

Abstract
Somatostatin receptor subtype 2 (sstr2) is a G-protein-coupled receptor (GPCR) that is overexpressed in neuroendocrine tumors. The homology model of sstr2 was built and was used to aid the design of new somatostatin analogues modified with phosphonate-containing cross-bridged chelators for evaluation of using them as PET imaging radiopharmaceuticals. The new generation chelators were conjugated to Tyr3-octreotate (Y3-TATE) through bioorthogonal, strain-promoted alkyne azide cycloaddition (SPAAC) to form CB-TE1A1P–DBCO–Y3-TATE (AP) and CB-TE1K1P–PEG4–DBCO–Y3-TATE (KP) in improved yields compared to standard direct conjugation methods of amide bond formation. Consistent with docking studies, the clicked bioconjugates showed high binding affinities to sstr2, with Kd values ranging from 0.6 to 2.3 nM. Selected isomers of the clicked products were used in biodistribution and PET/CT imaging. Introduction of the bulky dibenzocyclooctyne group in AP decreased clearance rates from circulation. However, the additional carboxylate group and PEG linker from the KP conjugate significantly improved labeling conditions and in vivo stability of the copper complex and ameliorated the slower pharmacokinetics of the clicked somatostatin analogues.
Introduction
Copper radionuclides, such as 60Cu, 61Cu, 62Cu, 64Cu, and 67Cu, have applications in nuclear medicine for SPECT, PET, and/or targeted radiotherapy. Copper-64, with a half-life of 12.7 h, and its unique decay profile (β+: 18%; β–: 38%; electron capture: 44%), is well suited for radiolabeling biomolecules.1 However, due to the presence of copper-chelating proteins in vivo, a challenge of developing copper-based radiopharmaceuticals is the in vivo stability.2 There is interest in developing new copper chelators for improved copper radiopharmaceuticals.3,4 Our laboratory has a long history of developing copper chelators, with CB-TE2A being one of the gold standards for forming kinetically stable chelates with Cu(II).5 Concomitant with its high in vivo stability is the challenge of radiolabeling under mild conditions. The phosphonate-pendant-armed cross-bridged chelator, CB-TE1A1P, can be labeled with Cu-64 at ambient temperature in high specific activity.6 However, when the carboxylate pendant arm in CB-TE1A1P was utilized for conjugation to the sstr2 analogue, Y3-TATE, the requirements for room temperature labeling were compromised, and the synthesis yield was very low.7 With an aim to further optimize peptide conjugates with phosphonate-based cross-bridged chelators, CB-TE1K1P was synthesized with a homolysine pendant arm replacing the carboxylate pendant arm of CB-TE1A1P, which conserved the carboxylate and phosphonate pendant arms after conjugation to biomolecules.8 Because of the challenges of conjugating the phosphonate-based chelators with biomolecules directly through peptide bond formation, including low yields and requirement of higher temperatures for labeling, click chemistry was applied as the bioconjugation method.
The Huisgen 1,3-dipolar cycloaddition, also called copper-catalyzed alkyne–azide cycloaddition (CuAAC) click chemistry,9 has been applied extensively in the development of radiopharmaceuticals since its introduction to this field.10,11 Struthers et al. reviewed the development of metal chelating systems using this strategy.12 Strain-promoted copper-free click chemistry (also called strain-promoted alkyne azide cycloaddition, SPAAC) is especially attractive for conjugating copper chelators, because there is no need to remove copper from the clicked products, which constitutes a significant problem for unprotected copper chelators.13 Baumhover et al. pioneered the application of this chemistry in copper-based radiopharmaceuticals by functionalizing DOTA and NOTA with monofluorocyclooctyne, which was then conjugated with an azide-modified peptide.14 Another advantage of using SPAAC chemistry is that the prelabeled clickable chelators can be conjugated to the biomolecules almost quantitatively at equal equivalency under mild and bioorthogonal conditions. This is especially important for labeling biomolecules using chelators that require harsh labeling conditions that the biomolecules cannot tolerate.
Modification of this new generation of cross-bridged chelators with the commercially available dibenzocyclooctynes (DBCO)15 allows facile conjugation of them to other biomolecules through SPAAC. However, a concern of this approach is the influence of the bulky and hydrophobic DBCO on the binding affinity, internalization, and pharmacokinetics of the clicked products. While the influence of the DBCO group is relatively small when conjugated with macromolecules such as viruses,16 nanoparticles,17,18 or antibodies, there is a high likelihood that DBCO will have a greater impact on the pharmacokinetics of smaller molecules, such as peptides and peptidomimetics. It was previously shown that a 18F-labeled peptide targeting integrin αvβ6 containing a DBCO prosthetic group had considerable lipophilicity that contributed to increased hepatobiliary clearance.19
We hypothesized that the higher hydrophilicity of the cross-bridged macrocyclic chelators with pendant carboxylate and/or methane phosphonate arms in combination with DBCO would retain the high target-to-background contrast of the clicked products. Recently, Chen et al. synthesized a 64Cu-labeled RGD peptide from a DBCO-modified, PEG-linked DiAmSar chelator which showed high contrast between tumor and nontargeted uptake in the U87MG human glioblastoma xenograft model.20 Inspired by these results, we set out to explore the effects of our newly developed DBCO-modified chelators on binding affinities and pharmacokinetics of the clicked somatostatin analogues and to demonstrate the potential utility of these chelators on 64Cu-based PET imaging of neuroendocrine tumors.
As radiolabeled somatostatin analogues showed promise for both imaging and therapy of neuroendocrine tumors,21 the sstr2 agonist Tyr3-octreotate (Y3-TATE) was chosen as the model peptide for this study.22,23 A 3-D atomic structure model of sstr2 based on the crystal structure of the opioid receptors was used to explore the effects of the DBCO-cross-bridged chelator moieties on the receptor targeting of the new bioconjugates.24 The synthesis, in silico, in vitro, ex vivo, and in vivo evaluation of 64Cu-CB-TE1A1P–DBCO–Y3-TATE (64Cu-AP) and 64Cu-CB-TE1K1P–PEG4–DBCO–Y3-TATE (64Cu-KP) derived from two DBCO-modified chelators are reported here (Figure 1). The results are compared with CB-TE1A1P directly conjugated to Y3-TATE, 64Cu-CB-TE1A1P–Y3-TATE (64Cu-1A1P), in a structure–pharmacokinetic relationship analysis.7
Figure 1.

Structures of Tyr3-octreotate (Y3-TATE) analogues.
Results
Homology Modeling of sstr2
To the best of our knowledge, there is no reported crystal structure for somatostatin receptor subtype 2 (sstr2). It is known that somatostatin receptors have about 40% sequence homology similarity to the opioid receptors.25 In the present work, we constructed 3D sstr2 structural model using our established sequence homology modeling approach26 based on the known crystal structures of opioid receptors to construct the 3D structures of sstr2. Subsequently, molecular dynamics (MD) and molecular mechanics (MM) calculations were performed using SYBYL-X 1.32. Three dimensional sstr2 structural and conformation evaluation analyses were performed using proSA-web for Z-scores and PROCHECK for Ramachandran plots (Figures S7 and S8, Supporting Information). Furthermore, root mean squared deviation (RMSD), superimposition of query and template structure, and visualization of generated ssrt2 structural models were performed using UCSF Chimera 1.8.1. The model with the lowest RMSD based on the nociceptin/orphanin FQ receptor (NOP, 4EA3) was selected as the candidate structure for further ssrt2 studies.
Molecular Docking
3D docking was performed by the Tripos Surflex-dock module with different binding poses or conformations of each ligand and sstr2 structural models generated. The binding pocket was rendered with the molecular surface generated by Sybyl MOLCAD.27 The docking poses and ligand/receptor interactions were analyzed by the PyMol program, including intermolecular H-bonds and hydrophobic and hydrophilic interactions within 4.0 Å.
