class of inhibitors of peptide sorption and acylation in PLGA. J Control Release. 2009;137:179–184. doi: 10.1016/j.jconrel.2009.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sophocleous AM, Desai KG, Mazzara JM, Tong L, Cheng JX, Olsen KF, Schwendeman SP. The nature of peptide interactions with acid end-group PLGAs and facile aqueous-based microencapsulation of therapeutic peptides. J Control Release. 2013;172:662–670. doi: 10.1016/j.jconrel.2013.08.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Reinhold SE, Desai KGH, Zhang L, Olsen KF, Schwendeman SP. Self-healing microencapsulation of biomacromolecules without organic solvents. Angew Chem Int Ed. 2012;51:10800–10803. doi: 10.1002/anie.201206387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Reinhold SE, Schwendeman SP. Effect of polymer porosity on aqueous self-healing encapsulation of proteins in PLGA microspheres. Macromol Biosci. 2013;13:1700–1710. doi: 10.1002/mabi.201300323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yamamoto M, Okada H, Ogawa Y, Miyagawa T. US4849228. Polymer, Production and Use Thereof. 1989
- 49.Khossravi D, Connors K. Solvent effects on chemical processes. 3. Surface tension of binary aqueous organic solvents. J Solution Chem. 1993;22:321–330. [Google Scholar]
- 50.Rouse JJ, Mohamed F, van der Walle CF. Physical ageing and thermal analysis of PLGA microspheres encapsulating protein or DNA. Int J Pharm. 2007;339:112–120. doi: 10.1016/j.ijpharm.2007.02.026. [DOI] [PubMed] [Google Scholar]
- 51.Gill HS, Prausnitz MR. Does needle size matter? J Diabetes Sci Technol. 2007;1:725–729. doi: 10.1177/193229680700100517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Buchholz SH. G Henke's Med-Math, Dosage Calculation, Preparation and Administration. 6th. Vol. 1996. Lippincott Williams & Wilkins; Philadelphia, PA: 2008. [Google Scholar]
- 53.Bydureon(R) AstraZeneca; 2013. package insert. [Google Scholar]
- 54.DeYoung MB, MacConell L, Sarin V, Trautmann M, Herbert P. Encapsulation of exenatide in poly-(d,l-lactide-co-glycolide) microspheres produced an investigational long-acting once-weekly formulation for type 2 diabetes. Diabetes Technol Ther. 2011;13:1145–1154. doi: 10.1089/dia.2011.0050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sartor O. Eligard: leuprolide acetate in a novel sustained-release delivery system. Urology. 2003;61:25–31. doi: 10.1016/s0090-4295(02)02396-8. [DOI] [PubMed] [Google Scholar]
- 56.Zoladex(R) AstraZeneca; 2013. package insert. [Google Scholar]
- 57.Floyd AG. Injectable emulsions and suspensions. In: Lieberman HA, Rieger MM, Banker GS, Dekker Marcel, editors. Pharmaceutical Dosage Forms, Disperse Systems. New York: 1996. [Google Scholar]
- 58.Rubin RR, Peyrot M, Kruger DF, Travis LB. Barriers to insulin injection therapy: patient and health care provider perspectives. Diabetes Educ. 2009;35:1014–1022. doi: 10.1177/0145721709345773. [DOI] [PubMed] [Google Scholar]
- 59.Arendt-Nielsen L, Egekvist H, Bjerring P. Pain following controlled cutaneous insertion of needles with different diameters. Somatosens Mot Res. 2006;23:37–43. doi: 10.1080/08990220600700925. [DOI] [PubMed] [Google Scholar]
- 60.Cox MC, Scripture CD, Figg WD. Leuprolide acetate given by a subcutaneous extended-release injection: less of a pain? Expert Rev Anticancer Ther. 2005;5:605–611. doi: 10.1586/14737140.5.4.605. [DOI] [PubMed] [Google Scholar]
- 61.Kinoshita H, Kawa G, Hiura Y, Chizaki R, Matsuda T. Effectiveness of skin icing in reducing pain associated with goserelin acetate injection. Int J Clin Oncol. 2010;15:472–475. doi: 10.1007/s10147-010-0095-0. [DOI] [PubMed] [Google Scholar]
- 62.Montgomery BS, Borwell JP, Higgins DM. Does needle size matter? Patient experience of luteinising hormone-releasing hormone analogue injection. Prostate Cancer Prostatic Dis. 2005;8:66–68. doi: 10.1038/sj.pcan.4500778. [DOI] [PubMed] [Google Scholar]
- 63.Langer R, Tirrell DA. Designing materials for biology and medicine. Nature. 2004;428:487–492. doi: 10.1038/nature02388. [DOI] [PubMed] [Google Scholar]
- 64.Pillai O, Panchagnula R. Polymers in drug delivery. Curr Opin Chem Biol. 2001;5:447–451. doi: 10.1016/s1367-5931(00)00227-1. [DOI] [PubMed] [Google Scholar]
- 65.Liechty WB, Kryscio DR, Slaughter BV, Peppas NA. Polymers for drug delivery systems. Annu Rev Chem Biomol. 2010;1:149–173. doi: 10.1146/annurev-chembioeng-073009-100847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhang Y, Chan HF, Leong KW. Advanced materials and processing for drug delivery: the past and the future. Adv Drug Deliv Rev. 2013;65:104–120. doi: 10.1016/j.addr.2012.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Grund S, Bauer M, Fischer D. Polymers in drug delivery—state of the art and future trends. Adv Eng Mater. 2011;13:B61–B87. [Google Scholar]
- 68.Park K, Mrsny RJ, editors. Controlled drug delivery: designing technologies for the future. Am Chem Soc. 2000;752:1–459. [Google Scholar]
- 69.Nair LS, Laurencin CT. Biodegradable polymers as biomaterials. Prog Polym Sci. 2007;32:762–798. [Google Scholar]
- 70.Karp JM, Langer R. Development and therapeutic applications of advanced biomaterials. Curr Opin Biotechnol. 2007;18:454–459. doi: 10.1016/j.copbio.2007.09.008. [DOI] [PubMed] [Google Scholar]
- 71.Park K, Mrsny RJ. Controlled drug delivery: designing technologies for the future. Am Chem Soc. 2000;752:2–12. [Google Scholar]
- 72.Long WA, Takov D, Tchernev K, et al. Q2 week controlled-release-interferon-alpha2b + ribavrin reduces flu-like symptoms >50% and provides equivalent efficacy in comparison to weekly PEGylated- interferon-alpha2b + ribavirin in treatment-naive-genotype-1-chronic-hepatitis-C: results from EMPOWER, a randomized-open-label-12-week-comparison in 133 patients. 45th Annual Meeting of the European Association for the Study of the Liver (EASL 2010); Vienna, Austria. 2010. [Google Scholar]
- 73.De Leede LG, Humphries JE, Bechet AC, Van Hoogdalem EJ, Verrijk R, Spencer DG. Novel controlled-release Lemna-derived IFN-alpha2b (Locteron): pharmacokinetics, pharmacodynamics, and tolerability in a phase I clinical trial. J Interf Cytokine Res. 2008;28:113–122. doi: 10.1089/jir.2007.0073. [DOI] [PubMed] [Google Scholar]
- 74.de Rio CL, George R, Kloepfer P, Ueyama Y, Youngblood B, Georgopoulos L, Arnold S, Hamlin RL. VASOMERATM, a novel vpac2-selective vasoactive intestinal peptide agonist, enhances contractility and decreases myocardial demand in dogs with both normal hearts and with pacing-induced dilated cardiomyopathy. J Am Coll Cardiol. 2013;61 [Google Scholar]
- 75.Woodruff MA, Hutmacher DW. The return of a forgotten polymer— polycaprolactone in the 21st century. Prog Polym Sci. 2010;35:1217–1256. [Google Scholar]