Docking of Y3-TATE with the defined sstr2 structure model revealed hydrogen bond interactions of the peptidomimetic with amino acid residues, including Asn196, Trp197, Tyr205, Asn276, Asn119, Gln88, and Thr212 of sstr2 (Figure 2). The binding site of Y3-TATE is located deep inside sstr2. Data from the docking model also revealed an unoccupied pocket next to the N-terminus of Y3-TATE, suggesting that substitution at this position should be well tolerated. The docking score, which correlates with the predicted binding affinity (pKd value) of the ligand to the receptor, was 12.9. Docking of 1A1P and AP with this model showed similar docking scores of 10.1. 1A1P interacts with amino acid residues Trp188, Ser192, Lys291, Asn276, Tyr302, Thr174, Val173, Ile177, and Phe208 of sstr2 through hydrogen bonds (Figure 3). The docking pose of AP binding to sstr2 indicates hydrogen bond interactions with Asn276, Cly216, Cys193, Gln126, Ile209, Phe127, Phe208, and Gln102 of sstr2 (Figure 4).
Figure 2.
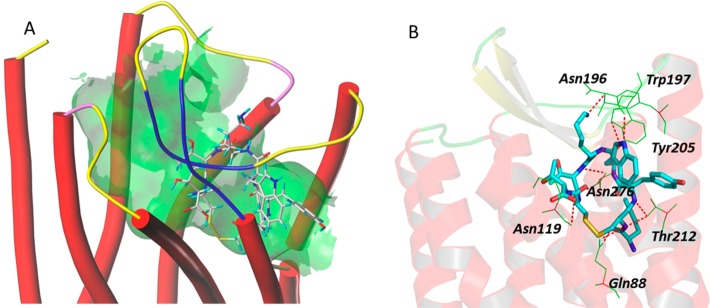
A: Overview of the docking mode of Y3-TATE in the binding pocket of sstr2. B: Molecular interactions between the Y3-TATE and specific amino acid residues of sstr2.
Figure 3.
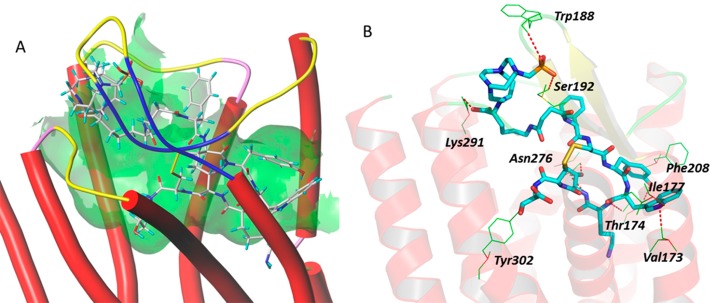
A: Overview of the docking mode of CB-TE1A1P–Y3-TATE (1A1P) in the binding pocket of sstr2. B: The detailed molecular interactions between 1A1P and specific amino acids of sstr2.
Figure 4.
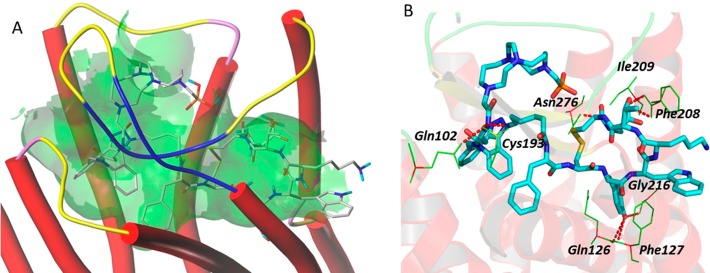
A: Overview of the docking mode of DBCO–CB-TE1A1P–N3-Y3-TATE (AP) in the binding pocket of sstr2. B: The detailed molecular interaction between the AP and specific amino acids in sstr2.
Docking of KP with the sstr2 model showed that KP fits the binding pocket even with the flexible PEG linker between the chelator and DBCO (Figure 5A), with a predicted score of 13.2. It interacts with amino acid residues Trp197, Ile195, Thr206, Gly123, Asp122, Asn276, Gln126, Thr212, and Lys291 of sstr2 through hydrogen bonds (Figure 5B).
Figure 5.
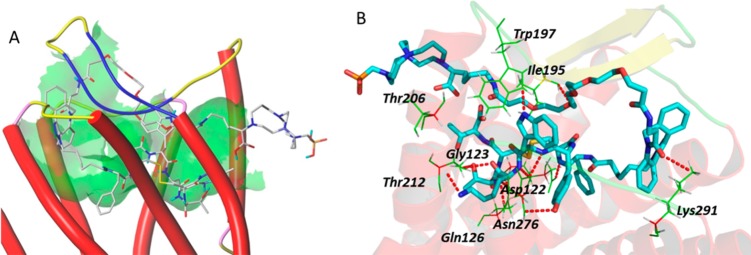
A: Overview of the docking mode of CB-TE1K1P–PEG4–DBCO–Y3-TATE (KP) in the binding pocket of sstr2. B: The detailed molecular interactions between KP and specific amino acids in sstr2.
Synthesis and Radiolabeling of Peptide Conjugates
The precursor Y3-TATE was synthesized via standard solid-phase Fmoc-based peptide synthesis on a microwave peptide synthesizer.28 After cleavage and HPLC purification, 30% of the product was isolated and was used for strain-promoted click chemistry with DBCO–CB-TE1A1P and DBCO–CB-TE1K1P (Figure S1, Supporting Information).
CB-TE1A1P–Y3-TATE (1A1P) was synthesized following a published procedure.7 Consistent with the previously published result, less than 10% of pure 1A1P was isolated after HPLC purification. CB-TE1A1P–DBCO–Y3-TATE (AP) was synthesized by mixing DBCO–CB-TE1A1P and N3-Y3-TATE in H2O and t -BuOH, with stirring at 40 °C for 1.5 h. Because of the lack of regioselectivity of SPAAC, three isomers were separated on a semiprep reversed-phase HPLC column. The total isolated yield of the three isomers was 72%. KP was synthesized by the same procedure described above for AP. Because of the lack of regioselectivity of SPAAC, two regioisomers were separated. The total isolated yield of the two regioisomers was 43%.
The radiolabeling conditions for the three chelator–peptide conjugates were optimized. The optimal buffer solution was 0.1 M NH4OAc (pH 8.1). All of the isomers of AP were labeled at 70 °C in 30 min, while both isomers of KP were labeled within 5 min at 70 °C. Radio-HPLC was used to validate the radiochemical purities and the labeling yields. The ease of labeling of KP may be attributed to the extra carboxylate pendant arm and/or the PEG linker that reduces the steric hindrance. The relatively mild labeling conditions achievable with these chelators is consistent with previously published phosphonate containing chelators such as CB-TE1A1P.7 For the purpose of comparison, 1A1P, AP-1, and KP-1 were all labeled at 90 °C for 5 min in the same S.A. in >98% radiolabeling yields and were used without further purification for in vitro and in vivo studies.
Cellular Internalization
Internalization studies were done with sstr2-transfected HCT116 cells (Figure S2, Supporting Information).29 All three isomers of 64Cu-AP were internalized rapidly within 2 h after administration of the radiotracers. The internalization was blocked with Y3-TATE administered 10 min before the tracer, indicating receptor-specific uptake of the agents. The two isomers of 64Cu-KP also showed receptor-specific uptake into sstr2-transfected HCT116 cells, albeit the internalization was not as rapid as 64Cu-AP. For 64Cu-AP-1, the maximum internalized tracer (45 ± 3%ID/mg) occurred after 2 h at 37 °C; the internalized 64Cu-AP-2 peaked at after 2 h at 37 °C (73 ± 7%ID/mg). Interestingly, 64Cu-AP-3 showed slower internalization which was not saturated after 4 h at 37 °C. The more rapid internalization of isomers of AP could be due to the favorable interactions between the DBCO with the hydrophobic core of sstr2. In contrast to the rapid internalization of the isomers of AP, the isomers of KP internalized more slowly, especially at early time points, which could be due to the hydrophilic PEG linker repelling the hydrophobic sstr2 core.