- 76.Ghassemi AH, van Steenbergen MJ, Barendregt A, Talsma H, Kok RJ, van Nostrum CF, Crommelin DJA, Hennink WE. Controlled release of octreotide and assessment of peptide acylation from poly(d, l-lactide-co-hydroxymethyl glycolide) compared to PLGA microspheres. Pharm Res. 2012;29:110–120. doi: 10.1007/s11095-011-0517-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kim S, Seong K, Kim O, Kim S, Seo H, Lee M, Khang G, Lee D. Polyoxalate nanoparticles as a biodegradable and biocompatible drug delivery vehicle. Biomacromolecules. 2010;11:555–560. doi: 10.1021/bm901409k. [DOI] [PubMed] [Google Scholar]
- 78.Yang SC, Bhide M, Crispe IN, Pierce RH, Murthy N. Polyketal copolymers: A new acid-sensitive delivery vehicle for treating acute inflammatory diseases. Bioconjug Chem. 2008;19:1164–1169. doi: 10.1021/bc700442g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Butler D. Translational research: crossing the valley of death. Nature. 2008;453:840–842. doi: 10.1038/453840a. [DOI] [PubMed] [Google Scholar]
- 80.Carmichael M. Why Don't More Medical Discoveries Become Cures? Newsweek USA. 2010 May 14; [Google Scholar]
- 81.Kim HK, Lee KH, Oh JG, Lee BY. Method for Preparing Microparticles With Reduced Initial Burst and Microparticles Prepared Thereby. EP2709594. 2014
- 82.Kamei S, Yamada M, Ogawa Y. US5575987. Method of Producing Sustained-release Microcapsules. 1996
- 83.Bouissou C, Rouse JJ, Price R, van der Walle CF. The influence of surfactant on PLGA microsphere glass transition and water sorption: remodeling the surface morphology to attenuate the burst release. Pharm Res. 2006;23:1295–1305. doi: 10.1007/s11095-006-0180-2. [DOI] [PubMed] [Google Scholar]
- 84.Kim HK, Chung HJ, Park TG. Biodegradable polymeric microspheres with “open/ closed” pores for sustained release of human growth hormone. J Control Release. 2006;112:167–174. doi: 10.1016/j.jconrel.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 85.Kim TH, Park TG. Critical effect of freezing/freeze-drying on sustained release of FITC-dextran encapsulated within PLGA microspheres. Int J Pharm. 2004;271:207–214. doi: 10.1016/j.ijpharm.2003.11.021. [DOI] [PubMed] [Google Scholar]
- 86.Jeyanthi R, Thanoo BC, Metha RC, DeLuca PP. Effect of solvent removal technique on the matrix characteristics of polylactide/glycolide microspheres for peptide delivery. J Control Release. 1996;38:235–244. [Google Scholar]
- 87.Viswanathan NB, Thomas PA, Pandit JK, Kulkarni MG, Mashelkar RA. Preparation of non-porous microspheres with high entrapment efficiency of proteins by a (water-in-oil)-in-oil emulsion technique. J Control Release. 1999;58:9–20. doi: 10.1016/s0168-3659(98)00140-0. [DOI] [PubMed] [Google Scholar]
- 88.Gopferich A, Alonso MJ, Langer R. Development and characterization of microencapsulated microspheres. Pharm Res. 1994;11:1568–1574. doi: 10.1023/a:1018901619230. [DOI] [PubMed] [Google Scholar]
- 89.Hinds KD, Campbell KM, Holland KM, Lewis DH, Piche CA, Schmidt PG. PEGylated insulin in PLGA microparticles. In vivo and in vitro analysis. J Control Release. 2005;104:447–460. doi: 10.1016/j.jconrel.2005.02.020. [DOI] [PubMed] [Google Scholar]
- 90.Wang S, Cui W, Bei J. Bulk and surface modifications of polylactide. Anal Bioanal Chem. 2005;381:547–556. doi: 10.1007/s00216-004-2771-2. [DOI] [PubMed] [Google Scholar]
- 91.Alcock R, Blair JA, O'Mahony DJ, Raoof A, Quirk AV. Modifying the release of leuprolide from spray dried OED microparticles. J Control Release. 2002;82:429–440. doi: 10.1016/s0168-3659(02)00165-7. [DOI] [PubMed] [Google Scholar]