Saturation Binding Assay and Log D Measurements
Saturation binding assays of 64Cu-AP, and 64Cu-KP were performed using a modified protocol because of high nonspecific binding of these bioconjugates to the 96-well plates. The Bmax, Kd, and log D values of these tracers are summarized in Table 1. There is an increasing trend of Bmax with increased isomer hydrophobicity, suggesting aggregation of the more hydrophobic isomers of the tracers (Figure S3, Supporting Information).
Table 1. Saturation Binding Assay and Log D of Different Isomers of 64Cu-AP and 64Cu-KP.
| tracers | Bmax (fmol/mg) | Kd (nM) | log Da | RT (min)b |
|---|---|---|---|---|
| 64Cu-AP-1 | 2740 ± 180 | 0.6 ± 0.2 | –1.8 ± 0.06 | 19.0 |
| 64Cu-AP-2 | 5680 ± 390 | 0.9 ± 0.2 | –1.7 ± 0.17 | 21.1 |
| 64Cu-AP-3 | 8930 ± 390 | 2.3 ± 0.3 | –1.7 ± 0.03 | 22.6 |
| 64Cu-KP-1 | 1910 ± 230 | 0.8 ± 0.3 | –2.3 ± 0.08 | 17.4 |
| 64Cu-KP-2 | 6230 ± 570 | 1.4 ± 0.3 | –1.8 ± 0.04 | 19.2 |
Log D was measured at pH 7.2.
See Experimental Section for detailed HPLC conditions.
The log D values of these tracers were determined by the traditional shake flask method using neutral PBS buffer and octanol. The log D values of three isomers of AP were −1.8 ± 0.06, −1.7 ± 0.17, −1.7 ± 0.03, respectively, while the log D of the two isomers of KP were −2.3 ± 0.08 and −1.8 ± 0.04, respectively. The values of log D showed the same trend as the retention times of these tracers on reversed-phase HPLC (Table 1).
Biodistribution
The most hydrophilic isomers of 64Cu-AP and 64Cu-KP were chosen for in vivo evaluations, based on their higher affinity to sstr2 and lower tendency for aggregation in buffered aqueous solution. The uptake of all tracers was high in sstr2-expressing tissues (tumor, pancreas). Co-injected Y3-TATE blocked 63%, 84%, and 95% of the tumor uptake of 64Cu-AP-1, 64Cu-KP-1, and 64Cu-1A1P, respectively, showing the high specificity of these probes for sstr2.
The tracers were excreted mainly through the kidneys, resulting in high kidney uptake at earlier time points. The higher kidney uptake of 64Cu-AP-1 might be a result of the additional positive charge of the molecule, which could be attracted by the negatively charged basement membrane and the podocytes in the kidneys. The higher liver uptake of 64Cu-AP-1 at 1 h postinjection (p.i.) reflected its greater hydrophobicity. At 24 h p.i., less than 1% ID/g activity remained in the liver for both 64Cu-AP-1 and 64Cu-KP-1, which suggests in vivo stability of the copper chelates (Figure 6). At 24 h p.i., 64Cu-KP-1 showed lower liver uptake than 64Cu-AP-1 and 64Cu-1A1P, suggesting that the additional carboxylate group further stabilized the copper chelate. There was no significant difference in the tumor-to-blood and tumor-to-muscle ratios for 64Cu-AP-1 and 64Cu-KP-1 at 1, 4, and 24 h p.i. (Figure 7); however, tumor-to-blood ratio of 64Cu-1A1P was significantly higher than both 64Cu-AP-1 and 64Cu-KP-1 at all time points. Tumor-to-muscle ratios of 64Cu-1A1P were significantly higher than both 64Cu-AP-1 and 64Cu-KP-1 at 1 and 24 h p.i. (P < 0.001); however, there was no significant difference at 4 h p.i. (P > 0.05). At 1 h p.i., the tumor-to-kidney ratio of 64Cu-1A1P was significantly higher than those of both 64Cu-AP-1 and 64Cu-KP-1 (P < 0.001). The tumor-to-kidney ratio of 64Cu-AP-1 and 64Cu-KP-1 were not significantly different (P > 0.05) at 1 h. At 4 h, 64Cu-1A1P and 64Cu-KP-1 had similar tumor-to-kidney ratios (P > 0.05), which were significantly higher than 64Cu-AP-1 (P < 0.05). At 24, tumor-to-kidney ratios were not significantly different for the three tracers. At 1 and 24 h, the tumor-to-liver ratios of the three tracers followed the trend, 64Cu-1A1P > 64Cu-KP-1 > 64Cu-AP-1. At 4 h, the tumor-to-liver ratio of 64Cu-KP-1 was significantly higher than for 64Cu-AP-1 (P < 0.05), while there was no significant difference between 64Cu-KP-1 and 64Cu-1A1P (P = 0.32, Figure 8).
Figure 6.

Biodistribution of 64Cu-1A1P, 64Cu-AP-1, and 64Cu-KP-1 in sstr2-transfected HCT116 tumor-bearing female nu/nu mice at 1 and 24 h (A), and at 4 h with and without 20 μg of Y3-TATE co-injected as a blocking agent (B). N = 4 for each time point.
Figure 7.

Tumor-to-organ ratios of 64Cu-1A1P, 64Cu-AP-1, and 64Cu-KP-1 with sstr2-transfected HCT116 tumor-bearing female nu/nu mice. ***: significantly different (P < 0.001). **: significantly different (0.001 < P < 0.01). *: significantly different (0.01 < P < 0.05). ns: no significant difference (P > 0.05). n = 4 mice per time point.
Figure 8.

Clearance profiles of 64Cu-1A1P (green dots), 64Cu-AP-1 (red dots), and 64Cu-KP-1 (blue dots). ***: significantly different (P < 0.001). **: significantly different (0.001 < P < 0.01). *: significantly different (0.01 < P < 0.05). ns: no significant difference (P > 0.05).
The blood clearance of 64Cu-AP-1 and 64Cu-KP-1 was similar from 1 to 4 h p.i., which was significantly slower than the blood clearance of 64Cu-1A1P (P < 0.001, Figure 8). By 4 h, all three tracers showed similar uptake in the blood (P = 0.16). The slower blood clearance of 64Cu-AP-1 was possibly due to the increased hydrophobicity of the tracer; while the slower blood clearance of 64Cu-KP-1 was also possibly due to the PEG linker. At 1 h, the liver uptake of 64Cu-AP-1 was significantly higher than that of 64Cu-KP-1 and 64Cu-1A1P (P < 0.001), while that of 64Cu-KP-1 and 64Cu-1A1P were similar. At 4 and 24 h, 64Cu-KP-1 showed lower uptake in the liver than 64Cu-AP-1 and 64Cu-1A1P (P < 0.01). At early time points, the kidney clearance rate followed the trend, 64Cu-1A1P > 64Cu-KP-1 > 64Cu-AP-1. At 1 h, all three tracers showed similar tumor uptake (P = 0.62). At 4 and 24 h, 64Cu-1A1P showed significantly higher tumor uptake than 64Cu-KP-1 and 64Cu-AP-1 (P > 0.01, Figure 8). Compared with 64Cu-AP-1, the addition of an extra carboxylate group and the PEG linker dramatically improved the pharmacokinetic profile of 64Cu-KP-1.