- 92.Fu K, Harrell R, Zinski K, Um C, Jaklenec A, Frazier J, Lotan N, Burke P, Klibanov AM, Langer R. A potential approach for decreasing the burst effect of protein from PLGA microspheres. J Pharm Sci. 2003;92:1582–1591. doi: 10.1002/jps.10414. [DOI] [PubMed] [Google Scholar]
- 93.Castellanos IJ, Flores G, Griebenow K. Effect of cyclodextrins on alpha-chymotrypsin stability and loading in PLGA microspheres upon S/O/W encapsulation. J Pharm Sci. 2006;95:849–858. doi: 10.1002/jps.20512. [DOI] [PubMed] [Google Scholar]
- 94.Huang X, Brazel CS. On the importance and mechanisms of burst release in matrix-controlled drug delivery systems. J Control Release. 2001;73:121–136. doi: 10.1016/s0168-3659(01)00248-6. [DOI] [PubMed] [Google Scholar]
- 95.Johnson OL, Cleland JL, Lee HJ, Charnis M, Duenas E, Jaworowicz W, Shepard D, Shahzamani A, Jones AJS, Putney SD. A month-long effect from a single injection of microencapsulated human growth hormone. Nat Med. 1996;2:795–799. doi: 10.1038/nm0796-795. [DOI] [PubMed] [Google Scholar]
- 96.Johnson OL, Jaworowicz W, Cleland JL, Bailey L, Charnis M, Duenas E, Wu CC, Shepard D, Magil S, Last T, Jones AJS, Putney SD. The stabilization and encapsulation of human growth hormone into biodegradable microspheres. Pharm Res. 1997;14:730–735. doi: 10.1023/a:1012142204132. [DOI] [PubMed] [Google Scholar]
- 97.Desai KGH, Schwendeman SP. Active self-healing encapsulation of vaccine antigens in PLGA microspheres. J Control Release. 2013;165:62–74. doi: 10.1016/j.jconrel.2012.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Desai KGH, Kadous S, Schwendeman SP. Gamma irradiation of active self-healing PLGA microspheres for efficient aqueous encapsulation of vaccine antigens. Pharm Res. 2013;30:1768–1778. doi: 10.1007/s11095-013-1019-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Estey T, Kang J, Schwendeman SP, Carpenter JF. BSA degradation under acidic conditions: a model for protein instability during release from PLGA delivery systems. J Pharm Sci. 2006;95:1626–1639. doi: 10.1002/jps.20625. [DOI] [PubMed] [Google Scholar]
- 100.Kang JC, Schwendeman SP. Comparison of the effects of Mg(OH)2 and sucrose on the stability of bovine serum albumin encapsulated in injectable poly(d, l-lactide-co-glycolide) implants. Biomaterials. 2002;23:239–245. doi: 10.1016/s0142-9612(01)00101-6. [DOI] [PubMed] [Google Scholar]
- 101.Sommer A, Rifkin DB. Interaction of heparin with human basic fibroblast growth factor: protection of the angiogenic protein from proteolytic degradation by a glycosaminoglycan. J Cell Physiol. 1989;138:215–220. doi: 10.1002/jcp.1041380129. [DOI] [PubMed] [Google Scholar]
- 102.Jiang WL, Schwendeman SP. Stabilization of tetanus toxoid encapsulated in PLGA microspheres. Mol Pharm. 2008;5:808–817. doi: 10.1021/mp800027f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hiemstra C, Zhong Z, Van Tomme SR, van Steenbergen MJ, Jacobs JJL, Otter WD, Hennink WE, Feijen J. In vitro and in vivo protein delivery from in situ forming poly (ethylene glycol)-poly (lactide) hydrogels. J Control Release. 2007;119:320–327. doi: 10.1016/j.jconrel.2007.03.014. [DOI] [PubMed] [Google Scholar]
- 104.Ding AG, Schwendeman SP. Determination of water-soluble acid distribution inpoly(lactide-co-glycolide) J Pharm Sci. 2004;93:322–331. doi: 10.1002/jps.10524. [DOI] [PubMed] [Google Scholar]
- 105.Mader K, Gallez B, Liu KJ, Swartz HM. Non-invasive in vivo characterization of release processes in biodegradable polymers by low-frequency electron paramagnetic resonance spectroscopy. Biomaterials. 1996;17:457–461. doi: 10.1016/0142-9612(96)89664-5. [DOI] [PubMed] [Google Scholar]
- 106.Mader K, Bittner B, Li Y, Wohlauf W, Kissel T. Monitoring microviscosity and microacidity of the albumin microenvironment inside degrading microparticles from poly(lactide-co-glycolide) (PLG) or ABA-triblock polymers containing hydrophobic poly(lactide-co-glycolide) A blocks and hydrophilic poly(ethyleneoxide) B blocks. Pharm Res. 1998;15:787–793. doi: 10.1023/a:1011939607573. [DOI] [PubMed] [Google Scholar]
- 107.Brunner A, Mader K, Gopferich A. pH and osmotic pressure inside biodegradablemicrospheres during erosion. Pharm Res. 1999;16:847–853. doi: 10.1023/a:1018822002353. [DOI] [PubMed] [Google Scholar]
- 108.Fu K, Pack DW, Klibanov AM, Langer R. Visual evidence of acidic environment within degrading poly(lactic-co-glycolic acid) (PLGA) microspheres. Pharm Res. 2000;17:100–106. doi: 10.1023/a:1007582911958. [DOI] [PubMed] [Google Scholar]
- 109.Shenderova A, Ding AG, Schwendeman SP. Potentiometric method for determination of microclimate pH in poly(lactic-co-glycolic acid) films. Macromolecules. 2004;37:10052–10058. [Google Scholar]
- 110.Ding AG, Shenderova A, Schwendeman SP. Prediction of microclimate pH inpoly(lactic-co-glycolic acid) films. J Am Chem Soc. 2006;128:5384–5390. doi: 10.1021/ja055287k. [DOI] [PubMed] [Google Scholar]
- 111.Hawkes SJ. Predicting retention data using the slope of the log adjusted retention time vs. carbon number plot for n-alkanes. Chromatographia. 1988;25:313–318. [Google Scholar]
- 112.Schwendeman SP, Liu Y. Simulation of microclimate pH distribution and kinetics inside degrading PLGA microspheres, poster present; 2013 AAPS Annual Meeting; San Antonio, TX. 2013. [Google Scholar]
- 113.Fu K, Pack D, Klibanov A, Langer R. Visual evidence of acidic environment within degrading poly(lactic-co-glycolic acid) (PLGA) microspheres. Pharm Res. 2000;17:100–106. doi: 10.1023/a:1007582911958. [DOI] [PubMed] [Google Scholar]
- 114.Li L, Schwendeman SP. Mapping neutral microclimate pH in PLGA microspheres. J Control Release. 2005;101:163–173. doi: 10.1016/j.jconrel.2004.07.029. [DOI] [PubMed] [Google Scholar]
- 115.Ding AG, Schwendeman SP. Acidic microclimate pH distribution in PLGA microspheres monitored by confocal laser scanning microscopy. Pharm Res. 2008;25:2041–2052. doi: 10.1007/s11095-008-9594-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Zhang Y, Sophocleous AM, Schwendeman SP. Inhibition of peptide acylation in PLGA microspheres with water-soluble divalent cationic salts. Pharm Res. 2009;26:1986–1994. doi: 10.1007/s11095-009-9914-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Zhang Y, Schwendeman SP. Minimizing acylation of peptides in PLGA microspheres. J Control Release. 2012;162:119–126. doi: 10.1016/j.jconrel.2012.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Shah RB, Schwendeman SP. A biomimetic approach to active self-healing micro-encapsulation of proteins in PLGA, poster present; AAPS Annual Meeting; Chicago, IL. 2013. [Google Scholar]
- 119.Johansen P, Merkle HP, Gander B. Physico-chemical and antigenic properties of tetanus and diphtheria toxoids and steps towards improved stability. Biochim Biophys Acta. 1998;1425:425–436. doi: 10.1016/s0304-4165(98)00097-x. [DOI] [PubMed] [Google Scholar]