Small Animal PET/CT Imaging
Small animal PET/CT imaging after administration of the three tracers in the sstr2-transfected HCT116 tumor-bearing mice was performed at 2 h p.i. In one group of mice, Y3-TATE was co-injected with the radiotracers to block the sstr2-specific binding of the tracers. At 2 h, the tumor SUVs for 64Cu-AP-1, 64Cu-KP-1, and 64Cu-1A1P were 4.6 ± 0.8 (n = 2), 3.2 ± 0.2 (n = 2), and 2.8 ± 0.4 (n = 2), respectively. Consistent with the biodistribution data, the PET/CT images of the tracers showed high tumor uptake at 2 h p.i. with more than 90% of the uptake blocked by co-injected Y3-TATE (Figures 9 and S4, Supporting Information).
Figure 9.
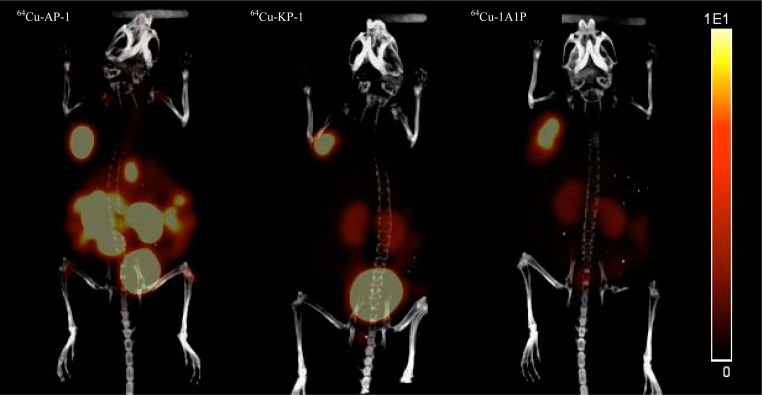
Small animal PET/CT imaging of 64Cu-AP-1 (SUV = 4.6 ± 0.8, n = 2), 64Cu-KP-1 (SUV = 3.2 ± 0.2, n = 2), and 64Cu-1A1P (SUV = 2.8 ± 0.4, n = 2) at 2 h p.i. The images of mice injected with the three tracers are scaled the same (from 0 to 10%ID/cc). Mice injected with unlabeled Y3-TATE to block specific uptake of the tracers had tumor SUVs for 64Cu-AP-1, 64Cu-KP-1, and 64Cu-1A1P of 0.47 ± 0.18, 0.21 ± 0.06, and 0.19 ± 0.02, respectively (Figure S4, Supporting Information).
Discussion
G-protein-coupled receptors are the most targeted proteins for currently used drugs. Among the five subtypes of somatostatin receptors which share common signaling pathways in inhibiting adenylate cyclase, modulating mitogen-activated protein kinase, and activating phosphotyrosine phosphatase, sstr2 has been most widely investigated due to its overexpression in many neuroendocrine tumors, its inhibition of cell growth regulation, suppressive effect on pancreatic cancer,30 and antiproliferative effect on medullary thyroid carcinoma.31 Because to the best of our knowledge there is no available crystal structure for sstr2, targeting this GPCR for drug discovery has revolved around modifications of its native ligands. Theoretical modeling has emerged as a powerful approach to simulate the native structures of GPCRs, which can aid in the rational design of ligands at the molecular level. Because somatostatin receptors are about 40% identical to the opioid receptors,25 we used the crystal structures of opioid receptors to construct the 3D structures of sstr2. After the structures were evaluated and stereochemistry analyzed, the most reliable model was selected for the ligand design based on molecule docking and validated with known sstr2 ligands (Figure S9, Supporting Information). The docking result of the agonist, Y3-TATE, showed that this ligand was located deep inside the receptor and revealed an unoccupied pocket next to the d-phenylalanine of Y3-TATE. Although it is known that substitution at this position is well tolerated, the size of the moiety that can be accommodated at this position has not been widely investigated. The somatostatin analogues were designed based on modifications at N-terminus of Y3-TATE to introduce different phosphonate-containing cross-bridged chelators through bioorthogonal SPAAC, which incorporates larger linkers than have previously been investigated.
The docking of the designed ligands (1A1P, AP, KP) showed binding affinity similar to that of sstr2 (docking scores higher than 10), suggesting that all ligands were worthy of further investigation. Notably, our docking studies revealed a common amino acid residue of sstr2, Asn276, which interacts with all of the peptidomimetics that were docked through hydrogen bonding. This is consistent with previous reports that indicated the importance of this amino acid residue on the selectivity of octreotide for sstr2.32 The consistency of the computational docking for the reported site-directed mutagenesis data indirectly validated our model and the docking method, which may be an effective tool for future sstr2 ligand design and structure–activity studies even in the absence of a crystal structure.
Neuroendocrine tumors have been clinically imaged with sstr2 specific ligands labeled with 111In, 68Ga, 18F, and 64Cu for diagnosis and selection of patients for peptide receptor radionuclide therapy.33177Lu- and 90Y-labeled sstr2 ligands had been successful used clinically and showed great promise as radiotherapeutic agents.2364Cu-labeled sstr2 specific ligands are unique in that they are potentially theranostics agents, allowing for diagnosis and therapy using the same radiotracer and providing accurate and direct dosimetry measurement to plan therapeutic doses of tracers for each individual patient. Alternatively, targeted radiotherapy could be achieved with 67Cu (100% β–, t1/2 = 62 h),34,35 which is advantageous in having a longer half-life, higher abundance of β–, and less radiation from gamma photons compared to 64Cu.
As the commonly used copper chelator, DOTA, suffers from significant in vivo dissociation of copper ion, better copper chelators have been designed to improve the in vivo stability of the copper complex. CB-TE2A-conjugated somatostatin analogue formed kinetically stable chelate with Cu(II).28,36 However, the extended labeling time at high temperature for 64Cu-labeled CB-TE2A conjugate is not optimal for future clinical use. More recently, we reported CB-TE1A1P–Y3-TATE that can be labeled under milder conditions.7 The caveat of this agent is that the synthesis is challenging, with long reaction time and low chemical yield.
Click chemistry is among the most efficient ways of conjugating chelators with biomolecules. Although clickable chelators have been synthesized and conjugated with biomolecules through Cu-catalyzed click chemistry, there are fewer reported biological evaluations on these bioconjugates.37 For copper-64-based radiopharmaceuticals, SPAAC has advantages over CuAAC, because SPAAC obviates the need to remove copper ion from the final product when unprotected copper chelators are applied.14,38 Commercially available DBCO derivatives have been used in SPAAC; however, DBCO may significantly change the binding affinity, solubility, and pharmacokinetics of the imaging probe.19 Presumably, the smaller the targeting molecule, the more severe its influence will be. Therefore, modeling was performed to determine whether modifications to the N-terminus with the bulky DBCO group would be tolerated. The in silico docking supported that the modifications were unlikely to cause drastic changes in binding affinities. A PEG linker was incorporated into KP to lessen the impact of the hydrophobic DBCO on the pharmacokinetics of the tracer.
The lack of regioselectivity of SPAAC must be considered when using this approach in the design of molecular imaging agents. Unlike the copper-catalyzed click chemistry which generally forms only a single 1,4-regioisomer,39 or Ru-catalyzed click chemistry which generally forms the 1,5-regioisomer,40 because of the lack of regioselectivity, SPAAC produces both 1,4- and 1,5-regioisomers, complicating the purification and potentially complicating future regulatory approval for translation to the clinic. In addition to the two regioisomers produced by SPAAC, the cross-bridged clam chelators have two enantiomers, producing a total of four possible isomers after conjugation to the peptidomimetic. Here we evaluated all the HPLC-separated isomers in vitro. Although we found that the more hydrophobic isomers bound more receptor sites (higher Bmax), they also easily aggregated in aqueous solution. Therefore, we chose the most hydrophilic tracers for in vivo studies to explore the feasibility of using DBCO-modified cross-bridged phosphonate-based chelators for imaging sstr2 by PET.
Another concern of introducing the bulky hydrophobic DBCO to the biomolecule is the likelihood of increased nonspecific binding and slower clearance profile of the bioconjugate. This was not a major problem for hydrophilic biomolecules such as galacto-RGD peptide.20 The log D values of the sstr2-targeted regioisomers, which reflect the hydrophilicity of the molecule, were also considered for choosing agents for in vivo evaluation. The most hydrophilic isomers of AP and KP were evaluated in PET/CT imaging and biodistribution studies.
Copper ion accumulation in the liver at later time points has been proposed to reflect the in vivo stability of copper complexes and their biomolecule conjugates.36 The lower liver uptake of 64Cu-KP-1 compared to 64Cu-AP-1 and 64Cu-1A1P at 24 h p.i. suggests that the extra carboxyl group of KP increased the in vivo stability. This was consistent with the in vivo studies comparing CB-TE2A with its analogue bearing an extra carboxyl group, CB-TE2A-PA.41 The chelator CB-TE1K1P has an advantage over the recently reported CB-TE2A-PA in that it can be labeled at much milder conditions (70 °C, 5 min vs 95 °C, 30 min). Future studies include comparing the in vivo stability of the CB-TE1K1P-based tracers with the CB-TE2A-PA-based tracers. Future plans are to investigate other nonclick chemistry type linking groups between CB-TE1K1P and receptor-targeted peptides.
Conclusion
Here we demonstrate the application of in silico analysis to predict the successful application of SPAAC to design sstr2-targeted agents that can be readily synthesized in high yields with phosphonate-based cross-bridged chelators for labeling with 64Cu under mild conditions. DBCO–CB-TE1A1P (AP) and DBCO–CB-TE1K1P (KP) were conjugated to azide-modified Y3-TATE efficiently in 72% and 43% isolated yields, which is in significantly higher yield than direct conjugation through peptide bond formation (7% isolated yield).7 PET/CT imaging of sstr2-positive tumors in mice showed specific tumor uptake and good contrast of tumor to background for both 64Cu-AP-1 and 64Cu-KP-1; however, 64Cu-1A1P, where the chelator is directly conjugated to Y3-TATE, showed higher tumor to nontumor ratios than the clicked bioconjugates. With the rapid development of the SPAAC reagents, further improvement on the pharmacokinetics of the bioconjugates derived from our new generation chelators could arise from using more hydrophilic strained alkynes as the coupling partner.44 These results demonstrate the potential for using SPAAC chemistry in conjugation of copper chelators to biomolecules for molecular imaging.
Experimental Section
Unless otherwise specified, all reagents and solvents were purchased from Sigma-Aldrich Chemical Co. (St. Louis, MO). Matrigel was purchased from BD Biosciences, Bedford, MA. Reactions were monitored by TLC on 0.25 mm silica gel glass plates containing F-254 indicator. Visualization by TLC was monitored by UV light, KMnO4, or radioactivity. Flash chromatography was performed using 200 mesh silica gel. 1H NMR spectra were recorded on a Bruker DRX 400 MHz NMR spectrometer (Billerica, MA). 13C NMR spectra were obtained at 100 MHz. ESI-MS were obtained on a Waters LCT-Premier XE LC-MS station (Milford, MA). 64CuCl2 was purchased from Washington University School of Medicine (St. Louis, MO) and University of Wisconsin—Madison (Madison, WI). Aqueous solutions were prepared using ultrapure water (resistivity, 18 MΩ). The Wang resin (loading, 0.61 mmol/g) and all Fmoc-protected amino acids were purchased from Chem-Impex International, Inc. (Wood Dale, IL). Reversed-phase high-performance liquid chromatography (HPLC) were performed either on a Waters 600E (Milford, MA) chromatography system with a Waters 991 photodiode array detector and an Ortec model 661 (EG&G Instruments, Oak Ridge, TN) radioactivity detector, or a Waters 1525 Binary HPLC pump (Milford, MA) with a Waters 2489 UV/visible detector and a model 105-S-1 (Carroll&Ramsey Associates; Berkeley, CA) radioactivity detector. HPLC samples were analyzed on an analytical C18 column (Phenomenex, Torrance, CA) and purified on a semipreparative C18 column (Phenomenex, Torrance, CA). H2O (0.1% TFA; solvent A) and acetonitrile (0.1% TFA; solvent B) were used as mobile phase. All final compounds were at least 95% pure by HPLC analysis. Radioactive samples were counted using an automated well-type gamma-counter (8000; Beckman, Irvine, CA). PET/CT data were acquired using an Inveon preclinical PET scanner (Siemens Medical Solutions, Erlangen, Germany).
Animal Model
All animal studies were conducted according to the procedures outlined by the University of Pittsburgh and Washington University Institutional Animal Care and Use Committees (IACUC). Human colorectal HCT116 cells (kindly provided by Dr. Bert Vogelstein, Johns Hopkins University) were transfected with sstr2 as previously described.29 Cell media (Iscove’s) was obtained from Gibco (Carlsbad, CA) and supplemented with 10% fetal bovine serum (FBS, Gibco) and 1 mM Zeocin (Gibco). Female, mice 4–6 weeks old athymic nude from Taconic (Hudson, NY) were injected with 0.8–1.2 million sstr2-transfected HCT116 tumor cells mixed with Matrigel and were allowed to grow for 9–12 days.
3D Protein Structure Prediction for sstr2
The known crystal structures of opioid receptors were used to construct the 3D structures of sstr2. These receptors and their structures used in our studies are nociceptin/orphanin FQ receptor (NOP, PDB entry: 4EA3, resolution: 3.01), μ-opioid receptor (MOR, PDB entry: 4DKL, resolution: 2.80), κ-opioid receptor (KOR, PDB entry: 4DJH, resolution: 3.01), and δ-opioid receptor (DOR, PDB entry: 4EJ4, resolution: 3.40). These structures were retrieved from the Protein Data Bank (http://www.pdb.org/pdb/) and were then prepared by SYBYL-X 1.3. The whole sequence of sstr2 was retrieved from the UniProtKB/Swiss-Prot (http://www.uniprot.org/uniprot/P30874).
Sstr2 belongs to class A family of GPCRs, which shares high sequence similarities with other GPCR structures. Sequence alignments of sstr2 and the known crystal structure of GPCR revealed that sstr2 has a moderate sequence identity similarity: ∼40% to nociceptin/orphanin FQ receptor (NOP, PDB entry: 4EA3, resolution: 3.01), ∼44% to μ-opioid receptor (MOR, PDB entry: 4DKL, resolution: 2.80), ∼41% to κ-opioid receptor (KOR, PDB entry: 4DJH, resolution: 3.01), 42% to δ-opioid receptor (DOR, PDB entry: 4EJ4, resolution: 3.40), ∼34% to chemokine receptor CXCR4 (PDB entry: 3ODU, resolution: 2.50), ∼28% todopamine D3 receptor (D3R, PDB entry: 3PBL, resolution: 2.89), 27% to human beta2-adrenergic receptor (PDB entry: 2RH1, resolution: 2.40), ∼28% to chemokine receptor CXCR1 (PDB entry: 2LNL, solid-state NMR), ∼22% to human A2A receptor (A2AAR, PDB entry: 2YDO, resolution: 3.00), ∼26% to human histamine H1 receptor (H1R, PDB entry: 3RZE, resolution: 3.10), ∼25% to sphingosine 1-phosphate receptor (S1P, PDB entry: 3V2W, resolution: 3.35), and ∼31% to human beta1-adrenergic receptor (PDB entry: 2Y00, resolution: 2.50).