- 120.Luck M, Pistel KF, Li YX, Blunk T, Muller RH, Kissel T. Plasma protein adsorption on biodegradable microspheres consisting of poly(d, l-lactide-co-glycolide), poly(l-lactide) or ABA triblock copolymers containing poly(oxyethylene). Influence of production method and polymer composition. J Control Release. 1998;55:107–120. doi: 10.1016/s0168-3659(98)00030-3. [DOI] [PubMed] [Google Scholar]
- 121.Castellanos IJ, Al-Azzam W, Griebenow K. Effect of the covalent modification with poly(ethylene glycol) on alpha-chymotrypsin stability upon encapsulation in poly(lactic-co-glycolic) microspheres. J Pharm Sci. 2005;94:327–340. doi: 10.1002/jps.20243. [DOI] [PubMed] [Google Scholar]
- 122.Daly SM, Przybycien TM, Tilton RD. Adsorption of poly(ethylene glycol)-modified ribonuclease A to a poly(lactide-co-glycolide) surface. Biotechnol Bioeng. 2005;90:856–868. doi: 10.1002/bit.20481. [DOI] [PubMed] [Google Scholar]
- 123.Gabizon A, Shmeeda H, Grenader T. Pharmacological basis of pegylated liposomal doxorubicin: impact on cancer therapy. Eur J Pharm Sci. 2012;45:388–398. doi: 10.1016/j.ejps.2011.09.006. [DOI] [PubMed] [Google Scholar]
- 124.Schwendeman SP, Reinhold SE, Kang J. US8017155. Immersing and Soaking Insoluble Pore Containing Polymer Into Encapsulating Solution Containing Active Ingredient Then Initiating Pore Closing Rearrangement by Adjusting Temperature and/or pH. 2011
- 125.Kyekyoon KK, Pack DW. Microspheres for Drug Delivery. Springer; New York, NY: 2006. [Google Scholar]
- 126.Xu QB, Hashimoto M, Dang TT, Hoare T, Kohane DS, Whitesides GM, Langer R, Anderson DG. Preparation of monodisperse biodegradable polymer microparticles using a microfluidic flow-focusing device for controlled drug delivery. Small. 2009;5:1575–1581. doi: 10.1002/smll.200801855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Veldhuis G, Gironès M, Bingham D. Monodisperse microspheres for parenteral drug delivery. Drug Deliv Technol. 2009;9:24–31. [Google Scholar]
- 128.Ramstack JM, Riley GI, Zale SE, Hotz JM, Johnson OFL. US6667061. Preparation of Injectable Suspensions Having Improved Injectability. 2003
- 129.Kontermann R. Therapeutic Proteins: Strategies to Modulate Their Plasma Half-lives. Wiley; 2012. [Google Scholar]
- 130.Fares F, Guy R, Bar-Ilan A, Felikman Y, Fima E. Designing a long-acting human growth hormone (hGH) by fusing the carboxyl-terminal peptide of human chorionic gonadotropin beta-subunitto the coding sequence of hGH. Endocrinology. 2010;151:4410–4417. doi: 10.1210/en.2009-1431. [DOI] [PubMed] [Google Scholar]
- 131.Yuen KC, Conway GS, Popovic V, Merriam GR, Bailey T, Hamrahian AH, Biller BM, Kipnes M, Moore JA, Humphriss E, Bright GM, Cleland JL. A long-acting human growth hormone with delayed clearance (VRS-317): results of a double-blind, placebo- controlled, single ascending dose study in growth hormone-deficient adults. J Clin Endocrinol Metab. 2013;98:2595–2603. doi: 10.1210/jc.2013-1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Heinemann L. The failure of Exubera: are we beating a dead horse? J Diabetes Sci Technol. 2008;2:518–529. doi: 10.1177/193229680800200325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Mankind Corporation Pipeline. 2014 [Google Scholar]
- 134.Park K, Kwon IC, Park K. Oral protein delivery: current status and future prospect. React Funct Polym. 2011;71:280–287. [Google Scholar]