Moreover, as one disulfide bridge/bond between Cys115 and Cys193 in sstr2 was suggested by the UniProtKB/Swiss-Prot (http://www.uniprot.org/uniprot/), we adjusted the alignments of sequence of the extracellular loop 2 (ECL2) part manually. For example, for the alignment of sstr2 and NOP (4EA3), we adjusted the Pro5.50 in sstr2 to align the Pro5.50 in NOP (4EA3). Meanwhile, we also assured that alignments of TMs and conserved motifs such as “D/ERY” in TM3, “CWFPV″in TM6, and “NPxxY” in TM7 were reasonable.
We used four crystal structures of opioid receptors to build the 3D structure of sstr2 by using Modeller9.12.1. The protocols in Modeller9.12 for constructing the 3D structures of sstr2 were the following: (1) downloaded these four structures from Protein Data Bank (PDB); (2) aligned the templates with the sequence of sstr2; (3) constructed a 3D structure of sstr2. Once the 3D models were generated, energy minimization was performed using SYBYL-X 1.32. Powell was used as the modeling method, with a gradient of 0.5 kcal/mol, 5000 maximum iterations, in MMFF94s force field with MMFF94 charges.
Structural evaluation and stereochemical analyses were performed by using proSA-web for Z-scores and PROCHECK for Ramachandran plots (Figures S7 and S8, Supporting Information). Root mean squared deviation (RMSD), superimposition of query and template structure, and visualization of generated models were performed using UCSF Chimera 1.8.1. The model with lowest RMSD was selected as the candidate structure for sstr2.
Molecular Docking
Molecular docking was performed using the Tripos Surflex-docking program to investigate the detailed interaction between our compounds and sstr2.22,32 The initial binding pocket of this peptide was subsequently characterized to be nearby Asn276 and Phe294 according to site-directed mutagenesis data. Molecular surface analysis was performed by MOLCAD to find a solvent-accessible cavity around these two key residues. Model validation was performed by docking with known sstr2 ligands and comparing the docking score with experimental Ki values (Figure 9S, Supporting Information).
Synthesis of DBCO–CB-TE1A1P, and DBCO–PEG4–CB-TE1K1P
CB-TE1A1P was synthesized as previously reported with minor modifications.6 The synthesis of DBCO–CB-TE1A1P, CB-TE1K1P, and DBCO–PEG4–CB-TE1K1P was performed following our recently published procedure.
Synthesis of N3-Y3-TATE
Y3-TATE on resin was synthesized by standard Fmoc solid-phase peptide synthesis on a CEM microwave peptide synthesizer (Matthews, NC). 6-Azidohexanoic acid was incorporated as the last “amino acid” by a standard peptide coupling protocol, with HBTO and DIEA used as activator and base, respectively. The peptide was cleaved with a cocktail comprising TFA/TIPS/PhOH/H2O (85:5:5:5), precipitated with diethyl ether, and purified with semipreparative HPLC under isocratic conditions (30% ACN in H2O, with 0.1% TFA). The isolated yield was 30%. ESI-MS [M + H]+: observed, 1188.2; calculated, 1188.5.
Synthesis of CB-TE1A1P–DBCO–Y3-TATE (AP)
N3-Y3-TATE (0.31 μmol) was mixed with DBCO–CB-TE1A1P (0.31 μmol) in 1 mL of t-BuOH/H2O (1:1). The mixture was heated at 40 °C for 1.5 h. Reversed-phase analytical HPLC of a small aliquot of the crude reaction mixture showed complete conversion. The crude reaction product was diluted with water and purified with reversed-phase semipreparative HPLC under isocratic conditions (30% ACN in H2O with 0.1% TFA). The isolated yield was 72%. Three peaks were resolved and separated. ESI-MS [M + H] +: observed, 1824.8; calculated, 1825.0.
Synthesis of CB-TE1K1P–PEG4–DBCO–Y3-TATE (KP)
N3-Y3-TATE (0.98 μmol) was mixed with CB-TE1K1P–PEG4–DBCO (0.75 μmol) in 1 mL of t-BuOH/H2O (1:1). The mixture was heated at 40 °C for 1.5 h, diluted with water, and purified by reversed-phase semipreparative HPLC, using isocratic conditions (30% ACN in H2O, with 0.1% TFA). HPLC showed complete conversion. Two peaks were resolved and separated. The isolated yield was 43%. ESI-MS [M + H] +: observed, 2216.4; calculated, 2216.8.
Radiolabeling of CB-TE1A1P–Y3-TATE (1A1P), CB-TE1A1P–DBCO–Y3-TATE (AP), and CB-TE1K1P–PEG4–DBCO–Y3-TATE (KP)
64Cu-1A1P, 64Cu-KP, or 64Cu-AP (1 nmol) was labeled with 64Cu(OAc)2 (30–37 MBq) by incubation in 100 μL of 0.1 M NH4OAc (pH 8.1) for 5 min at 90 °C. Radiochemical purity was confirmed by radio-TLC or radio-HPLC. The labeling kinetics of 64Cu-KP and 64Cu-AP at lower temperatures were also measured (Figures S5, Supporting Information).
Cell Internalization
Cell internalization assays were performed as previously described.7 Briefly, aliquots of sstr2-transfected HCT116 cell suspension were placed in 12-well plates. 64Cu-1A1P, 64Cu-KP, or 64Cu-AP was added to a final concentration of 4 nM. A 1000-fold excess of Y3-TATE was used as blocking agent to determine nonspecific binding and internalization. At 15, 30, 60, 120, 240 min after addition of radiotracers, the surface-bound and internalized radioactivity was measured with a gamma counter (Packard II gamma counter). Total protein concentration in the cell lysate was determined using the BCA Protein assay (Pierce Biotechnology, Rockford, IL). Results were normalized to the administered activity and content of protein.
Saturation Binding Assay
Cell membrane was prepared from sstr2-transfected HCT116 cells and was used for binding assays. Saturation binding assays were performed using freshly prepared cell membrane in Eppendorf tubes, following the previously described protocol with some modifications.45 Briefly, Eppendorf tubes were saturated with binding buffer containing 0.1% BSA for 30 min. 64Cu-1A1P, 64Cu-KP, or 64Cu-AP were added in increasing concentrations (0.1–10 nM), followed by the addition of 15 μg of sstr2-transfected HCT116 membranes. After 2 h incubation at room temperature, the pellets were precipitated and washed twice with 300 μL of binding buffer, followed by centrifuging at 7000 rpm for 4 min. The pellets were transferred to tubes for counting in a gamma counter (Packard II gamma counter).
Log D Measurements
Radiolabeled bioconjugates (5 μL) were added to 1 mL of PBS (pH 7.4) and 1 mL of octanol. The mixture was vortexed for 10 min and then centrifuged for 10 min. Both layers were counted in an automated gamma counter. The log D was calculated based on formula of log D = log([M]oct/[M]aq). Data are presented as mean of triplicate measurements.
Biodistribution
The biodistribution studies were carried out as previously described.28 The tracers (1.85 MBq) were injected into sstr2-transfected HCT116 tumor-bearing female nu/nu mice via the tail vein. The most hydrophilic isomers of 64Cu- AP and 64Cu- KP were evaluated and compared to 64Cu- 1A1P. At 1, 4, and 24 h p.i., mice were sacrificed, and selected organs were removed, weighed, and counted on a gamma counter (PerkinElmer Wizard2, Waltham, MA). A blocking study was conducted at 4 h postinjection to confirm the specificity of the tracers for binding to sstr2 by co-injecting 20 μg of Y3-TATE with the radiotracers.
Small Animal PET/CT Imaging
Small animal PET/CT imaging was performed as previously described.28 Briefly, the radiotracers (3.7 MBq) were injected via tail vein and imaged at 2 h p.i. A blocking study was performed by co-injecting 20 μg of Y3-TATE with the radiotracers. Static imaging was performed on an Inveon PET/CT scanner46,47 with 10 min PET scanning followed by 5 min CT. Inveon Research Workplace (IRW) from Siemens Healthcare Global was used for coregistration of PET/CT images and quantification of regions of interest (ROI). PET/CT images were reconstructed with maximum a posteriori (MAP), 3D ordered-subset expectation maximization (OSEM3D), 2D ordered-subset expectation maximization (OSEM2D), and filtered back projection (2DFBP). Standard uptake values (SUV) were generated by measuring ROI from PET/CT images and calculated with the formula: SUV = [nCi/ml] × [animal weight (g)]/injected dose [nCi].
Statistical Methods
All of the data were presented as mean ± standard deviation. Two-tailed unpaired t tests were performed using GraphPad Prism to generate P values. When comparing more than two columns, one-way ANOVA analysis with Tukey post-test was performed with α = 0.05 (95% confidence intervals), α = 0.01 (99% confidence intervals), and α = 0.001 (99.9% confidence intervals).
Acknowledgments
The authors thank Kathryn E. Day, Kimberly Ferrero, Nikki Fettig, Amanda Roth, and Margaret Morris for assistance with animal experiments. Small animal PET/CT imaging at UPCI was supported in part by P30CA047904 (UPCI CCSG). This project is supported by NCI grants R01CA093375 and R01 CA064475, and by NIDA grant R01DA025612.
Glossary
Abbreviations Used
- SPAAC
strain-promoted alkyne azide cycloaddition
- sstr2
somatostatin receptor subtype 2
- DBCO
dibenzocyclooctyne
- TLC
thin layer chromatography
- GPCR
G-protein-coupled receptor
- MD
molecular dynamics
- MM
molecular mechanics
- MSD
root mean squared deviation
- AP
CB-TE1A1P–DBCO–Y3-TATE
- KP
CB-TE1K1P–PEG4–DBCO–Y3-TATE
- 1A1P
CB-TE1A1P–Y3-TATE
- PEG
poly(ethylene glycol)
- OSEM3D
3D ordered-subset expectation maximization
- OSEM2D
2D ordered-subset expectation maximization
- PET
positron emission tomography
- RMAP
maximum a posteriori
- 2DFBP
filtered back projection
- ROI
region of interest
- SUV
standard uptake values
Supporting Information Available
Additional figures on the radiolabeling kinetics of the tracers, HPLC profiles of the peptidomimetics, internalization profiles, saturation binding assay profiles, and PET/CT images. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
∥ College of Pharmacy Third, Military Medical University, Chongqing 400038, China.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Shokeen M.; Anderson C. J. Molecular Imaging of Cancer with Copper-64 Radiopharmaceuticals and Positron Emission Tomography (PET). Acc. Chem. Res. 2009, 42, 832–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wadas T. J.; Wong E. H.; Weisman G. R.; Anderson C. J. Coordinating Radiometals of Copper, Gallium, Indium, Yttrium, and Zirconium for PET and SPECT Imaging of Disease. Chem. Rev. 2010, 110, 2858–2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Z.; Anderson C. J. Chelators for copper radionuclides in positron emission tomography radiopharmaceuticals. J. Labelled Compd. Radiopharm. 2014, 57, 224–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paterson B. M.; Roselt P.; Denoyer D.; Cullinane C.; Binns D.; Noonan W.; Jeffery C. M.; Price R. I.; White J. M.; Hicks R. J.; Donnelly P. S. PET imaging of tumours with a 64Cu labeled macrobicyclic cage amine ligand tethered to Tyr3-octreotate. Dalton Trans. 2014, 43, 1386–1396. [DOI] [PubMed] [Google Scholar]
- Wadas T. J.; Anderson C. J. Radiolabeling of TETA- and CB-TE2A-conjugated peptides with copper-64. Nat. Protoc. 2007, 1, 3062–3068. [DOI] [PubMed] [Google Scholar]
- Ferdani R.; Stigers D. J.; Fiamengo A. L.; Wei L.; Li B. T. Y.; Golen J. A.; Rheingold A. L.; Weisman G. R.; Wong E. H.; Anderson C. J. Synthesis, Cu(II) complexation, 64Cu-labeling and biological evaluation of cross-bridged cyclam chelators with phosphonate pendant arms. Dalton Trans. 2012, 41, 1938–1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y.; Ferdani R.; Anderson C. J. Preparation and Biological Evaluation of 64Cu Labeled Tyr3-Octreotate Using a Phosphonic Acid-Based Cross-Bridged Macrocyclic Chelator. Bioconjugate Chem. 2012, 23, 1470–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng D.; Ouyang Q.; Cai Z.; Xie X.-Q.; Anderson C. J. New cross-bridged cyclam derivative CB-TE1K1P, an improved bifunctional chelator for copper radionuclides. Chem. Commun. 2014, 50, 43–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolb H. C.; Finn M. G.; Sharpless K. B. Click Chemistry: Diverse Chemical Function from a Few Good Reactions. Angew. Chem., Int. Ed. 2001, 40, 2004–2021. [DOI] [PubMed] [Google Scholar]
- Marik J.; Sutcliffe J. L. Click for PET: Rapid preparation of [18F]fluoropeptides using CuI catalyzed 1,3-dipolar cycloaddition. Tetrahedron Lett. 2006, 47, 6681–6684. [Google Scholar]
- Zeng D.; Zeglis B. M.; Lewis J. S.; Anderson C. J. The Growing Impact of Bioorthogonal Click Chemistry on the Development of Radiopharmaceuticals. J. Nucl. Med. 2013, 54, 829–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Struthers H.; Mindt T. L.; Schibli R. Metal chelating systems synthesized using the copper(I) catalyzed azide-alkyne cycloaddition. Dalton Trans. 2010, 39, 675–696. [DOI] [PubMed] [Google Scholar]
- Sletten E. M.; Bertozzi C. R. Bioorthogonal Chemistry: Fishing for Selectivity in a Sea of Functionality. Angew. Chem., Int. Ed. 2009, 48, 6974–6998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumhover N. J.; Martin M. E.; Parameswarappa S. G.; Kloepping K. C.; O’Dorisio M. S.; Pigge F. C.; Schultz M. K. Improved synthesis and biological evaluation of chelator-modified α-MSH analogs prepared by copper-free click chemistry. Bioorg. Med. Chem. Lett. 2011, 21, 5757–5761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debets M. F.; van Berkel S. S.; Schoffelen S.; Rutjes F. P. J. T.; van Hest J. C. M.; van Delft F. L. Aza-dibenzocyclooctynes for fast and efficient enzyme PEGylation via copper-free (3 + 2) cycloaddition. Chem. Commun. 2010, 46, 97–99. [DOI] [PubMed] [Google Scholar]
- Hao J.; Huang L. L.; Zhang R.; Wang H. Z.; Xie H. Y. A mild and reliable method to label enveloped virus with quantum dots by copper-free click chemistry. Anal. Chem. 2012, 84, 8364–70. [DOI] [PubMed] [Google Scholar]
- Zeng D.; Lee N. S.; Liu Y.; Zhou D.; Dence C. S.; Wooley K. L.; Katzenellenbogen J. A.; Welch M. J. 64Cu Core-Labeled Nanoparticles with High Specific Activity via Metal-Free Click Chemistry. ACS Nano 2012, 6, 5209–5219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee D.-E.; Na J. H.; Lee S.; Kang C. M.; Kim H. N.; Han S. J.; Kim H.; Choe Y. S.; Jung K.-H.; Lee K. C.; Choi K.; Kwon I. C.; Jeong S. Y.; Lee K.-H.; Kim K. Facile Method To Radiolabel Glycol Chitosan Nanoparticles with 64Cu via Copper-Free Click Chemistry for MicroPET Imaging. Mol. Pharmacol. 2013, 10, 2190–2198. [DOI] [PubMed] [Google Scholar]
- Hausner S. H.; Carpenter R. D.; Bauer N.; Sutcliffe J. L. Evaluation of an integrin αvβ6-specific peptide labeled with [18F]fluorine by copper-free, strain-promoted click chemistry. Nucl. Med. Biol. 2013, 40, 233–239. [DOI] [PubMed] [Google Scholar]
- Chen K.; Wang X.; Lin W.-Y.; Shen C. K. F.; Yap L.-P.; Hughes L. D.; Conti P. S. Strain-Promoted Catalyst-Free Click Chemistry for Rapid Construction of 64Cu-Labeled PET Imaging Probes. ACS Med. Chem. Lett. 2012, 3, 1019–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jong M.; Breeman W. A. P.; Kwekkeboom D. J.; Valkema R.; Krenning E. P. Tumor Imaging and Therapy Using Radiolabeled Somatostatin Analogues. Acc. Chem. Res. 2009, 42, 873–880. [DOI] [PubMed] [Google Scholar]
- Patel Y. C. Somatostatin and Its Receptor Family. Front. Neuroendocrinol. 1999, 20, 157–198. [DOI] [PubMed] [Google Scholar]
- Kwekkeboom D. J.; Kam B. L.; van Essen M.; Teunissen J. J. M.; van Eijck C. H. J.; Valkema R.; de Jong M.; de Herder W. W.; Krenning E. P. Somatostatin receptor-based imaging and therapy of gastroenteropancreatic neuroendocrine tumors. Endocr. Relat. Cancer 2010, 17, R53–R73. [DOI] [PubMed] [Google Scholar]
- Roy A.; Kucukural A.; Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat. Protoc. 2010, 5, 725–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darlison M. G.; Richter D. Multiple genes for neuropeptides and their receptors: Co-evolution and physiology. Trends Neurosci. 1999, 22, 81–88. [DOI] [PubMed] [Google Scholar]
- Xie X.-Q.; Chen J.-Z.; Billings E. M. 3D structural model of the G-protein-coupled cannabinoid CB2 receptor. Proteins: Struct., Funct., Bioinf. 2003, 53, 307–319. [DOI] [PubMed] [Google Scholar]
- Chen J.-Z.; Wang J.; Xie X.-Q. GPCR Structure-Based Virtual Screening Approach for CB2 Antagonist Search. J. Chem. Inf. Model. 2007, 47, 1626–1637. [DOI] [PubMed] [Google Scholar]
- Sprague J. E.; Peng Y.; Sun X.; Weisman G. R.; Wong E. H.; Achilefu S.; Anderson C. J. Preparation and Biological Evaluation of Copper-64-Labeled Tyr3-Octreotate Using a Cross-Bridged Macrocyclic Chelator. Clin. Cancer Res. 2004, 10, 8674–8682. [DOI] [PubMed] [Google Scholar]
- Nguyen K.; Parry J. J.; Rogers B. E.; Anderson C. J. Evaluation of copper-64-labeled somatostatin agonists and antagonist in SSTr2-transfected cell lines that are positive and negative for p53: implications for cancer therapy. Nucl. Med. Biol. 2012, 39, 187–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benali N.; Cordelier P.; Calise D.; Pagès P.; Rochaix P.; Nagy A.; Estève J.-P.; Pour P. M.; Schally A. V.; Vaysse N.; Susini C.; Buscail L. Inhibition of growth and metastatic progression of pancreatic carcinoma in hamster after somatostatin receptor subtype 2 (sst2) gene expression and administration of cytotoxic somatostatin analog AN-238. Proc. Natl. Acad. Sci. U.S.A. 2000, 97, 9180–9185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tagliati F.; Zatelli M. C.; Bottoni A.; Piccin D.; Luchin A. Culler, M. D.; degli Uberti, E. C. Role of Complex Cyclin D1/Cdk4 in Somatostatin Subtype 2 Receptor-Mediated Inhibition of Cell Proliferation of a Medullary Thyroid Carcinoma Cell Line in Vitro. Endocrinology 2006, 147, 3530–3538. [DOI] [PubMed] [Google Scholar]
- Kaupmann K.; Bruns C.; Raulf F.; Weber H. P.; Mattes H.; Lubbert H. Two amino acids, located in transmembrane domains VI and VII, determine the selectivity of the peptide agonist SMS 201–995 for the SSTR2 somatostatin receptor. EMBO J. 1995, 14, 727–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maecke H. R.; Reubi J. C. Somatostatin Receptors as Targets for Nuclear Medicine Imaging and Radionuclide Treatment. J. Nucl. Med. 2011, 52, 841–844. [DOI] [PubMed] [Google Scholar]
- Qaim S. M. Decay data and production yields of some non-standard positron emitters used in PET. Q. J. Nucl. Med. Mol. Imaging 2008, 52, 111. [PubMed] [Google Scholar]
- Lederer C. M.; Shirley V. S., Eds. Table of Isotopes, 7th ed.; John Wiley & Sons: New York, 1978. [Google Scholar]
- Boswell C. A.; Sun X.; Niu W.; Weisman G. R.; Wong E. H.; Rheingold A. L.; Anderson C. J. Comparative in Vivo Stability of Copper-64-Labeled Cross-Bridged and Conventional Tetraazamacrocyclic Complexes. J. Med. Chem. 2004, 47, 1465–1474. [DOI] [PubMed] [Google Scholar]
- Lebedev A. Y.; Holland J. P.; Lewis J. S. Clickable bifunctional radiometal chelates for peptide labeling. Chem. Commun. 2010, 46, 1706–1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knör S.; Modlinger A.; Poethko T.; Schottelius M.; Wester H.-J.; Kessler H. Synthesis of Novel 1,4,7,10-Tetraazacyclodecane-1,4,7,10-Tetraacetic Acid (DOTA) Derivatives for Chemoselective Attachment to Unprotected Polyfunctionalized Compounds. Chem.—Eur. J. 2007, 13, 6082–6090. [DOI] [PubMed] [Google Scholar]
- Hein J. E.; Fokin V. V. Copper-catalyzed azide-alkyne cycloaddition (CuAAC) and beyond: New reactivity of copper(I) acetylides. Chem. Soc. Rev. 2010, 39, 1302–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boren B. C.; Narayan S.; Rasmussen L. K.; Zhang L.; Zhao H.; Lin Z.; Jia G.; Fokin V. V. Ruthenium-Catalyzed Azide–Alkyne Cycloaddition: Scope and Mechanism. J. Am. Chem. Soc. 2008, 130, 8923–8930. [DOI] [PubMed] [Google Scholar]
- Persson M.; Hosseini M.; Madsen J.; Jørgensen T. J. D.; Jensen K. J.; Kjaer A.; Ploug M. Improved PET imaging of uPAR expression using new (64)Cu-labeled cross-bridged peptide ligands: Comparative in vitro and in vivo studies. Theranostics 2013, 3, 618–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jewett J. C.; Bertozzi C. R. Cu-free click cycloaddition reactions in chemical biology. Chem. Soc. Rev. 2010, 39, 1272–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shokeen M.; Zheleznyak A.; Wilson J. M.; Jiang M.; Liu R.; Ferdani R.; Lam K. S.; Schwarz J. K.; Anderson C. J. Molecular Imaging of Very Late Antigen-4 (α4β1 Integrin) in the Premetastatic Niche. J. Nucl. Med. 2012, 53, 779–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Constantinescu C. C.; Mukherjee J. Performance evaluation of an Inveon PET preclinical scanner. Phys. Med. Biol. 2009, 54, 2885–2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemp B. J.; Hruska C. B.; McFarland A. R.; Lenox M. W.; Lowe V. J. NEMA NU 2-2007 performance measurements of the Siemens Inveon preclinical small animal PET system. Phys. Med. Biol. 2009, 54, 2359–2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.